

Abstract
Profiling RNA expression in a cell-specific manner continues to be a grand challenge in biochemical research. Bioorthogonal nucleosides can be utilized to track RNA expression; however, these methods currently have limitations due to background and incorporation of analogs into undesired cells. Herein, we design and demonstrate that uracil phosphoribosyltransferase can be engineered to match 5-vinyluracil for cell-specific metabolic labeling of RNA with exceptional specificity and stringency.
Profiling biomolecules in a cell-specific manner is a grand challenge in biochemical research. Chemical approaches toward this problem have expanded researchers’ ability to understand protein expression and dynamics within specific cell types1–5, better characterize cell cycle dynamics with modified DNA analogs6–7, and characterize metabolic flux with chemical reporters of metabolites and glycans8–10. These approaches have matured because of efforts focused on pairing chemically modified metabolic intermediates with enzymes to control their flux and incorporation into endogenous cellular biomolecules.
Few researchers have developed chemically modified nucleoside analogs to track RNA expression, but these reagents are not cell specific.11–13 We have recently expanded the chemical methods for cell-specific metabolic labeling of RNA, by chemically diversifying nucleobases to make them become either activated by enzymes or de-“caged” such that liberated nucleobases can eventually be incorporated into cellular RNA.14–16 However, each of these approaches has their own limitations.
“Caged” nucleobases are often protected by carbonyl groups which may be susceptible to hydrolysis and may not be stable enough to work in vivo.17 An alternative, and more actively pursued approach, is to utilize chemically modified nucleobases/nucleosides as metabolic intermediates that cannot be processed in normal cells. Pairing these with an enzyme that can convert inert into mediates into a form that can be processed for eventual RNA incorporation is being currently pursued. As an example, we and others recently worked to identify modified nucleosides (2’-azidouridine) to match with the enzyme UCK2.18–20 Despite the exciting observations it is widely appreciated that sugar-functionalized nucleoside analogs have limitations of deep tissue penetrance, crossing the plasma membrane, and the lack of permeability across the blood-brain barrier due to their hydrophilic nature.21–23
The potential problems with nucleoside analogs are in many cases not shared with nucleobases, which are much smaller in molecular weight and less hydrophilic.24–25 In parallel, our laboratory has worked to expand the substrate capacity of Toxoplasma gondii uracil phosphoribosyltransferase (TgUPRT) with modified 5-uracil analogs to produce modified 5’-phosphorylated uridines (Fig. 1, A). However, mammalian cells are able to salvage uracil analogs for eventual incorporation into cellular RNA, without expression of TgUPRT.26–27 Together, these studies suggest that tailoring cell-specific metabolic labeling efforts to decrease the background associated nucleobase analogs leave critical components to be optimized, but present a unique opportunity to expand the chemical repertoire of analogs to achieve high-stringency and cell-specific RNA labeling.
Figure 1. UPRT-dependent metabolic labeling of RNA.
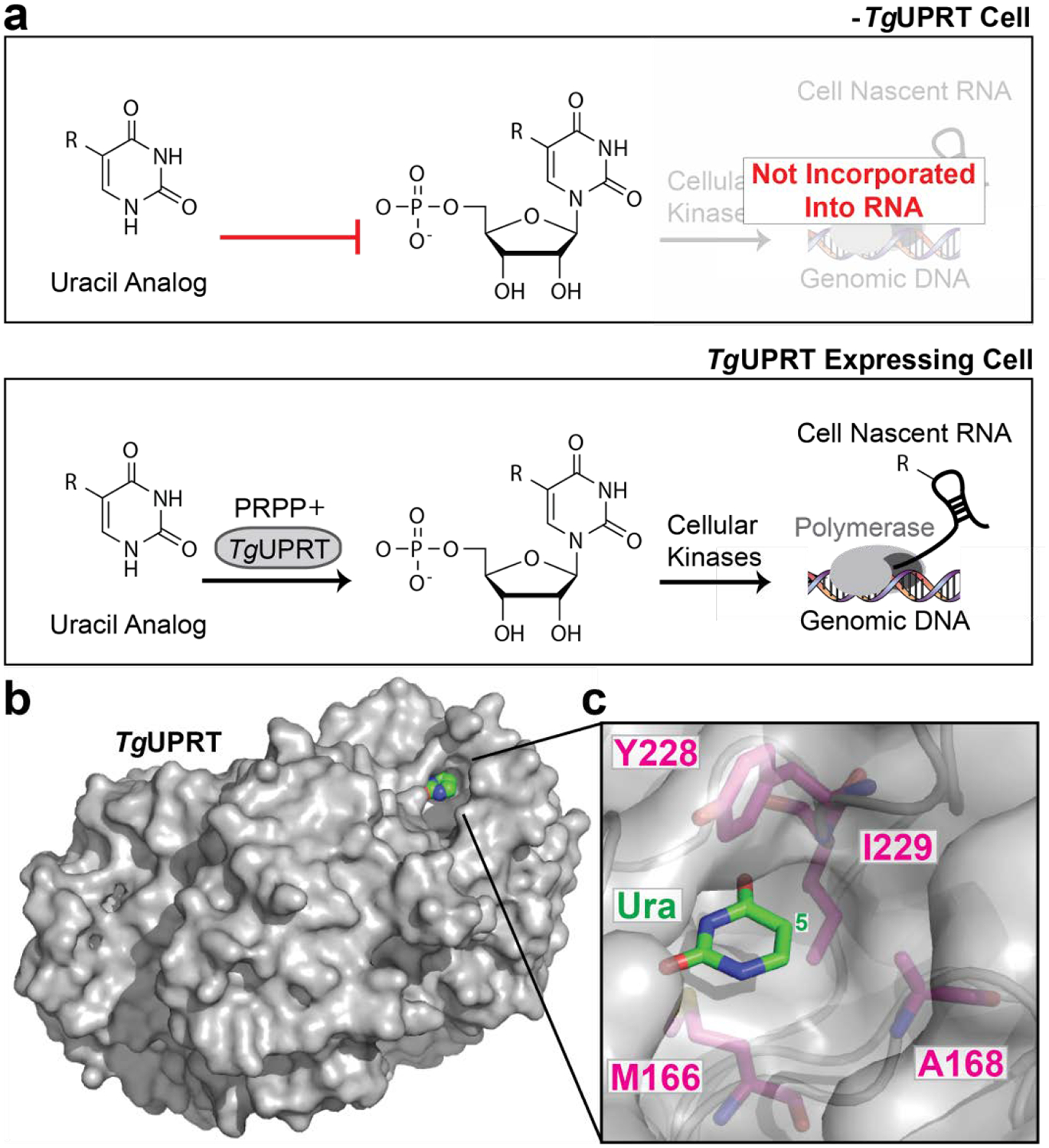
a. Schematic of (−) TgUPRT versus TgUPRT expressing cell that enable cell-specific metabolic labeling of RNA. b. Crystal structure of TgUPRT enzyme (PDB 1bd4). c. Close-up view of TgUPRT active site. Positions chosen for mutagenesis are labeled.
As mentioned, previous work in our laboratory investigated the ability of 5-modified uracil analogs to be eventually incorporated into cellular RNA in a TgUPRT-dependent manner. We observed that 5-ethynyluracil (Fig. S1) was capable of such transformations [and later demonstrated in (–)TgUPRT cells]. Consistent with our previous results, the most widely adopted uracil analog, 4-thiouracil also has a low level of background incorporation in (–)TgUPRT cells (Fig. S2).18 However, uracil analogs with bulkier functional groups, such as vinyl and methylazido were not, presumably due to steric clashes with the TgUPRT active site.27 Inspection of the TgUPRT crystal structure in complex with uracil supports this notion as the uracil nucleobase is tightly surrounded by many amino acid residues which likely clash with bulkier functional groups at the 5-position on uracil (Fig 1, B,C).
A commonly used strategy for fitting bulky substrates into the active sites of enzymes is to create a corresponding ‘hole’ to the bulky ‘bump’ of large functional groups (‘bump-and-hole’).16 This approach has been employed to pair nucleotide kinase enzymes with nucleobases, but these modified nucleosides can be toxic to cells20 and have limited tissue penetrance in vivo. This strategy has been used successfully for many classes of enzymes; therefore, we applied this method to screen for TgUPRT mutants with different bulky modified uracil analogs. Herein, we report an analysis of TgUPRT mutants (Table S1) and corresponding uracil analogs that provide a binary stringency for cell-specific metabolic RNA labeling.
We began by inspecting the active site of TgUPRT and identified several active site residues that could be amenable to mutation (Fig. 1, B, C; Fig. 2, A). Single, double and triple mutations were cloned and transiently transfected into HEK293T cells. Forty hours post-transfection, each uracil analog was added at 200 μM final concentration for 5 hours. RNA was subsequently isolated and appended with biotin using either copper-catalyzed azide-alkyne cycloaddition (CuAAC), with biotin-conjugated alkyne or azide (for azido- or alkynyl-uracil analogs, respectively). For analog 2, we utilized inverse electron-demand Diels-Alder with a biotin-conjugated tetrazine (IEDDA), a well-established reaction used with RNA, DNA, and other biomolecules for conjugation.28–30 Biotinylation was assayed using streptavidin-HRP dot blots (Fig. 2, B). We observed background incorporation of 1 at this moderate concentration, as we had previously (Fig. 2, C, D).15 Notably, at higher concentrations (1 mM) we did not observe evidence of incorporation of 2 (Fig. S3), but very robust incorporation of 1.
Figure 2. In-cell screening of TgUPRT mutants matched with bioorthogonal analogs.
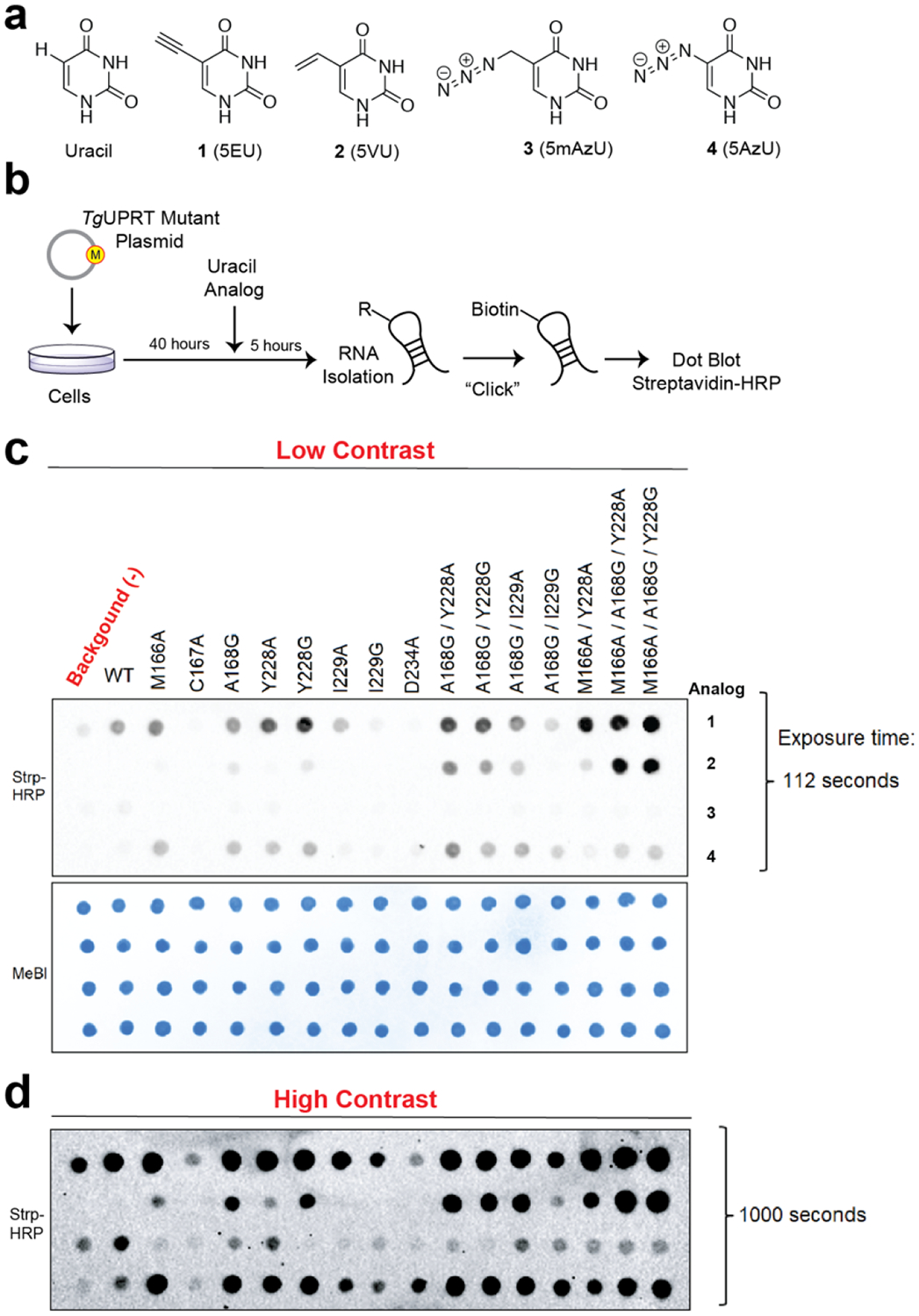
a. Chemical structures of uracil and uracil analogs used herein. b. Schematic of in-cell screening experiments. HEK293T cells were transfected with TgUPRT plasmids containing various mutations. Uracil analogs were added at 200 μM and incubated for 5 hours. Following RNA isolation, biotinylation was performed and incorporation of analog was determined by streptavidin dot blot. c. Dot blot screening for RNA incorporation of four different uracil analogs by fifteen TgUPRT mutants. d. Longer exposure of the dot blot shown in panel c. MeBl = methylene blue staining served as loading control.
From the comparison of uracil analog incorporation in RNA of cells containing the wild-type (WT) or mutant TgUPRT to (−)TgUPRT (un-transfected) cells, we identify several mutants that seemed to be compatible with most C-5 modified uracil analogs and that the triple mutants (3xMT), M166A/A168G/Y228A and M166A/A168G/Y228G enabled robust incorporation of 2 (5-vinyluracil, 5VU). Cell viability measurement also demonstrated that this pair (M166A/A168G/Y228A, and 2) exhibits no significant differences to untreated cells (Fig. S4), consistent with our recent evaluations with 5-vinyluridine analogs.28
In previous work we demonstrated that background labeling of uracil and uracil-like compounds (e.g. 1) was due to the expression of uridine monophosphate synthase (UMPS).18 However, UMPS overexpression did not result in 2 incorporation into RNA (Fig. S5). These results further suggest that 2 is not a viable substrate in endogenous enzyme pathways for the eventual incorporation into RNA.
Following these exciting observations, we aimed to obtain a more quantitative understanding of the differences between enzyme mutants and their enzyme kinetics with the analogs (Supplementary Information and Fig. S6). To do so, we expressed and purified five representative mutants [single (M166A, A168G, Y228A), double (A168G/Y228A) and triple (M166A/A168G/Y228A)] and the WT TgUPRT (Supplementary Information; Fig. 3, A; Table S2 and S3). 3 showed no detectable activity with any mutants. 4 was reactive with only three TgUPRT mutants (M166A, A168G/Y228A and M166A/A168G/Y228A) but has no activity with the WT, A168G and Y228A mutants). We also observed reactivity of uracil for all five mutants but at different level of specific activity, which suggests these enzymes are not orthogonal to host cell metabolism but are selective for the chemically modified uracil analogs described here.
Figure 3. In vitro analysis of phosphoribosyltransferase activity of TgUPRT variants with different uracil analogs.
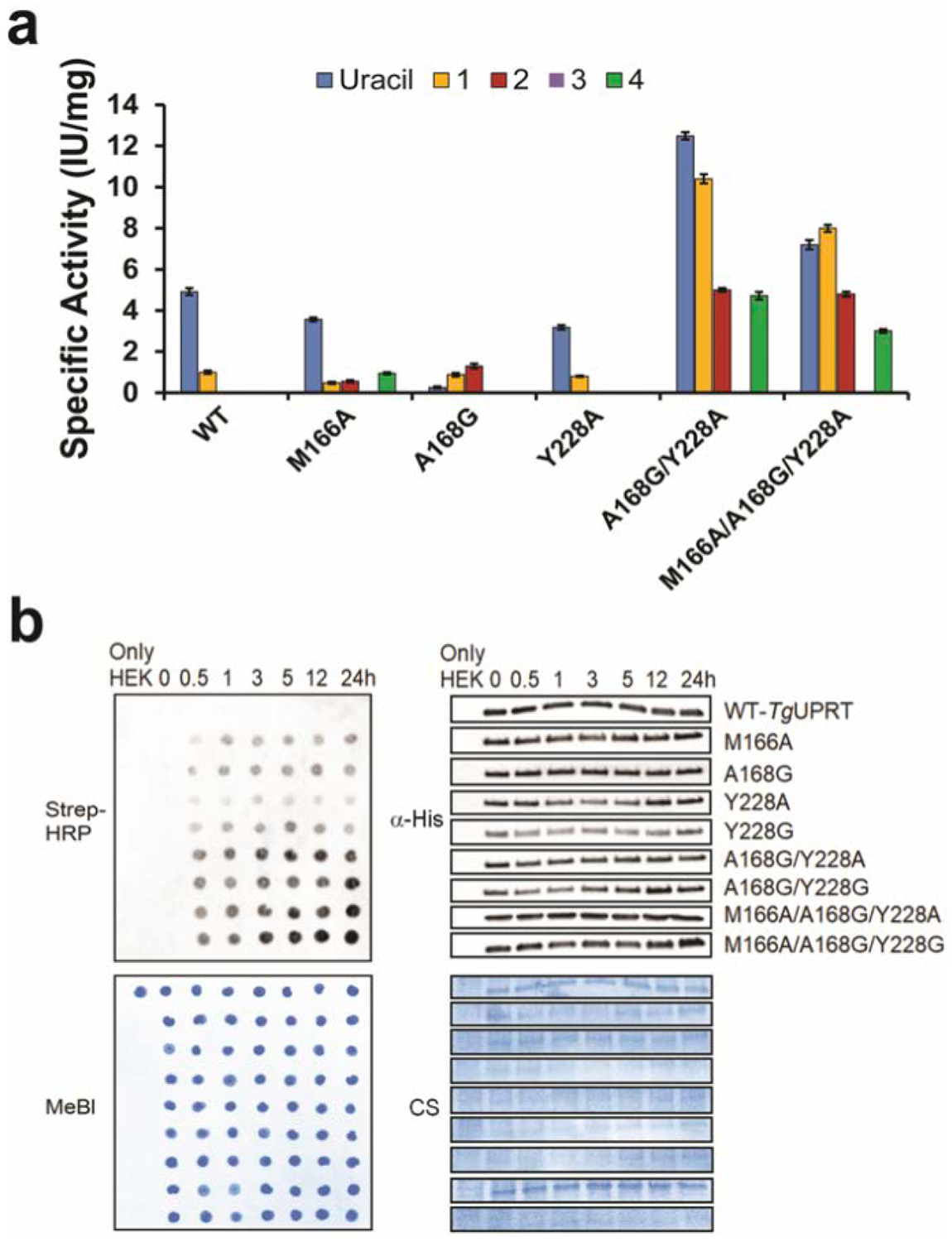
a. Specific activity of TgUPRT variants with different uracil analogs (1 to 4 as shown in Fig. 2A). n = 3 technical replicates. Error bars are standard deviation of the mean b. Time course analysis of 2 (5VU) analog incorporation into RNA by streptavidin dot blot (left panels) along with immunoblot analysis (right panels) of corresponding 6xHis-TgUPRT protein levels. α-His = anti-His antibody for immunoblot. Strep-HRP = Streptavidin conjugated Horseradish peroxidase was used for assessment of biotin levels resulting from clicked RNA. MeBl = Methylene blue stain and CS = Coomassie staining served as a loading control of RNA and proteins, respectively.
Consistent with our in-cell screening, compound 2 has undetectable activity in WT but increased dramatically with all mutants except Y228A, whereas compound 1 was reactive with WT enzymes, while also exhibiting increased reactivity with the mutants (10 IU/mg). Using dot blot analysis to evaluate the level of 5-vinyluracil incorporation into RNA, we also observed the analog incorporation in as little as 0.5 hr with TgUPRT mutants, which is faster and more robust than we previously reported (Fig. 3, B). Overall, these results further support the specificity of our designed mutants and also demonstrate the increased metabolic incorporation and efficiency of the triple mutant 3xMT-TgUPRT/2 pair.
The above experiments demonstrate the specificity of the 3xMT-TgUPRT/2 pair; however, we aimed to better understand if these observations would enable cell-specific imaging of RNA incorporation of 2 and enrichment of RNA molecules. We began with an imaging experiment where 2 or DMSO was incubated with untransfected or transfected 3xMT-TgUPRT (M166A/A168G/Y228A) HEK293T cells for 24 hours at 1 mM concentration (Supplementary Information). As shown in Fig. 4, A and Fig. S 7, we observed fluorescent signal only in cells with the presence of both 2 and the 3xMT-TgUPRT mutant. However, cells in the experiments treated with either 2 or transfected with mutant TgUPRT alone show no signal.
Figure 4. Characterizing the stringency of the 2 (5VU)-mutant TgUPRT pair.
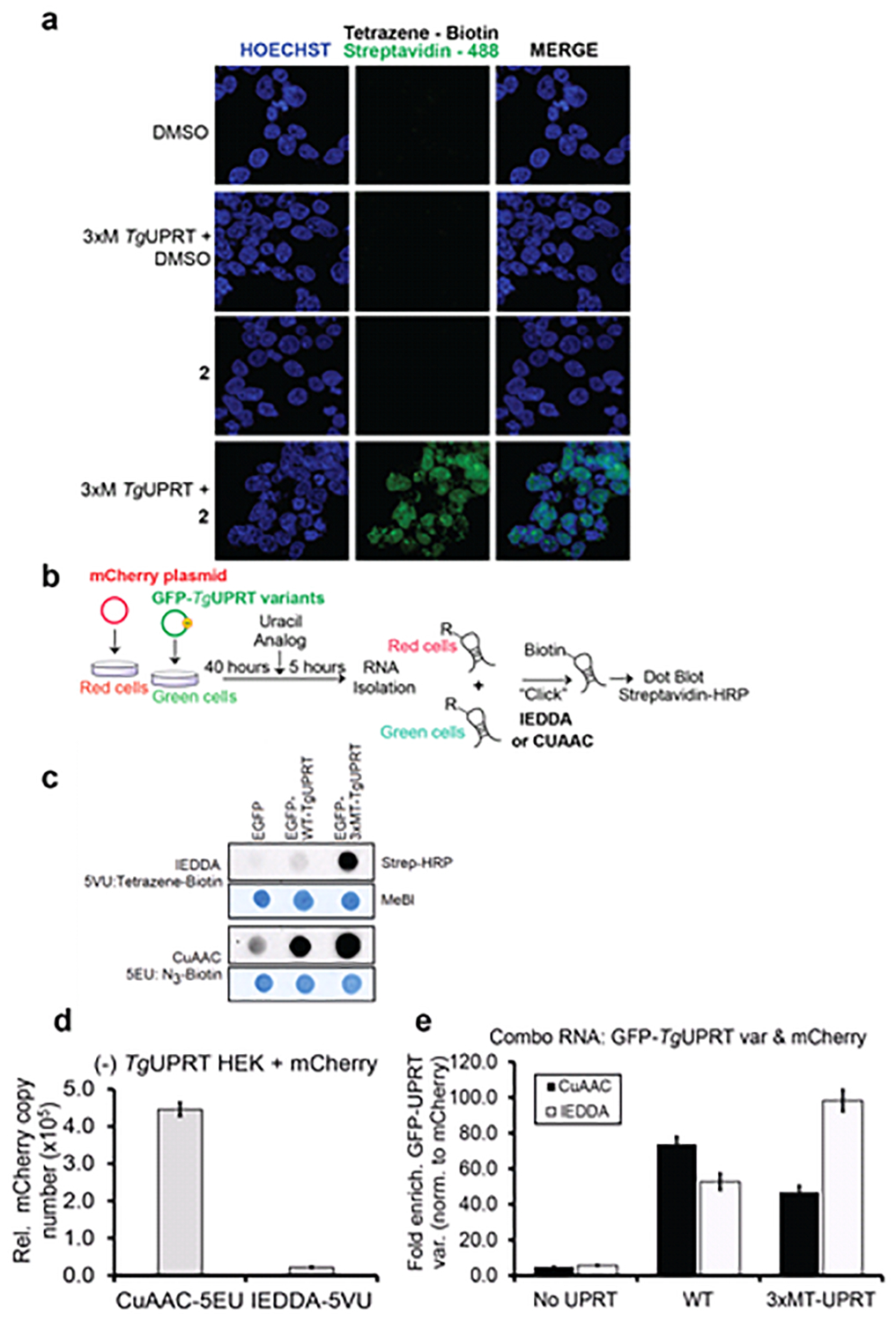
a. Microscopy analysis RNA incorporation of 2 in a mutant TgUPRT-dependent manner. Cells were transfected with TgUPRT variants and incubated with 2 at 1 mM final concentration for 24h. 2 incorporation was imaged using two-step labeling: IEDDA using Tetrazine-biotin then followed by Alexa488-streptavidin. b. Schematic of experiment to assess specificity of 2 in cells transfected with mCherry (No UPRT) or GFP, GFP-TgUPRT variants treated with both 200 μM 1 and 400 μM 2. c. Dot blot analysis of RNA isolated from 1-2 treated cells underwent either CuAAC or IEDDA click reaction. Streptavidin-HRP was used for assessment of biotin levels resulting from clicked RNA, and methylene blue (MeBl) staining served as loading control. d. QPCR analysis of enriched CuAAC- or IEDDA-biotinylated mCherry cDNA from (−)TgUPRT HEK293T cells. e. QPCR analysis of enriched CuAAC or IEDDA-biotinylated GFP cDNA from (−)TgUPRT (No UPRT), WT-, and 3xMT-TgUPRT (M166A/A168G/Y228A) transfected HEK293T cells
We next aimed to understand the stringency of RNA enrichment with the two analogs and the expression of the 3xMT-TgUPRT enzyme. 1 or 2 was incubated with un-transfected or transfected 3xMT-TgUPRT (M166A/A168G/Y228A) cells for 5 hours at 1 mM concentration. RNA was isolated from cells and CuAAC- or IEDDA-biotinylated transcripts were subsequently enriched. GAPDH mRNA copy number was quantified using RT-qPCR of enriched RNAs. As shown in Fig. S8, we were able to enrich GAPDH mRNA from untransfected cells treated with 1, but with negligible copies of GAPDH mRNA enriched in cells treated with 2 alone. GAPDH mRNA was enriched above 1X109 copies when 1 or 2 was added to cells transfected with 3xMT-TgUPRT. These results suggest that 2 and 3xMT-TgUPRT is a suitable pair for enrichment of mRNAs with negligible background due to spurious uracil analog incorporation into RNA.
To further demonstrate the specificity of RNA tagging, we performed co-culture experiments. In these experiments, cells expressing transfected mCherry (off-target cells) were co-cultured with cells expressing fusion of GFP-TgUPRT variants (target cells) or GFP without TgUPRT (negative control target cells) and treated with 200 μM of 1 and 400 μM of 2. Total RNA from the co-culture was subjected to biotinlyation via: (a) CuAAC click reaction or (b) IEDDA ligation for assessment of 1 or 2 incorporation in RNA, respectively. Dot blot analysis of the co-culture demonstrated again that 1 was incorporated into the co-culture RNA (and presumably both cells) in the absence of UPRT as well as both variants (GFP-WT-TgUPRT and GFP-3xMT-TgUPRT). In contrast, 2 was only incorporated to high levels in the presence of 3xMT-TgUPRT (Fig. 4, C).
Finally, to test enrichment specificity of 3xMT-TgUPRT and 2, we repeated the above experiment and isolated total RNA. Enrichment and profiling of mCherry mRNA enrichment from the negative control cells ((−) TgUPRT) demonstrated the copy number of enriched CuAAC-biotinylated 1-mCherry is almost 19-fold higher than the enriched IEDDA-biotinylated 2-mCherry transcripts, indicating the incorporation of 2 in RNA is much less than 1 in (−)TgUPRT cells (Fig. 4, D). We finally compared the enrichment of GFP transcripts in the combo RNA derived from green and red cells as described above, and compared the ratio of copy numbers of GFP or GFP-TgUPRT variants (signal) to mCherry (noise), GFP/mCherry (Fig. 4, E). A higher copy number of enriched mCherry (noise) would result lower GFP/mCherry ratio and vice versa. In GFP or mCherry only expressing cells (No UPRT), which is a condition for testing and establishing the background baseline of this metabolic labeling approach, CuAAC bar is slightly lower than IEDDA, indicating more 1-tagged mCherry was enriched than 2-tagged mCherry. When comparing enriched biotinylated GFP mediated by WT-TgUPRT, we observed 2 incorporation into RNA at 50-fold higher than noise. However, such ratio of GFP/mCherry does not improve over the ratio of signal-to-noise resulted from CuAAC-biotinylated 1 and the WT-TgUPRT bioorthogonal pair. Consistently, the enrichment of IEDDA-biotinylated 2-tagged GFP transcripts mediated by 3xMT-A-TgUPRT is 100-fold higher than the background and has 50% improvement over the signal-to-noise ratio of CuAAC-biotinylated 1-labeled GFP. These results clearly demonstrate the background RNA incorporation of 1, which can result in enrichment of transcripts in off-target cells. In stark contrast, our new 3xMT-TgUPRT/2 pair is highly stringent and can be used to enrich RNAs specifically from target cells.
Herein, we have demonstrated that the nucleobase analog 5-vinyluracil (2) is likely not an amenable substrate for endogenous enzymes and incorporation into cellular RNA. We also demonstrate that in contrast to 5-ethynyluracil (1), 2 is not a substrate for the main enzyme responsible for uracil analog background in cellular RNA, UMPS. Finally, through in-cell enzyme mutant screening we identified that mutant UPRT enzymes with more open active sites are able to produce 5’-phosphorylated 5-vinyluracil (2) in vitro and this enzymatic activity translates in cells to enable incorporation of 2 into cellular RNA. Imaging and enrichment RT-qPCR experiments further support the stringency of utilizing a bump-hole approach for the control of cell-specific metabolic labeling. Overall, our results put forth a novel nucleobase-enzyme pair for highly-stringent and cell-specific metabolic labeling of RNA. We anticipate these results will expand the scope of such experiments, which are appreciated to have reduced background issues due to endogenous enzymatic and metabolic activities. Future goals ongoing in the lab are to extend these findings to living animal settings. These experiments are currently being pursued in our lab and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
We thank members of the Spitale lab for their careful reading and critique of the manuscript. RNA research in the Spitale lab is supported by startup funds from the University of California, Irvine, the NIH Director’s New Innovator Award (1DP2GM119164 RCS). RCS is a Pew Biomedical Scholar. S.B. is supported by an NIH training grant 5T32CA009054-40.
Footnotes
(Supporting Information. Experimental methods, synthetic schemes and spectra, for all compounds are available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Landgraf P; Antileo ER; Schuman EM; Dieterich DC, BONCAT: metabolic labeling, click chemistry, and affinity purification of newly synthesized proteomes. Methods Mol Biol 2015, 1266, 199–215. [DOI] [PubMed] [Google Scholar]
- 2.Krogager TP; Ernst RJ; Elliott TS; Calo L; Beranek V; Ciabatti E; Spillantini MG; Tripodi M; Hastings MH; Chin JW, Labeling and identifying cell-specific proteomes in the mouse brain. Nat Biotechnol 2018, 36 (2), 156–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ernst RJ; Krogager TP; Maywood ES; Zanchi R; Beranek V; Elliott TS; Barry NP; Hastings MH; Chin JW, Genetic code expansion in the mouse brain. Nat Chem Biol 2016, 12 (10), 776–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrett RM; Liu HW; Jin H; Goodman RH; Cohen MS, Cell-specific Profiling of Nascent Proteomes Using Orthogonal Enzyme-mediated Puromycin Incorporation. ACS Chem Biol 2016, 11 (6), 1532–6. [DOI] [PubMed] [Google Scholar]
- 5.Li Z; Zhu Y; Sun Y; Qin K; Liu W; Zhou W; Chen X, Nitrilase-Activatable Noncanonical Amino Acid Precursors for Cell-Selective Metabolic Labeling of Proteomes. ACS Chem Biol 2016, 11 (12), 3273–3277. [DOI] [PubMed] [Google Scholar]
- 6.Triemer T; Messikommer A; Glasauer SMK; Alzeer J; Paulisch MH; Luedtke NW, Superresolution imaging of individual replication forks reveals unexpected prodrug resistance mechanism. Proc Natl Acad Sci U S A 2018, 115 (7), E1366–E1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neef AB; Pernot L; Schreier VN; Scapozza L; Luedtke NW, A Bioorthogonal Chemical Reporter of Viral Infection. Angew Chem Weinheim Bergstr Ger 2015, 127 (27), 8022–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hubbard SC; Boyce M; McVaugh CT; Peehl DM; Bertozzi CR, Cell surface glycoproteomic analysis of prostate cancer-derived PC-3 cells. Bioorg Med Chem Lett 2011, 21 (17), 4945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rabuka D; Forstner MB; Groves JT; Bertozzi CR, Noncovalent cell surface engineering: incorporation of bioactive synthetic glycopolymers into cellular membranes. J Am Chem Soc 2008, 130 (18), 5947–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang PV; Prescher JA; Hangauer MJ; Bertozzi CR, Imaging cell surface glycans with bioorthogonal chemical reporters. J Am Chem Soc 2007, 129 (27), 8400–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jao CY; Salic A, Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci U S A 2008, 105 (41), 15779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng Y; Beal PA, Synthesis and evaluation of an alkyne-modified ATP analog for enzymatic incorporation into RNA. Bioorg Med Chem Lett 2016, 26 (7), 1799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nainar S; Beasley S; Fazio M; Kubota M; Dai N; Correa IR Jr.; Spitale RC, Metabolic Incorporation of Azide Functionality into Cellular RNA. Chembiochem 2016, 17 (22), 2149–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hida N; Aboukilila MY; Burow DA; Paul R; Greenberg MM; Fazio M; Beasley S; Spitale RC; Cleary MD, EC-tagging allows cell type-specific RNA analysis. Nucleic Acids Res 2017, 45 (15), e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abud EM; Ramirez RN; Martinez ES; Healy LM; Nguyen CHH; Newman SA; Yeromin AV; Scarfone VM; Marsh SE; Fimbres C; Caraway CA; Fote GM; Madany AM; Agrawal A; Kayed R; Gylys KH; Cahalan MD; Cummings BJ; Antel JP; Mortazavi A; Carson MJ; Poon WW; Blurton-Jones M, iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94 (2), 278–293 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Islam K, The Bump-and-Hole Tactic: Expanding the Scope of Chemical Genetics. Cell Chem Biol 2018, 25 (10), 1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu H; Li J; Wu D; Qiu Z; Zhang Y, Chemistry and biological applications of photo-labile organic molecules. Chem Soc Rev 2010, 39 (2), 464–73. [DOI] [PubMed] [Google Scholar]
- 18.Nainar S; Cuthbert BJ; Lim NM; England WE; Ke K; Sophal K; Quechol R; Mobley DL; Goulding CW; Spitale RC, An optimized chemical-genetic method for cell-specific metabolic labeling of RNA. Nat Methods 2020, 17 (3), 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang D; Zhang Y; Kleiner RE, Cell- and Polymerase-Selective Metabolic Labeling of Cellular RNA with 2’-Azidocytidine. J Am Chem Soc 2020, 142 (34), 14417–14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y; Kleiner RE, A Metabolic Engineering Approach to Incorporate Modified Pyrimidine Nucleosides into Cellular RNA. J Am Chem Soc 2019, 141 (8), 3347–3351. [DOI] [PubMed] [Google Scholar]
- 21.Xie R; Dong L; Du Y; Zhu Y; Hua R; Zhang C; Chen X, In vivo metabolic labeling of sialoglycans in the mouse brain by using a liposome-assisted bioorthogonal reporter strategy. Proc Natl Acad Sci U S A 2016, 113 (19), 5173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinogradov SV, Polymeric nanogel formulations of nucleoside analogs. Expert Opin Drug Deliv 2007, 4 (1), 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balimane PV; Sinko PJ, Involvement of multiple transporters in the oral absorption of nucleoside analogues. Adv Drug Deliv Rev 1999, 39 (1–3), 183–209. [DOI] [PubMed] [Google Scholar]
- 24.Tomorsky J; DeBlander L; Kentros CG; Doe CQ; Niell CM, TU-Tagging: A Method for Identifying Layer-Enriched Neuronal Genes in Developing Mouse Visual Cortex. eNeuro 2017, 4 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gay L; Miller MR; Ventura PB; Devasthali V; Vue Z; Thompson HL; Temple S; Zong H; Cleary MD; Stankunas K; Doe CQ, Mouse TU tagging: a chemical/genetic intersectional method for purifying cell type-specific nascent RNA. Genes Dev 2013, 27 (1), 98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basnet H; Tian L; Ganesh K; Huang YH; Macalinao DG; Brogi E; Finley LW; Massague J, Flura-seq identifies organ-specific metabolic adaptations during early metastatic colonization. Elife 2019, 8:e43627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen K; Fazio M; Kubota M; Nainar S; Feng C; Li X; Atwood SX; Bredy TW; Spitale RC, Cell-Selective Bioorthogonal Metabolic Labeling of RNA. J Am Chem Soc 2017, 139 (6), 2148–2151. [DOI] [PubMed] [Google Scholar]
- 28.Kubota M; Nainar S; Parker SM; England W; Furche F; Spitale RC, Expanding the Scope of RNA Metabolic Labeling with Vinyl Nucleosides and Inverse Electron-Demand Diels-Alder Chemistry. ACS Chem Biol 2019, 14 (8), 1698–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rieder U; Luedtke NW, Alkene-tetrazine ligation for imaging cellular DNA. Angew Chem Int Ed Engl 2014, 53 (35), 9168–72. [DOI] [PubMed] [Google Scholar]
- 30.Knall AC; Slugovc C, Inverse electron demand Diels-Alder (iEDDA)-initiated conjugation: a (high) potential click chemistry scheme. Chem Soc Rev 2013, 42 (12), 5131–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.