

Abstract
Purpose of review
FOXOs are transcription factors that regulate downstream target genes to counteract to cell stress. Here we review the function and regulation of FOXO transcription factors, the mechanism of FOXO3 activation in the kidney, and the role of FOXO3 in delaying the development of chronic kidney disease (CKD).
Recent findings
Progressive renal hypoxia from vascular dropout and metabolic perturbation is a pathogenic factor for the initiation and development of CKD. Hypoxia and low levels of α-ketoglutarate generated from the TCA cycle inhibit prolyl hydroxylase domain (PHD)-mediated prolyl hydroxylation of FoxO3, thus reducing FoxO3 protein degradation via the ubiquitin proteasomal pathway, similar to HIF stabilization under hypoxic conditions. FoxO3 accumulation and nuclear translocation activate two key cellular defense mechanisms, autophagy and antioxidative response in renal tubular cells, to reduce cell injury and promote cell survival. FoxO3 directly activates the expression of Atg proteins, which replenishes core components of the autophagic machinery to allow sustained autophagy in the chronically hypoxic kidney. FoxO3 protects mitochondria by stimulating the expression of superoxide dismutase 2 (SOD2), as tubular deletion of FoxO3 in mice results in reduced SOD2 levels and profound mitochondrial damage.
Summary
Knowledge gained from animal studies may help understand the function of stress responsive transcription factors that could be targeted to prevent or treat CKD.
Keywords: autophagy, chronic kidney disease, FOXO transcription factors, oxidative stress, renal hypoxia
INTRODUCTION
Our incomplete understanding of the pathobiology in the initiation and progression of chronic kidney disease (CKD) limits the development of effective measures to prevent and treat CKD. Molecular characterization of mouse kidneys during the course of CKD development indicates predominant activation of transcription factors, many of which overlap with those of transcriptomes of human transplanted kidneys [1▪▪,2]. Transcription factors are known to play pivotal roles in response to injury and adaption to stress. Classic pathophysiological studies and recent molecular analyses indicate that hypoxia and metabolic stress occur in the kidneys with CKD changes [1▪▪,3,4]. The present review will highlight a previously unrecognized hypoxia-mediated molecular regulation and function of the FoxO3 transcription factor in renal tubular defense against the development of CKD.
FUNCTION AND REGULATION OF FOXO TRANSCRIPTION FACTORS
The forkhead box (FOX) genes belong to an evolutionarily ancient family of transcription factors that participate in a wide range of essential functions including regulation of organ development, cell proliferation, senescence, and metabolic homeostasis. The first FOX gene was discovered in Drosophila, in which gene mutation resulted in a fork-headed appearance in the fly [5]. There are 44 FOX genes in the mouse genome and more than 50 FOX genes in the human genome, divided into 19 subfamilies classified as FOXA through FOXS. The proteins encoded by FOX genes share a well conserved winged-helix DNA-binding domain, or forkhead domain, that binds to the promoter of a large number of target genes as a monomer to activate gene expression [6].
Among FOX transcription factors, FOXO is one of the most studied subfamilies because of the critical role in the regulation of metabolism, oxidative stress response, and cell-cycle progression. There are four mammalian FOXO genes, FOXO1, FOXO3, FOXO4, and FOXO6, which are located on different chromosomes in humans. FOXO2 and FOXO3 are identical and FOXO5 is the ortholog of FOXO3 in fish. In invertebrates, there is only one FOXO gene, daf-16 in Caenorhabditis elegans and dFOXO in Drosophila. All FOXO transcription factors bind to a consensus DNA sequence (5’-TTGTTTAC-3’) in the promoter region of their target genes, which may explain functional redundancy [7]. However, FOXO isoforms also exhibit distinct functions, partially because of their tissue expression patterns. Although FOXO6 is largely restricted to neural cells, FOXO1, FOXO3, and FOXO4 are expressed ubiquitously. Mouse knockout studies have revealed an essential role of FoxO family members. FoxO1-null mice die on the embryonic day 10.5 because of incomplete vascular formation. FoxO3-null mice have normal organ development but are infertile because of premature germ line stem cell differentiation leading to early ovarian failure. FoxO4-null mice are indistinguishable from their littermate controls [8,9]. Although initial studies in FoxO6-null mice show normal learning but impaired memory consolidation [10], recent studies indicate that FOXO6 also mediates insulin actions via a different mechanism than other FOXO family members, suggesting that FOXO6 is evolutionally divergent from other FOXO isoforms [11,12].
FOXOs are generally regarded as stress responsive transcription factors that counteract three major stress conditions: metabolic stress, oxidative stress, and growth factor deficiency. In C. elegans, daf-16 is notable for regulating the entry into a hibernation-like state or Dauer stage under adverse conditions [13] and responsible for lifespan extension. Similarly, optimal FOXO activity prolongs lifespan in humans and dysregulation of this pathway contributes to the two most common diseases in aging, cancer and diabetes [7,14,15]. The major upstream regulators of FOXOs are mediated through the PI3K/AKT and cellular stress pathways including JNK and AMPK signaling [16]. Under stress conditions such as starvation, or in the absence of growth factors, FOXO proteins translocate to the nucleus to trigger gene expression for downstream stress response. Conversely, under nutrient and growth factor abundant conditions or in cancer cells where PI3K/AKT pathway is aberrantly activated, AKT phosphorylates FOXO proteins at three conserved serine and threonine residues, which creates a docking site for the 14-3-3 protein that exports FOXOs from nucleus to the cytoplasm where FOXOs are inactivated. The exception is that FOXO6 subcellular localization is not significantly affected by the PI3K/AKT signaling [17].
The activity of FOXOs are mainly regulated by posttranslational modifications, although micro-RNAs (miRNAs) have been shown to affect FOXO expression. These posttranslational modifications include phosphorylation, acetylation, methylation, and ubiquitination, which influence the subcellular localization, protein stability, and transcriptional activity of FOXOs and their coregulators. Please refer to excellent reviews on the topic [16,18]. Specific to FOXO3 and kidney disease, we have recently demonstrated that FoxO3 protein abundance can also be regulated by prolyl hydroxylation (Fig. 1). Here we will focus on the function and regulation of FOXO3 during the development of CKD.
FIGURE 1.
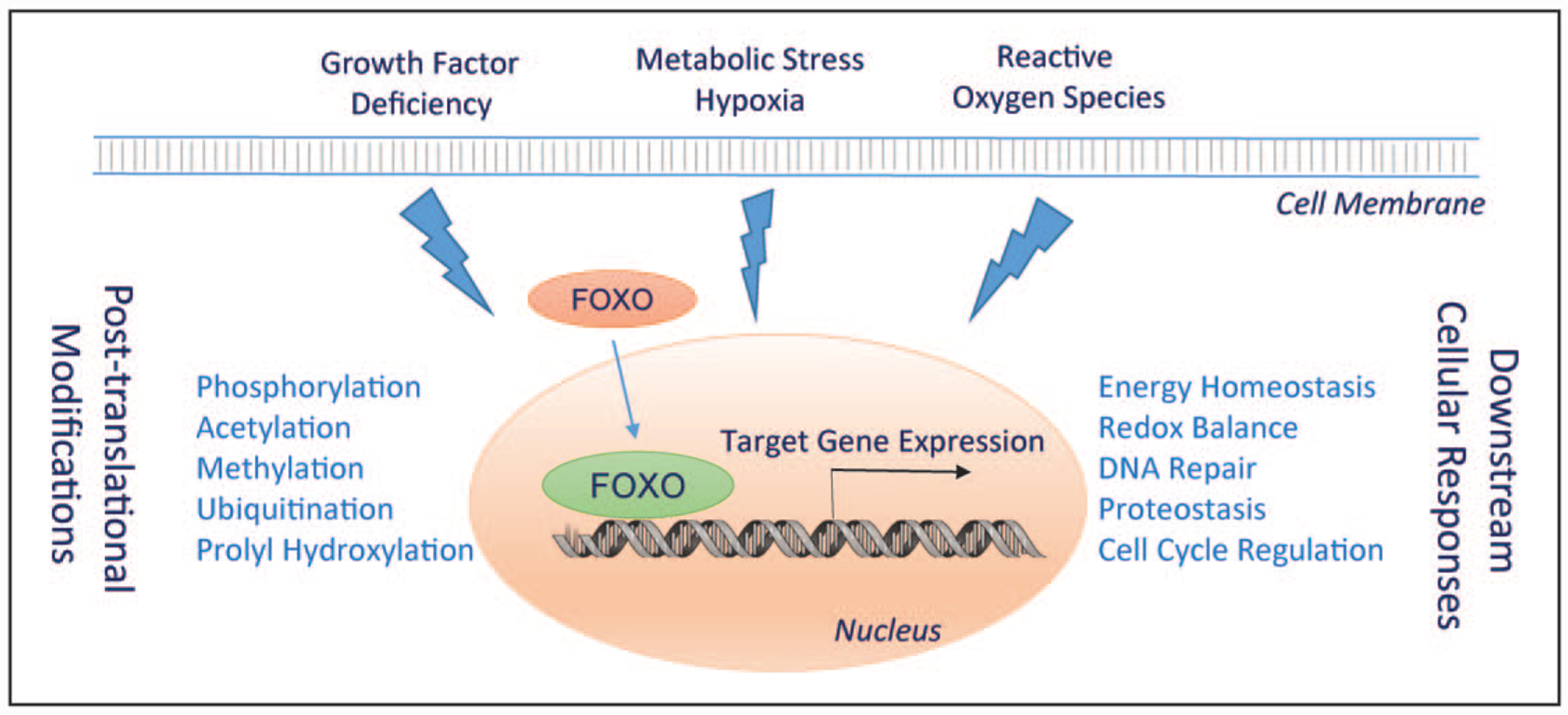
Stress induced activation of FOXO transcription factors. Growth factor deficiency, metabolic stress/hypoxia, and high level of reactive oxygen species stimulate stress-sensitive signaling pathways leading to posttranslational modifications and activation of FOXO. FOXO stimulates the expression of target genes for cell adaptation and defense.
HYPOXIA IS A PATHOGENIC FACTOR FOR CHRONIC KIDNEY DISEASE AND ACTIVATOR FOR FOXO3
Epidemiological studies indicate that the incidence of AKI is increasing [19] and AKI can lead to the development of CKD if renal repair is unsuccessful [20,21]. Cellular and molecular mechanisms of incomplete renal repair following AKI are not fully understood, but could result from severe tubular injury or multiple episodes of AKI leaving fewer competent precursors to enter the proliferative state and/or an altered transcriptional profile limiting proliferative capacity in surviving tubular cells [22▪▪]. Following ischemic injury, pericytes migrate away from the capillary wall to the interstitial space and transition into myofibroblasts [23]. Peritubular capillaries without mural cells regress [24,25]. The transformation of pericytes into myofibroblasts could explain, at least in part, capillary rarefaction, interstitial fibrosis, and renal hypoxia [26,27]. Additionally, tubule-derived vascular endothelial growth factor A (Vegfa) is required to maintain capillary network after birth [28]. Tubules that failed in repair may have low Vegf secretion causing further compromise in peritubular capillaries, which feeds back to cause more tubular hypoxia thus leading to a self-perpetuating, vicious cycle in the pathogenesis of CKD.
Cells can sense and respond to hypoxia and nutrient deficiency. Hypoxia-inducible factors (HIFs) are key transcription factors regulating metabolic reprogramming in normal and cancer cells. Under normoxic conditions, the HIF-α subunits are hydroxylated at two proline residues by HIF prolyl hydroxylase domain proteins PHD1–3, which are O2- and α-ketoglutarate-dependent dioxygenases. Proline hydroxylation allows HIF binding to the pVHL-E3 ubiquitin ligase complex and HIF degradation via the ubiquitin proteasomal system (UPS) [29▪▪]. Of the three PHD isoforms that function as oxygen sensors, PHD2 is the primary prolyl hydroxylase for the α subunit of the HIF proteins [30,31], and PHD1 and PHD3 may hydroxylate other substrates. Using bio-chemical assays, Zheng et al. demonstrate that FoxO3, but not FOXO1, is hydroxylated by PHD1 at Pro426 and Pro437, which disrupted the binding with USP9x deubiquitinase, thus promoting FoxO3 degradation via UPS. The prolyl hydroxylation reaction is confirmed in a breast cancer cell line and 293 embryonic kidney cells [32].
In normal mouse kidneys, nuclear FoxO3 is mainly expressed in the distal nephron and collecting ducts, with only 6% of proximal tubular cells expressing detectable nuclear FoxO3. However, FoxO3 activation with nuclear translocation is significantly increased in the hypoxic tubules following ischemic or obstructive injury [33▪,34]. Exposing renal tubular epithelial cells to a hypoxic environment with 1% O2 or hypoxia-mimetic dimethyloxalylglycine (DMOG) that inhibits PHD enzymes reduces prolyl hydroxylation of FoxO3, prevents its degradation through UPS and leads to FoxO3 protein accumulation. Starving cells by withdrawing amino acids and glucose also increases FoxO3 protein abundance. Conversely, supplementing starved cells with a cell permeable analogue of α-ketoglutarate, dimethyl α-ketoglutarate (DMKG), results in a dramatic decrease in FoxO3 protein, likely because of enhanced prolyl hydroxylation that facilitates FoxO3 degradation. Using an antibody to OH-FoxO3 at Pro437, we detect OH-FoxO3 in renal epithelial cell cultures under normoxic conditions (21% O2). The OH-FoxO3 increases substantially when ubiquitin proteasomal degradation is inhibited. Furthermore, knocking-down studies suggest that all three PHD isoforms that are known to be expressed in the kidney can catalyze prolyl hydroxylation of FoxO3 [33▪]. We conclude that, similar to HIF proteins, FOXO3 is subjected to posttranslational modification in an oxygen sensitive manner and hypoxia activates the FoxO3 transcription factor (Fig. 2A). Interestingly, FoxO3 activation under hypoxic conditions is partially dependent on Hif1, as deletion of Hif-1α attenuated the increase of FoxO3 mRNA and protein, suggesting a molecular nexus in kidney stress response.
FIGURE 2.
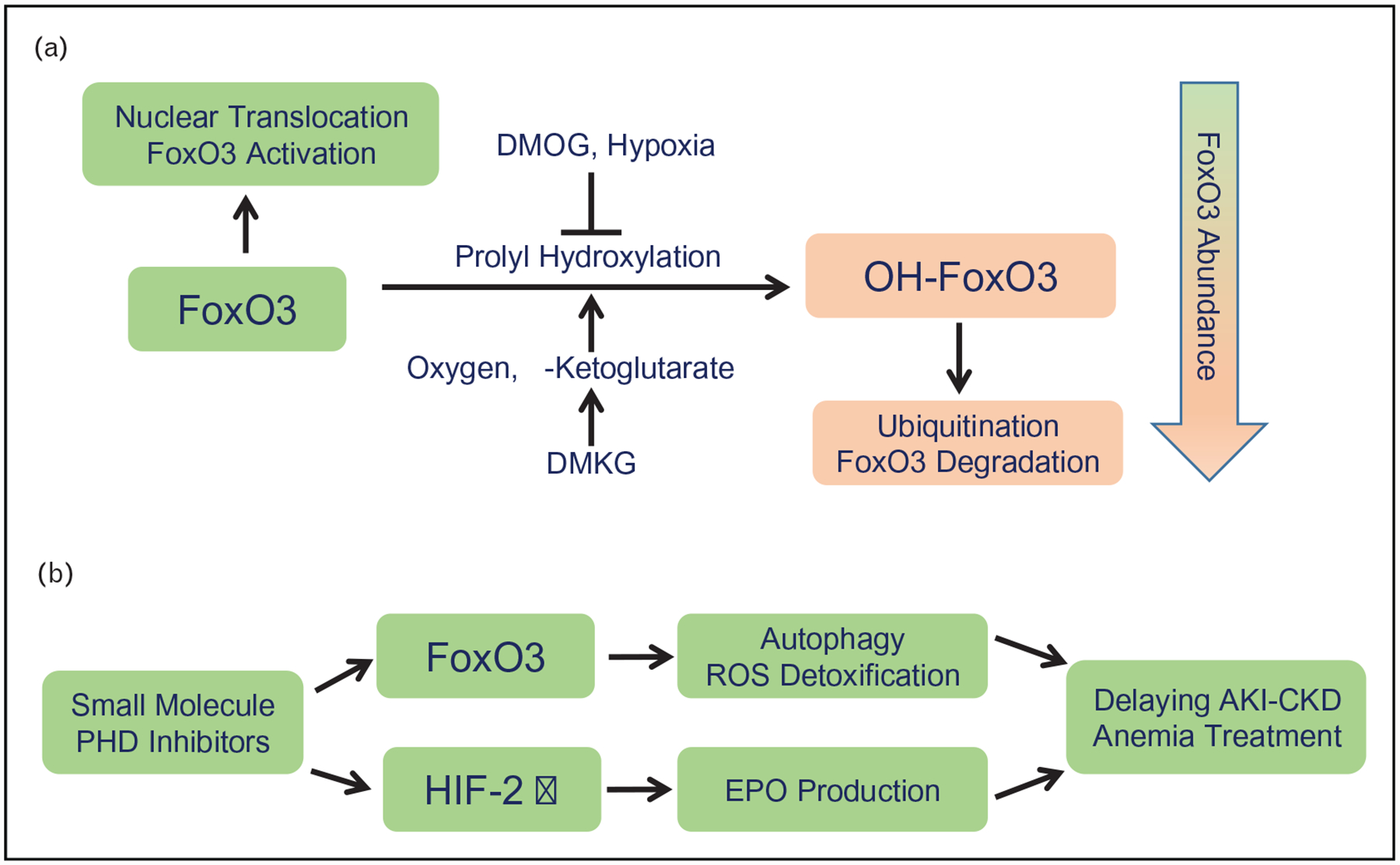
(A) Hypoxia results in increased FoxO3 protein abundance. Prolyl hydroxylation of FoxO3 depends on molecular oxygen and α-ketoglutarate. OH-FoxO3 is then degraded through the ubiquitin proteasomal system. Dimethyl α-ketoglutarate (DMKG), a cell permeable analogue of α-ketoglutarate enhances prolyl hydroxylation. Conversely, hypoxia or hypoxia-mimetic dimethyloxalylglycine (DMOG) inhibits this reaction leading to FoxO3 accumulation and activation of autophagy and ROS detoxification. ROS, reactive oxygen species. (B) The new orally active small molecule inhibitors of PHD enzymes may have a tubular protective effect by stimulating FoxO3 activation in addition to stabilizing the a subunit of HIF2 to increase erythropoietin (EPO) production for the treatment of anemia of chronic kidney failure.
FOXO3 ACTIVATION COUNTERACTS STRESS SIGNALS DURING AKI TO CHRONIC KIDNEY DISEASE TRANSITION
Sensing environmental changes, such as growth factor deficiency and stress from nutrient scarcity, hypoxia and reactive oxygen species, multiple upstream pathways are activated and converge on FOXO3, which activates a plethora of stress responsive genes to adapt the changing environment. Many of the direct target genes are conserved in human, mouse, C. elegans and Drosophila [35]. FOXO3 is one of the rare genes that have consistently been shown to be associated with lifespan extension across the species [36,37,38▪]. The antiaging capability of FOXO3 is attributed to its effects in suppressing tumors, reducing oxidative stress, stimulating autophagy, and regulating cell metabolism and the cell cycle in a wide variety of tissues. Our current knowledge of FOXO3 function in the kidney is limited. FoxO3 null mice have no apparent kidney structural abnormalities [9], suggesting that FoxO3 is not required for nephrogenesis in the nutrient and growth factor abundant environment. However, following caloric restriction, FoxO3 can be activated in a histone deacetylase Sirt1-dependent manner. FoxO3 then activates a core autophagy protein Bnip3, stimulating mitophagy that serves to protect the aging kidney [39].
During the development of CKD, interstitial fibrosis and vascular dropout reduce oxygen and nutrient delivery, causing metabolic and oxidative stress to the kidney. Hypoxia, nutrient deficiency, and reactive oxygen species (ROS) are powerful stimuli for autophagy [40–42], which is a lysosomal degradation pathway that targets nonessential or damaged intracellular constituents for energy reutilization required for cell survival and cell quality control under stress [42,43]. Using the new autophagy reporter line that expresses RFP-EGFP-LC3 fusion protein that allows us to study autophagic capacity and flux in real time, we tracked the autophagic process in the kidneys following ischemia-reperfusion injury (IRI). We showed that epithelial autophagy was dynamically regulated. Autophagy was activated in the early phase post-IRI and resolved as mTOR was activated during tubular repair. Autophagy returned to the baseline level 1 week post-IRI [44]. However, epithelial autophagy re-appeared 2–4 weeks later and persisted in regions of low capillary density and tissue hypoxia. Coin-ciding with the autophagic stress response, FoxO3 was activated in tubular epithelial cells via hypoxia-induced protein stabilization [33▪], which agrees with our previous finding that FoxO3 is also activated in the hypoxic kidney injured by urinary tract obstruction [34]. FoxO3 activation increases the capacity of epithelial autophagy by stimulating early steps of autophagic flux but does not enhance lysosomal fusion and degradation at the terminal clearance of the autophagic process [33▪,34]. Unlike transient autophagy that requires modification and assembly of preformed autophagy-related proteins (Atgs), protracted hypoxia and metabolic perturbation in the kidneys with CKD changes elicit a persistent autophagic response. FoxO3 stimulated mRNA expression of Atgs leads to replenishment of core components of the autophagic machinery that would otherwise be exhausted.
Conditional deletion of FoxO3 in all tubular cells during the AKI to CKD transition reveals a renal protective effect of FoxO3, as FoxO3 deletion leads to profound CKD pathological changes in the mutant kidney following IRI. The mice also exhibit accelerated renal functional decline with higher serum creatinine and urinary albumin excretion. At the subcellular level, mitochondrial swelling, loss of cristae and rupture of inner and outer membranes are frequently detected in the mutant kidneys. These signs of injury are concurrent with reduced epithelial autophagy and lower expression of superoxide dismutase 2 (SOD2), a mitochondrial scavenging enzyme that is a direct downstream target of FoxO3. Stimulation of autophagy and reduction of oxidative injury serve an underlying mechanism of renal protection by FoxO3.
TARGETING FOXO3 FOR POTENTIAL CHRONIC KIDNEY DISEASE TREATMENT
FoxO3 activation as a counteractive mechanism to stress is conserved in worms, flies, mice, and humans. We show that in human kidneys biopsied for AKI or AKI-on-CKD, vascular dropout, and increased nuclear FOXO3 expression coexist with autophagosomes and autolysosomes in injured proximal tubules [33▪]. The association of FOXO3 activation with the autophagic response suggests that, like what have learned from mouse studies, FOXO3 may regulate stress adaptation in human kidneys as well. Given the importance of prolyl hydroxylation in FOXO3 activation in the context of renal hypoxia, targeting PHD enzymes to stabilize FOXO3 protein could represent a new therapeutic approach.
PHD isoforms have diverse patterns of tissues expression and may exhibit substrate preference. All three isoforms are expressed in rodent kidneys [45]. Although PHD2 is likely to be the predominant enzyme for prolyl hydroxylation of the α-subunit of HIF-1, PHD3 has been shown to be the primary regulator for HIF-2α [30,31,46]. All three PHD isoforms appear to be involved in FoxO3 prolyl hydroxylation [33▪]. The substrate list for PHDs is growing as more research results become available [47,48▪]. Therefore, new generations of orally active small molecule PHD inhibitors used to treat anemia of renal failure [49–53] via stabilizing HIF-2 that stimulates the expression of erythropoietin in FoxD1-derived stromal cells in the kidney [54], may offer additional therapeutic benefits by FOXO3-mediated renal protection (Fig. 2B). Given a diverse pattern of expression of PHD isoforms, these new inhibitors may also create unexpected extra-renal consequences. Animal studies to test direct tubular effects of PHD inhibitors are under way.
It is important to note FoxOs interact with other transcription factors and co-regulators, and cross talk with other signaling pathways that include p53, Myc, Wnt/β-catenin, and mTOR complexes [16]. These interactions may serve as a positive reinforcement or negative feedback in a context-dependent fashion. For example, FoxO3 promotes metabolic adaptation to hypoxia and increases mitochondrial detoxification of ROS by antagonizing Myc function [55,56]. On the other hand, an interesting output of FOXO activation is downstream activation of PI3K/AKT, which feeds back to inhibit FOXO activation in leukemia cells [57] or in the liver during insulin signaling [58,59]. It is conceivable that FOXO may integrate diverse stress signals and regulate cell survival, apoptosis, or senescence depending on the level of stress, degree of cells injury, and state of cell proliferation.
CONCLUSION
FOXO3 is activated in response to metabolic and oxidative stress to reduce tubular injury and promote cell survival. However, altered immune response, inflammation and growth factor signaling are also among other pathogenic factors for the development of CKD [1▪▪,2,60▪]. A more in-depth understanding of the pathogenesis of CKD and FOXO signaling and function during the difference phase of kidney injury, and effective or futile repair is prerequisite before harnessing FOXO for preventing and treating kidney disease.
KEY POINTS.
Hypoxia and metabolic derangement in the kidney trigger stress response in renal tubular epithelial cells.
The stress responsive transcription factor FoxO3 can be activated under hypoxic conditions via hypoxia-induced inhibition of prolyl hydroxylation and subsequent degradation.
FoxO3 activation stimulates autophagy and ROS detoxification in renal tubular cells, thus reducing cell injury and promoting cell survival.
A more in-depth understanding of CKD pathogenesis and molecular regulation and function of FOXOs in the disease kidney is required before targeting FOXOs for potential therapy.
Acknowledgements
We thank Dr Qais Al-Awqati for helpful suggestions and discussions, and Holly Hurst for professional proofreading.
Financial support and sponsorship
This work was supported by the NIH grant R01DK 107653.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
- 1.Cippa PE, Sun B, Liu J, et al. Transcriptional trajectories of human kidney injury progression. JCI Insight 2018; 3:pii: 123151. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪▪This is the first study to use RNA-seq and machine learning for unbiased transitional profiling of human transplanted kidneys with acute ischemia-reperfusion injury and subsequent transition to chronic changes. The information may help predict clinical outcomes of the transplanted kidneys.
- 2.Liu J, Kumar S, Dolzhenko E, et al. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2017; 2:pii: 94716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hostetter TH, Olson JL, Rennke HG, et al. Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol 1981; 241:F85–93. [DOI] [PubMed] [Google Scholar]
- 4.Nath KA, Croatt AJ, Hostetter TH. Oxygen consumption and oxidant stress in surviving nephrons. Am J Physiol 1990; 258(5 Pt 2):F1354–F1362. [DOI] [PubMed] [Google Scholar]
- 5.Weigel D, Jurgens G, Kuttner F, et al. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell 1989; 57:645–658. [DOI] [PubMed] [Google Scholar]
- 6.Hannenhalli S, Kaestner KH. The evolution of Fox genes and their role in development and disease. Nat Rev Genet 2009; 10:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 2008; 20:126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hosaka T, Biggs WH 3rd, Tieu D, et al. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A 2004; 101:2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.John GB, Gallardo TD, Shirley LJ, Castrillon DH. Foxo3 is a PI3K-dependent molecular switch controlling the initiation of oocyte growth. Dev Biol 2008; 321:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salih DA, Rashid AJ, Colas D, et al. FoxO6 regulates memory consolidation and synaptic function. Genes Dev 2012; 26:2780–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calabuig-Navarro V, Yamauchi J, Lee S, et al. Forkhead Box O6 (FoxO6) depletion attenuates hepatic gluconeogenesis and protects against fat-induced glucose disorder in mice. J Biol Chem 2015; 290:15581–15594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim DH, Lee B, Lee J, et al. FoxO6-mediated IL-1beta induces hepatic insulin resistance and age-related inflammation via the TF/PAR2 pathway in aging and diabetic mice. Redox Biol 2019; 24:101184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kenyon CJ. The genetics of ageing. Nature 2010; 464:504–512. [DOI] [PubMed] [Google Scholar]
- 14.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005; 24:7410–7425. [DOI] [PubMed] [Google Scholar]
- 15.Greer EL, Brunet A. FOXO transcription factors in ageing and cancer. Acta Physiol (Oxf) 2008; 192:19–28. [DOI] [PubMed] [Google Scholar]
- 16.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev 2013; 14:83–97. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs FM, van der Heide LP, Wijchers PJ, et al. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem 2003; 278:35959–35967. [DOI] [PubMed] [Google Scholar]
- 18.Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci 2014; 39:159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewington AJ, Cerda J, Mehta RL. Raising awareness of acute kidney injury: a global perspective of a silent killer. Kidney Int 2013; 84:457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. New Engl J Med 2014; 371:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol 2015; 26:1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang-Panesso M, Kadyrov FF, Lalli M, et al. FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J Clin Invest 2019; 129:5501–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪▪ The authors developed a Kim1-GFPCreERt2 knockin mouse line to perform lineage tracing and demonstrated unequivocally that dedifferentiated proximial tubular cell proliferation accounted for tubular repair. Transcriptome analysis showed signatures of successful and maladpative repair.
- 23.Kramann R, Schneider RK, DiRocco DP, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015; 16:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu CF, Chiang WC, Lai CF, et al. Transforming growth factor beta-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am J Pathol 2013; 182:118–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka T, Nangaku M. Angiogenesis and hypoxia in the kidney. Nat Rev Nephrol 2013; 9:211–222. [DOI] [PubMed] [Google Scholar]
- 26.Lin SL, Chang FC, Schrimpf C, et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathology 2011; 178:911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kramann R, Humphreys BD. Kidney pericytes: roles in regeneration and fibrosis. Semin Nephrol 2014; 34:374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimke H, Sparks MA, Thomson BR, et al. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J Am Soc Nephrol 2015; 26:1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schodel J, Ratcliffe PJ. Mechanisms of hypoxia signalling: new implications for nephrology. Nat Rev Nephrol 2019; 15:641–659. [DOI] [PubMed] [Google Scholar]; ▪▪ This comprehensive review co-authored by the Nobel laureate Sir Peter J. Ratcliffe provides up-to-date information on the hypoxia-induced signaling with an emphasis on HIF and EPO pathways.
- 30.Berra E, Benizri E, Ginouves A, et al. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J 2003; 22:4082–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeda K, Ho VC, Takeda H, et al. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol 2006; 26:8336–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng X, Zhai B, Koivunen P, et al. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev 2014; 28:1429–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Kang H, Zhang Q, et al. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J Clin Invest 2019; 130:2374–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪▪ This is the first study to demonstrate hypoxia-induced FoxO3 prolyl hydroxylation as a new mechanism of FoxO3 activation in the kidney. The authors provide clear evidence that FoxO3 activation stimulates autophagy and ROS detoxification. These stress responses protect kidneys and delay the development of CKD.
- 34.Li L, Zviti R, Ha C, et al. Forkhead box O3 (FoxO3) regulates kidney tubular autophagy following urinary tract obstruction. J Biol Chem 2017; 292:13774–13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Webb AE, Kundaje A, Brunet A. Characterization of the direct targets of FOXO transcription factors throughout evolution. Aging Cell 2016; 15:673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willcox BJ, Donlon TA, He Q, et al. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A 2008; 105:13987–13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grossi V, Forte G, Sanese P, et al. The longevity SNP rs2802292 uncovered: HSF1 activates stress-dependent expression of FOXO3 through an intronic enhancer. Nucleic Acids Res 2018; 46:5587–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanese P, Forte G, Disciglio V, et al. FOXO3 on the road to longevity: lessons from SNPs and chromatin hubs. Comput Struct Biotechnol J 2019; 17:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ FOXO3 gene variants have a strong association with human longevity. The authors propose a putative aging chromatin hub in which FOXO3 plays a central role to modulate the physical connection and activity of neighboring genes involved in aging.
- 39.Kume S, Uzu T, Horiike K, et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest 2010; 120:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, posttranscriptional, and posttranslational regulation of autophagy. Trends Cell Biol 2015; 25:354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014; 157:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell 2014; 159:1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728–741. [DOI] [PubMed] [Google Scholar]
- 44.Li L, Wang ZV, Hill JA, Lin F. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol 2014; 25:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schodel J, Klanke B, Weidemann A, et al. HIF-prolyl hydroxylases in the rat kidney: physiologic expression patterns and regulation in acute kidney injury. Am J Pathol 2009; 174:1663–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem 2004; 279:38458–38465. [DOI] [PubMed] [Google Scholar]
- 47.Lee S, Nakamura E, Yang H, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell 2005; 8:155–167. [DOI] [PubMed] [Google Scholar]
- 48.Strowitzki MJ, Cummins EP, Taylor CT. Protein hydroxylation by hypoxia-inducible factor (HIF) hydroxylases: unique or ubiquitous? Cells 2019; 8:pii: E384. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ This review discusses alternative targets for PHDs and FIH and introduces techniques to identify these targets.
- 49.Holdstock L, Cizman B, Meadowcroft AM, et al. Daprodustat for anemia: a 24-week, open-label, randomized controlled trial in participants with chronic kidney disease. Clin Kidney J 2019; 12:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meadowcroft AM, Cizman B, Holdstock L, et al. Daprodustat for anemia: a 24-week, open-label, randomized controlled trial in participants on hemodialysis. Clin Kidney J 2019; 12:139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsubakihara Y, Akizawa T, Nangaku M, et al. A 24-week anemia correction study of daprodustat in Japanese dialysis patients. Ther Apher Dial 2019; 24:108–114. [DOI] [PubMed] [Google Scholar]
- 52.Chen N, Hao C, Liu BC, et al. Roxadustat treatment for anemia in patients undergoing long-term dialysis. New Engl J Med 2019; 381: 1011–1022. [DOI] [PubMed] [Google Scholar]
- 53.Chen N, Hao C, Peng X, et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. New Engl J Med 2019; 381:1011–1022. [DOI] [PubMed] [Google Scholar]
- 54.Kobayashi H, Liu Q, Binns TC, et al. Distinct subpopulations of FOXD1 stroma-derived cells regulate renal erythropoietin. J Clin Invest 2016; 126:1926–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferber EC, Peck B, Delpuech O, et al. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ 2012; 19:968–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jensen KS, Binderup T, Jensen KT, et al. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J 2011; 30:4554–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hui RC, Gomes AR, Constantinidou D, et al. The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Mol Cell Biol 2008; 28:5886–5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ide T, Shimano H, Yahagi N, et al. SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat Cell Biol 2004; 6:351–357. [DOI] [PubMed] [Google Scholar]
- 59.Puig O, Tjian R. Transcriptional feedback control of insulin receptor by dFOXO/FOXO1. Genes Dev 2005; 19:2435–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu H, Lai CF, Chang-Panesso M, Humphreys BD. Proximal tubule translational profiling during kidney fibrosis reveals proinflammatory and long non-coding RNA expression patterns with sexual dimorphism. J Am Soc Nephrol 2019; 31:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ This is the first study to demonstrate differences in gender response to kidney injury and CKD progression based on a large-scale transcriptomic map of the proximal tubules. The authors discovered a sexual dimorphism in proinflammatory and long nonconding RNA expression patterns.