

Abstract
Inflammasomes are supramolecular complexes that play key roles in immune surveillance. This is accomplished by activating inflammatory caspases, which lead to the proteolytic maturation of IL-1β and pyroptosis. Here, we show that NLRP3- and pyrin-mediated inflammasome assembly, caspase activation, and IL-1β conversion occur at the microtubule-organizing center (MTOC). Furthermore, the dynein adaptor HDAC6 is indispensable for the microtubule transport and assembly of these inflammasomes, shown by chemical inhibition and targeted deletion in immortalized and primary macrophages and in mice. Because HDAC6 can transport ubiquitinated pathological aggregates to the MTOC for aggresome formation and autophagosomal degradation, its role in NLRP3 and pyrin inflammasome activation also offers an inherent mechanism to downregulate these inflammasomes by autophagy. This work suggests an unexpected parallel between the formation of physiological and pathological aggregates.
One Sentence Summary
The NLRP3 and pyrin inflammasomes use an HDAC6-dependent aggresome-like mechanism for their activation at the microtubule-organizing center (MTOC).
Inflammasomes play important roles in cytosolic host defense (1–5). Architecturally, canonical inflammasomes are composed of an upstream sensor, an adaptor, and the downstream caspase-1 (6). By contrast, in non-canonical inflammasomes, when bacterial lipopolysaccharide (LPS) gains access to the cytosol, it directly engages and activates caspase−4 and −5 in humans and caspase-11 in mice (1). The nucleotide-binding domain (NBD), leucine-rich repeat (LRR), and pyrin domain (PYD)-containing protein 3 (NLRP3) constitutes an extensively studied inflammasome sensor, which can be activated by diverse stimuli including the bacterial pore-forming toxin nigericin, extracellular ATP, and various particulates such as monosodium urate crystals (MSU), alum, and silica (2–4). Activated NLRP3 recruits the apoptosis-associated speck-like protein containing a CARD (ASC), which in turn recruits caspase-1 (Fig. 1A). Absent in melanoma 2 (AIM2) and pyrin are sensors for two other ASC-dependent inflammasomes. AIM2 is activated by cytosolic dsDNA, and pyrin can be stimulated by Rho-glucosylation activity of Clostridium difficile toxin B (TcdB) (2, 7) (Fig. 1A). The NLR family CARD-containing protein 4 (NLRC4) can form an inflammasome with or without ASC upon complex formation with an NLR family apoptosis inhibitory protein (NAIP), which directly senses bacterial flagellin or type III secretion system proteins (1–5). The formation of inflammasomes leads to proximity-induced caspase dimerization, activation, and autoprocessing. Caspase-1 cleaves pro-IL-1β and pro-IL-18 to generate the mature cytokines, and caspase-1, −4, −5, and −11 can proteolytically activate gasdermin D (GSDMD) to form membrane pores for cytokine release and pyroptosis (2–4, 8–10). Dysregulated inflammasome activity has been implicated in numerous human diseases including hereditary autoinflammatory syndromes and common conditions such as gout, diabetes, atherosclerosis, Alzheimer’s disease, and colorectal cancer (2–5).
Fig. 1. NLRP3 and pyrin inflammasomes, but not the AIM2 inflammasome, co-localize with the MTOC.
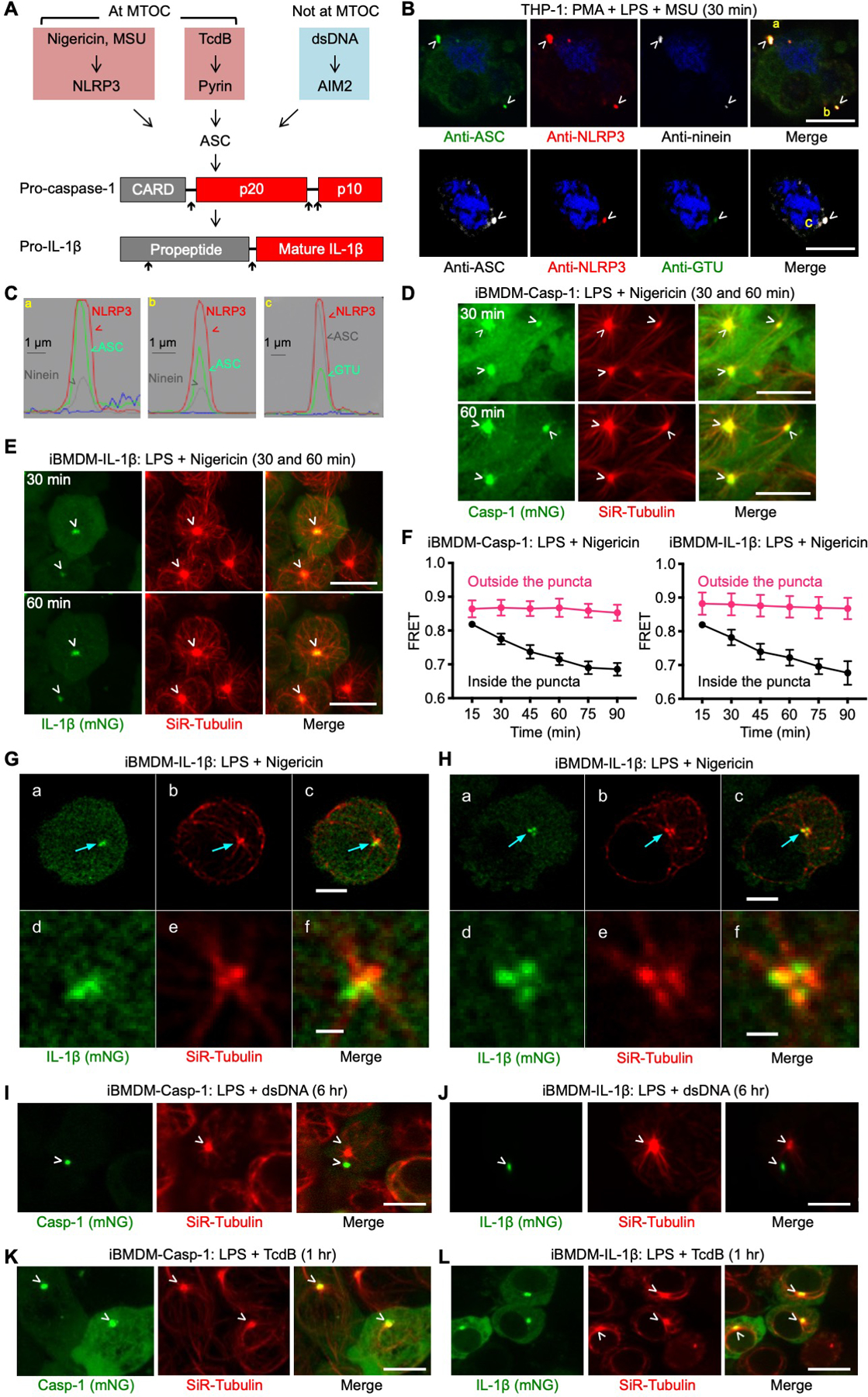
(A) The NLRP3, pyrin, and AIM2 inflammasome pathways triggered by nigericin or MSU, TcdB, and dsDNA, respectively. As shown below, NLRP3 and pyrin inflammasome puncta localize at the MTOC. Inflammasome activation culminates in pro-caspase-1 and pro-IL-1β processing. Upward arrows indicate processing sites. (B) Immunofluorescence images showing the co-localization of NLRP3 and ASC puncta with the centrosomal markers ninein and GTU in THP-1 cells. Blue represents nuclear staining by Hoechst 33342. (C) Line scan of intensity distribution profiles of puncta a, b, and c from (B). (D–E) Live-cell images of iBMDM-Casp-1 (D) and iBMDM-IL-1β (E) at 30 min (top panel) and 60 min (bottom panel) post-nigericin stimulation, showing co-localization of inflammasome puncta (depicted by mNeonGreen) with the MTOC (depicted by SiR-Tubulin staining that labels the microtubule network). (F) FRET analysis of caspase-1 cleavage and IL-1β processing at MTOC as a function of time for areas inside and outside the puncta in iBMDM-Casp-1 (left) and iBMDM-IL-1β (right) cells. FRET was calculated by dividing the FRET channel fluorescence intensity (donor excitation with acceptor emission) with mTurquoise2 channel fluorescence intensity (donor excitation with donor emission). Values are mean±SD for n=10–15 cells. (G–H) Recruitment of IL-1β to a region in proximity to the MTOC imaged using 3D lattice light-sheet microscopy (LLSM). iBMDM-IL-1β cells stained with SiR-Tubulin were exposed to nigericin for 12 min (G), and 23 min (H). (a–c) Representative images deconvolved using the Richardson–Lucy algorithm corresponding to a single optical plane section. The arrows highlight the MTOC and the nearby locations where IL-lβ was recruited. (d–f) Enlarged images of the regions indicated by the arrows. (I–J) Lack of co-localization of AIM2 inflammasome puncta with the MTOC in iBMDM-Casp-1 (I) and iBMDM-IL-1β (J) cells activated by dsDNA for 6 hours. (K–L) Co-localization of pyrin inflammasome puncta with the MTOC in iBMDM-Casp-1 (K) and iBMDM-IL-1β (L) cells activated by TcdB toxin for 1 hour. Images are representative of three or more independent experiments and arrowheads indicate puncta or MTOC (B, D–E, G–L). Scale bars: 10 μm (B, D–E), 5 μm (Ga–c, Ha–c, I–L), and 1 μm (Gd–f, Hd–f).
NLRP3 and pyrin inflammasomes, but not the AIM2 inflammasome, localize at the microtubule-organizing center
An intriguing observation has been that many inflammasomes assemble into a single major perinuclear punctum in each activated cell (11, 12). The singularity and the perinuclear nature of an inflammasome punctum led us to investigate its connection if any, with the centrosome, which also acts as the microtubule-organizing center (MTOC) in eukaryotic cells and is perinuclear and punctate in appearance. We used both the human monocytic cell line THP-1 and immortalized mouse bone marrow-derived macrophages (iBMDMs), which endogenously express NLRP3 and other inflammasome components. NLRP3 and ASC co-localized with the centrosomal markers ninein and γ-tubulin (GTU) (13) upon priming by LPS and activation by MSU or nigericin (Fig. 1B&C, fig. S1A–C). When THP-1 cells were stimulated with MSU, 88.5% (62/70 cells) showed co-localization between ASC and ninein. By contrast, only 1.4% (1/70 cells) lacked apparent co-localization. The location of the remaining ASC puncta could not be determined due to lack of corresponding ninein staining. Similarly, 80% (40/50) of MSU-stimulated THP-1 cells exhibited ASC co-localization with GTU, 4% (2/50 cells) showed no co-localization, and the remaining 16% (8/50) showed no visible GTU staining. NEK7, a member of the NIMA-related kinase (NEK) family, which predominantly resides at the MTOC and is required for NLRP3 inflammasome activation (14–16), also co-localized with ASC and GTU (fig. S1D). The specificity of anti-NLRP3 and anti-ASC antibodies was thoroughly validated (fig. S1E–F, S2). Thirty minutes after nigericin stimulation, we observed robust caspase-1 processing (fig. S3A–B) and IL-1β secretion (fig. S3C).
In addition to NLRP3 and ASC, IL-1β also localized at the inflammasome puncta (fig. S3D&E). Labeling of active caspase-1 by FAM-FLICA™ (17) showed that in 9% (2/22) and 18% (4/22) of cells respectively, active caspase-1 completely and partially co-localized with the ASC puncta (fig. S3F, Examples 1 and 2). In most cells (73%, 16/22), active caspase-1 surrounded the ASC punctum (fig. S3F, Example 3), suggesting that caspase-1 is first activated at an inflammasome punctum by dimerization and then released into the cytosol upon auto-cleavage from the N-terminal CARD pro-domain. Thus, NLRP3 inflammasome puncta at the MTOC may serve as a major site for both caspase-1 activation and IL-1β conversion.
We attempted to visualize NLRP3 inflammasome activation using live-cell imaging of fluorescent protein-fused ASC. However, unlike wild-type (WT) cells, nigericin stimulation induced iBMDM cell line stably expressing ASC-mRuby3 and THP-1 cell line expressing ASC-TagRFP-T to form puncta distinctly away from the MTOC marked by SiR-Tubulin, a live-cell fluorogenic probe for microtubules (fig. S4A–C, movie S1). In contrast, iBMDM cell line stably expressing Flag-ASC showed that the main puncta formed upon nigericin stimulation, visualized by anti-Flag immunofluorescence, coincided with the MTOC (fig. S4D), suggesting that the GFP family of tags may have led to non-physiological localization of ASC. Therefore we did not use ASC for live cell imaging of inflammasomes.
We then followed the NLRP3 inflammasome cascade to pro-caspase-1 and pro-IL-1β. We performed live-cell confocal microscopy on iBMDM stable cell lines expressing pro-caspase-1 (iBMDM-Casp-1) or pro-IL-1β (iBMDM-IL-1β) fused to a fluorescence resonance energy transfer (FRET) pair, mTurquoise2 (mTur) at the N-terminus and mNeonGreen (mNG) at the C-terminus, to track both their location and cleavage (fig. S5A). Both pro-caspase-1 and pro-IL-1β formed puncta at the MTOC upon nigericin stimulation (Fig. 1D–E, fig. S5B–C, movies S2–S3). For some cells, we also observed signal trails from the inflammasome puncta that matches the microtubule staining pattern (Fig. 1D, 60 min), suggesting trafficking of inflammasome complexes on the microtubule network. 3D lattice light-sheet microscopy (LLSM) (18) of nigericin stimulated iBMDM-IL-1β cells at 12 min and 23 min post treatment, respectively, further revealed that IL-1β was recruited to a region in close proximity to but not overlapping with the MTOC (Fig. 1G–H, movie S4). For both iBMDM-IL-1β and iBMDM-Casp-1 cells, FRET signals inside the puncta were high at the beginning, consistent with tethered FRET pairs, but decreased significantly with time, consistent with proteolytic processing (Fig. 1F). By contrast, the FRET signals outside the puncta stayed high (Fig. 1F). Thus, it is upon recruitment to the MTOC that pro-caspase-1 and pro-IL-1β are processed and activated.
To activate the AIM2 inflammasome, iBMDM-Casp-1 and iBMDM-IL-1β cells were transfected with dsDNA. Unlike NLRP3 stimulation, the puncta induced by AIM2 activation did not co-localize with the MTOC despite the similar dependence on ASC as NLRP3 (Fig. 1I–J). By contrast, when iBMDM-Casp-1 and iBMDM-IL-1β cells were stimulated with Clostridium difficile toxin B (TcdB) to activate the pyrin inflammasome, activation-induced puncta co-localized with the MTOC (Fig. 1K–L, movies S5–S6). Similarly, anti-pyrin, anti-ASC, and anti-ninein IF confirmed the MTOC localization of the pyrin inflammasome (fig. S6).
Microtubule retrograde transport by HDAC6 is important for NLRP3 and pyrin inflammasome activation
A major cellular mechanism for clearance of ubiquitinated protein aggregates, which are too large to be effectively degraded by the ubiquitin proteasome pathway, requires retrograde transport by the dynein adaptor and α-tubulin-deacetylase HDAC6 to form aggresomes at the MTOC for degradation by autophagy (19–23). The aggresome pathway is thought to play an important role in the regulation of protein aggregation diseases such as Alzheimer’s disease and age-related macular degeneration (19, 21). Viruses including the influenza A virus are known to subvert the host aggresome pathway by forming viral inclusions at the MTOC in an HDAC6-dependent manner in order to facilitate replication and assembly (24, 25). The MTOC localization of NLRP3 and pyrin inflammasome puncta, their aggresome-like meshwork morphology (26), and ubiquitination of NLRP3 inflammasome components (27–30) suggest a possible link between inflammasomes and aggresomes.
To test the effects of HDAC6 and microtubule retrograde transport inhibition on NLRP3 function, we pre-treated LPS-primed iBMDM-Casp-1 and iBMDM-IL-1β cells with microtubule, dynein, and HDAC6 inhibitors (31–33). Nigericin stimulation in iBMDM-Casp-1 and iBMDM-IL-1β cells pre-treated with these inhibitors invariably failed to induce the generation of puncta. By contrast, untreated nigericin-stimulated cells readily formed puncta (fig. S7A–B). Inhibitor pre-treatment of WT iBMDMs markedly decreased nigericin-induced caspase-1 processing (Fig. 2A), propidium iodide (PI) permeability (Fig. 2B), and IL-1β release (Fig. 2C, fig. S7C) without affecting NF-κB dependent priming as measured by TNFα secretion (fig. S8A). Dynamitin (also known as p50) is a subunit of the dynactin complex important in dynein function, whose overexpression disrupts dynein-dependent transport (36). Stable expression of mRuby3-fused dynamitin in iBMDM-Casp-1 and iBMDM-IL-1β cells severely compromised nigericin-induced punctum formation without affecting the MTOC-localization of NEK7 (fig. S7A–B, S8B, movie S7), and caused defective caspase-1 processing, PI permeability, and IL-1β secretion (fig. S8C–E). Thus, the microtubule–dynein machinery and the HDAC6 adaptor both appear to be essential for NLRP3 inflammasome activation.
Fig. 2. Microtubule retrograde transport is required for the activation of NLRP3 and pyrin inflammasomes, but not the AIM2 inflammasome.
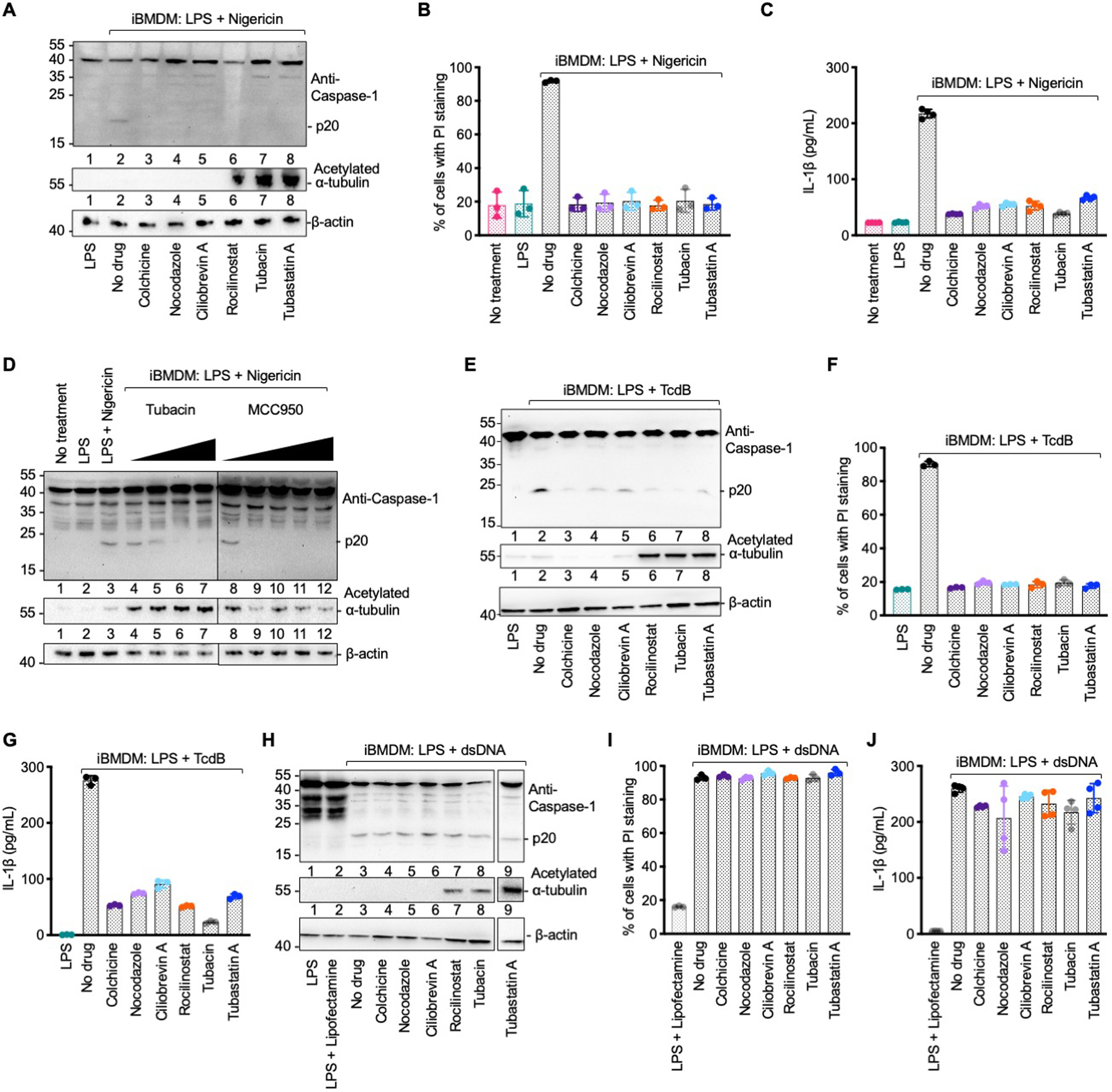
(A–C) NLRP3 inflammasome activation under various inhibition conditions analyzed by caspase-1 processing (p20) (A), quantification of propidium iodide (PI) permeability by flow cytometry (B), and secreted IL-1β quantified by ELISA (C). Colchicine and nocodazole: microtubule polymerization inhibitors; Ciliobrevin A: dynein ATPase inhibitor; Rocilinostat, tubacin and tubastatin A: HDAC6 inhibitors. (D) Caspase-1 processing (p20) upon NLRP3 inflammasome activation with pre-treatment of increasing concentrations of tubacin (left to right: 5 μM, 10 μM, 20 μM and 40 μM) or the NLRP3 inhibitor MCC950 (left to right: 0.1 μM, 0.5 μM, 1 μM, 10 μM and 20 μM). Anti-acetylated α-tubulin and anti-β-actin immunoblots are shown for tubulin acetylation and as the loading control, respectively. (E–G) Pyrin inflammasome activation under various pharmacological conditions analyzed by caspase-1 processing (p20) (E), PI permeability (F), and secreted IL-1β (G). (H–J) AIM2 inflammasome activation under various pharmacological conditions analyzed by caspase-1 processing (p20) (H), PI permeability (I) and secreted IL-1β (J). Data are presented as the mean±SD for 3–4 wells from three or more independent experiments.
Our data contradict a previous notion that increased α-tubulin acetylation upon NLRP3 stimulation is responsible for dynein-dependent, microtubule-mediated assembly of the NLRP3 inflammasome (34). Treatment with HDAC6 inhibitors tubastatin A, rocilinostat and tubacin markedly increased α-tubulin acetylation; however, instead of increasing NLRP3 inflammasome activation, it inhibited it (Fig. 2A, 2D). A control NLRP3 inhibitor MCC950 that interferes with NLRP3 oligomerization (35) did not change α-tubulin acetylation but potently impeded NLRP3 activation (Fig. 2D). Thus, retrograde transport by HDAC6 on the microtubule network aids NLRP3 and pyrin inflammasome activation independent of α-tubulin acetylation.
We investigated the effects of pharmacological inhibition and dynamitin overexpression in other inflammasomes. Consistent with the co-localization of the pyrin inflammasome with the MTOC, treatment by microtubule, dynein, and HDAC6 inhibitors or dynamitin expression hindered TcdB-induced punctum formation (fig. S9A–D), and compromised TcdB-induced caspase-1 processing, PI permeability, and IL-1β release (fig. S8C–E, Fig. 2E–G). It is possible that the pyrin inflammasome is even more sensitive to microtubule transport inhibitors than the NLRP3 inflammasome. This is reflected in previous studies in which lower concentrations of colchicine (1–4 μM) inhibited the pyrin inflammasome alone (37, 38), whereas higher concentrations of colchicine (5–15 μM) like in the present study (10 μM) affected both the pyrin and NLRP3 inflammasome (34). In contrast to the NLRP3 and pyrin inflammasomes, neither pharmacological treatment nor dynamitin expression inhibited dsDNA-induced AIM2 inflammasome punctum formation (fig. S10A–D), caspase-1 processing, cell death, or IL-1β secretion (Fig. 2H–J, fig. S8C–E), consistent with its lack of co-localization with the MTOC. Thus, HDAC6 is important for the activation NLRP3 and pyrin inflammasomes, but not the AIM2 inflammasome.
HDAC6 targeting compromises NLRP3 inflammasome activation
To further interrogate the role of HDAC6 in inflammasome activation, we generated an HDAC6-deficient iBMDM cell line (Hdac6−/−) using the CRISPR/Cas9 system (39). Genomic PCR confirmed that the targeting resulted in an internal deletion of the Hdac6 gene (fig. S11A) and lack of HDAC6 protein expression (Fig. 3A). Normal physiology of Hdac6−/− iBMDMs was reflected in the unperturbed centrosome structure (fig. S11B), NEK7 distribution at the centrosome (fig. S11C), and transferrin uptake (fig. S11D–E). HDAC6 deficiency also did not affect the expression of NLRP3, pyrin, ASC, pro-caspase-1, or pro-IL-1β upon LPS priming (fig. S11F–G). However, the absence of HDAC6 resulted in markedly reduced caspase-1 cleavage (Fig. 3B), PI permeability (Fig. 3C), IL-1β secretion (Fig. 3D), and punctum formation (movies S8–9) upon nigericin stimulation.
Fig. 3. HDAC6 is required for NLRP3 inflammasome activation.
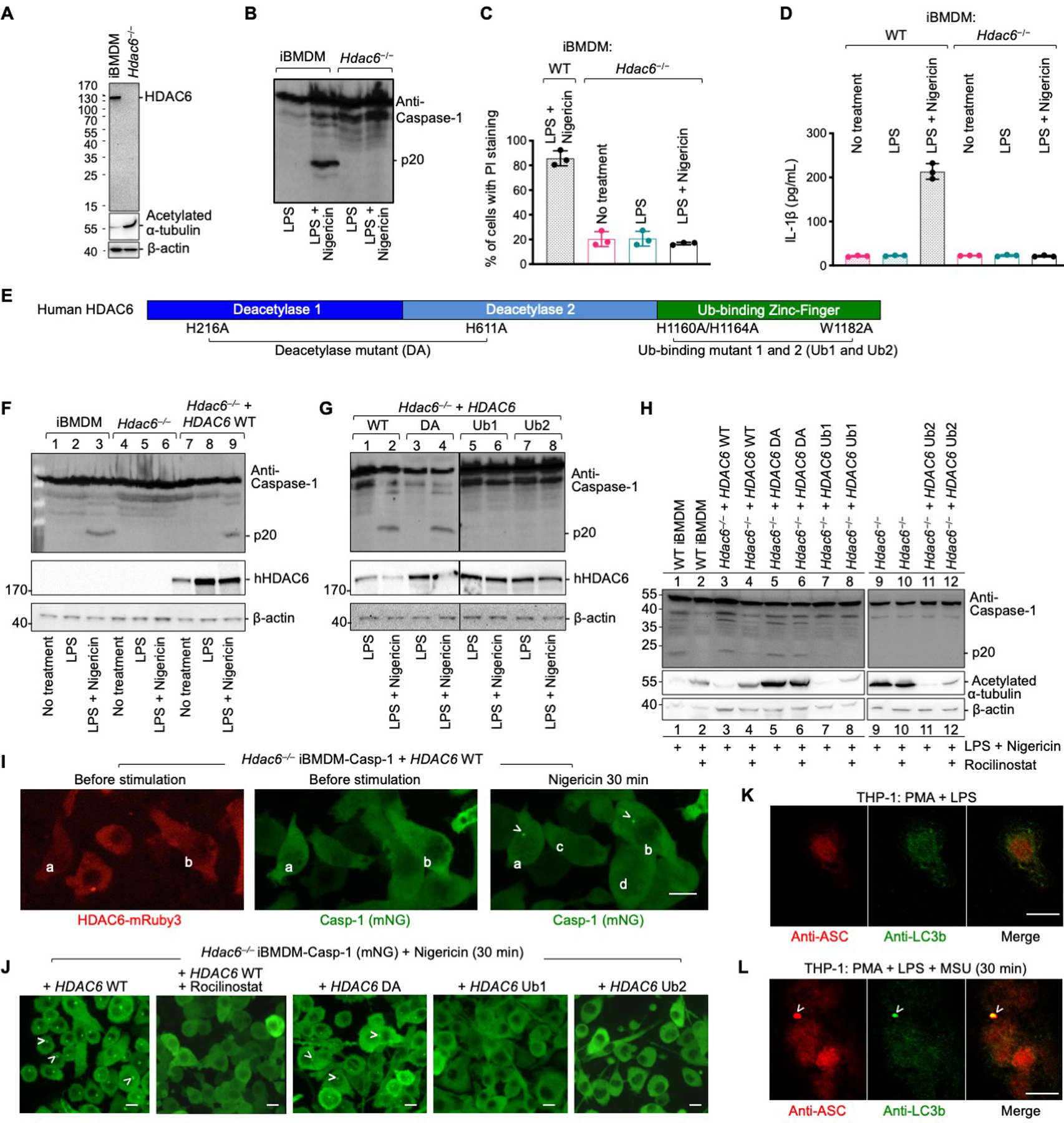
(A) Immunoblotting shows the absence of HDAC6 protein in CRISPR/Cas9 Hdac6−/− iBMDMs as compared to WT iBMDMs. Loading control was provided by the anti-β-actin antibody. The loss of HDAC6 leads to an increase in acetylated α-tubulin depicted using anti-acetylated α-tubulin antibody. (B–D) Compromised NLRP3 inflammasome activation in Hdac6−/− iBMDMs challenged with nigericin, shown for caspase-1 processing (B), PI permeability (C), and secreted IL-1β (D). Data are presented as the mean±SD for triplicate wells from three or more independent experiments (C, D). (E) Domain architecture of human HDAC6 with important mutations (DA, Ub1 and Ub2) labeled. DA: H216A/H611A on catalytic residues, deacetylase mutant; Ub1: mutations H1160A/H1164A on zinc-coordinating residues; Ub2: mutation W1182A on the surface that binds ubiquitin. (F) Rescue of nigericin-mediated caspase-1 processing in Hdac6−/− iBMDMs by reconstituting with WT human HDAC6. (G) Analysis of nigericin-mediated caspase-1 processing in Hdac6−/− iBMDMs reconstituted with WT HDAC6, and the DA, Ub1 and Ub2 mutants. (H) Sensitivity to rocilinostat in Hdac6−/− iBMDMs reconstituted with WT HDAC6, but not the DA mutant, as depicted by inhibition of p20 processing. (I) Rescue of nigericin-induced punctum formation in Hdac6−/− iBMDM-Casp-1 cells transfected with WT HDAC6-mRuby3. Arrowheads indicate puncta. Cells containing puncta had HDAC6 expression (a and b), whereas cells that do not contain puncta did not have HDAC6 expression (c and d). (J) Rescue of nigericin-induced punctum formation in Hdac6−/− iBMDM-Casp-1 cells stably reconstituted with WT and DA mutant of HDAC6-mRuby3, but not with Ub1 and Ub2 mutants of HDAC6-mRuby3. Arrowheads indicate puncta. HDAC6 WT reconstituted cells failed to form puncta upon pretreatment by rocilinostat. (K–L) Inflammasome puncta formation and its link to autophagy analyzed by immunofluorescence of ASC and the autophagy marker LC3b before (K) and after (L) NLRP3 inflammasome stimulation. Arrowheads indicate puncta. Images are representative of three or more independent experiments. Scale bars: 10 μm.
To test the possibility that HDAC6 might have a role in ligand accessibility for NLRP3 stimuli that require endocytosis, we utilized several additional NLRP3 ligands. These included ATP—which directly engages its cell surface P2X7 receptor, and should not depend on cellular uptate—as well as silica and alum, which are NLRP3-activating particulates and may require ligand accessibility. HDAC6 deficiency impaired caspase-1 cleavage, PI permeability, and IL-1β secretion after both ATP (fig. S12A) and silica and alum stimulation (fig. S12B). Additionally, puncta were formed upon treatment with all these ligands, whereas few puncta were observed in similarly treated Hdac6−/− iBMDMs (fig. S13A–C). Thus, HDAC6 dependency exists for a broad range of NLRP3 activators regardless of their exact mechanism of action.
To corroborate the Hdac6−/− studies, we knocked down the expression of HDAC6 at the RNA and protein levels using small interfering RNA (siRNA) and the Trim-Away system (40), respectively. Seventy-two hours after iBMDMs were transfected with siRNA, HDAC6 was knocked down (fig. S14A), and iBMDMs exhibited markedly reduced nigericin-stimulated caspase-1 activation (fig. S14A), cell death (fig. S14B), and IL-1β secretion (fig. S14C). Similarly, electroporation of anti-HDAC6 antibody into TRIM21-iBMDMs resulted in substantial HDAC6 degradation after 24 hours (fig. S14D), and reduced caspase-1 processing (fig. S14D), PI permeability (fig. S14E), and IL-1β secretion (fig. S14F) after nigericin stimulation. By contrast, a previous study using small hairpin RNA (shRNA) targeting HDAC6 showed modestly enhanced NLRP3 inflammasome activation in iBMDMs in comparison with a scrambled shRNA (41). However, no untreated iBMDMs were used as a control and it is unclear whether the shRNAs had off-target effects. Thus, these data further support that HDAC6 plays an important role in NLRP3 inflammasome activation.
HDAC6 ubiquitin-binding domain but not deacetylase activity is required for NLRP3 activation
We reconstituted Hdac6−/− iBMDMs with human WT HDAC6 and its deacetylase catalytic mutant H216A/H611A (DA) (42) (Fig. 3E, fig. S15A). WT HDAC6 rescued caspase-1 activation upon nigericin stimulation (Fig. 3F). Surprisingly, the deacetylase activity was not required to support NLRP3 inflammasome activation as the HDAC6 DA also restored NLRP3 inflammasome activation as shown by caspase-1 processing (Fig. 3G), PI permeability (fig. S15B), and IL-1β secretion (fig. S15C). The effects of small molecule HDAC6 inhibitors on inflammasome activation may be due to inability of inhibitor-bound HDAC6 to interact with dynein (42, 43). Indeed, WT HDAC6-reconstituted Hdac6−/− iBMDMs were sensitive to HDAC6 inhibitor rocilinostat. By contrast, HDAC6 DA-reconstituted Hdac6−/− iBMDMs were insensitive to rocilinostat (Fig. 3H) because the inhibitor binding requires an intact active site. Intriguingly, the α-tubulin deacetylase activity of HDAC6 is also not required for HDAC6- and microtubule-mediated chemotaxis of T–lymphocytes (44), whereas misfolded protein-induced aggresome formation appeared to require the catalytic activity (22). Thus, HDAC6 supports NLRP3 inflammasome activation by its scaffolding role in dynein-mediated transport without the need for its catalytic activity.
The C-terminal region of HDAC6 contains a zinc-finger domain for ubiquitin interaction (20). We reconstituted Hdac6−/− iBMDMs with the zinc-coordinating mutant H1160/H1164A (Ub1) and ubiquitin-binding site mutant W1182A (Ub2) (Fig. 3E, fig. S15A). Both mutants failed to rescue NLRP3 inflammasome activation shown by caspase-1 processing (Fig. 3G), PI permeability (fig. S15B), and IL-1β secretion (fig. S15C). Live-cell imaging of Hdac6−/− iBMDM-Casp-1 cells transiently transfected with mRuby3-fused WT human HDAC6 showed a correlation between HDAC6 expression and punctum formation. Only cells with detectable HDAC6 expression formed puncta (Fig. 3I, movie S10). Furthermore, Hdac6−/− iBMDM-Casp-1 and iBMDM-IL-1β cells stably reconstituted with WT and DA mutant of mRuby3-HDAC6 rescued punctum formation, whereas those transfected with the zinc-finger mutants did not (Fig. 3J, fig. S15D, movies S11–S18). Rocilinostat inhibited punctum formation in WT HDAC6 reconstituted Hdac6−/− iBMDM-Casp-1 cells (Fig. 3J). Furthermore, NLRP3 activation by ATP, silica, and alum, shown by PI permeability (fig. S16) and punctum formation (fig. S17), had the same dependence on the ubiquitin-interacting domain. Thus, HDAC6’s ubiquitin-binding function is essential for its role in the NLRP3 inflammasome.
Inflammasome puncta at the MTOC are subjected to autophagic regulation
Because HDAC6-mediated aggresome formation at the MTOC facilitates autophagosomal degradation, we hypothesized that inflammasome puncta are also subject to this regulation. Before nigericin stimulation, there was no co-localization of ASC with the microtubule-associated protein 1 light chain 3b (LC3b), a standard autophagosomal marker (Fig. 3K). By contrast, at 30 min post-stimulation, ASC-containing inflammasome puncta were highly enriched in LC3b, suggesting induction of autophagy (Fig. 3L). We previously showed by anti-ASC immunogold electron microscopy (EM) that dense perinuclear structures represent inflammasome puncta (26). Here we observed that the single perinuclear inflammasome punctum was surrounded by double-membrane bilayers, which are suggestive of autophagosome formation (fig. S18). LLSM analysis showed adjacency between an inflammasome punctum and the MTOC per se (Fig. 1G–H). Thus, inflammasome puncta may be selectively engulfed without affecting the MTOC. Consistent with these findings, reduction or deficiency of autophagic proteins LC3b, beclin-1, p62, and Atg16L1 can enhance inflammasome activation and maturation of IL-1β and IL-18 (30, 45, 46). Furthermore, IL-1β secretion was enhanced by the pharmacological blockade of autophagosome formation by 3-methyladenine (3-MA) (fig. S7C). As controls, a pan-caspase inhibitor (Z-VAD-FMK) and a caspase-1 inhibitor (YVAD-CHO) both reduced IL-1β secretion (fig. S7C). Thus, inflammasome puncta formed at the MTOC are regulated by autophagy.
HDAC6 deficiency compromised activation of the pyrin inflammasome, but not AIM2, NLRC4 and non-canonical inflammasomes
Consistent with the MTOC localization and the effects of HDAC6 inhibitors, pyrin inflammasome activation induced by TcdB was compromised by knocking out Hdac6. This was evident in terms of caspase-1 cleavage, PI staining, IL-1β secretion, and punctum formation (Fig. 4A–C, movies S19–20). By contrast, dsDNA-induced AIM2 inflammasome activation in Hdac6−/− iBMDMs was similar to WT controls (Fig. 4E–H). Reconstitution of WT and mutant human HDAC6 into Hdac6−/− iBMDMs revealed that pyrin inflammasome activation required its intact zinc-finger domain but not its deacetylase activity, as shown by caspase-1 cleavage (Fig. 4A), PI staining (Fig. 4B), IL-1β secretion (Fig. 4C), and punctum formation (Fig. 4D).
Fig. 4. HDAC6 is required for pyrin inflammasome activation, but not AIM2 inflammasome activation.
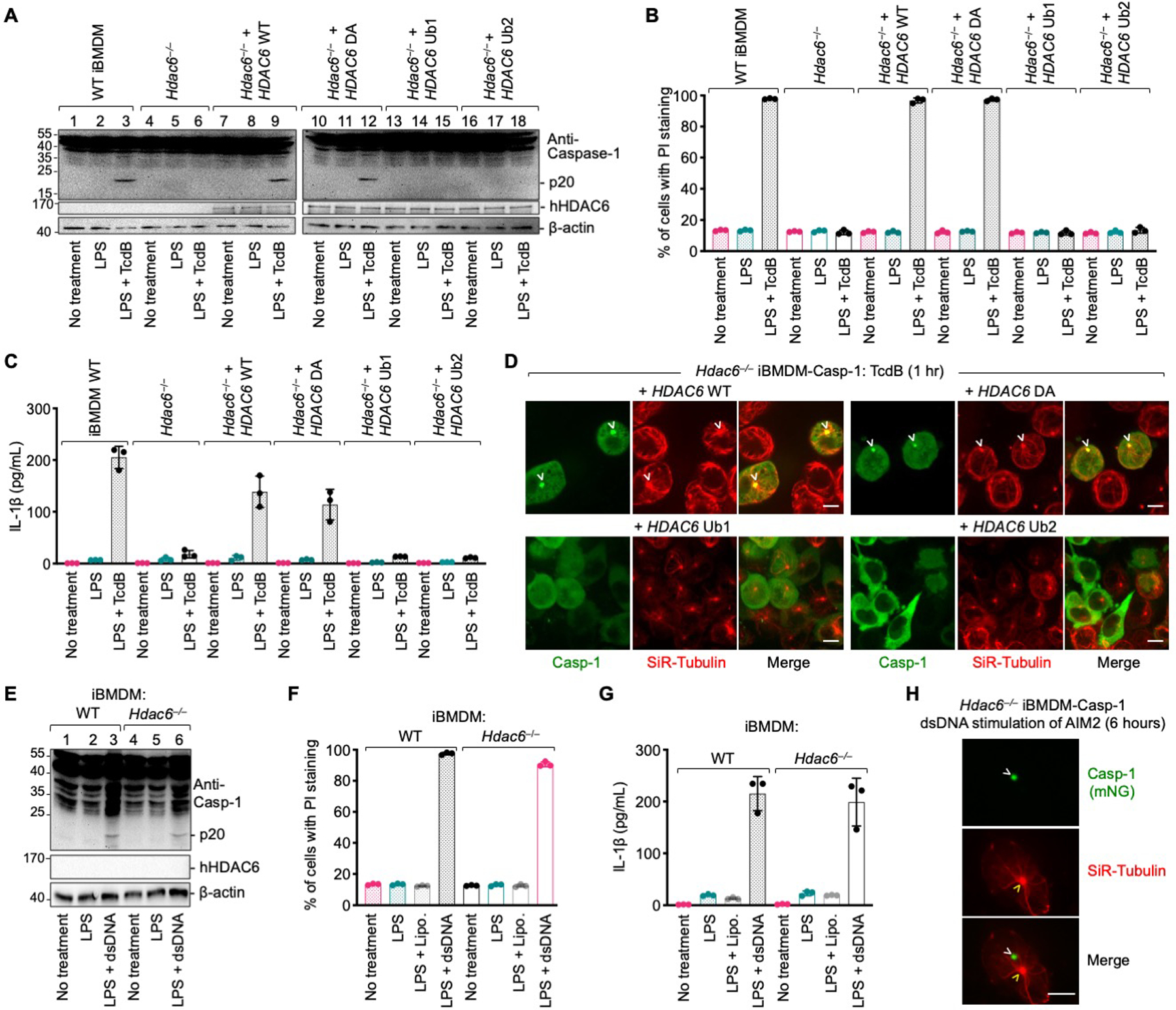
(A–D) Requirement of HDAC6 and its ubiquitin binding ability in TcdB-induced pyrin inflammasome activation, shown by caspase-1 processing (p20) (A), PI permeability (B), IL-1b secretion (C), and punctum formation (D). DA: H216A/H611A on catalytic residues, deacetylase mutant; Ub1: mutations H1160A/H1164A on zinc-coordinating residues; Ub2: mutation W1182A on the surface that binds ubiquitin. (E–H) Lack of HDAC6-dependence in dsDNA-induced AIM2 inflammasome activation, shown by caspase-1 processing (p20) (E), PI permeability (F), IL-1b secretion (G), and punctum formation (H). Arrowheads indicate puncta or MTOC. Data are presented as the mean±SD for triplicate wells from three or more independent experiments (B, C, F, G). Images are representative from three or more independent experiments (D, H). Scale bars: 5 μm.
Because the LPS-induced non-canonical inflammasome and the NLRC4 inflammasome are known to induce secondary NLRP3 activation (47–49), we generated CRISPR/Cas9 KO of Hdac6 in the Nlrp3−/− background. The non-canonical inflammasome engages mouse caspase-11 in iBMDMs but does not engage caspase-1 for IL-1β processing (50, 51). PI staining indicated equivalent membrane disruption upon LPS transfection in Nlrp3−/− and Nlrp3−/−Hdac6−/− iBMDMs (Fig. 5A). The percentage of cells with caspase-11 activity was similar in Nlrp3−/− and Nlrp3−/− Hdac6−/− iBMDMs (Fig. 5B). For the NLRC4 inflammasome, we used Legionella pneumophila flagellin fused to the N-terminal domain of Bacillus anthracis lethal factor (FlaTox) (52). Inactive FlaTox that only engages TLR5 but not NAIP5 was used as a negative control. FlaTox induced comparable caspase-1 processing and PI permeability in Nlrp3−/− and Nlrp3−/−Hdac6−/− iBMDMs (Fig. 5C–D), supporting that HDAC6 is dispensable for activation of the flagellin–NAIP5–NLRC4 inflammasome. NLRC4 inflammasome activation in Nlrp3−/− and Nlrp3−/−Hdac6−/− iBMDMs resulted in the formation of ASC puncta, but these puncta did not localize at the MTOC (Fig. 5E). Thus, HDAC6 only plays a role in inflammasomes formed at the MTOC, as shown for NLRP3 and pyrin inflammasomes, but not for AIM2 and NLRC4 inflammasomes or the non-canonical inflammasome (Fig. 5F). These conclusions were confirmed in mouse primary BMDMs (fig. S19).
Fig. 5. HDAC6 deficiency does not affect non-canonical and NLRC4 inflammasome activation in Nlrp3−/− background.
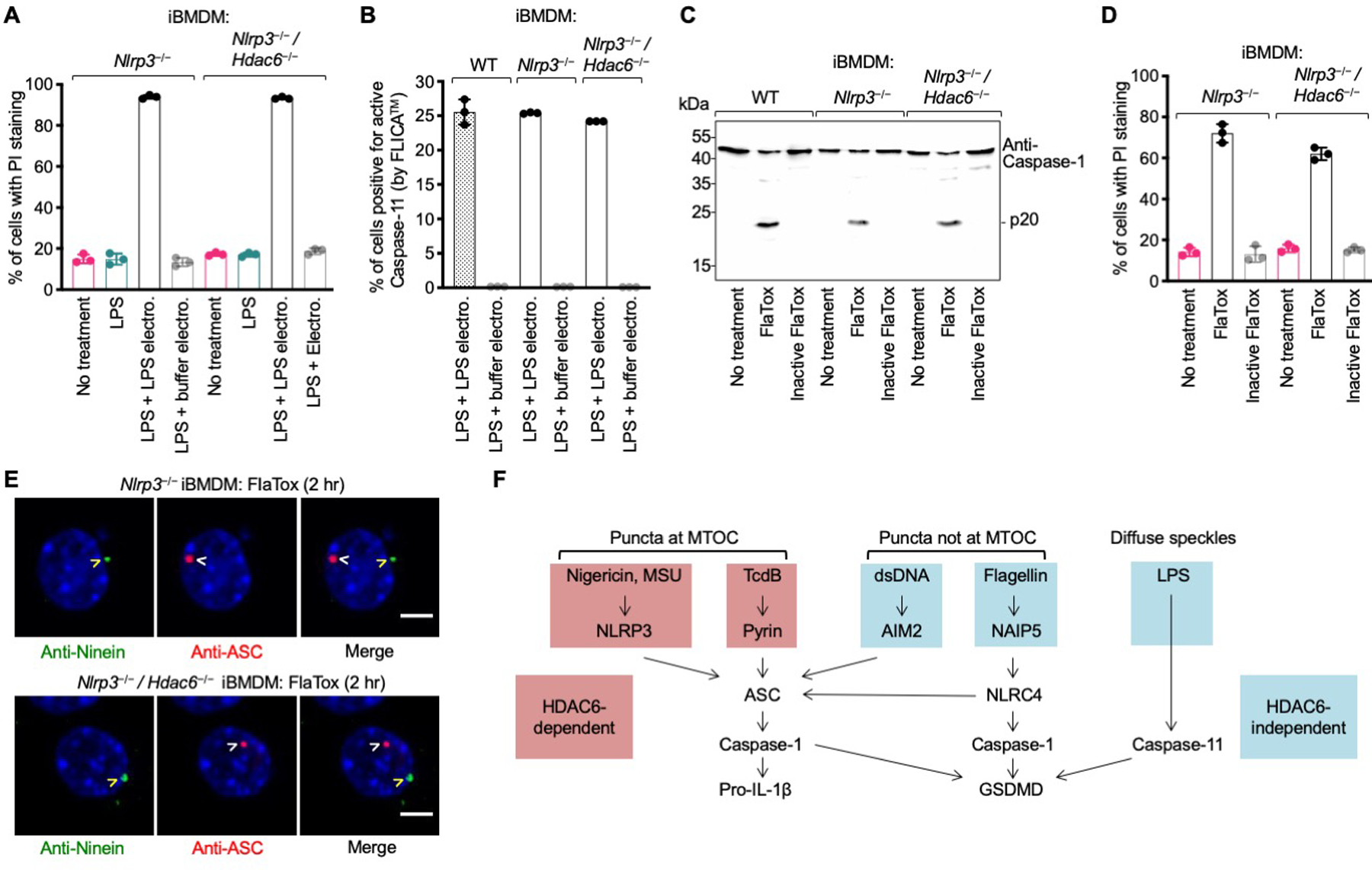
(A–B) Non-canonical inflammasome activated by intracellular delivery of LPS (electroporation) quantified by PI permeability (A) and FAM-FLICA™ substrate cleavage by active caspase-11 (B). (C–D) NLRC4 inflammasome activation triggered by active FlaTox (inactive FlaTox served as a control) analyzed for caspase-1 processing (p20) (C), and PI permeability (D). Data are presented as the mean±SD for triplicate wells from three or more independent experiments. (E) Immunofluorescence analysis of NLRC4 punctum formation in Nlrp3−/− (top) and Nlrp3−/−Hdac6−/− (bottom) iBMDMs upon treatment with FlaTox. Blue represents nuclear staining by Hoechst 33342. The NLRC4 inflammasome punctum represented by ASC staining is distinctly apart from the centrosomal marker ninein. Arrowheads depict puncta or MTOC. Images are representative of three or more independent experiments. Scale bars: 5 μm. (F) Summary of location of punctum formation and HDAC6-dependence in the different inflammasomes.
HDAC6 is required for transport of trans-Golgi network (TGN)-localized NLRP3
Diverse NLRP3 stimuli have been shown to disperse the TGN, which recruits NLRP3 into multiple small speckles before ASC engagement (53). We observed partial co-localization between the TGN marker TGN38 and multiple NLRP3 speckles in Asc−/− iBMDMs after nigericin stimulation (Fig. 6A). Surprisingly, in WT iBMDMs, TGN38 redistributed into a single punctum at the MTOC which co-localized with the NLRP3 single punctum (Fig. 6B). This suggests that NLRP3 is transported by the dynein machinery while on TGN-derived vesicles. By contrast, in Hdac6−/− iBMDMs, NLRP3 showed multiple speckles similar to TGN pattern as observed in ASC−/− iBMDMs (Fig. 6C), confirming that HDAC6 is required for the transport. NEK7 is required for NLRP3 activation (14–16), likely by bridging NLRP3 subunits in an inflammasome disk (54). However, NEK7 did not co-localize with NLRP3 at the TGN before HDAC6-mediated microtubule transport (Fig. 6D). This suggests that such transport may be required in NLRP3 activation in order to bring NLRP3 to the MTOC where NEK7 resides. Subsequently, activated NLRP3 may nucleate ASC filament formation, which in turn recruits and activates caspase-1 to execute inflammasome effector functions.
Fig. 6. HDAC6 mediates microtubule transport of trans-Golgi network (TGN)-localized NLRP3.
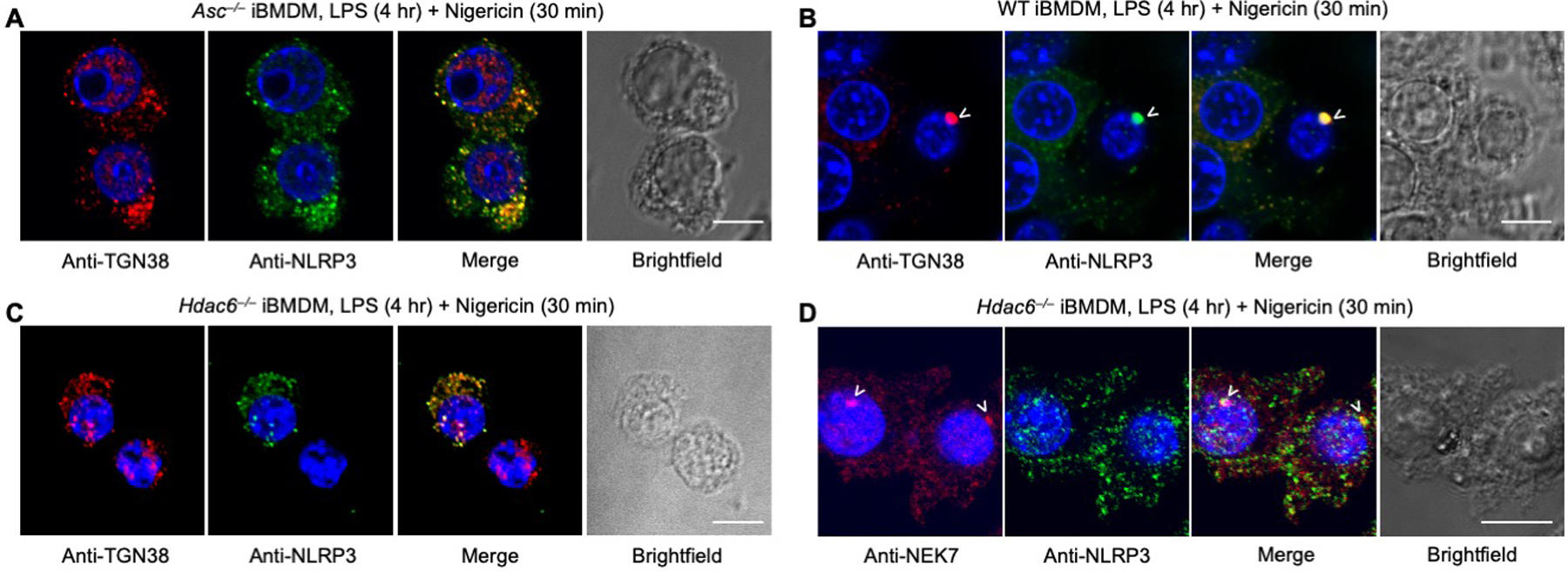
(A–C) Immunofluorescence analysis of TGN38 and NLRP3 in Asc−/− iBMDMs (A), WT iBMDMs (B) and Hdac6−/− iBMDMs (C). (D) Immunofluorescence analysis of NEK7 and NLRP3 distribution in Hdac6−/− iBMDMs. Images are representative of three or more independent experiments containing ~100 cells. Arrowheads depict puncta. Scale bars: 10 μm.
HDAC6 is required for LPS- and MSU-induced inflammation in mice
To gain insights into the role of HDAC6 in vivo, we investigated an LPS-induced endotoxic shock model in mice using Hdac6 deficiency and the HDAC6 inhibitor tubastatin A, as well as the NLRP3 inhibitor MCC950 (35) because this model is known to engage secondary NLRP3 activation (55, 56) (Fig. 7A–F, fig. S20A–H). Serum IL-1β levels were reduced in mice challenged with LPS but treated with tubastatin A or MCC950 in comparison to those challenged with LPS alone (Fig. 7B, D). IL-18 levels were also significantly reduced by tubastatin A or MCC950 treatment (fig. S20A, C, D). Harvested lung tissue showed less damage, such as septal mononuclear cell and lymphocyte infiltration, alveolar macrophage and neutrophil infiltration, and alveolar edema, in both the tubastatin A- and MCC950-treated groups, with a significantly reduced overall acute lung injury (ALI) score (Fig. 7E, F, fig. S20F). Substantially decreased LPS-induced secretion of IL-1β and IL-18 and acute lung injury were also recapitulated in Hdac6−/− mice compared to WT mice (Fig. 7C, fig. S20B, E). Collectively, these data support the requirement of HDAC6 in NLRP3 inflammasome activation in vivo.
Fig. 7. HDAC6 is required for NLRP3 inflammasome activation in mice.
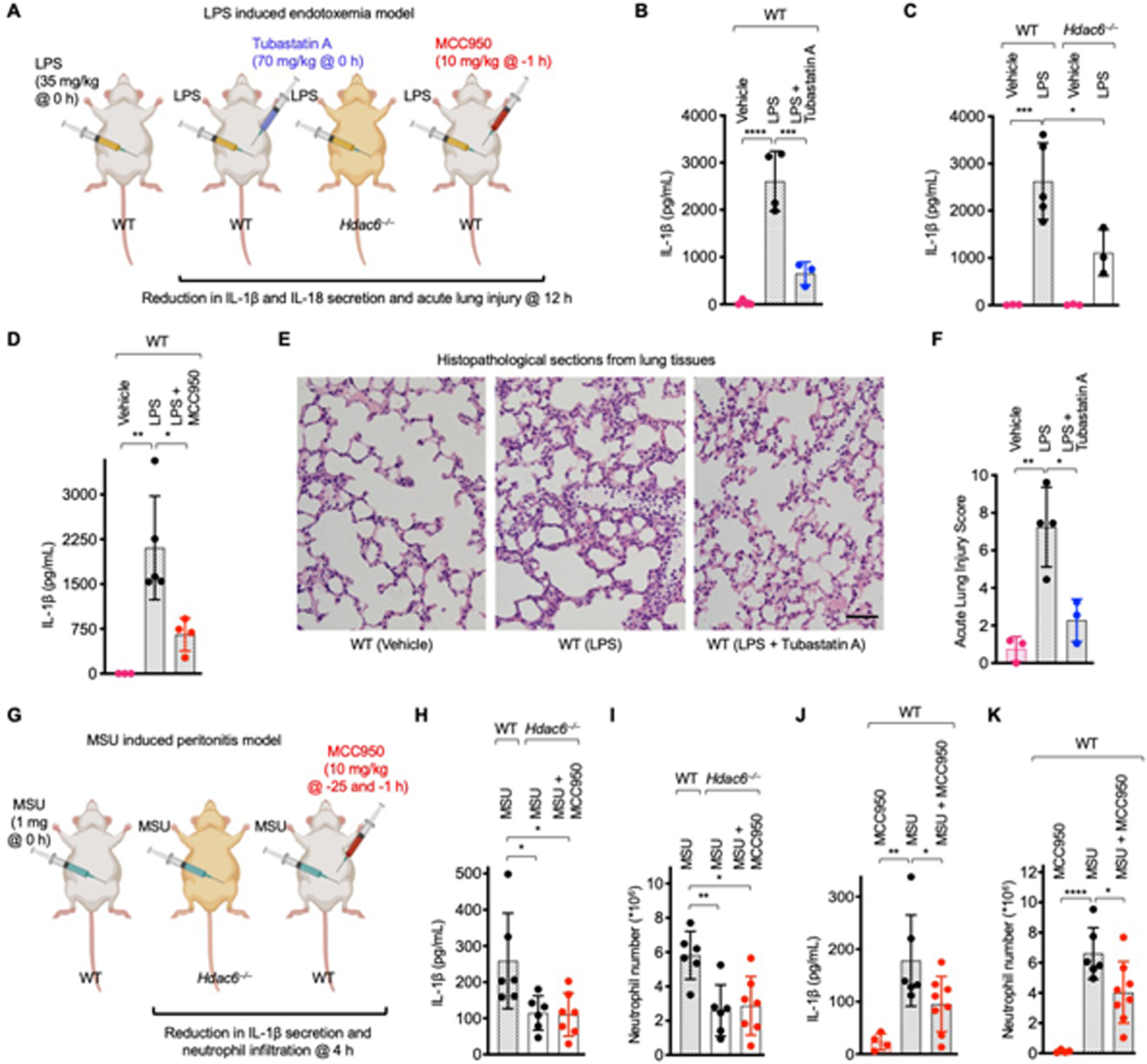
(A–F) A mouse model of lethal LPS-induced endotoxemia. (A) Experimental layout with indicated timing and dose. (B–D) Effects of tubastatin A (HDAC6 inhibitor) (B), Hdac6−/− (C), or MCC950 (NLRP3 inhibitor) (D) on IL-1β secretion. Values are mean±SD (n=3–5/group). (E) Effects of tubastatin A on acute lung injury (ALI). Representative histopathological images from harvested lung tissues are shown. Scale bar: 50 μm. (F) Quantified lung injury depicted by defined clinical parameters in ALI score. ALI scores are mean±SD (n=3–4/group). (G–K) A mouse model of MSU-induced peritonitis. (G) Experimental layout with indicted timing and dose. (H–I) Effects of Hdac6−/− or Hdac6−/− + MCC950 on peritoneal IL-1β production (H) and neutrophil recruitment (I) upon MSU challenge. Values are mean±SD (n=6–7/group). (J–K) Effects of MCC950 on peritoneal IL-1β production (J) and neutrophil recruitment (K) upon MSU challenge. Values are mean±SD (n=5–8/group). For (B–D, F, H–K), one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test was performed for data analysis as used previously (64, 65). *: P≤0.05, **: P≤0.01, ***: P≤0.001 and ****: P≤0.0001.
To test a direct NLRP3 activation model, we used the bona fide NLRP3 stimulus MSU to induce peritonitis in mice (Fig. 7G). Consistent with the role of HDAC6 in the NLRP3 inflammasome, IL-1β and IL-18 secretion and neutrophil number in peritoneal lavage were decreased in Hdac6−/− mice as compared to WT mice (Fig. 7H, I, fig. S20G). Treatment of WT mice with MCC950 exerted similar effects on IL-1β secretion and neutrophil recruitment as Hdac6 deficiency (Fig. 7J, K). In comparison with LPS, MSU only induced minimal IL-18 activation, and MCC950 did not significantly reduce the level of IL-18 (fig. S20I). Additionally, MCC950 treatment in Hdac6−/− mice did not further suppress MSU-induced IL-1β and IL-18 secretion and neutrophil recruitment (Fig. 7H, I, fig. S20J). Thus, the MSU-induced peritonitis model further demonstrated that NLRP3 inflammasome activation is dependent on HDAC6 in vivo.
Discussion
In summary, our studies informed the striking mechanistic parallel between NLRP3 and pyrin inflammasome assembly and HDAC6-mediated transport of pathological aggregates to the MTOC for aggresome formation and degradation. They provide unique insights into the ever-evolving understanding on the complex process of inflammasome activation. They especially offer a framework to integrate the previously implicated role of the microtubule in inflammasome biology, including the importance of the centrosomal protein NEK7 and the microtubule-affinity regulating kinase 4 (MARK4) (14, 15, 57). The correlation between MTOC localization and sensitivity to microtubule transport disruption among inflammasomes further demonstrates the specificity of the mechanism, and points to the importance of the physiological sites of assembly for their activation and regulation in cells (Fig. 5F). Notably, the microtubule polymerization inhibitor colchicine is an approved drug used for both gout, an NLRP3 inflammasome disease, and familial Mediterranean fever (FMF), a pyrin inflammasome disease. Furthermore, the intermediate filament vimentin, an aggresome component, was shown to regulate NLRP3 inflammasome activation (22, 55). The additional role of the MTOC in inflammasome biology extends its previously established role in directional secretion by T cells at the immune synapse in adaptive immunity (58) to innate immunity.
Our data support a model of NLRP3 and pyrin inflammasome activation that depends on regulated ubiquitination (27–30) and engagement of the dynein adaptor HDAC6 and possibly cargo adaptors such as p62. We do not yet know exactly which inflammasome components need to be ubiquitinated. Most likely NLRP3 is not the inflammasome component recognized by the HDAC6-dynein machinery as the small NLRP3 clusters formed in the absence of ASC are not transported to assemble into one large punctum (53). ASC expression, however, leads to formation of a single punctum per cell (59) and both NLRP3 and pyrin are ASC-dependent inflammasomes. In this regard, critical ubiquitination sites on ASC and IL-1β have been mapped (27, 29). Because NLRP3 is already partially aggregated on the TGN, we hypothesize that small speckles of partially assembled inflammasomes are transported on TGN vesicles.
Why do some inflammasomes form at the MTOC? It is possible that the accumulated focal concentrations promote inflammasome assembly and caspase-1 activation. In the case of NLRP3, its MTOC targeting may ensure engagement of the upstream activator NEK7, a centrosomal kinase (14–16, 54). We further resolved that IL-1β conversion also occurs at the MTOC, which although surprising, is consistent with the recent finding that caspase-1 activity is most dominant at the puncta (60). Therefore, site-specific higher-order inflammasome complexes act as supramolecular organizing centers (SMOCs) (61) to orchestrate inflammasome signaling. The flip side of MTOC localization may be the facilitation of autophagosome formation as a built-in checkpoint mechanism for inflammasome destruction to avoid unrestrained activation. The pericentriolar region is enriched in lysosomes (62), and the MTOC acts as a hub to promote fusion of autophagosomes with lysosomes to accelerate inflammasome degradation. Thus, our data suggest the dual activating and inhibiting roles played by MTOC localization to achieve balanced inflammasome regulation. Although further mechanistic details of this model remain to be elucidated, our studies clearly identified HDAC6 as a potential therapeutic target for inflammasome-centric diseases.
Materials and methods
Cloning and mutagenesis
The cDNA for human IL1B was provided as a generous gift from Dr. J. Yuan, Harvard Medical School. To construct the FRET reporters for IL-1β and caspase-1, the full-length inserts were amplified and ligated into the mTurquoise2-C1 plasmid (a gift from M. Davidson, Addgene no: 54842) between the XhoI and BamHI sites to yield mTurquoise2-IL-1β and mTurquoise2-Casp-1 with a SGLRSRG linker sequence. Next, these constructs were sub-cloned into the pLV-eGFP lentiviral expression plasmid (a gift from P. Tsoulfas, Addgene no: 36083) between the XbaI and BamHI sites to yield mTurquoise2-IL-1β-eGFP and mTurquoise2-Casp-1-eGFP fusion proteins. Finally, eGFP was replaced with mNeonGreen (mNG) with an engineered linker sequence of TGSGS between the AgeI and SalI sites using the following primers (Integrated DNA Technologies) to generate the mTurquoise2-IL-1β-mNG and mTurquoise2-Casp-1-mNG constructs: forward primer of 5’-GATGCAACCGGTAGCGGCTCAATGGTGAGCAAGGGCGAG-3’, and reverse primer of 5’-GATGCAGTCGACTTACTTGTACAGCTCGTC-3’.
The mRuby3-Dynamitin (p50) fusion construct was produced by first inserting mRuby3 into the pLV-eGFP plasmid between XbaI and BamHI sites. Then the full-length Dynamitin insert was amplified and ligated between the AgeI and SalI sites from the cDNA (a gift from I. Cheeseman, Addgene no: 37388) using the following primers (Integrated DNA Technologies): forward primer of 5’-GATGCAACCGGTGGCGGCCGCTC-3’, and reverse primer of 5’-GATGCAGTCGACTCACTTTCCCAGCTTC-3’.
The HDAC6-Flag-mRuby3 fusion construct was made by first sub-cloning mRuby3 into the pLV-eGFP plasmid between AgeI and SalI sites to yield pLV-mRuby3, followed by amplifying HDAC6-Flag from the cDNA (P. Matthias) and ligating between XbaI and AgeI sites to yield HDAC6-Flag-mRuby3. The following primers were used (Integrated DNA Technologies): forward primer of 5’-GATGCATCTAGAATGACCTCAACCGGCC-3’, and reverse primer of 5’-GATGCAACCGGTCTTGTCATCGTCGTCC-3’.
The HDAC6 deacetylase mutant (DA, H216A/H611A) was produced similarly as above except with the plasmid pcDNA-HDAC6.DC-Flag (a gift from T.-P. Yao, Addgene no: 30483). The ubiquitin-binding mutants Ub1 (H1160A/H1164A) and Ub2 (W1182A) were produced by site-directed mutagenesis using the QuikChange Lightning Multi kit (Agilent Technologies) using the following primers (Integrated DNA Technologies).
Ub1 forward primer: 5’-GTCGTTACATCAATGGCGCCATGCTCCAAGCCCATGGAAATTCTGGAC-3’;
Ub1 reverse primer: 5’-GTCCAGAATTTCCATGGGCTTGGAGCATGGCGCCATTGATGTAACGAC-3’;
Ub2 forward primer: 5’-CATCGACCTGTCAGCCCGTGTTACTACTGTCAGG-3’;
Ub2 reverse primer: 5’-CCTGACAGTAGTAACACGCGGCTGACAGTCGATG-3’.
The γ-tubulin eGFP (GTU-eGFP) construct was produced similarly as above except that it was cloned in the plasmid pLV-eGFP (a gift from P. Tsoulfas, Addgene no: 36083). Prior to cloning, the existing XbaI site was mutated to NheI site in the plasmid. The human GTU insert was amplified using mRuby-Gamma-Tubulin-17 as a template (a gift from M. Davidson, Addgene no: 55864) using the following primers (Integrated DNA Technologies): forward primer of 5’-TTTTGCTAGCTGCCACCATGCCGAGGGAAATCATCA-3’, and reverse primer of 5’-TTTTACCGGTTTCTGCTCCTGGGTGCCC-3’.
pENTR221 mASC-tagRFP-T was generated by a BP gateway recombination reaction of pDONR221 with the PCR product attB1_mASC-tagRFP-T_attB2, catalyzed by BP clonase II (Thermo Fisher) according to the manufacturer’s recommendations. Mouse ASC and tagRFP-T (63) coding sequences are connected by the linker SGGRSSGSGSTSGSG. pRRL mASC-tagRFP-T puro was generated by three fragment gateway recombination of pRRL R4-R3 Puro with pENTR L4-R1 pMT (mouse metallothionein I promoter), pENTR221 mASC-EGFP, and pENTR R2-L3 STOP MCS using LR Clonase II Plus enzyme (Thermo Fisher). Vector were kind gifts of S. Lindquist (Whitehead Institute for Biomedical Research, Cambridge, MA, USA).
Human ASC was subcloned into the pLV-mRuby3 plasmid (described previously) between the XbaI and AgeI sites using the following primers (Integrated DNA Technologies): forward primer of 5’-GATGCATCTAGAATGGGGCGCGCGCGCG-3’, and reverse primer of 5’-GATGCAACCGGTGGATCCGCTTCCGCTCCGCTCCAGGTCC-3’. This resulted in pLV-ASC-mRuby3 with a short linker GSGSTGSAS between ASC and mRuby3. The full-length human Flag-ASC cDNA was generated by inserting the full-length human ASC-flag tagged at N-terminus in the EcoRI and XhoI sites of plenti-CAG-IRES-GFP (a gift from W. Kaelin, Addgene no: 69047) using the following primers (Integrated DNA Technologies): forward primer of 5’-TGTGCTGTCTCATCATTTGGCAAAGAATTCGACTACAAAGACGATGACGACAAG-3’, and reverse primer of 5’-CCAGTAACGTTAGGGGGGGGGCGGAATTGATCCCGCTCGAGTCAGCTCCGCTCCAG GTCCTCCACC-3’.
pET15b LFn-Fla and LFn-Fla 3A fusion constructs were a gift from R. Vance (Addgene no: 84871 and 84872, respectively).
Cell culture and transfection
Immortalized bone marrow–derived macrophages (iBMDMs) from WT C57BL/6 mice was gift from Prof. Jonathan Kagan (Boston Children’s Hospital, Boston, MA, USA). Immortalized Asc−/−, Nlrp3−/−, Casp-1/11−/− deficient macrophages were a gift from Prof. K. A. Fitzgerald (University of Massachusetts Medical School, Worcester, MA, USA). Immortalized Nek7−/− macrophages were a gift from Prof. Gabriel Nuñez of University of Michigan.
HEK293T and iBMDMs were grown in Dulbecco’s Modified Eagle’s medium (DMEM), with L-glutamine (Thermo Fisher Scientific, Cat. no: 10569–004), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Cat. no: 16000–044). Human monocytic THP-1 cells were maintained in Roswell Park Memorial Institute medium (RPMI), with L-glutamine (Thermo Fisher Scientific, Cat. no: 11835–055), supplemented with 10% fetal bovine serum and 0.05 mM 2-mercaptoethanol (SigmaAldrich, Cat. no: D2650–100ML). Newly thawed HEK-Blue™ IL-1β cells were first grown in growth medium containing DMEM with L-glutamine, supplemented with heat inactivated 10% fetal bovine serum, 50 U/mL penicillin, 50 μg/mL streptomycin (Thermo Fisher Scientific, Cat. no: 15140–163), and 100 μg/mL Normocin™ (Invivogen, Cat. no: ant-nr-1). After two passages, the cells were grown in selective medium containing DMEM with L-glutamine, supplemented with heat inactivated 10% fetal bovine serum, 100 μg/mL of Zeocin (Invivogen, Cat. no: ant-zn-1), 200 μg/mL hygromycin B (Invivogen, Cat. no: ant-hg-1), 50 U/mL penicillin, 50 μg/mL streptomycin, and 100 μg/mL Normocin™. For preparation of L929-conditioned media, 5 × 105 L929 cells were grown in a 75-cm2 flask containing 50 mL DMEM media. Supernatant was collected on day 7 and was filtered through a 0.45 μm filter. The 25 mL aliquots of (L929-conditioned medium) were stored at −80 °C.
All cells were maintained at 37 °C with 5% CO2. For inflammasome activation studies, THP-1 cells were treated overnight with 300 ng/mL phorbol myristate acetate (PMA), (Sigma-Aldrich, Cat. no: P8139–5MG), followed by 4 hour priming with 1 μg/mL lipopolysaccharides (LPS) (Invivogen, Cat. no: tlrl-b5lps). iBMDMs were primed for 4 hours with LPS. The following activators were used for inflammasome studies: 200 μg/mL Monosodium Urate (MSU) Crystals (Santa Cruz Biotechnology, Cat. no: sc-202711), 20 μM nigericin (Sigma-Aldrich, Cat. no: N7143–5MG), 5 mM ATP (Sigma-Aldrich, Cat. No: FLAAS-1VL), 100 μg/mL alum (Invivogen, Cat. No: tlrl-alk), 100 μg/mL Nano-SiO2 (Invivogen, Cat. No: tlrl-sio) for NLRP3 inflammasome activation, 0.5 μg/mL TcdB, for pyrin inflammasome activation, 1 μg/mL dsDNA (Sigma-Aldrich, Cat. no: P0883–25UN) for AIM2 inflammasome activation, for the indicated time intervals, active and inactive FlaTox (2 μg/mL) in combination with protective antigen (2 μg/mL) (List Biological Laboratories, Inc, Cat. No: 171E) for NLRC4 inflammasome activation.
For live-cell and-fixed cell confocal microscopy, cells were grown in microwell dishes (MatTek corporation, Cat. no: P35G-1.5–14-C). Transfection was carried out at 60% confluency using PEI (linear PEI, MW 25,000, Polysciences, Inc., Cat. no: 23966–2), or FuGENE 6 (Promega, Cat. no: E2691), according to manufacturer’s guidelines.
Generation of stable cell lines
To generate stable cell lines, on day 0, the lentivirus was produced using HEK293T cells by co-transfecting 1 μg of pLV plasmid containing the gene, 750 ng psPAX2 packaging plasmid, and 250 ng pMD2.G envelope plasmid (both plasmids were a gift from D. Trono, Addgene no: 12260 and 12259, respectively). The transfected cells were incubated overnight. The following day (day 1), the media was removed, and replenished with 1 mL of fresh medium. The cells were incubated for another day. On day 2, the supernatant containing the virus was filtered using a 0.45 μm filter (Pall Corporation, Cat. no.: 4184), and used directly to infect iBMDMs using a spinfection protocol to increase the efficacy. Spinfection was performed at 2,500 X g for 90 min at room temperature using 8 μg/mL of polybrene (Santa Cruz Biotechnology, Cat. no: sc-134220). Post spinfection, cells were further incubated for the expression of marker genes to identify the positive clones. Positive clones were selected either by cell sorting or antibiotic selection, and colonies were expanded from single clones. Positive clones were extensively validated by PCR, western blotting and immunofluorescence microscopy.
Flow-cytometry cell sorting
Cell sorting was performed using a FACSAria II cell sorter from Becton Dickinson equipped with FacsDiva version 8.03. The instrument was setup with 100 μm nozzle 20 psi and the samples were introduced to the system at the lowest flow rate to minimize shear stress. The sorted populations were gated to exclude double, dead and auto fluorescent cells. The sort was done with a purity precision mode.
Pharmacological inhibition
We pre-treated LPS-primed iBMDMs for 1 hour with HDAC6 inhibitors 10 μM Tubastatin A (Sigma-Aldrich, Cat. no: SML0044), 30 μM Rocilinostat (Selleckchem, Cat. no: S8001), or 20 μM, or 5–40 μM Tubacin (Enzo Life Sciences, Cat. no: BML-GR362–0500), 10 μM microtubule polymerization inhibitors Colchicine (Sigma-Aldrich, Cat. no: C9754–100MG) or 10 μM Nocodazole (Sigma-Aldrich, Cat. no: M1404–2MG), or with 0.1–20 μM NLRP3 inhibitor MCC950 (CP-456773) (Cayman Chemicals Cat. no: 210826–40-7 and Sigma-Aldrich, Cat. no: PZ0280). Cytoplasmic dynein-dependent microtubule transport was inhibited using 25 μM Ciliobrevin A (TOCRIS bioscience, Cat. no. 4529). One-hour post drug pre-treatment, activation was carried out either with nigericin (30 min), dsDNA (6 hours), or TcdB (1 hour). iBMDMs were pretreated with 5 mM of 3-MA (Invivogen, Cat. no: tlrl-3ma) for 6 hours to block autophagy.
Measurement of membrane disruption by propidium iodide assisted flow cytometry
Membrane permeability was measured by propidium iodide (PI) exclusion assay. Untreated and treated cells were collected, including dead floating cells in the medium, washed two times in PBS 1X before re-suspension in 2 μg/mL PI (ImmunoChemistry TECHNOLOGIES, Cat. no: 638). The percentage of cells which took up PI was measured by flow cytometry (BD Biosciences FACSAria II).
Detection of IL-1β by reporter HEK-Blue™ IL-1β cells
Procedures were carried out according to manufacturer’s instruction (Invivogen). Briefly, HEK-Blue™ IL-1β cells grown to 60% confluence were seeded on a flat-bottom 96-well plate at ~50,000 cells per well. These cells were incubated overnight with 50 μL of activated THP-1 or iBMDM supernatant along with corresponding controls at 37 °C in 5% CO2. The supernatants from the induced HEK-Blue™ IL-1β cells were collected for the soluble embryonic alkaline phosphatase assay (SEAP) using QUANTI-Blue (Invivogen, Cat. no: rep-qb1). In this colorimetric assay, 150 μL of resuspended QUANTI-Blue was incubated with 50 μL of induced HEK-Blue™ IL-1β cell supernatant in a flat-bottom 96-well plate at 37 °C from 30 min to 3 hours. SEAP levels were read at 650 nm. The graphs were plotted using GraphPad prism 7 software.
IL-1β detection by enzyme linked immunosorbent assay (ELISA)
Detection kits for human IL-1β (BD biosciences, Cat. no: 557953) and mouse IL-1β (Affymetrix Ebioscience, Cat. no: 88–7013) were used at specified temperature according to manufacturer’s instruction. Corning Costar 9018 ELISA plates were coated overnight (4 °C) with 100 μL/well of capture antibody in 1X coating buffer. After overnight incubation, wells were aspirated and washed thrice with wash buffer. These procedures were carried out at room temperature. Wells were blocked for an hour using 1X ELISA/ELISPOT diluent. 100 μL of sample or standard was used in each well, and incubated for 2 hours. Wells were washed five times and incubated with 100 μL/well of detection antibody for an hour. Unbound detection antibody was removed by washing five times, and the wells were incubated with 100 μL/well of avidin horseradish peroxidase (HRP) for 30 min. Unbound avidin HRP was removed by washing seven times, and the wells were incubated with 100 μL/well of 1X TMB solution for 15 min. The reaction was terminated using 50 μL/well of 2N sulfuric acid, and plates were read at 450 nm.
TNFα detection by ELISA
Detection kit for mouse TNFα (Invitrogen, Cat. no: BMS607HS) was used at specified temperature according to manufacturer’s instruction. Microwell strips were washed twice with 400 μL wash buffer with 15 seconds in-between incubation. During the washing the wells were tapped on absorbent pad to remove excess buffer. 50 μL of sample or standard was used in each well, and to it 50 μL of biotin conjugate was added. Incubation was carried out for 2 hours at room temperature (RT) on a microplate shaker set at 200 rpm. Afterwards, the wells were washed 6 times as specified above. Wells were incubated with 100 μL of streptavidin-HRP for 1 hour at RT on a microplate shaker. The wells were washed six times and incubated with 100 μL of amplification diluent I for 15 min on a microplate shaker. Wells were washed again six times and incubated with 100 μL amplification diluent II for 30 min on a microplate shaker set at 200 rpm. After another six washes, the wells were incubated with 100 μL TMB solution in dark for 10–20 min. The reaction was terminated using 100 μL/well of stop solution, and plates were read at 450 nm. The statistical analysis and the graphs were plotted using GraphPad prism 8 software.
IL-18 detection by enzyme linked immunosorbent assay (ELISA)
IL-18 was detected using standard ELISA technique using both pre-coated commercial kit according to manufacturer’s guidelines (Thermofisher, Cat. No: BMS618–3) as well as by self-coated 96 well plates. For the latter, 500 ng/mL of mouse anti-IL-18 capture antibody (MBL, Cat. No: D047–3) was used to coat plate at room temperature for 2 hours in 0.1 M carbonate buffer (pH 9.6). After one-time wash, wells were blocked for 1 hour using casein buffer (0.2% in PBS). 100 μL of sample or standard (MBL, Cat. No: B002–5) was used in each well and incubated overnight at 4 ℃. Wells were washed 4 times and incubated with biotinylated detecting antibody (MBL, Cat. No: D048–6) for 3 hours at room temperature. After 4 times wash, wells were incubated with HRP for 1.5 hours at room temperature. Unbound avidin HRP was removed by washing five times, and the wells were incubated with TMBX (Surmodics, Cat. No: TMBX-1000–01) solution for 5 min. The reaction was stopped using equal volume of 0.4% NaF, and plates were read at 620 nm.
Spinning-disk confocal live-cell microscopy
Sorted cells were grown in 35mm petri dish (10mm microwell, No. 1.5 coverglass, MatTek corporation, Cat. no: P35G-1.5–14-C) and mounted in a 20/20 Technologies Bionomic microscope stage heated chamber warmed to 37 °C. DMEM medium without phenol red (Thermo Fisher Scientific, Cat. no: 21063–045) was used during image acquisition, with a layer of mineral oil on top of the medium to prevent evaporation. All images were collected with a Yokogawa spinning disk confocal on a Nikon Ti inverted microscope equipped with Plan Apo phase 3 oil-immersion 40X (1.3 numerical aperture) and oil-immersion 60X (1.4 numerical aperture) lenses. The Perfect Focus System was in place for continuous maintenance of focus. IL-1β and caspase-1 mNG fluorescence was excited with the 491 nm line (selected with a 488/10 filter, Chroma #53044) from a 100mW cobalt diode laser and collected with a quadruple band pass dichroic mirror (Chroma #89100bs) and a 525/50 emission filter (Chroma #53051). Human HDAC6-mRuby3 and p50 dynamitin-mRuby3 fluorescence was excited with 561 nm line 200 mW cobalt diode laser and collected with a quadruple band pass dichroic mirror (Chroma #89100bs) and a 620/60 emission filter. SiR-Tubulin fluorescence was excited with 642 nm line from a 101mW cobalt diode laser and collected with a quadruple band pass dichroic mirror (Chroma #89100bs) and a 700/75 emission filter (Chroma #ET700/75m). Before acquisition, cells were primed with LPS for 4 hours followed by activation using nigericin, MSU and ATP for 30 min, alum and nano-SiO2 for 6 hours, TcdB for 1 hour, or with dsDNA for 6 hours. SiR-Tubulin incubation was carried out for 4 hours (Cytoskeleton, Inc. CY-SC006, SiR-Tubulin 1 μM, verapamil 10 μM) to stain the microtubule network in live cells.
Images were acquired with a Hamamatsu ORCA ER cooled CCD camera controlled with MetaMorph 7 software. Images were collected using an exposure time of 700 ms and 2 X 2 binning, with illumination light shuttered between acquisitions. At each time point, z-series optical sections were collected with a step size of 1 μm for the indicated time intervals, using a Prior Proscan focus motor. Gamma, brightness, and contrast were adjusted on displayed images (identically for comparative image sets) using MetaMorph 7 software.
Confocal laser-scanning microscopy
For imaging fixed cells, THP-1 or iBMDM cells were seeded at a density of 2 × 105/mL. 8–12 hours post-transfection, HEK293T cells were washed once with 1X BRB80 buffer, fixed and analyzed by immunofluorescence microscopy. THP-1 and iBMDMs, post-activation, were washed once with 1X BRB buffer, fixed and labeled with antibodies as mentioned in immunolabeling and antibodies section. Confocal sections were obtained using Olympus FV-1000 laser-scanning confocal Microscope (Olympus America, Waltham, MA) with an Olympus Fluoview version 3.0 software, using 405 nm excitation /425–475 nm emission, 488 nm excitation / 500–545 nm emission, 559 nm excitation / 575–620 nm emission and 635 nm excitation / 655–755 nm emission for blue, green, red and far-red detections, respectively.
512 × 512 or 1024 × 1024 images were collected at 4 μsec/pixel using a 20X (0.75 NA, air objective), 40X (0.95 NA, air objective), 40X (1.15 NA, water immersion objective), or 60X (1.2 NA, water immersion objective) lenses. The images were identically acquired and processed using Adobe Photoshop software. Distribution of fluorescence intensities and co-localization were analyzed by Velocity 6.2.1 software.
Lattice light-sheet microscopy (LLSM)
iBMDM-IL-1β cells stably expressing mTurquoise2-pro-IL-1β-mNG were plated on 5-mm round glass coverslips, and stained with SiR-Tubulin. These coverslips were picked with forceps, and placed in the sample bath of the 3D LLSM (18). The sample was imaged in two ways. First, it was imaged in a time series in 3D using a dithered multi-Bessel lattice light-sheet by stepping the sample stage at 500 nm intervals in the s-axis equivalent to ~261 nm translation in the z-axis; thus, each 3D image took 450 ms to acquire for a total of 90 time points. Each 3D stack corresponded to a pre-deskewed volume of ~30 μm x 50 μm x 4 μm (300 × 512 × 15 pixels). The cell was excited with a 488-nm laser (~300 mW operating power with an illumination of ~400 μW at the back aperture) and a 642-nm laser (~100 mW operating power with an illumination of ~200 μW at the back aperture) to acquire 15 imaging planes, each exposed for ~14.8 ms and recorded with two Andor iXon 897 EMCCD cameras. Second, the sample was imaged at a single time point in 3D using a dithered multi-Bessel lattice light-sheet by stepping the sample stage at 250 nm intervals in the s-axis equivalent to ~130 nm translation in the z-axis; the 3D image took 10 s to acquire. The stack corresponded to a pre-deskewed volume of ~30 μm x 50 μm x 13 μm (300 × 512 × 100 pixels). The cells were excited with a 488 nm laser (~300 mW operating power with an illumination of ~400 μW at the back aperture) and a 642 nm laser (~15 mW operating power with an illumination of ~30 μW at the back aperture) to acquire 100 imaging planes, each exposed for ~49.8 ms and recorded with two Andor iXon 897 EMCCD cameras. The inner and outer numerical apertures (NAs) of excitation were 0.35 and 0.4, respectively.
Immunolabeling and antibodies
THP-1 or iBMDM cells were first primed for 4 hours with LPS, and then activated with nigericin, TcdB, or dsDNA for various times. Fixative, permeabilization, and blocking buffers were prepared in Brinkley Buffer 80 (BRB80), and kept at 37 °C prior to use. BRB80 buffer was prepared freshly using 80 mM PIPES, 1 mM MgCl2, and 1 mM EGTA, titrated to pH 6.8 with saturated solution of KOH.
Cells were fixed in 3.7% paraformaldehyde for 1–5 min at room temperature. Afterwards, cells were washed twice using BRB80 with 5-min intervals between washes. Permeabilization was carried out for 5 min at room temperature using 0.15% Triton X-100 (in 1X BRB80). Washing was carried out to remove permeabilization buffer. Cells were then blocked for 1 hour at room temperature using blocking buffer (3% gelatin from cold water fish skin prepared in 1X BRB80). Cells were incubated with primary (3 hours to overnight) and secondary (1 hour) antibodies. The nucleus was stained using Hoechst 33342 (Invitrogen, Cat. no: H3750). In between, extensive washing steps were carried out to remove unbound antibodies and stains.
To stain for active caspase-1, the carboxyfluorescein (FAM) labeled inhibitor of caspase-1 (FAM-FLICA™) kit (ImmunoChemistry TECHNOLOGIES, Cat. no: 97) was used according to the manufacturer’s instructions. Post NLRP3 activation, the supernatant from macrophages was removed, and was replaced with medium containing 1X reconstituted fluorescently labeled inhibitor of active caspase-1 (FAM-FLICA™), and incubated for 1 hour. Unbound fluorescent label was aspirated, and replaced with regular media. This washing step using regular media was carried out for an hour at 37 °C at 5% CO2. Post washing, live cells were fixed in 3.7% paraformaldehyde and stained as described above.
The following primary antibodies were used: anti-ASC (N-15), 1:1,000 (rabbit polyclonal, Santa Cruz Biotechnology Cat. no: sc-22514-R); anti-ASC (clone 2EI-7), 1:1,000 (mouse monoclonal, EMD Millipore, Cat. no: 04–147); anti-ASC (D2W8U), 1:1,000 (rabbit monoclonal, Cell Signaling Technology, Cat. no: 67824); anti-GTU, 1:500 (mouse monoclonal, Abcam Cat. no: ab11316); anti-ninein (F-5), 1:2,000 (goat polyclonal, Santa Cruz Biotechnology, Cat. no: sc-376420); anti-pro-IL-1β, 1:1,000 (rabbit polyclonal, Abcam, Cat. no: ab104279); anti-pro-IL-1β, 1:1,000 (goat polyclonal, R&D systems, Cat. no: AF-401-NA); anti-TGN38, 1:1,1000 (rabbit polyclonal, Novus Biologicals, Cat. no: NBP1–03495SS); anti-IL-1β (H-153), 1:1,000 (rabbit polyclonal, Santa Cruz Biotechnology, Cat. no: sc-7884); anti-NLRP3, 1:100 (goat polyclonal, Abcam, Cat. no: ab4207); mouse anti-caspase-1 (p20), 1:2,000 (mouse monoclonal, Adipogen, Cat. no: AG-20B-0042-C100); human anti-caspase-1 (p20), 1:2,000 (mouse monoclonal, Adipogen, Cat. no: AG-20B-0048-C100); human HDAC6 (D2E5), 1:1000 (rabbit monoclonal, Cell Signaling Technology, Cat. no: 7558S); mouse HDAC6 (D21B10), 1:1000 (rabbit monoclonal, Cell Signaling Technology, Cat. no: 7612S); mouse anti-NEK7 (B-5), 1:1000 (mouse monoclonal, Santa Cruz Biotechnology, Cat. no: sc-393539); Anti-FLAG M2 1:500 (mouse monoclonal, Sigma-Aldrich, Cat. no: A2220); Rabbit immunoglobulin fraction (Normal) (Agilent Dako, Cat. no: X0903); Goat IgG, polyclonal isotype control (Abcam, Cat no: ab37373); Mouse IgG1 isotype control (R&D, Cat no: MAB002); Anti-β-actin 1:5,000 (mouse monoclonal, Santa Cruz Biotechnology, Cat. no: 47778); Anti-acetylated α-tubulin (mouse monoclonal, Sigma-Aldrich, Cat. no: T7451); anti-pyrin (rabbit monoclonal, Abcam, Cat no: ab214772).
The secondary antibodies included: Alexa Fluor 488, 1:500 (goat anti-mouse, Thermo Fisher Scientific, Cat. no: A10680); Alexa Fluor 647, 1:2,000 (goat anti-rabbit, Thermo Fisher Scientific, Cat. no: A21244); Alexa Fluor 568, 1:1,000 (donkey anti-goat, Thermo Fisher Scientific, Cat. no: A11057); Alexa Fluor 568, 1:1,000 (goat anti-rabbit, Thermo Fisher Scientific, Cat. no: A11036); Alexa Fluor 647, 1:1,000 (goat anti-mouse, Thermo Fisher Scientific, Cat. no: A21236); anti-rabbit HRP conjugated 1:10,000 (Cell Signaling Technology, Cat. no: 7074S); and anti-mouse HRP conjugated 1:10,000 (Cell Signaling Technology, Cat. no: 7076S).
For the caspase-1 (p20) analysis the whole cell lysate was prepared in 1X SDS sample buffer as described previously (64). Lysates were resolved on 12.5% SDS–PAGE and western blotted using the mouse caspase-1 specific antibodies shown above.
Affinity purification of NEK7 rabbit polyclonal antibodies
NEK7 antibody was a kind gift from Dr. K. Rhee. The NEK7 antibody was affinity purified by incubation of the antiserum with a purified full-length NEK7 blotted on a nitrocellulose strip (SDS-PAGE separated and transferred on a nitrocellulose membrane). Elution was carried out on a shaker at 50 °C at 500 rpm (Mixer HC, USA Scientific) using 100 mM glycine, pH 2.5. The eluted antibody fraction was neutralized by 1.5 M Tris-HCl, pH 8.8.
Generation of HDAC6 CRISPR knockout in mouse iBMDM
Two independent HDAC6 CRISPR knockout cells were generated. First HDAC6−/− was generated by using two gRNAs, which targets the first coding exon of mouse HDAC6. The sequences for mouse HDAC6 gRNAs are as follows: gRNA#1, sense strand: 5’-AGTAGAAGAATCTTGGCCGG-3’, antisense strand: 5’-CCGGCCAAGATTCTTCTACT-3’; gRNA#2, sense strand: 5’-GTGGGTGATTTTTCTGGGGAA-3’, antisense strand: 5’-TTCCCCAGAAAAATCACCCAC-3’.
Second HDAC6−/− was generated by using two gRNAs, which targets the 4th and 18th coding exon of mouse HDAC6. The sequences for mouse HDAC6 gRNAs are as follows: gRNA#3, sense strand: 5’-CCTGAGACAAGAGTGCCAGT-3’, antisense strand: 5’-ACTGGCACTCTTGTCTCAGGC-3’; gRNA#4, sense strand: 5’-CAGCGCATCTTACGCATCATC-3’, antisense strand: 5’-ATGATGCGTAAGATGCGCTG-3’.
To clone the gRNA sequences into LentiCRISPR vector V2 (a gift from F. Zhang, Addgene no: 52961), the plasmid was cut and dephosphorylated with FastDigest BsmBI (Fermentas, Cat. no: FD0454), and FastAP (Fermentas, Cat. no: E0651) at 37 °C for 2 hours. Oligonucleotides for the mouse HDAC6 gRNA sequences (Integrated DNA Technologies) were annealed by heating to 95 °C for 5 min and cooling to 25 °C at 1.5 °C/min. The sequence of the oligonucleotides used for cloning into the LentiCRISPR vector V2 are the following: forward primer for gRNA#1: 5’-CACCGAGTAGAAGAATCTTGGCCGG-3’, reverse primer for gRNA#1: 5’-AAACCCGGCCAAGATTCTTCTACTC-3’ and forward primer for gRNA#2: 5’-CACCGGTGGGTGATTTTTCTGGGGAA-3’, reverse primer for gRNA#2: 5’-AAACTTCCCCAGAAAAATCACCCAC-3’, forward primer for gRNA#3: 5’-CACCGCCTGAGACAAGAGTGCCAGT-3’, reverse primer for gRNA#3: 5’-AAACACTGGCACTCTTGTCTCAGGC-3’ and forward primer for gRNA#4: 5’-CACCGCAGCGCATCTTACGCATCATC-3’, reverse primer for gRNA#4: 5’-AAACATGATGCGTAAGATGCGCTGC-3’. Using T7 ligase (Enzymatics, Cat. no: L602L), annealed oligos were ligated into gel purified vectors time at 25 °C for 5 min. Cloned transfer plasmids were amplified using an endotoxin-free mini-prep kit (Qiagen, Cat. no: 27104). LentiCRISPR vector V2 containing HDAC6 gRNA was used to generate the virus in HEK293T cells followed by infection of WT mouse iBMDM cell line. Positive infected cells were isolated by Puromycin selection (inserted into LentiCRISPR vector V2) at 10 μg/mL (Thermo Fisher Scientific Cat. no: A1113803). Colonies were expanded from single clones and were selected for the puromycin resistance. The Hdac6−/− iBMDM were analyzed by PCR on genomic DNA and was further validated at protein and functional level.
Genomic PCR
The genomic DNA was extracted using the QiaAmp DNA mini kit (Qiagen, Cat. no: 13323, Qiagen) according to the manufacturer’s instructions, and the knockout phenotype was confirmed by PCR on the first coding exon using following primers (Integrated DNA Technologies).
Forward primer: 5’-GTGGGGTCTGAAGGATTCTGA-3’.
Reverse primer: 5’- TCTTGTCTCAGGGTTCAGATC-3’.
For the second HDAC6−/− iBMDM, the following primers are used to verify the positive clones.
Forward primer: 5’- TGATTGTAGTCCAGAGTGAA-3’.
Reverse primer: 5’- AGCAGGTAGGATGAGACA-3’.
Transferrin-647 uptake assay
For uptake assays, the WT and HDAC6−/− cells were grown on 35-mm dishes, serum starved for 30 min in DMEM + 0.5% BSA containing media, and incubated with 25 μg/mL of transferrin-Alexa Fluor 647 conjugate (Invitrogen, Cat no: T23366) at 4 °C for 10 min. Unbound transferrin-647 was removed by brief washing, and the transferrin uptake was analyzed by confocal laser scanning microscopy.
siRNA knockdown of HDAC6 in iBMDM
The HDAC6 knockdown experiments was performed using negative control siRNA targeting a non-mammalian sequence (Thermo Fisher Scientific, Cat. no: AM4611) and two siRNAs targeting mouse HDAC6 (Thermo Fisher Scientific, Cat. no: 67164 and 158920 for siRNA #1 and #2 respectively). 1.0 X 106 iBMDM or cells were first counted in the Neubauer chamber, washed with PBS 1X and centrifuged at 218 X g for 10 min. The pellet was resuspended in 100 μL of Mouse Macrophage Nucleofector® Solution (Lonza, Cat. no: VPA-1009) in which 4 μg of siRNA was added (control siRNA or HDAC6 siRNA) prior to perform the nucleofection. The nucleofection was performed with the Nucleofector™ 2b Device (Lonza, Cat. no: AAB-1001) using the Y-001 program. The electroporated cells were incubated for 72 hours before being processed for further experiments.
HDAC6 knockdown in iBMDM using TRIM21 ubiquitin ligase
iBMDM cells (3.0 × 106) stably overexpressing mouse pSMPP-mCherry-TRIM21 (a gift from L. James, Addgene no: 104971) were washed with PBS 1X and centrifuged at 218 X g for 10 min. The pellet was resuspended in 100 μL of Mouse Macrophage Nucleofector® Solution in the presence of 2 μg of anti-mouse HDAC6 antibody (Cell Signaling Technology, Cat. no: 7612) or 2 μg of isotype control IgG antibody (Agilent Dako, Cat. no: X0903) using the Y-001 program. The electroporated cells were incubated for 24 hours before being processed for further experiments.
Flow cytometry assisted Caspase-11 FLICA assay
For Caspase-11 FLICA assay (ImmunoChemistry TECHNOLOGIES, Cat. no: 97), upon priming with LPS as described above, 1.0 X 106 buffer electroporated and LPS electroporated WT, NLRP3−/− or NLRP3−/−/HDAC6−/− iBMDM cells, were incubated with medium containing 1X reconstituted fluorescently labeled inhibitor of active caspase (FAM-FLICA™), and incubated for an hour. Cells were than washed three times with 1X Apoptosis Wash Buffer and centrifuged at 218 X g for 10 min before being resuspended in regular DMEM medium and analyzed with a flow cytometer.
Mice
Male and female C57BL/6J WT and HDAC6−/− mice (8–10 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME). Animals were housed in animal facility for 3 days before any procedures. All animal experiments were performed in compliance with guidelines approved by the institutional Animal Care and Use Committee in the University of Michigan (Protocol No.: PRO00008861).
In vivo mouse model of endotoxic shock and treatment
Three experiments were performed. First, WT mice were randomly divided into 3 groups to test the effect of the HDAC6 inhibitor tubastatin A: (1) WT + dimethyl sulfoxide (DMSO) vehicle (1 μl/g mouse body weight), (2) WT + lipopolysaccharides (LPS, 35 mg/kg in PBS; L2630; Sigma-Aldrich) + vehicle, and (3) WT + LPS + tubastatin A (HDAC6 inhibitor, 70 mg/kg) + vehicle. tubastatin A and vehicle were administered immediately after the intraperitoneal (i.p.) LPS challenge. Second, WT and HDAC6−/− mice were randomly divided into 4 groups to test the effect of HDAC6 deletion: (1) WT + vehicle, (2) WT + LPS + vehicle, (3) HDAC6−/− + vehicle, and (4) HDAC6−/− + LPS + vehicle. LPS and vehicle were similarly administered by the i.p. route. Third, WT mice were randomly divided into 3 groups to test the effect of the NLRP3 inhibitor MCC950: (1) WT + DMSO vehicle (1 μl/g mouse body weight), (2) WT + LPS (35 mg/kg in PBS; L2630; SigmaAldrich) + vehicle, and (3) WT + LPS + MCC950 (10 mg/kg) + vehicle. MCC950 and vehicle were administered 1 hour before the i.p. LPS challenge. For all experiments, blood was collected at 12 hours after the LPS injection. Serum was prepared by centrifugation of the blood at 500 X g at 4 °C for 20 min, and stored at −80 °C for further use. Lung tissues were harvested for acute lung injury (ALI) scoring. Circulating levels of IL-1β and TNFα were measured using the mouse IL-1β/IL-1F2 DuoSet ELISA (DY401; R&D Systems) and mouse TNFα DuoSet ELISA (DY410–05; R&D Systems).
Histological analysis of acute lung injury
The lung tissues were fixed in formalin, and transfer to 70% ethanol, prior to being embedded in paraffin and sliced into 5-μm sections. Hematoxylin–eosin staining was performed according to standard procedures. The ALI scoring was performed by a pathologist blinded to the treatment assignment of the samples. In brief, ALI was classified into six categories based on the parameters of 1) septal mononuclear cell/lymphocyte infiltration, 2) septal hemorrhage and congestion, 3) neutrophils, 4) alveolar macrophages, 5) alveolar hemorrhage, and 6) alveolar edema. The severity of each category was graded from 0 (minimal) to 3 (maximal) and the total score was calculated by adding the scores in each of these categories.
In vivo mouse model of mono-sodium urate (MSU) induced peritonitis
Peritonitis was induced by intraperitoneal injection of 1 mg of MSU crystals (tlrl-msu-25; Invivogen) in 0.2 ml sterile phosphate-buffered saline (PBS), with or without administration of MCC950. MCC950 was administered with an interval of 24 hour (i.p. 10 mg/kg). MSU crystals were challenged 1 hour after the second treatment. Control mice were administered vehicle at the same time points. After 4 hours, mice were euthanized by CO2 exposure. Peritoneal cavities were flushed with 5 ml cold PBS. The lavage fluids were centrifuged at 94 X g for 5 min. Cell pellets were collected, and neutrophil (CD45, Ly6G) (103128, 108417; BioLegend) recruitment in peritoneal cavity was analyzed by flow cytometry. Levels of IL-1β were measured using ELISA (MLB00C; R&D Systems) after the lavage fluids supernatant was concentrated using Amicon Ultra-0.5 Centrifugal Filter (UFC501096; Millipore).
Bone marrow isolation and BMDM differentiation
Bone marrow isolation and differentiation was performed in sterile and aseptic conditions in laminar flow hood as described by J. Weischenfeldt and B. Porse (65). For isolation of bone marrow mice was sacrificed by cervical dislocation. The abdomen and hind legs were sterilized with 70% ethanol. The incision on the midline of the abdomen was made to expose the hind leg. Using scissors, the muscles were removed from the bones, and the bones were cut at both ends to free the channel. The bones were flushed with PBS 5 mL syringe and a 25 gauge needle. The collected bone marrow in dish was pipetted up and down gently to bring the cells into a single-cell suspension. The suspension was passed through a 70-μm sterile cell strainer (Fischer Scientific cat no: 22363548). Cells were sedimented at 400 X g, 5 min at RT. The cells were carefully collected in a 100 × 20 mm sterile petri dishes containing 10 mL of conditioned media (DMEM +10% FBS, Pen/Strep and 10% L929). On day 3, the dishes were supplemented with 10 mL more conditioned media (total 20 mL volume), and the majority of cells were found to be adherent to the dish. The further growth was carried out till day 6, when the cells were split for the experiments carried out on day 7.
FRET analysis
For FRET channel images, the 447 laser was used in conjunction with the 535/30 emission filter. All images were collected with a Yokogawa spinning disk confocal on a Nikon Ti inverted microscope equipped with Plan Apo phase 3 oil-immersion 60X lens (1.4 numerical aperture). The Perfect Focus System was in place for continuous maintenance of focus. Image processing was mainly carried out using MATLAB 2014b. Images were imported from the original files and sorted into channels. All metadata were extracted and saved. Dark current camera noise was corrected for using images acquired each session. Shading or “flat field” correction based on empty fields from each sample was applied to correct for uneven illumination patterns. A background mask was generated by thresholding at a value 3 standard deviations above background, where the background intensity distribution is estimated by fitting the ‘left half’ of a Gaussian function (the portion below its mean) to the left shoulder of the image intensity histogram. This mask was then used to find and subtract the average background intensity on a frame-by-frame basis. For all FRET calculations the data were pre-filtered with a 3 X 3 pixel Gaussian filter. Since the tension sensor module is a single-chain construct, FRET can be calculated by dividing the FRET channel fluorescence intensity (donor excitation with acceptor emission) with the mTurquoise2 channel fluorescence intensity (donor excitation with donor emission). To minimize artifacts from division of small integers, only pixels that have a value above three times the background standard deviation of the current frame were used. FRET data were visualized using an inverted heat map. The quantification of the loss of FRET inside and outside the puncta (as control) was performed using FIJI software on the inverted heat map.
Embedding in resin (plastic sections) for transmission electron microscopy (TEM)
iBMDM cells post nigericin activation were processed for plastic embedding as follows. Cells were incubated in fixative for 1 hour at room temperature. A 2X fixative mixture was added in a 1:1 ratio to the media in the dish containing the cells. This avoids shock and allows gentle fixation. Fresh fixative was prepared using 1.25% paraformaldehyde, 2.5% glutaraldehyde, and 0.03% picric acid in 0.1 M sodium cacodylate buffer, pH 7.4. Post fixation, cells were washed three times in 0.1 M sodium cacodylate buffer. This followed by incubation with 1% osmium tetroxide/1.5% potassium ferrocyanide for 1 hour at room temperature. Cells were then washed in water (three times), and incubated in aqueous 1% uranyl acetate for 30 min. This was followed by another three rounds of washing with water.
Dehydration steps were carried out twice in grades of alcohol (70% ethanol for 15 min / 90% ethanol for 15 minutes/100% ethanol for 15 min. Samples were then placed in propyleneoxide for 1 hour and infiltration was carried out with Epon mixed 1+1 with propyleneoxide for 2–3 hours at room temperature. Samples were moved to the embedding mold filled with freshly mixed Epon and allowed to polymerize for 24–48 hours at 60 °C. Ultrathin sections (~60 nm) were cut on a Reichert Ultracut-S microtome, picked up on to copper grids, stained with lead citrate. The grids from above mentioned EM procedures were examined in a JEOL 1200EX 80 KeV TEM and images were recorded with an AMT 2k CCD camera (Harvard Medical School core facility).
Protein expression and purification
pET15b LFn-Fla and LFn-Fla 3A fusion constructs were transfected into Escherichia coli BL21 (DE3) cells and were grown in LB medium supplemented with Ampicillin. Cells were grown at 37 °C until the optical density (600 nm) reached at 0.8. Protein expression was induced with 0.2 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 18 °C overnight. Cells were then harvested and re-suspended in lysis buffer (20 mM Tris-HCl pH 7.4 and 150 mM NaCl, 10 mM imidazole, and 5 mM β-mercaptoethanol). The proteins were purified by affinity chromatography using Ni-NTA beads (Qiagen). The proteins were further purified to homogeneity by size exclusion chromatography in a buffer containing lysis buffer 20 mM Hepes pH 7.4, 150 mM NaCl, 2 mM Dithiothreitol on a Superdex-200 column (GE Healthcare Life Sciences). Protein purified to homogeneity was analyzed by SDS–PAGE.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Shah (Harvard Medical School), T. Mitchison (Harvard Medical School) and R. Vale (UCSF) for discussions, R. Mazitschek (Harvard Medical School) for discussions on HDAC inhibitors, G. Nuñez (University of Michigan) for Nek7−/− iBMDMs and for cDNA encoding human caspase-1, J. Yuan (Harvard Medical School) for cDNA encoding human IL-1β, M. Dong (Boston Children’s Hospital) for recombinant full-length TcdB toxin, P. Matthias (Friedrich Miescher Institute for Biomedical Research) for human HDAC6 and mouse Hdac6 plasmids, Y. Zhang and J. Lieberman (Boston Children’s Hospital) for anesthetizing and dissecting out the bones from mice, K. Rhee (Seoul National University) for NEK7 antibody, HCIA/HHMI summer institute, Woods Hole, and H. Leung (Optical Microscopy Core Facility of Program in Cellular and Molecular Medicine, Boston Children’s Hospital) for help with laser scanning confocal microscopy, J. Waters and T. Lambert (Nikon Imaging Center, Harvard Medical School) for help with live-cell microscopy, R. Mathieu (Flow Cytometry Research Facility, Boston Children’s Hospital) for help with cell sorting and cell analysis, and M. Ericsson (Electron Microscopy Core Facility, Harvard Medical School) for help with transmission electron microscopy.
Funding: This work was supported by the National Institutes of Health (HD087988 and AI124491 to H.W., MIRA award GM130386 to T.K., and T32 training fellowship to A.V.H.), Biogen (SRA to T.K.), and the Joint Institute of University of Michigan and Peking University Health Science Center (U068874 to Y.L.). T.K. acknowledges support from the Janelia Visitor Program and E. Betzig, E. Marino, T. Liu, G. Upadhyayula, and W. Legant for help and advice in constructing and installing the lattice light-sheet microscope. Construction of the lattice light-sheet microscope was supported by grants from Biogen and Ionis Pharmaceuticals to T.K.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in this paper are available in the main text and the supplementary materials.
REFERENCES AND NOTES
- 1.Broz P, Dixit VM, Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Rathinam VA, Vanaja SK, Fitzgerald KA, Regulation of inflammasome signaling. Nat Immunol 13, 333–332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamkanfi M, Dixit VM, Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Strowig T, Henao-Mejia J, Elinav E, Flavell R, Inflammasomes in health and disease. Nature 481, 278–286 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Back M, Hansson GK, Anti-inflammatory therapies for atherosclerosis. Nature reviews. Cardiology 12, 199–211 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Yin Q, Fu TM, Li J, Wu H, Structural Biology of Innate Immunity Ann Rev Immunology 33, 393–416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu H et al. , Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Martinon F, Burns K, Tschopp J, The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10, 417–426 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Liu X et al. , Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding J et al. , Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Hornung V et al. , AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu J, Fernandes-Alnemri T, Alnemri ES, Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J Clin Immunol 30, 693–702 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pihan GA, Centrosome dysfunction contributes to chromosome instability, chromoanagenesis, and genome reprograming in cancer. Front Oncol 3, 277 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He Y, Zeng MY, Yang D, Motro B, Nunez G, NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi H et al. , NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17, 250–258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmid-Burgk JL et al. , A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem 291, 103–109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bedner E, Smolewski P, Amstad P, Darzynkiewicz Z, Activation of caspases measured in situ by binding of fluorochrome-labeled inhibitors of caspases (FLICA): correlation with DNA fragmentation. Exp Cell Res 259, 308–313 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Aguet F et al. , Membrane dynamics of dividing cells imaged by lattice light-sheet microscopy. Mol Biol Cell 27, 3418–3435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyttinen JM et al. , Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing research reviews 18C, 16–28 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Ouyang H et al. , Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J Biol Chem 287, 2317–2327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pandey UB et al. , HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Kawaguchi Y et al. , The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Kopito RR, Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10, 524–530 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Wileman T, Aggresomes and pericentriolar sites of virus assembly: cellular defense or viral design? Annu Rev Microbiol 61, 149–167 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Banerjee I et al. , Influenza A virus uses the aggresome processing machinery for host cell entry. Science 346, 473–477 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Lu A et al. , Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duong BH et al. , A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 42, 55–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodgers MA et al. , The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med 211, 1333–1347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan K et al. , MAVS Promotes Inflammasome Activation by Targeting ASC for K63-Linked Ubiquitination via the E3 Ligase TRAF3. J Immunol 194, 4880–4890 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Shi CS et al. , Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13, 255–263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang XX, Wan RZ, Liu ZP, Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur J Med Chem 143, 1406–1418 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Florian S, Mitchison TJ, Anti-Microtubule Drugs. Methods Mol Biol 1413, 403–421 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Firestone AJ et al. , Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 484, 125–129 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Misawa T et al. , Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14, 454–460 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Coll RC et al. , A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21, 248–255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burkhardt JK, Echeverri CJ, Nilsson T, Vallee RB, Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J Cell Biol 139, 469–484 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park YH, Wood G, Kastner DL, Chae JJ, Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 17, 914–921 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Gorp H et al. , Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc Natl Acad Sci U S A 113, 14384–14389 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu PD, Lander ES, Zhang F, Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clift D et al. , A Method for the Acute and Rapid Degradation of Endogenous Proteins. Cell 171, 1692–1706 e1618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hwang I, Lee E, Jeon SA, Yu JW, Histone deacetylase 6 negatively regulates NLRP3 inflammasome activation. Biochem Biophys Res Commun 467, 973–978 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Miyake Y et al. , Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat Chem Biol 12, 748–754 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Hideshima T et al. , Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A 102, 8567–8572 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cabrero JR et al. , Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Mol Biol Cell 17, 3435–3445 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakahira K et al. , Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12, 222–230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saitoh T et al. , Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Chen KW et al. , Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol 3, eaar6676 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Ruhl S, Broz P, Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol 45, 2927–2936 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Man SM et al. , Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A 111, 7403–7408 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kayagaki N et al. , Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Shi J et al. , Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014). [DOI] [PubMed] [Google Scholar]
- 52.von Moltke J et al. , Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490, 107–111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Chen ZJ, PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharif H et al. , Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570, 338–343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.dos Santos G et al. , Vimentin regulates activation of the NLRP3 inflammasome. Nature communications 6, 6574 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang Y et al. , Inhibition of peptidylarginine deiminase alleviates LPS-induced pulmonary dysfunction and improves survival in a mouse model of lethal endotoxemia. Eur J Pharmacol 833, 432–440 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X et al. , MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nature communications 8, 15986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huse M, Le Floc’h A, Liu X, From lipid second messengers to molecular motors: microtubule-organizing center reorientation in T cells. Immunol Rev 256, 95–106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Proell M, Gerlic M, Mace PD, Reed JC, Riedl SJ, The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem J 449, 613–621 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boucher D et al. , Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med 215, 827–840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kagan JC, Magupalli VG, Wu H, Supramolecular organizing centres: location-specific higher-order signalling complexes that control innate immunity. Nat Rev Immunol 14, 821–826 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ren PH et al. , Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 11, 219–225 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shaner NC et al. , Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods 5, 545–551 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jakobs C, Bartok E, Kubarenko A, Bauernfeind F, Hornung V, Immunoblotting for active caspase-1. Methods Mol Biol 1040, 103–115 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Weischenfeldt J, Porse B, Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc 2008, pdb prot5080 (2008). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.