

Abstract
Human HRAS, KRAS, and NRAS genes encode four isoforms of Ras, a p21 GTPase. Mutations in KRAS account for the majority of RAS-driven cancers. The KRAS has two splice variants, K-Ras4A and K-Ras4B. Due to their reversible palmitoylation, K-Ras4A and N-Ras have bimodal signaling states. K-Ras4A and K-Ras4B differ in four catalytic domain residues (G151R/D153E/K165Q/H166Y) and in their disordered C-terminal hypervariable region (HVR). In K-Ras4A, the HVR is not as strongly positively charged as in K-Ras4B (+6e vs +9e). Here, we performed all-atom molecular dynamics simulations to elucidate isoform-specific differences between the two splice variants. We observe that the catalytic domain of GDP-bound K-Ras4A has a more exposed nucleotide binding pocket than K-Ras4B, and the dynamic fluctuations in switch I and II regions also differ; both factors may influence guanine–nucleotide exchange. We further observe that like K-Kas4B, full-length K-Ras4A exhibits nucleotide-dependent HVR fluctuations; however, these fluctuations differ between the GDP-bound forms of K-Ras4A and K-Ras4B. Unlike K-Ras4B where the HVR tends to cover the effector binding region, in K-Ras4A, autoinhibited states are unstable. With lesser charge, the K-Ras4A HVR collapses on itself, making it less available for binding the catalytic domain. Since the HVRs of N- and H-Ras are weakly charged (+1e and +2e, respectively), autoinhibition may be a unique feature of K-Ras4B.
Graphical Abstract
INTRODUCTION
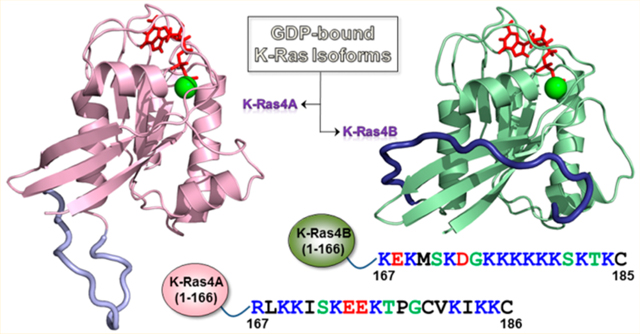
In humans, HRAS, NRAS, and KRAS proto-oncogenes encode four isoforms of the Ras protein (H-Ras, N-Ras, K-Ras4A, and K-Ras4B). Ras is a member of the family of small monomeric p21 GTPases1–3 and superfamily of guanine nucleotide-binding proteins. Ras plays a central role in cellular signal transduction pathways, acting as a critical regulator of receptor tyrosine kinase-mediated signaling to orchestrate diverse cellular activities, including proliferation, differentiation, apoptosis, and senescence.4,5 The N-terminal lobe (residues 1–86), also called the effector lobe, contains two conserved switch regions: switch I (residues 30–38) and switch II (residues 60–76), which together modulate Ras binding to effectors and regulators,6 including Raf (switch I) and phosphatidylinositol 3-kinase (PI3K) (switch I and II).7,8 The C-terminal lobe (residues 87–166), also called the allosteric lobe, contains three α-helices (α3, α4, and α5) and two β-strands (β5 and β6). The catalytic domain (also known as the G-domain, residues 1–166) of Ras proteins shares more than 90% sequence identity across isoforms and is characterized by a three-layer αβα sandwich architecture.8–10 The C-terminal hypervariable region (HVR, residues 167–188/189), the primary source of variation across Ras isoforms, is involved in Ras membrane association and localization.11–13 The functional characteristics of each Ras isoform in a cellular context,14,15 coupled with the fact that Ras–effector interactions are mediated by an effector lobe having 100% sequence identity across Ras isoforms, indicate that HVR-mediated Ras compartmentalization and membrane localization may be responsible for isoform-specific activities.16–19 In addition to the C-terminal farnesylated cysteine that is present on all Ras isoforms after post-translational processing, other upstream cysteine residues can be reversibly palmitoylated in H-Ras, N-Ras, and K-Ras4A, acting as secondary localization signals for targeting Ras to the membrane.11,18 H-Ras has two palmitoyl moieties, whereas N-Ras and K-Ras4A each have one. The HVR of K-Ras4B is strongly positively charged, whereas that of K-Ras4A (+6e) is positively charged but not as strongly as K-Ras 4B (+9e) (Figure 1). The HVRs of N-Ras and H-Ras are weakly charged (+1e and +2e, respectively). In K-Ras4B, the polybasic lysine motif is able to act as a secondary localization signal due to its favorable interactions with the negatively charged phospholipid head groups of acidic plasma membranes.20–23 We have proposed that K-Ras4A has two distinct functional states: a palmitoylated state in which it acts analogously to N-Ras, and a depalmitoylated state in which it is K-Ras4B-like, although due to the lesser charge the population time of the interaction with acidic membranes will likely be shorter, resulting in less sustained signaling.24 We suggested that the reversibility of palmitoylation effectively makes K-Ras4A as well as N-Ras exist in bimodal signaling states which may take place under different oncogenic cell/tissue conditions.
Figure 1.

Multiple sequence alignment of the amino acids in the K-Ras4A and K-Ras4B proteins. In the sequence, hydrophobic, polar/glycine, positively charged, and negatively charged residues are colored black, green, blue, and red, respectively. The nonidentity of residues in the alignment is indicated by underlined text. In the hypervariable region (HVR) sequences, a purple box denotes the palmitoylated cysteine in K-Ras4A, and an orange-box indicates the farnesylated cysteines in both K-Ras4A and K-Ras4B. The catalytic domain structures of K-Ras4A with highlighted effector lobe (left) and allosteric lobe (right) are shown. Four residues designated for the K-Ras4A catalytic domain are marked.
The Ras genes are the most frequently mutated in human cancer and together account for approximately 30% of all cancers.25,26 KRAS mutations account for nearly 86% of all Rasmediated cancers.5,27 K-Ras4A and K-Ras4B are splice variants of the KRAS gene. They arise from alternative mRNA splicing of the fourth coding exon (exon 5) of the K-Ras mRNA transcript.28,29 Whereas the sixth exon encodes the C-terminal hypervariable region of K-Ras4B, it remains untranslated in K-Ras4A. This leads to two K-Ras proteins having 188 and 189 residues, respectively, and differing in only four catalytic domain residues (151, 153, 165, and 166) and in their HVRs. Prior studies hypothesized that K-Ras4A is a minor species relative to K-Ras4B.30–33 However, a recent study utilizing splice variant-specific primers and antibodies has demonstrated that K-Ras4A is expressed in colorectal and bladder-derived tumor cell lines and in primary human colorectal adenocarcinoma tissues at levels equal to or greater than K-Ras4B, in terms of both mRNA transcript and protein expression.20 In this study, K-Ras4A was expressed in 30 different human cancer cell lines from various tissue types, accounting for 10–50% of KRAS expression.20 Coupled with the fact that both K-Ras splice variants are conserved across mammalian genomes,14 it was hypothesized that K-Ras4A and K-Ras4B have distinct physiological activities. Prior studies demonstrated that the ratio of K-Ras4A to 4B expression is commonly altered in colorectal cancers in favor of K-Ras4B, which was attributed to a potential tumor-suppressive effect of the K-Ras4A isoform in colon adenomas that favors K-Ras4B expression.34 Furthermore, both splice variants are coexpressed in cancer cells31,35 and differentially expressed in murine and human tissues.14,36 However, K-Ras4B alone is apparently insufficient, and in vivo lung oncogenesis may also be mediated by K-Ras4A.37,38 Administration of oncogenic (G12 V) K-Ras4A protein to cells in vitro induces cell transformation more efficiently than G12 V K-Ras4B.31,38 G12 V K-Ras4B activates Raf-1 2.3-fold more effectively than G12 V K-Ras4A,39 and each isoform exhibits unique activities: only oncogenic K-Ras4A induces anchorage-independent cellular growth and proliferation in vitro, and only oncogenic K-Ras4B induces cell migration and metastasis.34,38 Despite such observations, K-Ras4A signaling in cancer still remains largely unclear, and there is currently no detailed mechanistic understanding of its functional behavior.20 Recently, we proposed that the statistics of Ras isoforms in cancers are incomplete and may not accurately reflect isoform behavior.24 We pointed out that prenylated Ras isoforms may exist in two states: palmitoylated and depalmitoylated. Thus, K-Ras4A acting in K-Ras4B-driven cancers may be in a depalmitoylated state, whereas in its palmitoylated state it may act in N-Ras driven cancers. In the first state it is only farnesylated, analogous to K-Ras4B, and in the second state it is farnesylated and palmitoylated, analogous to N-Ras. We have observed that in the GDP-bound state the HVR of K-Ras4B acts to autoinhibit signaling by covering the effector binding site; however, it exposes the effector binding site in the GTP-bound state, which promotes effector binding and signaling.40,41 Here, we investigate the generality of this autoregulation mechanism which is also relevant to drug discovery.
Recently, Ras conformational ensembles, allostery, and signaling have been reviewed.42 The conformational dynamics of the K-Ras4B, H-Ras, and N-Ras isoforms have been investigated,8,43–47 but similar studies have not been done on K-Ras4A. Here, we employ all-atom MD simulations to assess to what extent residue differences modulate the conformational dynamics of K-Ras4A versus K-Ras4B in both GDP- and GTP-bound states and probe the possible impact of such dynamical behavior. Our studies on the catalytic domains and full-length structures of each isoform reveal dynamical differences that may influence activation and interactions with downstream effectors.
MATERIALS AND METHODS
Coordinates of the K-Ras4A Catalytic Domain.
Since no crystal structure of K-Ras4A is currently available, two crystal structures of the K-Ras4B isoform were obtained from the Protein Data Bank (PDB) in both the GDP-bound (PDB ID: 4EPT) and GNP-bound (PDB ID: 3GFT) states and subsequently used to model the catalytic domain of the K-Ras4A protein. Specific residue modifications were made to each structure to reflect the sequence of the K-Ras4A G-domain, as follows: G151R, D153E, K165Q, and H166Y. The structure of 4EPT contains the point mutation C118S, and the structure of 3GFT also contains a point mutation, Q61H. Both were replaced with the wild-type residues. In 3GFT, the GNP was replaced by GTP. Energy minimization using the steepest descent (SD) and adopted-basis Newton–Raphson (ABNR) methods was subsequently performed. The energy minimization, all edits, and the construction of missing hydrogen coordinates from the PDB files were performed using an input script written for the molecular simulation program CHARMM.48 The same CHARMM input script also generated the corresponding protein structure files and coordinate files for use in later steps. For comparison purposes, structures of the K-Ras4B catalytic domain were also prepared using CHARMM.
Preparing the Isolated K-Ras4A HVR Peptide for the Full-Length K-Ras4A Simulations.
The 23-residue K-Ras4A HVR peptide was constructed de novo using CHARMM48 and visualized with VMD.49 The CHARMM input script specified the amino acid sequence of the peptide, calculated its energy based on peptide bonds, dihedral angles, and torsions, constructed Cartesian coordinates for the peptide based on internal coordinates, and performed an energy minimization of the resulting peptide structure using 100 iterations of the SD method. During the MD simulations, the initially created 23-residue peptide was gradually relaxed after solvation. Using the same protocols as in our previous work,40,41,50 ensembles of conformations in an aqueous environment were obtained after a 50 ns production run, and these water-relaxed conformations were used as initial configurations in the full-length K-Ras4A simulations.
Generating Initial Configurations of Full-Length K-Ras4A Using Replica-Exchange Molecular Dynamics Simulations.
The previously generated K-Ras4A HVR peptide was covalently connected to Y166 in both the GDP- and GTP-bound catalytic domains, followed by several hundred iterations of energy minimization. 10 ns replica-exchange molecular dynamics (REMD) simulations of the full-length K-Ras4A in the GDP- and GTP-bound states were conducted. Twelve replicas of full-length K-Ras4A were simulated at specific starting temperatures, starting at a minimum temperature of 300 K and a maximum temperature of 340 K, incremented gradually for each replica. During the simulation, temperatures were exchanged between trajectories according to a Metropolis criterion. The first seven low-temperature replica trajectories were analyzed to determine ensemble configurations with favorable interaction energies between the catalytic domain and the HVR. From this process, five initial configurations were obtained for each nucleotide-bound state, for a total of 10 systems.
Atomistic Molecular Dynamics Simulations of the Catalytic Domain and Full-Length K-Ras4A.
The initial configurations of the GDP- and GTP-bound K-Ras4A1–166 (catalytic domain) and K-Ras4A1–189 (full-length) were solvated by the modified TIP3P water model. The solvated systems were subsequently minimized (2 fs/step) using the conjugate gradient method with the catalytic domain backbone held rigid for 50 000 time steps, followed by a dynamics cycle of 100 000 steps. The unit cell dimension for the systems typically was ∼119 Å3 and contained ∼167 000 atoms. The systems were subsequently neutralized, with the number of counterions needed to neutralize the GDP-bound and GTP-bound K-Ras4A systems varying in each simulation. All systems were neutralized with a number of ions, Na+ and Cl−, and contained a single coordinating Mg2+ at the nucleotide-binding site. The neutralized systems were minimized (2 fs/step) using the conjugate gradient method with the protein backbone held rigid for 50 000 time steps, followed by a dynamics cycle of 100 000 time steps. Harmonic restraints were then placed on the heavy atoms (k = 5 kcal/mol/Å2/atom) and gradually relaxed to k = 0 with a full PME calculation and a constant temperature (Nosé–Hoover) thermostat/barostat at 310 K. Following pre-equilibration, a 100 ns production run was performed with the NAMD 2.9 code51 and CHARMM version 3648 on the Biowulf cluster at the NIH. A total of 1.4 μs of simulations, including four catalytic domain and ten full-length K-Ras4A trajectories, were performed. Periodic boundary conditions with a full Ewald electrostatics calculation were used, specifying a local interaction distance of 12 Å and a pair list inclusion distance of 14 Å. In all minimization and dynamics steps, SHAKE52 was used to constrain the motion of bonds involving hydrogen atoms. Trajectories were written every 10 ps. Subsequent analysis was performed with CHARMM. Averages were taken after 30 ns, discarding initial transient trajectories.
RESULTS
Altered Nucleotide Binding Pocket Topology of the K-Ras4A Catalytic Domain.
To investigate specific structural differences between the catalytic domains of K-Ras4A and K-Ras4B, averaged catalytic domain structures of each isoform were computed for each system and superimposed (Figures 2A and 2B). The structural alignment points to dynamic fluctuations in the switch I and II regions that differ between GDP-bound K-Ras4A and K-Ras4B. However, in the GTP-bound states, K-Ras4A and K-Ras4B exhibit nearly identical conformational dynamics profiles. To further elucidate the functional differences in the nucleotide binding pocket, the averaged catalytic domain structures of each isoform were represented as protein surface diagrams (Figure 2C–F). A visual comparison of the structures suggested that the nucleotide binding pocket in K-Ras4A-GDP was more exposed to solvent relative to that of K-Ras4B-GDP, whereas in the GTP-bound state there appeared to be no significant change. To verify this observation, the average solvent accessible surface areas (SASA) of the nucleotide binding pocket for each isoform were computed (Figure S1 of the Supporting Information). The SASA for the binding pocket in K-Ras4A-GDP is indeed larger than K-Ras4B-GDP (101.4 Å2 vs 78.6 Å2, respectively), whereas the pocket is essentially identical for each isoform in the GTP-bound state.
Figure 2.

Superposed average structures of K-Ras isoforms in the (A) GDP-bound and (B) GTP-bound states are shown. The structures have been oriented with respect to the Ras effector lobe (residues 1–86). Protein surface diagrams of K-Ras illustrating nucleotide-specific binding pocket exposure in the catalytic domain of (C) K-Ras4A-GDP, (D) K-Ras4B-GDP, (E) K-Ras4A-GTP, and (F) K-Ras4B-GTP are also shown. The phosphate-binding loop residues (10–17) are colored in cyan, the switch I residues (25–40) are colored in tan, and the switch II residues (57–75) are colored lavender. The bound nucleotide is shown without hydrogen atoms to facilitate visualization.
To evaluate the dynamic changes in the nucleotide-binding pocket for the GDP-bound state, distances for three selected Cα atom pairs were computed as a function of time (Figure S2). The atom-pair distances from the Cα atom of P-loop residue G12 to the Cα atom of switch I residue P34, and from the Cα atom of P-loop residue G13 to the Cα atom of switch I residue E31, reflect the distance between the P-loop and switch I. The distance from the Cα atom of P-loop residue G12 to the Cα atom of switch II residue G60 reflects the distance between the P-loop and switch II. For K-Ras4B-GDP, the Cα atom-pair distances between G12 of the P-loop and P34 of switch I and between G12 of the P-loop and G60 of switch II were significantly decreased relative to K-Ras4A-GDP, corroborating the observation that the K-Ras4A-GDP binding pocket was more exposed (Figure 2). To investigate the impact of this difference on the conformational changes of switch I and II of the K-Ras isoforms, the probability distributions for two atom-pair distances were computed: the atom-pair distance d1 is defined by the distance from the Cα atom of switch II residue G60 to the Pβ atom of GDP/GTP, and d2 is defined by the distance from the Cα atom of switch I residue T35 to the Pβ atom of GDP/GTP. The probability distributions were converted into a three-dimensional surface plot and projected onto a two-dimensional contour plot, representing the potential of mean force (PMF) (Figure 3). The PMF can be calculated from the occupancy probability of visiting each grid point on the plane of the two atom-pair distances using the equation , where kB is the Boltzmann constant, T is the simulation temperature, and ρ(d1,d2) is the occupancy probability of the reaction coordinates d1 and d2. For K-Ras4A-GDP, an energy-minimum basin is located at d1 ∼ 10.2 Å and d2 ∼ 9.0 Å, while it is located at d1 ∼ 9.2 Å and d2 ∼ 8.8 Å for K-Ras4B-GDP. Larger distances from the nucleotide suggest that K-Ras4A-GDP has more opened conformations of the switch I and II loops, exposing the nucleotide. An additional subenergy minimum basin located at d1 ∼ 9.1 Å and d2 ∼ 11.8 Å suggests that K-Ras4A-GDP exhibits significant fluctuations in the switch I loop. However, no significant fluctuations of the switch I and II loops were observed in the GTP-bound state of both K-Ras4A and K-Ras4B.
Figure 3.

Three-dimensional potential of mean force, ΔGPMF, and the projection onto a two-dimensional subspace representing the relative free energy profile based on the calculation of the probability distributions for two atom pair distances, d1 (defined by the distance from G60 Cα atom to GDP/GTP Pβ atom) and d2 (defined by the distance from T35 Cα atom to GDP/GTP Pβ atom) for (A) K-Ras4A-GDP, (B) K-Ras4B-GDP, (C) K-Ras4A-GTP, and (D) K-Ras4B-GTP.
The differences in the nucleotide binding pocket solvent accessibility were probed through three-dimensional water density maps generated for the K-Ras isoforms (Figure S3). In the GDP-bound state, K-Ras4A has greater water occupancies near the nucleotide than K-Ras4B, further supporting the observation that the K-Ras4A binding pocket is more exposed relative to that of K-Ras4B. Taken together, these findings imply that K-Ras4A may have a slightly greater intrinsic rate of GDP dissociation relative to K-Ras4B, which may point to a greater signaling contribution from this isoform. To determine the source of the differences in water residence times between the K-Ras isoforms, further analysis was conducted to determine the contribution of crystallographic water molecules, present in the original crystal structures used for simulation, toward the observed results (Figure 4). To elucidate the contribution of these water molecules, water density maps for the two nucleotide-bound states of each isoform were overlaid on their original crystal structures, and locations at which crystal water molecule positions coincided with regions of high water density were marked with blue spheres. In the GDP-bound state, the observed differences in K-Ras4A relative to K-Ras4B are due to high-residency water molecules (>70% residence time) present during the simulations that were also present in the original crystal structure (Figure 4A,B, with the region of differences circled in red). Although the same crystallographic water molecules were originally present in both structures, their positions were found to coincide with certain regions of high water density in simulations of K-Ras4A-GDP, which were not observed in simulations of K-Ras4B-GDP. Thus, the observation of longer residence times of crystallographic water molecules near the nucleotide in K-Ras4A-GDP, relative to those same water molecules in K-Ras4B-GDP, supports the hypothesis that K-Ras4A-GDP may have a more exposed nucleotide-binding pocket than K-Ras4B-GDP. In the GTP-bound state, the observed differences in water residence times between K-Ras4A and K-Ras4B also appear to be due to crystallographic water molecules that were originally present in the crystal structure and in the simulation of both K-Ras4A and K-Ras4B (Figures 4C and 4D, with region of differences circled in red). The fact that these water molecules were found to coincide with regions of high water density in GTP-bound K-Ras4B, but not K-Ras4A, suggests that K-Ras4B may have a more exposed nucleotide-binding pocket than K-Ras4A in the GTP-bound state, although this is not reflected in the computed SASA or average structures obtained in previous analyses. Further analyses were conducted to determine whether the presence of these high-residency water molecules was influenced by additional interactions with surface residues of the binding pocket. We found that in the GDP-bound state of K-Ras4A, two water molecules having greater than 70% residence time form interactions with the backbone carbonyl of Y32 (Figure S4). However, these interactions are absent in GDP-bound K-Ras4B, suggesting that the orientation of Y32 in GDP-bound K-Ras4A may be a contributing factor to the increased residence time of hydration water in this isoform. Considered in conjunction with the greater binding pocket exposure of GDP-bound K-Ras4A, this result may also provide an explanation for the differences in residence times of hydration waters between the two isoforms. Taken together, this analysis suggests that the differences in nucleotide binding pocket exposure between the two isoforms in each nucleotide bound state can be explained by differences in the observed residence times of crystallographic water molecules in each of the structures, which may be mediated by additional surface residue interactions.
Figure 4.

Three-dimensional water density maps have been overlaid on the original crystal structures of GDP-bound K-Ras4B (PDB ID: 4EPT) and GTP-bound K-Ras4B (PDB ID: 3GFT). GDP-bound (A) K-Ras4A and (B) K-Ras4B structures as well as GTP-bound (C) K-Ras4A and (D) K-Ras4B structures are shown. Blue spheres denote positions at which the water densities, as obtained from simulations, overlap with the position of crystallographic water molecules. Differences in the water density figures between K-Ras4A and K-Ras4B are denoted with red dotted circles.
Altered Nucleotide-Specific Conformations between K-Ras Isoforms.
Approximate amide NMR chemical shift perturbations (CSPs) of K-Ras isoforms were computed using the SHIFTX2 server53 to examine whether the dynamic differences reflect changes in the local environment of residues (Figure 5). We observed large CSPs between the isoforms in the GDP-bound state, mainly in switch I and switch II residues, as well as some residues in helix 3 (100–105), which are absent in the GTP-bound state. An examination of the dynamic cross-correlation map (DCCM) of both isoforms also indicates differences in anticorrelated residues in the switch I and switch II regions between the isoforms when GDP-bound, but a lack of such observed differences between isoforms when GTP-bound (Figure S5). Taken together, these findings suggest that additional conformational states are being populated in K-Ras4A-GDP relative to K-Ras4B-GDP due to dynamic fluctuations in the switch regions. These additional observed GDP-bound conformational states may suggest differential efficiency of activation of K-Ras4A or activation by alternative Ras guanine–nucleotide exchange factors (GEFs). The numerous GEFs that can bind to Ras and Ras superfamily proteins have considerable structural variation due to diverse domain composition, but all contain a conserved cell division cycle 25 (CDC25) homology domain that functions to catalyze Ras nucleotide exchange and a Ras exchange motif (REM) involved in stabilizing Ras binding.54–56 The interaction between Ras and the ubiquitous GEF son of sevenless homologue 1 (Sos1) is known to occur via a protein–protein interface involving close interactions of switch I, switch II, and residues of helix 3 (residues 95–105), with Ras residues Y64, M67, and Y71 forming a critical hydrophobic anchor on the surface of Sos.57,58 An altered GEF involvement hypothesis might be justified in part by experimental data which demonstrate GEF specificity to the different Ras isoforms: Ras guanyl-releasing protein 2 (RASGRP2) catalyzes nucleotide exchange in N-Ras and K-Ras4B, but not H-Ras, and Ras-specific guanine nucleotide-releasing factor (RASGRF) catalyzes nucleotide exchange in H-Ras, but not N-Ras or K-Ras4B.59,60 Alternatively, these experimental observations may reflect different expression patterns, i.e. cell/tissue specificity, or different preferred membrane types/compositions for different Ras isoforms, since otherwise it is challenging to understand these variabilities among the highly related Ras isoforms with respect to specific GEFs. Sos1 is able to catalyze nucleotide exchange on N-Ras, H-Ras, and K-Ras, catalyzing nucleotide exchange most frequently in H-Ras, followed by N-Ras and then K-Ras.14
Figure 5.

Approximate amide chemical shift perturbations of the time-averaged structures of K-Ras4A over K-Ras4B in the (A) GDP-bound and (B) GTP-bound states, determined computationally using the SHIFTX2 server.
Allosteric Signaling Effects from K-Ras4A Residues Cause Fluctuations in Switch I and II.
On the basis of the observation of differences in the dynamic fluctuations of K-Ras4A in the GDP-bound state relative to K-Ras4B, we hypothesized that the residue differences between the catalytic domains of the two isoforms in the C-terminal region may affect the conformation of the switch regions allosterically. To test this hypothesis and identify the signal propagation pathway through the protein, a dynamical network analysis was conducted using the NetworkView plugin61,62 in VMD. The structure of the protein is represented as a network graph with nodes and edges. Each residue of the protein corresponds to a node, and edges are drawn between nodes which are in contact with one another (i.e., one residue is located within 4.5 Å of another for at least 75% of the MD simulation trajectory). The length of each edge is determined from a computation of the pairwise dynamic cross-correlation between the two nodes obtained from a MD simulation trajectory, such that an edge distance between two nodes i and j is given by , where Cij is the dynamic cross-correlation value. When identifying a signaling pathway between two residues in a protein, the reported length of the pathway is given as the sum of all the edge distances along the path connecting the two nodes. The dynamical network analysis reports the shortest signaling pathway and the number of other pathways between the two residues that are possible (suboptimal pathways). In this analysis, all suboptimal pathways within a distance of d = 20 of the shortest path were computed for signaling networks between residues in the switch I and switch II regions of K-Ras (residues 25–40 and 57–75, respectively) and the four catalytic domain residues that differ between K-Ras4A and K-Ras4B (151, 153, 165, and 166). The results of the dynamical signaling network analysis are presented in Table 1. The combination of a shorter signaling pathway, signifying greater residue cross-correlations, and a greater number of suboptimal paths suggests a stronger allosteric signal between the two residues in K-Ras4A relative to the K-Ras4B isoform. From this analysis it can be observed that both Q165 and Y166 in K-Ras4A have strong allosteric signaling effects on the switch I and switch II regions of Ras (with the effects of Q165 seen primarily in switch I), which are not observed in the corresponding K165 and H166 residues of K-Ras4B. Figure 6 shows an example of the difference in the signaling networks, illustrating the signaling pathways between Q165 and D33 in GDP-bound K-Ras4A and K-Ras4B. The figure represents the weights of each edge in the allosteric path between the two residues through line thickness, from which it can be observed that the signaling pathway in K-Ras4A, which propagates through helix α1, is stronger than the corresponding path that propagates through β2 in K-Ras4B. Taken together, these results suggest that the differences in the dynamic fluctuations of switch I and switch II in K-Ras4A-GDP relative to K-Ras4B-GDP may be caused by allosteric signaling effects from residues Q165 and Y166, either individually or in combination.
Table 1.
Allosteric Network Analysis between Switch I and II and C-Terminal Residues in K-Ras4A and K-Ras4B (151, 153, 165, and 166)a
| 151 |
153 |
165 |
166 |
|||||
|---|---|---|---|---|---|---|---|---|
| residue | K-Ras4A-GDP | K-Ras4B-GDP | K-Ras4A-GDP | K-Ras4B-GDP | K-Ras4A-GDP | K-Ras4B-GDP | K-Ras4A-GDP | K-Ras4B-GDP |
| 25 | 237(1) | 218(3) | 252(1) | 167(2) | 479(44) | 406(36) | 501(80) | 425(39) |
| 26 | 236(1) | 231(6) | 251(1) | 180(4) | 478(44) | 419(29) | 500(80) | 438(28) |
| 27 | 255(2) | 250(9) | 270(2) | 199(6) | 497(51) | 438(34) | 519(90) | 457(31) |
| 28 | 267(2) | 276(11) | 282(2) | 225(7) | 509(51) | 464(37) | 531(90) | 483(33) |
| 29 | 280(4) | 311(14) | 295(4) | 260(8) | 522(80) | 499(40) | 544(143) | 518(35) |
| 30 | 288(6) | 366(14) | 303(6) | 315(8) | 530(104) | 554(40) | 552(182) | 573(35) |
| 31 | 296(16) | 396(18) | 311(16) | 345(9) | 538(167) | 584(43) | 560(280) | 603(37) |
| 32 | 298(17) | 409(18) | 313(17) | 358(9) | 540(178) | 597(48) | 562(296) | 616(39) |
| 33 | 323(27) | 448(18) | 338(27) | 397(9) | 565(249) | 630(35) | 587(411) | 625(3) |
| 34 | 466(27) | 479(22) | 481(27) | 428(10) | 597(89) | 606(6) | 619(166) | 624(20) |
| 35 | 500(27) | 522(40) | 515(27) | 471(19) | 587(89) | 563(3) | 609(166) | 581(12) |
| 36 | 505(4) | 517(25) | 520(4) | 466(12) | 529(89) | 561(17) | 551(166) | 556(3) |
| 37 | 484(2) | 465(2) | 499(2) | 414(2) | 508(60) | 491(16) | 530(110) | 486(3) |
| 38 | 449(2) | 437(2) | 464(2) | 386(2) | 473(60) | 463(16) | 495(110) | 458(3) |
| 39 | 424(1) | 410(1) | 439(1) | 359(1) | 448(36) | 440(10) | 470(64) | 435(2) |
| 40 | 363(1) | 355(1) | 378(1) | 304(1) | 387(19) | 413(44) | 409(35) | 428(24) |
| 57 | 317(4) | 339(2) | 332(4) | 325(6) | 403(30) | 420(9) | 407(5) | 419(3) |
| 58 | 331(1) | 378(4) | 346(1) | 366(15) | 392(11) | 379(3) | 413(22) | 397(8) |
| 59 | 410(1) | 444(4) | 425(1) | 432(15) | 471(11) | 445(3) | 492(22) | 463(8) |
| 60 | 504(1) | 485(4) | 519(1) | 473(15) | 565(11) | 486(3) | 586(22) | 504(8) |
| 61 | 532(1) | 508(4) | 547(1) | 496(15) | 582(14) | 509(17) | 591(22) | 525(142) |
| 62 | 562(1) | 527(4) | 577(1) | 515(15) | 552(12) | 504(143) | 561(22) | 506(94) |
| 63 | 593(1) | 534(12) | 608(1) | 522(23) | 553(16) | 481(64) | 562(24) | 483(41) |
| 64 | 628(1) | 530(10) | 643(1) | 518(18) | 553(30) | 477(59) | 562(31) | 479(37) |
| 65 | 647(30) | 517(6) | 607(33) | 505(16) | 494(16) | 464(45) | 503(17) | 466(30) |
| 66 | 587(24) | 521(13) | 547(27) | 509(25) | 434(11) | 465(86) | 443(14) | 467(65) |
| 67 | 578(18) | 516(6) | 538(19) | 504(16) | 425(9) | 455(27) | 434(13) | 457(20) |
| 68 | 582(37) | 499(4) | 542(41) | 487(15) | 429(12) | 446(18) | 438(22) | 448(14) |
| 69 | 573(22) | 514(4) | 533(24) | 502(15) | 420(10) | 447(36) | 429(14) | 449(30) |
| 70 | 553(12) | 534(13) | 513(13) | 522(41) | 400(5) | 433(14) | 409(11) | 435(12) |
| 71 | 542(21) | 531(14) | 502(23) | 510(31) | 389(6) | 414(6) | 398(15) | 416(6) |
| 72 | 538(43) | 539(37) | 498(46) | 509(36) | 385(10) | 413(12) | 394(20) | 415(12) |
| 73 | 510(21) | 516(19) | 470(23) | 482(19) | 357(6) | 386(6) | 366(15) | 388(6) |
| 74 | 480(11) | 484(11) | 440(12) | 450(11) | 327(4) | 354(4) | 336(10) | 356(4) |
| 75 | 436(10) | 455(8) | 396(11) | 421(8) | 283(2) | 325(2) | 292(5) | 327(2) |
The length of the shortest path between the residues is noted, followed by the number of suboptimal paths identified between the two residues in parentheses. Highlighted in bold are those residues for which the shortest path length is shorter in K-Ras4A than in K-Ras4B and in which the number of suboptimal paths is greater in K-Ras4A than in K-Ras4B. Residues in the switch II region are denoted in bold.
Figure 6.

Differences in the allosteric signaling network between residues 165 (α5 helix) and 33 (switch I region) in catalytic domain structures of (A) GDP-bound K-Ras4A and (B) GDP-bound K-Ras4B. All suboptimal signaling pathways are drawn in red, and the optimal path is drawn in blue. The weight of the connection between each node (residue) in the pathways, a metric obtained from dynamic cross-correlation values, is represented by the thickness of each line.
Dynamic Fluctuations of the Switch I and II Loops Induce Distinct Conformational States of K-Ras4A.
To quantitatively characterize the major molecular motions and local fluctuations in the conformations sampled by the catalytic domain of K-Ras4A, a principal component analysis (PCA) was carried out for each GDP- and GTP-bound system (Figure S6) using the Bio3D package implemented in R.63,64 In the PCA studies, a hierarchical clustering of conformers was implemented based on a difference matrix computed from the first three principal components (PCs). The results indicate that for both GDP- and GTP-bound K-Ras4A three distinct conformational substates were sampled, with each substate being differentially populated. In the GDP-bound state of K-Ras4A (Figure 7A), state I is the predominant GDP-bound conformer, accounting for 65.31% of the structures sampled. State II, a minor conformer, differs from this highly populated state by adopting a different switch I conformation, whereas state III, accounting for 22.85% of the conformers, adopts a different switch II conformation. In state II the altered switch I conformation results in a displacement of F28, which normally stabilizes the guanine base, by nearly 7.5 Å relative to its orientation in states I and III (Figure S7A). The displacement of F28 determined through PCA may be mediated by backbone fluctuations and not by extensive rotameric fluctuations of the F28 side chain (Figure S7B). This conformational change may result in a loss of the stabilizing interaction in this substate, which could facilitate enhanced nucleotide dissociation. The distinct cluster positions on the subspace defined by PC1 and PC2 suggest that state I might be an intermediate conformation in the transition between states II and III. Given that all three populations are sampled throughout the course of the simulation, it is reasonable that the energetic barrier between the states is low enough that such a transition could occur. In the GTP-bound state of K-Ras4A (Figure 7B), state I accounts for 56.31% of the conformers. State II accounts for 31.01%, with a different switch II conformation, whereas state III, a minor conformer, deviates in the conformation of switch I and switch II. The orientations of residues T35 and F28 are unaffected by these conformational changes in all three conformers, suggesting that the different substates of K-Ras4A-GTP may not have different effector binding affinities. Taken together, these results highlight the flexibility of the switch I and switch II loops in K-Ras4A and indicate that the dynamic fluctuations in these loops result in distinct conformational states that may affect K-Ras activity.
Figure 7.

Three distinct conformers of (A) GDP-bound and (B) GTP-bound K-Ras4A are shown, as obtained from hierarchical clustering based on principal component analysis. For ease of comparison, the conformers can be denoted as state 1 (lavender), state 2 (green), and state 3 (gold). Percentages of the conformers in different states are marked.
HVR Fluctuations of Full-Length K-Ras4A Differ from K-Ras4B.
To model the full-length K-Ras4A conformation with the HVR covalently connected to the catalytic domain, all-atom MD simulations on full-length K-Ras4A (residues 1–186) were conducted. Preliminary replica-exchange molecular dynamics (REMD) simulations were performed to sample various initial configurations for both GDP- and GTP-bound states. Five different initial configurations of the full-length protein from the REMD simulations were extracted and used to perform five independent simulations for each nucleotide-bound state. To validate our selections of the starting conformations, PCA was carried out for each conformational ensemble derived from their respective initial structures in the REMD simulations (Figure S8). The PCA plots demonstrate that the conformational ensembles among the different replicas overlap sufficiently to allow for selection of the starting structures from these ensembles. In the GDP-bound state, the HVR appears to form intrachain contacts, yielding collapsed chains (Figure 8A). The P178 residue in the middle of the HVR may induce a kink in the HVR backbone chain. However, in the GTP-bound state, the HVR exhibits extended conformations with transient α-helices (Figure 8B). For K-Ras4B, the HVR in the GDP-bound state covers the effector binding region, thus autoinhibiting K-Ras4B signaling.40 In contrast, the HVR of GTP-bound K-Ras4B is extended away from this site, exposing it to effector binding. As Figure 1 shows, the HVR sequences differ between K-Ras4A and K-Ras4B. The higher charge in K-Ras4B prevents the HVR from collapsing on itself, a state that we observe only when Ser181 is phosphorylated.50 This is not the case for the HVR of K-Ras4A, as shown in Figure 8A. The fluctuating collapsed HVR of K-Ras4A may still hinder the effector’s approach to the binding site, but unlike the tight high affinity interaction observed in K-Ras4B,40 the interaction in K-Ras4A is unstable. In GTP-bound K-Ras4A, the collapsed HVR is still observed in conformers 1 and 4 and partly in conformer 5; however, conformers 2 and 3 resemble the K-Ras4B behavior, with residual secondary structure. Thus, the autoinhibition mechanism by the HVR which operates in K-Ras4B does not operate well in the collapsed HVR of K-Ras4A. To further characterize the effect of the conformation of the HVR on the accessibility of the nucleotide-binding pocket, the average radius of gyration of the HVR was computed as a function of the average solvent-accessible surface area for each configuration (Figure S9). This analysis revealed that in the GDP-bound state, a more collapsed chain seems to correspond to a binding pocket that is less solvent accessible. Conversely, in the GTP-bound state, a smaller radius of gyration appears to result in a more exposed binding pocket, and a larger radius of gyration results in a less accessible binding pocket. Thus, whereas the HVR does not significantly appear to impact the solvent-accessible surface area of GDP-bound K-Ras4A, it appears to render the GTP-bound state more solvent accessible.
Figure 8.

Ensemble configurations of full-length (A) GDP-bound and (B) GTP-bound K-Ras4A structures, based on sampling every 10 ns of their 100 ns molecular dynamics trajectories, show representative molecular motions of the catalytic domain and HVR over the course of the simulations.
To observe the effects of the HVR fluctuations on the catalytic domain residues, the residue root-mean-square fluctuations (RMSFs) of full-length K-Ras4A versus the catalytic domain only were computed. The catalytic domain conformations were very stable with RMSD values of ∼2.0 Å for the truncated and full-length K-Ras4A proteins. In the full-length K-Ras4A, the HVR exhibited large fluctuations, causing overall RMSD values to vary between 6 and 8 Å. To obtain the K-Ras4A catalytic domain structure from the K-Ras4B crystal structure, we substituted four amino acids at the positions 151, 153, 165, and 166 into the K-Ras4A sequence. In the catalytic domain of K-Ras4A-GDP, these substitutions had no significant effect on the stability of the protein: the backbone RMSF values of residues 151, 153, 165, and 166 differed by less than 0.04 Å relative to the same residues in K-Ras4B. In the catalytic domain of K-Ras4A-GTP, the backbone RMSF values of those residues differed by ∼0.01, ∼0.06, ∼0.3, and ∼1.6 Å, respectively, relative to the same residues in K-Ras4B. An examination of the catalytic domain residues in the full-length simulations compared to the residues in the simulation conducted on the catalytic domain only suggests that the presence of the HVR in the GDP-bound state of K-Ras4A further increases the flexibility of residues in switch I (residues 25–40) and switch II (residues 57–75), by nearly 2 Å for the majority of the residues in these regions (Figure 9A,B). The presence of the HVR affects the conformational fluctuations of the switch I and switch II regions even though there are no significant interactions between the HVR with the catalytic domain. A similar result is observed in the GTP-bound state, with the structural variability of the switch II region in the catalytic domain simulations further increased by nearly 4 Å in the full-length simulations of GTP-bound K-Ras4A (Figure 9C,D).
Figure 9.

Residual root-mean-square fluctuation (RMSF) profiles of catalytic domain residues from independent simulations of full-length K-Ras4A are presented relative to the RMSF profile obtained from the simulation of only the catalytic domain of K-Ras4A, in the cases of GDP-bound (A) K-Ras4A1–166 and (B) K-Ras4A1–186 and GTP-bound (C) K-Ras4A1–166 and (D) K-Ras4A1–186.
DISCUSSION
Despite the fact that the sequence of the original transforming gene in the murine sarcoma virus identified by Kirsten and colleagues differs from human K-Ras4A in only seven residues,65 the focus of scientific research on K-Ras has largely been on the K-Ras4B isoform due to numerous reports suggesting K-Ras4A as a minor splice variant. However, the fact that K-Ras4A is evolutionarily conserved across vertebrate species,14 coupled with the observation that it is differentially expressed in human tissues,29,31 suggests that this isoform may have a distinct functional role or that it may have been the first Ras isoform to evolve, a hypothesis that may explain why it is able to perform a broader range of activities, albeit not with the same efficiency as other isoforms.24 Our studies aimed to investigate the effects of C-terminal residue differences in the K-Ras4A isoform on its conformational dynamics relative to K-Ras4B to obtain insight into K-Ras4A functional activities.
Our results from simulations of the GDP-bound K-Ras4A catalytic domain suggest conformational changes in the nucleotide-binding pocket topology relative to GDP-bound K-Ras4B that render this pocket more exposed to solvent. The differential solvent exposure of the nucleotide-binding pocket may affect intrinsic GDP dissociation between the two isoforms by facilitating nucleotide release. Studies on Ras indicate that there is not necessarily a clear correlation between intrinsic or catalyzed guanine nucleotide dissociation and solvent exposure of the nucleotide binding pocket, and that it depends critically on residue interactions proximal to the nucleotide binding site that stabilize the guanine nucleotide and its coordinating Mg2+ ion.66,67 Results from the principal component analysis conducted on GDP-bound K-Ras4A did reveal a substate in which the stabilizing interaction between F28 and the guanine base may be disrupted, but the population of that state was minor.
Analyses of the contribution of crystallographic water molecules toward the differences in solvent accessibility of the isoforms indicated that waters exhibiting differences in residence times between the GDP-bound structures of K-Ras4A and K-Ras4B were crystallographic waters. Coupled with the observation that the switch I region of K-Ras4A has broader conformational sampling than the switch I region of K-Ras4B in the GDP-bound state, these findings suggest that conserved water molecules may play a role in the fluctuations of the switch regions. In support of this, prior MD simulations of Ras suggested the possibility of conserved water molecules having a role in the allosteric modulation of the switch II region.68 These water molecules may participate in transient hydrogen bonding with nearby residues and be involved in water-mediated signaling networks that could allosterically modulate the fluctuations of the switch residues. On this note, our observation of an allosteric network between the effector and allosteric lobes of Ras is supported by a number of computational and experimental studies that demonstrate isoform-specific dynamic residue interaction pathways between the nucleotide-binding site and allosteric lobe residues of Ras, which may also be implicated in modulating the conformation of the switch regions.44,45,69
We have found that the HVR of K-Ras4B covers the effector binding site of the GDP-bound catalytic domain, thereby autoinhibiting signaling, but is released in the GTP-bound state.40 This interaction is almost 100-fold tighter in the GDP-bound state relative to the GTP-bound protein. The interaction could be important, since HVR binding interferes with Ras-Raf interaction, slows down nucleotide exchange, and inhibits Ras signaling in tumor cells.40,41 This suggests that the HVR can play essential roles in regulation of K-Ras signaling and that targeting this surface with inhibitory synthetic molecules for the therapy of KRAS-dependent tumors may be feasible. The sequences of the two isoforms are identical in the effector binding site but differ in their HVR. This raises the question of whether the autoregulation mechanism observed in K-Ras4B can also take place in K-Ras4A. We observed that as in K-Ras4B, the HVR of K-Ras4A exhibits nucleotide-dependent behavior; however, the pattern of interactions and distinction between the GDP- and GTP-bound states differ in comparison to K-Ras4B. Rather than cover the effector binding site when GDP-bound, the HVR forms intra-HVR interactions that appear to collapse the peptide chain; when GTP-bound, it tends to sample conformations that are extended away from the catalytic domain. However, the tendency is not strong, and both collapsed and extended conformations are observed. This suggests that the access to the effector binding site in K-Ras4A may be hindered to some extent, but access is possible. The interaction between the catalytic domain and the HVR appears weak. The difference between the K-Ras4B and K-Ras4A behaviors can be attributed to the HVR sequence, with the high positive charge of the K-Ras4B HVR acting to preclude chain collapse.
Recently, we suggested that K-Ras4A may have two functional states resembling K-Ras4B and N-Ras.24 The HVR of K-Ras4A, which has a lesser net positive charge than the HVR of K-Ras4B, would be expected to also have a lesser, albeit still favorable, electrostatic interaction with acidic membranes and may also interact with zwitterionic membranes. On the other hand, its palmitoylated state may promote anchoring into neutral membranes, like N-Ras, whose HVR is farnesylated and palmitoylated. This led us to suggest that it evolved first; being able to serve in both roles confers an evolutionary advantage. In its K-Ras4B-like depalmitoylated state it may bind calmodulin, like K-Ras4B, and contribute to full activation of PI3K.70,71 At the same time, its lack of interaction with the effector binding site in the GDP-bound state suggests that inhibition strategies proposed to target K-Ras4B by enhancing the HVR interaction may not work and strategies to substitute the HVR by higher affinity HVR analogues40 may not be K-Ras specific. Of note, simulations supported by NMR chemical shift data of oncogenic full length K-Ras4B showed that the HVR of both the GDP- and GTP-bound states adopts a more open conformation, with a larger attenuation of the HVR-catalytic domain interactions observed in the GTP-bound state for mutations far away from the HVR binding site, like the G12 and Q61 mutations and those at the interaction site (E37K).41
CONCLUSIONS
The presence of the K-Ras4A HVR, in both the GDP- and GTP-bound states, increases the intrinsic fluctuations of switch I and switch II residues observed in simulations of the catalytic domain alone. Recent NMR spectroscopy of full-length H-Ras (1–185) and farnesylated H-Ras (1–189) investigated the impact of HVR farnesylation on Ras dynamics.72 The NMR chemical shift perturbation indicated that the presence of the HVR in H-Ras primarily induces changes in residues within and proximal to the α5 helix, including the β-sheets (β3, β4, and β5) that comprise the central core of the protein. No perturbations were found to be induced in residues of the switch regions or the nucleotide-binding pocket (i.e., P-loop residues) in either nucleotide-bound state, as our study observed. However, these results were for the H-Ras HVR, which differs from the HVR of K-Ras4A and which may have an alternate signal propagation pathway to the catalytic domain that would differentially influence the orientation and specific interactions of the catalytic domain with downstream effectors.13
This work represents the first interrogation of the dynamics and energetics of the full-length K-Ras4A protein on a molecular level. The next steps should involve K-Ras4A interactions with the cell membrane accounting for its bimodality, dimer formation focusing on the dimerization interfaces,73 and comparisons of the behavior of K-Ras4A with K-Ras4B, bimodal N-Ras, and H-Ras.
Supplementary Material
ACKNOWLEDGMENTS
This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under Contract HHSN261200800001E. This research was supported [in part] by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. All simulations have been performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.5b11110.
Figures S1–S9 including the average solvent accessible surface areas (SASA), the time series distances between selected residues, the three-dimensional water density maps, the interactions of high-residence hydration waters, the dynamic cross-correlation maps, the principal component analysis, the conformational states, torsional angles, and the radius of gyration of the HVR (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Malumbres M; Barbacid M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [DOI] [PubMed] [Google Scholar]
- (2).Adjei AA Blocking oncogenic Ras signaling for cancer therapy. J. Natl. Cancer Inst. 2001, 93, 1062–1074. [DOI] [PubMed] [Google Scholar]
- (3).Lacal JC; Srivastava SK; Anderson PS; Aaronson SA Ras p21 proteins with high or low GTPase activity can efficiently transform NIH/3T3 cells. Cell 1986, 44, 609–617. [DOI] [PubMed] [Google Scholar]
- (4).Quinlan MP; Settleman J. Isoform-specific ras functions in development and cancer. Future Oncol. 2009, 5, 105–116. [DOI] [PubMed] [Google Scholar]
- (5).van Hattum H; Waldmann H. Chemical biology tools for regulating RAS signaling complexity in space and time. Chem. Biol. 2014, 21, 1185–1195. [DOI] [PubMed] [Google Scholar]
- (6).Nussinov R; Tsai CJ; Muratcioglu S; Jang H; Gursoy A; Keskin O. Principles of K-Ras effector organization and the role of oncogenic K-Ras in cancer initiation through G1 cell cycle deregulation. Expert Rev. Proteomics 2015, 12, 669–682. [DOI] [PubMed] [Google Scholar]
- (7).Gysin S; Salt M; Young A; McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer 2011, 2, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lukman S; Grant BJ; Gorfe AA; Grant GH; McCammon JA The distinct conformational dynamics of K-Ras and H-Ras A59G. PLoS Comput. Biol. 2010, 6, e1000922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Vetter IR; Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [DOI] [PubMed] [Google Scholar]
- (10).Karnoub AE; Weinberg RA Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ahearn IM; Haigis K; Bar-Sagi D; Philips MR Regulating the regulator: post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Laude AJ; Prior IA Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J. Cell Sci. 2008, 121, 421–427. [DOI] [PubMed] [Google Scholar]
- (13).Abankwa D; Gorfe AA; Inder K; Hancock JF Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1130–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Castellano E; Santos E. Functional specificity of ras isoforms: so similar but so different. Genes Cancer 2011, 2, 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Arozarena I; Calvo F; Crespo P. Ras, an actor on many stages: posttranslational modifications, localization, and site-specified events. Genes Cancer 2011, 2, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Prior IA; Muncke C; Parton RG; Hancock JF Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003, 160, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Henis YI; Hancock JF; Prior IA Ras acylation, compartmentalization and signaling nanoclusters (Review). Mol. Membr. Biol. 2009, 26, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Prior IA; Hancock JF Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol. 2012, 23, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Aran V; Prior IA Compartmentalized Ras signaling differentially contributes to phenotypic outputs. Cell. Signalling 2013, 25, 1748–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tsai FD; Lopes MS; Zhou M; Court H; Ponce O; Fiordalisi JJ; Gierut JJ; Cox AD; Haigis KM; Philips MR K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Schubbert S; Shannon K; Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [DOI] [PubMed] [Google Scholar]
- (22).Jang H; Banerjee A; Chavan TS; Lu S; Zhang J; Gaponenko V; Nussinov R. The higher level of complexity of K-Ras4B activation at the membrane. FASEB J. 2016, DOI: 10.1096/fj.1015-279091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Banerjee A; Jang H; Nussinov R; Gaponenko V. The disordered hypervariable region and the folded catalytic domain of oncogenic K-Ras4B partner in phospholipid binding. Curr. Opin. Struct. Biol. 2016, 36, 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nussinov R; Tsai CJ; Chakrabarti M; Jang H. A new view of Ras isoforms in cancers. Cancer Res. 2016, 76, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Roberts PJ; Der CJ Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [DOI] [PubMed] [Google Scholar]
- (26).Thumar J; Shahbazian D; Aziz SA; Jilaveanu LB; Kluger HM MEK targeting in N-RAS mutated metastatic melanoma. Mol. Cancer 2014, 13, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cox AD; Fesik SW; Kimmelman AC; Luo J; Der CJ Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discovery 2014, 13, 828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Carta C; Pantaleoni F; Bocchinfuso G; Stella L; Vasta I; Sarkozy A; Digilio C; Palleschi A; Pizzuti A; Grammatico P; et al. Germline missense mutations affecting KRAS isoform B are associated with a severe Noonan syndrome phenotype. Am. J. Hum. Genet. 2006, 79, 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pells S; Divjak M; Romanowski P; Impey H; Hawkins NJ; Clarke AR; Hooper ML; Williamson DJ Developmentally-regulated expression of murine K-ras isoforms. Oncogene 1997, 15, 1781–1786. [DOI] [PubMed] [Google Scholar]
- (30).Jackson JH; Li JW; Buss JE; Der CJ; Cochrane CG Polylysine domain of K-ras 4B protein is crucial for malignant transformation. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 12730–12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Plowman SJ; Berry RL; Bader SA; Luo F; Arends MJ; Harrison DJ; Hooper ML; Patek CE K-ras 4A and 4B are coexpressed widely in human tissues, and their ratio is altered in sporadic colorectal cancer. J. Exp. Clin. Cancer Res. 2006, 25, 259–267. [PubMed] [Google Scholar]
- (32).Normanno N; Tejpar S; Morgillo F; De Luca A; Van Cutsem E; Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [DOI] [PubMed] [Google Scholar]
- (33).Stone JC Regulation and function of the RasGRP family of Ras activators in blood cells. Genes Cancer 2011, 2, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Luo F; Ye H; Hamoudi R; Dong G; Zhang W; Patek CE; Poulogiannis G; Arends MJ K-ras exon 4A has a tumour suppressor effect on carcinogen-induced murine colonic adenoma formation. J. Pathol. 2010, 220, 542–550. [DOI] [PubMed] [Google Scholar]
- (35).Butz JA; Roberts KG; Edwards JS Detecting changes in the relative expression of KRAS2 splice variants using polymerase colonies. Biotechnol. Prog. 2004, 20, 1836–1839. [DOI] [PubMed] [Google Scholar]
- (36).Wang Y; You M; Wang Y. Alternative splicing of the K-ras gene in mouse tissues and cell lines. Exp. Lung Res. 2001, 27, 255–267. [DOI] [PubMed] [Google Scholar]
- (37).To MD; Wong CE; Karnezis AN; Del Rosario R; Di Lauro R; Balmain A. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat. Genet. 2008, 40, 1240–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Patek CE; Arends MJ; Wallace WA; Luo F; Hagan S; Brownstein DG; Rose L; Devenney PS; Walker M; Plowman SJ; et al. Mutationally activated K-ras 4A and 4B both mediate lung carcinogenesis. Exp. Cell Res. 2008, 314, 1105–1114. [DOI] [PubMed] [Google Scholar]
- (39).Voice JK; Klemke RL; Le A; Jackson JH Four human ras homologs differ in their abilities to activate Raf-1, induce transformation, and stimulate cell motility. J. Biol. Chem. 1999, 274, 17164–17170. [DOI] [PubMed] [Google Scholar]
- (40).Chavan TS; Jang H; Khavrutskii L; Abraham SJ; Banerjee A; Freed BC; Johannessen L; G TS; Gaponenko V; Nussinov R; et al. High-affinity interaction of the K-Ras4B hypervariable region with the Ras active site. Biophys. J. 2015, 109, 2602–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lu S; Banerjee A; Jang H; Zhang J; Gaponenko V; Nussinov R. GTP binding and oncogenic mutations may attenuate hypervariable region (HVR)-catalytic domain interactions in small GTPase KRAS4B, exposing the effector binding site. J. Biol. Chem. 2015, 290, 28887–28900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lu S; Jang H; Muratcioglu S; Gursoy A; Keskin O; Nussinov R; Zhang J. Ras conformational ensembles, allostery and signaling. Chem. Rev. 2016, in press. [DOI] [PubMed] [Google Scholar]
- (43).Ma J; Karplus M. Ligand-induced conformational changes in ras p21: a normal mode and energy minimization analysis. J. Mol. Biol. 1997, 274, 114–131. [DOI] [PubMed] [Google Scholar]
- (44).Gorfe AA; Grant BJ; McCammon JA Mapping the nucleotide and isoform-dependent structural and dynamical features of Ras proteins. Structure 2008, 16, 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Grant BJ; Gorfe AA; McCammon JA Ras conformational switching: simulating nucleotide-dependent conformational transitions with accelerated molecular dynamics. PLoS Comput. Biol. 2009, 5, e1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kobayashi C; Saito S. Relation between the conformational heterogeneity and reaction cycle of Ras: molecular simulation of Ras. Biophys. J. 2010, 99, 3726–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kapoor A; Travesset A. Differential dynamics of RAS isoforms in GDP- and GTP-bound states. Proteins: Struct., Funct., Genet. 2015, 83, 1091–1106. [DOI] [PubMed] [Google Scholar]
- (48).Brooks BR; Brooks CL III; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Humphrey W; Dalke A; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14 (33–38), 27–38. [DOI] [PubMed] [Google Scholar]
- (50).Jang H; Abraham SJ; Chavan TS; Hitchinson B; Khavrutskii L; Tarasova NI; Nussinov R; Gaponenko V. Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. J. Biol. Chem. 2015, 290, 9465–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- (53).Han B; Liu Y; Ginzinger SW; Wishart DS SHIFTX2: significantly improved protein chemical shift prediction. J. Biomol. NMR 2011, 50, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bos JL; Rehmann H; Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [DOI] [PubMed] [Google Scholar]
- (55).Vigil D; Cherfils J; Rossman KL; Der CJ Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Rojas JM; Oliva JL; Santos E. Mammalian son of sevenless Guanine nucleotide exchange factors: old concepts and new perspectives. Genes Cancer 2011, 2, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Boriack-Sjodin PA; Margarit SM; Bar-Sagi D; Kuriyan J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [DOI] [PubMed] [Google Scholar]
- (58).Lu S; Jang H; Zhang J; Nussinov R. Inhibitors of Ras-SOS interactions. ChemMedChem. 2015, DOI: 10.1002/cmdc.201500481. [DOI] [PubMed] [Google Scholar]
- (59).Jones MK; Jackson JH Ras-GRF activates Ha-Ras, but not N-Ras or K-Ras 4B, protein in vivo. J. Biol. Chem. 1998, 273, 1782–1787. [DOI] [PubMed] [Google Scholar]
- (60).Clyde-Smith J; Silins G; Gartside M; Grimmond S; Etheridge M; Apolloni A; Hayward N; Hancock JF Characterization of RasGRP2, a plasma membrane-targeted, dual specificity Ras/Rap exchange factor. J. Biol. Chem. 2000, 275, 32260–32267. [DOI] [PubMed] [Google Scholar]
- (61).Sethi A; Eargle J; Black AA; Luthey-Schulten Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6620–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Eargle J; Luthey-Schulten Z. NetworkView: 3D display and analysis of protein.RNA interaction networks. Bioinformatics 2012, 28, 3000–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Grant BJ; Rodrigues AP; ElSawy KM; McCammon JA; Caves LS Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [DOI] [PubMed] [Google Scholar]
- (64).Skjaerven L; Yao XQ; Scarabelli G; Grant BJ Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinf. 2014, 15, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Shimizu K; Birnbaum D; Ruley MA; Fasano O; Suard Y; Edlund L; Taparowsky E; Goldfarb M; Wigler M. Structure of the Ki-ras gene of the human lung carcinoma cell line Calu-1. Nature 1983, 304, 497–500. [DOI] [PubMed] [Google Scholar]
- (66).Ford B; Boykevisch S; Zhao C; Kunzelmann S; Bar-Sagi D; Herrmann C; Nassar N. Characterization of a Ras mutant with identical GDP- and GTP-bound structures. Biochemistry 2009, 48, 11449–11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Ford B; Skowronek K; Boykevisch S; Bar-Sagi D; Nassar N. Structure of the G60A mutant of Ras: implications for the dominant negative effect. J. Biol. Chem. 2005, 280, 25697–25705. [DOI] [PubMed] [Google Scholar]
- (68).Prakash P; Sayyed-Ahmad A; Gorfe AA The role of conserved waters in conformational transitions of Q61H K-ras. PLoS Comput. Biol. 2012, 8, e1002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Buhrman G; Holzapfel G; Fetics S; Mattos C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4931–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Nussinov R; Muratcioglu S; Tsai CJ; Jang H; Gursoy A; Keskin O. The key role of calmodulin in KRAS-driven adenocarcinomas. Mol. Cancer Res. 2015, 13, 1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Nussinov R; Muratcioglu S; Tsai CJ; Jang H; Gursoy A; Keskin O. K-Ras4B/calmodulin/PI3Kα: A promising new adenocarcinomaspecific drug target? Expert Opin. Ther. Targets 2016, DOI: 10.1517/14728222.14722016.11135131. [DOI] [PubMed] [Google Scholar]
- (72).Thapar R; Williams JG; Campbell SL NMR characterization of full-length farnesylated and non-farnesylated H-Ras and its implications for Raf activation. J. Mol. Biol. 2004, 343, 1391–1408. [DOI] [PubMed] [Google Scholar]
- (73).Muratcioglu S; Chavan TS; Freed BC; Jang H; Khavrutskii L; Freed RN; Dyba MA; Stefanisko K; Tarasov SG; Gursoy A; et al. GTP-dependent K-Ras dimerization. Structure 2015, 23, 1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.