

Abstract
Protein glycosylation is involved in many biological processes and physiological functions. Despite the recent advances in LC-MS/MS methodologies, the profiling of site-specific glycosylation is one of the major analytical challenges of glycoprotein analysis. Herein, we first reported the separation of glycopeptide isomers on porous graphitic carbon (PGC)-LC was significantly improved by elevating the separation temperature under basic mobile phases. These findings permitted the isomeric separation of glycopeptides resulting from highly specific enzymatic digestions. The selectivity for different glycan types were studied using bovine fetuin, asialofetuin, IgG, ribonuclease B and alpha-1 acid glycoprotein (AGP) by PGC-LC-MS. Comprehensive structural isomeric separation of glycopeptides was observed by high resolution MS and confirmed by MS/MS. The specific structures of the glycopeptide isomers were identified and confirmed through exoglycosidase digestions. The glycosylation analysis of human AGP revealed the potential use of PGC-LC-MS for extensive glycoprotein analysis for biomarker discovery. This newly developed separation technique was shown as a reproducible and useful analytical method to study site-specific isomeric glycosylation.
Graphical Abstract
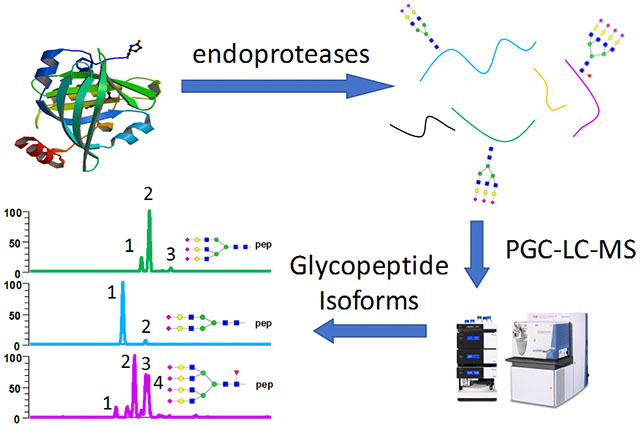
Introduction
Protein glycosylation is one of the most common posttranslational modifications (PTMs) of proteins, and more than 50% of proteins are found to be glycosylated.1 Glycoproteins are involved in many biological processes including cell-cell signaling, protein stability, protein localization, and immune response.2–4 In recent years, protein glycosylation was reported to play a key role in diseases such as inflammatory diseases,5–7 rheumatoid arthritis,8,9 Alzheimer’s disease,10,11 neuronal diseases,12,13 and cancer.14–16 Despite the importance of glycoproteins, the elucidation of glycans attached to each glycosylation site (glycosylation micro-heterogeneity) and the separation of structural glycopeptide isomers remain major challenges in glycoprotein analysis.17 Therefore, powerful analytical methods are needed for understanding the roles of glycoproteins in disease development and progression.
Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) is one of the most common and powerful analytical tools for analysis of glycans and glycoproteins.18 The site-specific glycosylation information is usually acquired from the analysis of intact glycopeptides or glycoproteins. Single glycoproteins can have more than one glycosylation site and/or other PTMs, which greatly complicates the analysis.19,20 To reduce the complexity of analysis, the general method is to digest glycoproteins with proteases such as trypsin and analyze the resulting glycopeptides containing individual glycosylation sites.21,22
Glycan isomers have been widely reported to play important roles in the disease diagnosis process23–25. However, site-specific analysis of isomeric glycopeptides remains challenging due to the difficulties of isomeric separation for glycopeptides. Reversed-phase liquid chromatography (RPLC) with a C18 stationary phase has been widely used in glycopeptide separation because of the relatively high hydrophobicity of the peptide moieties.26–29 The major advantage of RPLC in glycopeptides separation is its compatibility with proteomic research.22 However, no isomeric separation for glycopeptides was reported until a recent manuscript by Ji et al.30 The separation of sialylated N- and O-linked glycopeptide isomers derived from erythropoietin and alpha-1 acid glycoprotein (AGP) were successfully achieved under an elevated temperature. Nevertheless, the method was relatively time-consuming and the separation for non-sialylated species was not reported.
An alternative way to separate glycopeptides is to use porous graphitic carbon (PGC) as a stationary phase in liquid chromatography. PGC has been shown as a useful stationary phase for LC-MS for years.31 The application of PGC-LC has been reported as one of the major methods to achieved structural isomeric separation of detached glycans.32 However, PGC-LC has also been reported to have difficulties in eluting hydrophobic glycopeptides, especially those with highly sialylated glycan moieties.33,34 Several studies tried to overcome this issue by using nonspecific proteases.35–38 The advantage of using nonspecific proteases was the creation of glycopeptides with small peptide backbone (<4~6 amino acids). These glycopeptides were more compatible with PGC-LC and generated more useful MS/MS spectra than those with large peptide moieties. However, due to the low specificity of the nonspecific proteases, multiple glycopeptides with different peptide backbones were produced simultaneously from a single glycosylation site39(e.g. pronase). Consequently, it limited the sensitivity and accuracy of the quantification of glycosylation. In addition, even with small peptide backbone, compromises of isomeric selectivity were also observed.37
In this study, we first investigated the mobile phases compositions on PGC-LC. Elevating the temperature to 75°C was also proved to enhance isomeric separation of glycopeptides significantly. Chromatographic behaviors of glycopeptides with complex as well as neutral glycans were studied under the optimal conditions. The comprehensive study of glycoproteins micro-heterogeneity attained in a single PGC-LC run by using a combination of site-specific proteolytic digestion which reduced peptide sizes. Structure elucidation of each isoform were performed by exoglycosidase digestion experiments.
Materials and methods
Chemicals
Dithiothreitol (DTT), iodoacetamide (IAA), ammonium bicarbonate (ABC), ammonium acetate (AA), ammonium hydroxide, ribonuclease B from bovine pancreas (RNase B), fetuin from fetal calf serum (bovine fetuin), asialofetuin from fetal calf serum (asialofetuin) and alpha-1 acid glycoprotein from human plasma (AGP) were purchased from Sigma-Aldrich (St. Louis, MO). Murine IgG1 (Intact mAb Mass Check Standard) was obtained from Waters Corporation (Milford, MA). α2–3 neuraminidase, α2–3,6,8,9 neuraminidase, β1–3 galactosidase, β1–4 galactosidase and β1–3,4 galactosidase were acquired from New England Biolabs (Ipswich, MA). Mass spectrometry grade Trypsin/Lys-C mixture, sequencing grade chymotrypsin and sequencing grade endoprotease Glu-C (Glu-C) were obtained from Promega (Madison, WI). HPLC grade water was acquired from Mallinckrodt Chemicals (Phillipsburg, NJ). HPLC grade acetonitrile was purchased from Fisher Scientific (Fair Lawn, NJ).
Endoprotease digestion of proteins
Multiple digestion strategies were used to create various glycopeptides. The digestion conditions were adapted from the work by Heck et al.40 For the tryptic digestion, RNase B, bovine fetuin, asialofetuin, IgG and AGP were reduced by adding 100mM of dithiothreitol (DTT) to reach a final concentration of 5mM and then incubated for 45min at 60°C. The mixture was alkylated by adding 200mM iodoacetamide to a final concentration of 20mM and then incubated in the dark for 45min. The reaction was quenched with another addition of 5mM DTT. The reduced and alkylated proteins were subjected to proteolytic digestion by the addition of Trypsin/Lys-C mixture (Promega, Madison, WI, 1:25 (w/w) enzyme to protein ratio) and incubated at 37.5°C for 18 hours.
For the combination digestion experiment of trypsin and Glu-C or chymotrypsin, the reduction, alkylation and tryptic digestion of the corresponding glycoproteins were performed following the aforementioned protocol. After the overnight incubation with the trypsin/Lys-C mixture, endoprotease Glu-C or chymotrypsin was added to obtain a 1:25 enzyme to protein ratio. The mixture was incubated at 37.5°C for another 18 hours. Samples were dried in a speed vacuum and stored at −20°C prior to further usage. It is noteworthy to point out that the combination of enzymes was chosen based on the product peptide length to ensure a suitable hydrophobicity for C18 clean up and PGC elution.
Exoglycosidase digestion of glycopeptides
For the exoglycosidase digestions, the endoprotease digested glycoproteins were resuspended to 1 μg/μl aliquots and were treated following the protocols of each enzyme. Briefly, for the 2–3Neu, 2–3,6,8,9Neu, 1–3Gal and 1–4 Gal digestion, the glycopeptide samples were diluted to 45 μl with water, and 5 μl of 10X glycobuffer 1 (5 mM CaCl2, 50 mM sodium acetate, pH 5.5) was introduced prior to adding 5 μl of the corresponding exoglycosidases. In the case of 1–3,4Gal digestion, 10X glycobuffer 2 (50 mM sodium acetate, pH 4.5) were used instead. The digestion mixture of bovine fetuin was incubated at 37°C for 1 hour. The IgG and asialofetuin were digested with galactosidases at 37°C overnight. Negative controls for all enzymatic digestion experiments were generated by adding 5 μl of water instead of the enzymes and were subjected to the same workflows.
The exoglycosidase treated samples were off-line desalted using TopTip™ C18 cartridge (Glygen Corp., Columbia, MD), due to the reason that PGC is sensitive to pH and salt changes41. Briefly, the desalting columns were pre-conditioned by adding 50 μl of elution buffer 1 (60% ACN and 0.1% formic acid) and spun at 1.0×103 g in the centrifuge 3 times. The columns were rinsed with 50 μl loading buffer (0.1% formic acid in water) for 3 times before the samples were loaded using the same spinning speed. The sample eluent was collected and re-applied to the column to make sure that all peptides and glycopeptides were bound onto the C18 cartridge at 0.5×103 g. After washing the cartridge with loading buffer 3 times, the desalted products were eluted by applying elution buffer 1 3 times and elution buffer 2 (100% ACN and 0.1% formic acid) 2 times. The eluent was vacuum dried and kept at −20°C for storage. The samples were re-suspended in water before LC-MS injection.
LC-MS/MS analysis
LC-MS/MS was performed using Vanquish UHPLC system coupled to Q-Exactive (Thermo Scientific, San Jose, CA) and Dionex Ultimate 3000 UHPLC system (Dionex, Sunnyvale, CA) interfaced to Exactive equipped with a HESI source. The optimized LC conditions for isomeric separation on PGC were achieved on a Hypercarb™ columns (150 mm × 1 mm I.D, 5 μm particle sizes, Thermo Scientific, San Jose) at a flow rate of 0.1 ml/min unless further noted. Mobile phase A contained 25 mM ammonium acetate with 1.25% (v/v) ammonium hydroxide (pH 9.9) while mobile phase B consisted of 50% ACN with 25 mM ammonium acetate and 1.25% (v/v) ammonium hydroxide. The LC gradient of mobile phase B ramped from 0% to 100% over 35 min, and was kept at 100% for 20 min. The mobile phase B was then set at 0% for 30 mins to equilibrate the column and get prepared for the next injection.
The Q-Exactive mass spectrometer was operating in the positive ion-mode with the ESI voltage set to 3900V with sheath gas, auxiliary gas and sweep gas flow rates set to 35, 10 and 1 μl /min, respectively. Resolution was set to 120,000 for full MS scans. 5 μg of the starting material were injected for bovine fetuin, RNase B and AGP samples on Q-Exactive with Vanquish UHPLC. The Exactive mass spectrometer was applying 3.5 kV as spray voltage and 15 and 3 μl /min for sheath gas aux gas flow rate, respectively. No sweep gas was used on Exactive. The reolution was kept at 60,000 for Exactive runs. 500 μg of original bovine fetuin was used for reproducibility tests per injection on Ultimate 3000 with Exactive. 500 μg of IgG, and asialofetuin was also injected on Ultimate 3000 with Exactive. The MS/MS experiment was conducted on a LTQ Orbitrap Velos mass spectrometer, with a 30% normalized activation energy and a 15 ms activation time. The activation Q value was set to 0.250.
Data analysis
The LC-MS/MS raw data was processed using Xcalibur™ 2.1 (Thermo Scientific, San Jose). The EICs for glycopeptides were generated with mass tolerance of 10 ppm. Symbols of monosaccharides as well as the sialic acid and galactose linkage isomers were depicted in Scheme 1.
Scheme 1.
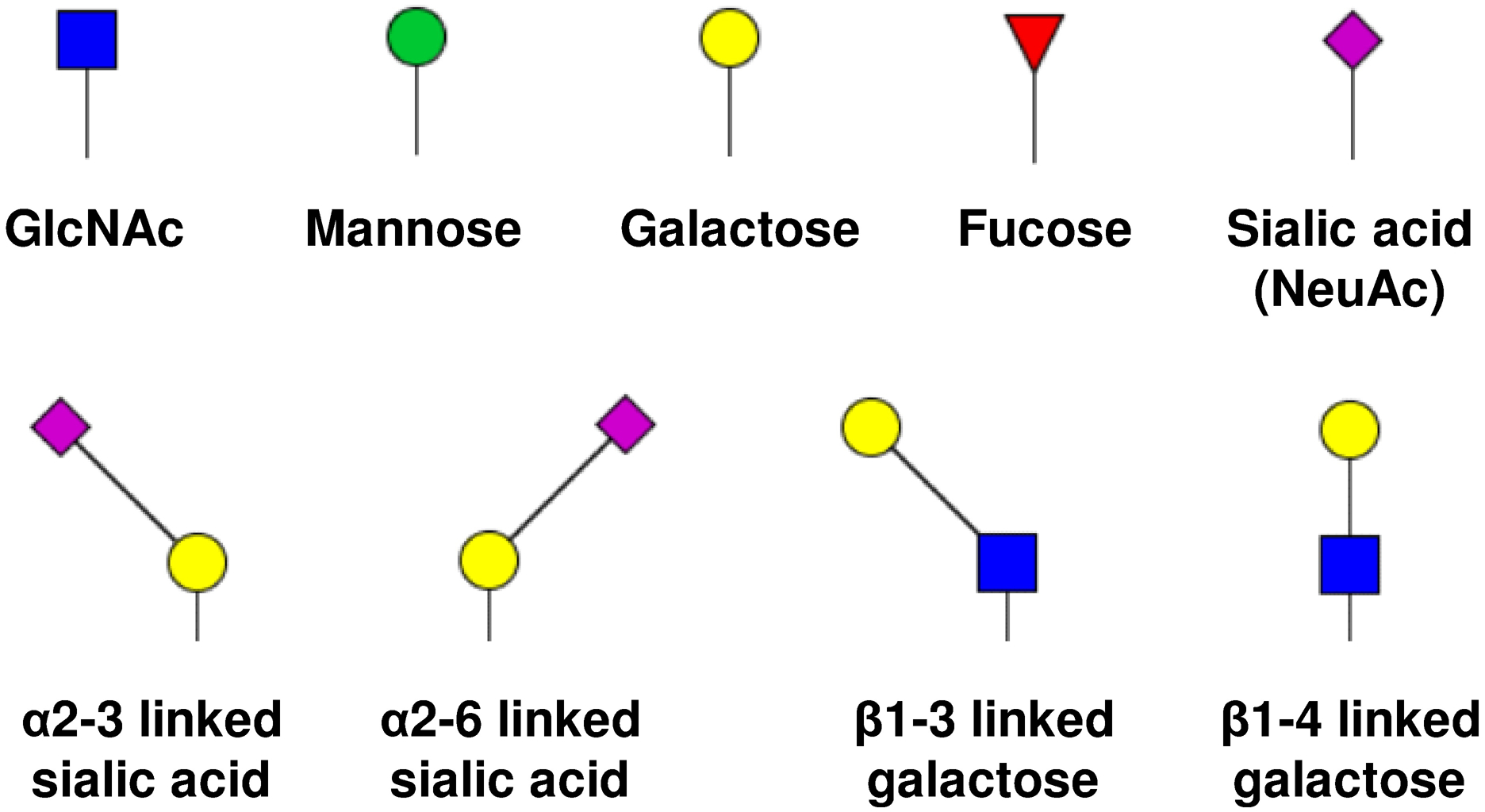
Symbols of monosaccharides as well as sialic acid and galactose linkage isomers.
Result and Discussion
Setting up the conditions of glycopeptides isomeric separation on PGC-LC-MS
Separation temperature has been widely reported to be critical in RP-LC analysis which affects both retention and selectivity.42 RP-LC separation of sialylated glycopeptide isomers has been achieved under 60°C.30 Meanwhile, we have recently shown the isomeric separation of permethylated glycans on PGC-LC at the high column temperature.43–46 Inspired by these studies, the separation temperature on PGC-LC was first kept at 75°C to develop the mobile phase compositions by using tryptic glycopeptides derived from bovine fetuin as analyte. The conventional mobile phases for glycopeptide separation on C18 columns were applied to PGC columns. As shown in Fig S1a–S1d, several tryptic peptides were identified under acidic conditions. However, glycopeptides were eluted with poor resolution (Fig S1e) or were not even eluted (Fig S1f) under the acidic conditions, possibly due to the strong interaction between analytes and stationary phase.47 Therefore, the study for a better composition of mobile phases is needed.
Unlike RP-LC, in which the hydrophobic interaction is mainly responsible for the separation, PGC offers the ability to separate both hydrophobic and hydrophilic analytes due to the unique retention mechanisms.48 In brief, for the hydrophobic analytes on PGC, such as permethylated glycans, hydrophobic interaction plays the most important role. However, in the case of hydrophilic analytes, the polar retention effect (PREG) on graphite is significantly influencing the separation.48 Though not completely understood, the PREG effect is known to be related to the polarizability of the analyte, which is also relevant to the charge states of glycopeptide analytes. Moreover, the carboxyl groups on sialic acid residues increase the complexity of the charge states of the glycopeptides of interest. Therefore, the pH of mobile phases significant effects the elution of glycopeptides. As mobile phases were switched to the conditions described in the methods section, the resolution dramatically increased for the late eluting analytes. Fig S2a–S2d depicts the elution profiles of the same peptides as in Fig S1a–S1d under basic conditions. Though the retention times for peptides were later in basic conditions, the resolution was higher in the case of QDGQFSVLFTK. Most importantly, the glycopeptides (Fig S2e and S2f) were eluted and were resolved as isomeric peaks. In addition, these peptide traces (Fig S2a–S2d) remained a single peak, indicating the separation was not prompted by the peptide conformers. In Fig S2g, peptide backbones were observed as one peak after deglycosylation treatment by PNGase F, which also proved that the separation of the corresponding glycopeptides was driven by the glycan isoforms.
Base line separations were achieved for all glycopeptides; however, the separation mechanism is not yet fully understood. A hypothesis is that in the pH 2.8 solutions, the charge states were not uniform among the analyte molecules, which resulted in peak broadening. This problem was overcome with the pH 9.9 solutions since both peptide and glycan moieties were deprotonated (i.e. NH2 for amine groups and COO− for carboxyl groups). Similar resolution can possibly be achievable via acidifying mobile phases to a point that all groups were carrying positive charges, however, the use of such acids would dramatically decrease the ionization efficiency due to the charge neutralization effect introduced by anion additives.49 Besides, the retention time decreased noticeably for the glycopeptides under basic conditions. This is probably due to the fact that negatively charged species tend to form ion-pairs with ammonium additives in mobile phases, which neutralized charges on sialic acids and thus decreased the retention.
The influence of salt and solvent pH on the ionization efficiency was further assessed. To compare the ESI efficiencies between acid and basic mobile phases, tryptic digested fetuin sample was first dried and resuspended in the corresponding acidic and basic mobile phases. The analyte solutions were direct-infused through the ESI source to mimic the LC-MS conditions. Over 100 spectra were acquired and averaged in each case, as shown in Fig S3, the intensities were comparable to when the solvent is basic despite the fact that fewer number of free protons was in the solvent. This is largely caused by the electrochemical reaction taking place in the ESI process, which oxidized water and generated the protons.50,51 Thus, the analytes were able to carry positive charges. However, a drawback of adding salts to the mobile phases is the forming of adduct ions. As depicted in Fig S4, both sodiated and ammoniated ions were detected with high abundance. This could possibly be improved by adding a post-column infusion flow with modifiers, as it has been proved that the adducts forms are depended on the mobile phase compositions.52
The temperature effects on PGC column were thoroughly investigated. Fig 1 showed the temperature study of the glycopeptide LCPDCPLLAPL156NDSR-A2G2S2 derived from tryptic digestion of bovine fetuin. The separation efficiency and resolution were substantially improved as the column temperature increased from 25°C to 75°C. At 25°C, only one peak was observed (Fig 1a); while at 50°C (Fig 1b) peak width as well as retention time increased and were eventually resolved into 3 peaks at 75°C (Fig 1c). Retention time also increased with higher temperature, suggesting that interaction with column stationary phase was enhanced by elevating temperature. Similar phenomenon has been reported in separation of acidic oligosaccharides53 and permethylated glycans54 on PGC-LC. However, increasing the temperature to 100°C resulted in a significant decrease in signal intensity yet had no obvious resolution improvement. Therefore, the optimum separation temperature of glycopeptides was set at 75°C.
Figure 1.
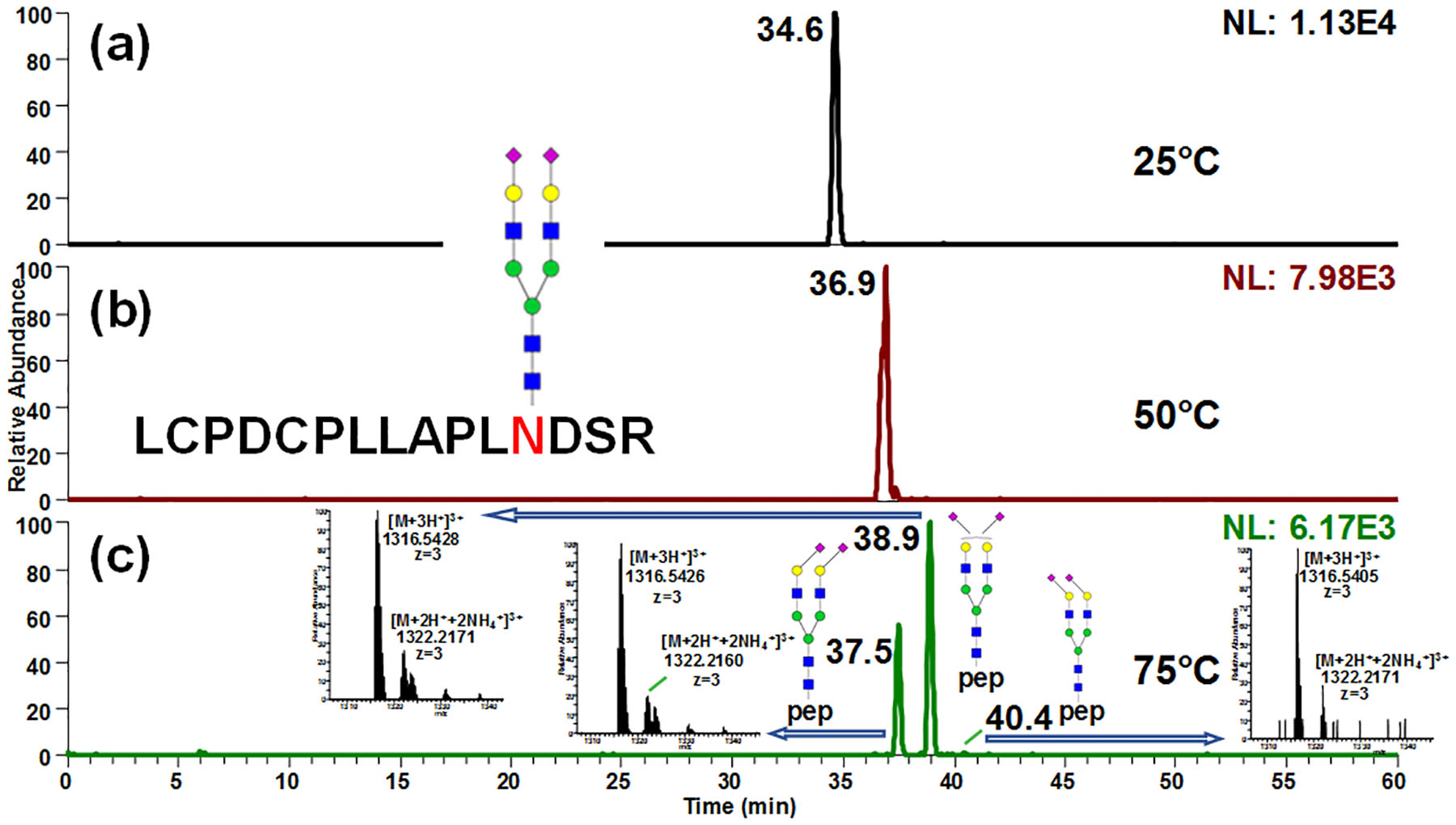
Separation of 156Asn glycopeptide from bovine fetuin on PGC-LC-MS at (a) 25°C, (b) 50°C and (c) 75°C. Isomeric separation was obtained at elevated temperature. Retention time also increased with higher temperature, suggesting that interaction with column stationary phase was enhanced by elevating temperature. Insets in (c) depict the full MS spectra of each isomer.
Prior to the structural elucidation, the reproducibility of the method and the stability of PGC column under basic conditions were verified. The same fetuin digest sample was applied as standards and was analyzed using the same column across different time periods. The retention times for each isoform detected in the fetuin digest were listed in Table S1, and the relative standard deviation was calculated. Fig S5 depicted the EICs of A2G2S2 on 156Asn over 15 runs. The first 10 runs were conducted back to back and runs 11–15 were made back to back on a different day. The averaged retention times for the two peaks were 40.1 ± 0.2 min and 42.0 ± 0.3 min for the 10 continuous runs. For the reproducibility on different days (i.e. runs 1–5 and runs 11–15), the RSDs were both less than 1% for both isoforms. The averaged RSDs for A3G3S3 and A3G3S4 were also showed to be less than 1% (Table S1). Thus, the separation technique is stable and reproducible.
Extensive profiling of sialylated glycopeptide isomers derived from fetuin on PGC-LC-MS
Since the glycopeptides not only possess the hydrophilic glycan moieties, the relative hydrophobic peptide backbones would also interfere with the elution by having hydrophobic interactions when the backbone is getting longer. Tryptic digestion of fetuin creates three peptide backbones, which are RPTGEVYDIEIDTLETTCHVLDPTPLA99NCSVR (32 amino acids), LCPDCPLLAPL156NDSR (15 amino acids) and VVHAVEVALATFNAES176NGSYLQLVEISR (28 amino acids). Only site 156Asn was able to elute directly. Due to the large peptide backbone, the glycosylation profiles on sites 99Asn and 176Asn remained challenging to study. An alternative way to reduce the retention caused by peptide backbone is introducing another protease such as Glu-C to perform a combination digestion. After trypsin and Glu-C digestion, the isomeric separation of all three N-glycopeptides from bovine fetuin can be observed (Fig 2). Among which, sites 156Asn (Fig 2a) and 99Asn (Fig 2b) had similar isoform distributions when comparing them to site 176Asn (Fig 2b). To address the elution order and assign the structures of the glycan isoforms, exoglycosidase digestion experiments were conducted.
Figure 2.
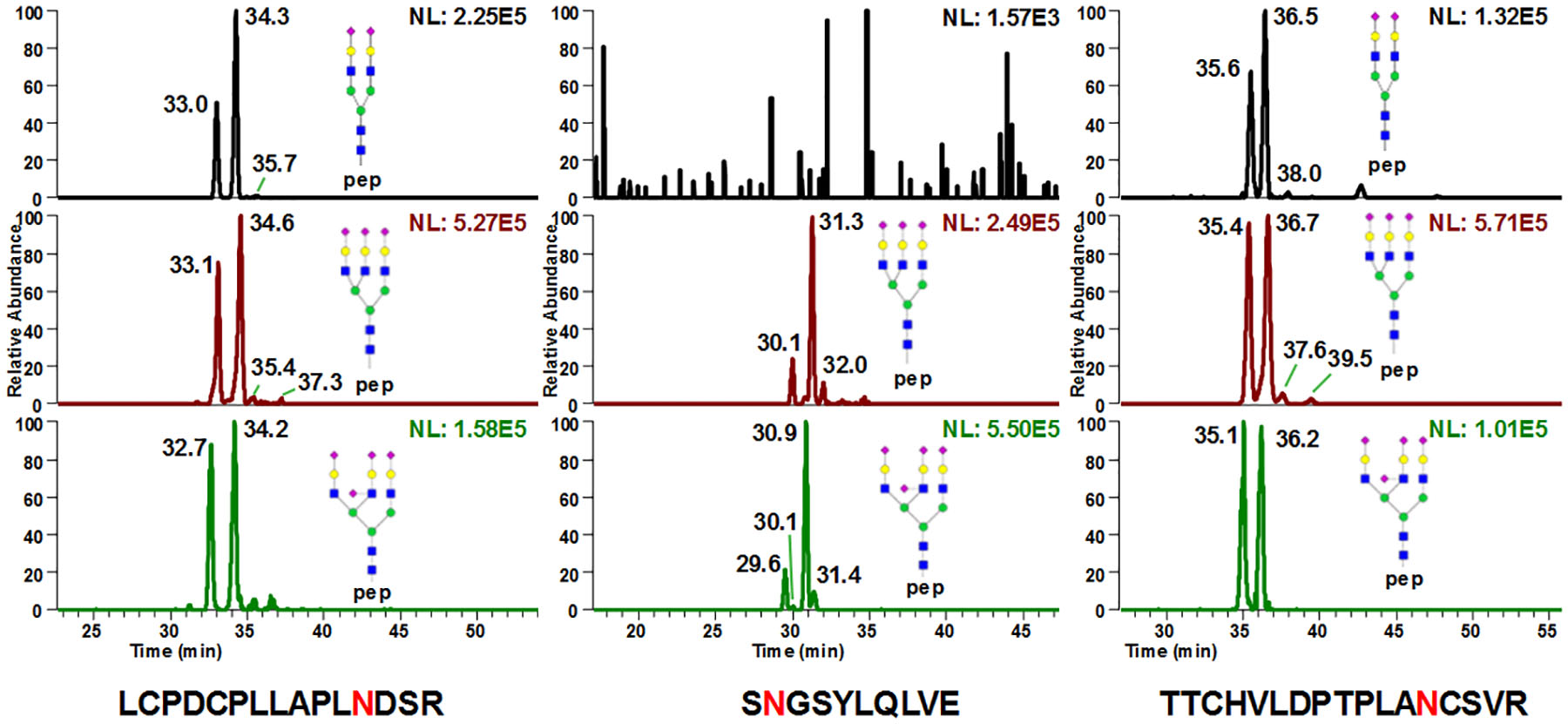
Site specific isomeric separation of glycopeptides (a) 156Asn, (b) 176Asn and (c) 99Asn derived from bovine fetuin digested with trypsin and Glu-C. All three N-glycopeptides from bovine fetuin can be observed. Among which, sites 156Asn (Fig 2a) and 99Asn (Fig 2b) have similar isoform distributions when comparing them to site 176Asn (Fig 2b).
Fig 3 depicts the EICs of A2G2S2, A2G2S1 and A2G2 glycopeptides in non-treated (Fig 3a–3c), α2–3 neuraminidase treated (Fig 3d–3f) and α2–3,6,8,9 neuraminidase treated samples (Fig 3g–3i). The glycopeptides eluted at 37.3 min and 39.2 min (Fig 3a) showed sensitivity for α2–3 neuraminidase, which disappeared after enzymatic digestion (Fig 3d), indicating these two peaks contained at least one sialic acid that was α2–3 linked to a galactose. Upon α2–3 neuraminidase digestion, A2G2S1 can be detected (Fig 3e), which was not originally in sample (Fig 3b). Since the peak at 35.5 min in Fig 3a remained intact after α2–3 neuraminidase digestion (Fig 3d), both the sialic acid linkages can be assigned α2–6. Comparing the intensities between digestion products in Fig 3e and Fig 3f, the 37.3 min peak in Fig 3a can be assigned to have both a α2–3 linked and α2–6 linked sialic acid, while the 39.2 min peak was associated with two α2–3 linked sialic acid. This conclusion is also in consensus with the previous NMR study55, quantitative analysis of reduced N-glycan isomers from bovine fetuin18, as well as the separation of permethylated N-glycans from fetuin54. It clearly shows that on backbone LCPDCPLLAPLNDSR, glycopeptides with α2–6 linked sialic acids have less retention than the ones with α2–3 linked sialic acids. This elution order of sialic acid linkages is also in agreement with the studies of released N-glycans by Altmann et al.53 and Thaysen-Andersen et al.18 MS/MS profiles of these isoforms were also investigated. As depicted in Fig S6, the Y-ions were detected in the MS/MS spectra of the A2G2S2 isomers. However, no significant differences in the fragment peaks were detected.
Figure 3.
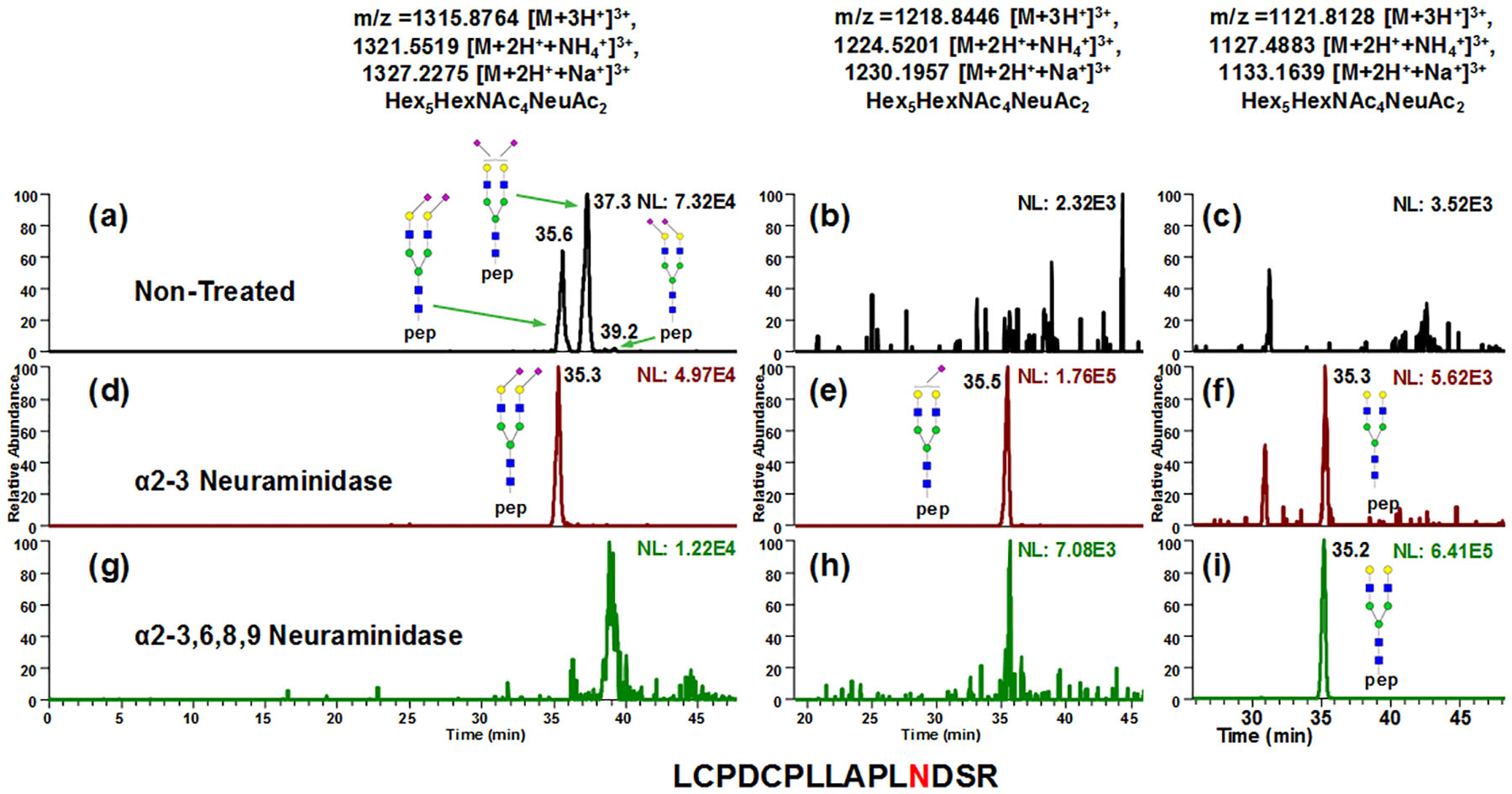
EICs of A2G2S2, A2G2S1 and A2G2 glycopeptides in non-treated (a-c), α2–3 neuraminidase treated (d-f) and α2–3,6,8,9 neuraminidase treated samples (g-i). The peaks at 37.3 min and 39.2 min in (a) showed sensitivity for α2–3 neuraminidase, which disappeared after enzymatic digestion (d), indicating these two peaks were containing at least one sialic acid that were α2–3 linked to galactose. Upon α2–3 neuraminidase digestion, A2G2S1 can be detected (e), which were not originally in sample (b). Since the 35.5 min peak in (a) remained after α2–3 neuraminidase digestion (d), both the sialic acid linkages can be assigned α2–6. Comparing the intensities between (e) and (f), the 37.3 min peak in (a) can be assigned to have both α2–3 linked and α2–6 linked sialic acid, while the 39.2 min peak is associated with two α2–3 linked sialic acid. This conclusion is also in consensus with the previous NMR study55 quantitative analysis of reduced N-glycan isomers from bovine fetuin18, as well as the separation of permethylated N-glycans from fetuin54.
Additionally, O-linked glycopeptides were not detected with the present method due to the length of peptide backbone of 271N, 280Asn, 282Asn and 296Asn. The low occupancy on site 334Asn and 341Asn also limited the characterization of O-linked glycopeptides in bovine fetuin.
Structural elucidation of neutral glycopeptide isomers on PGC-LC-MS
Interestingly, in the α2–3,6,8,9 neuraminidase treated sample, doublets were also observed for A3G3 on site 156Asn (Fig S7a), however, no isomeric separations were noticed on 176Asn and 99Asn (Fig S7b and S7c) though the peak broadening happened in the latter case, which is possibly due to the charge state change after desialylation. Nonetheless, Fig S7a showed promising results of isomerically separating the glycopeptides with neutral structures, which was in accordance with the previous glycan studies.44,55 Therefore, further study of the capability of PGC for separating neutral glycopeptides was conducted using asialofetuin, IgG and RNase B. Fig 4 demonstrates the chromatographic behavior of A3G3, A3G2, A3G1 and A3 glycopeptides in non-treated (top traces), β1–3 galactosidase treated (middle traces) and β1–4 galactosidase treated asialofetuin (bottom traces). LC flow rate was decreased to 0.05 ml/min to improve chromatographic behavior. Comparing between A3G3 in non-treated samples (Fig 4a) and β1–3 galactosidase treated samples (Fig 4b), the glycopeptide eluted at 43.9 min remained non-reactive to β1–3 galactosidase, indicating that the 43.9 min peak has all galactose β1–4 linked. The corresponding digestion product of the 44.7 min peak in Fig 4a can be identified in Fig 4c with a minor amount of overly digested product observed in Fig 4d. Meanwhile, with β1–4 galactosidase treatment, both peaks in (Fig 4a) were found to be digested (Fig 4e). Despite minor amount of under digested product (Fig 4f and 4g), the β1–3 linked A3G1 can be identified (Fig 4g). The linkage difference in different enzymatic digestion products resulted in retention time discrepancy between 42.0 min (Fig 4d) and 42.8 min (Fig 4g). The major product of digestion was A3 (Fig 4h), which also proved that the assignment of isomeric structures in (Fig 4a) was correct.
Figure 4.
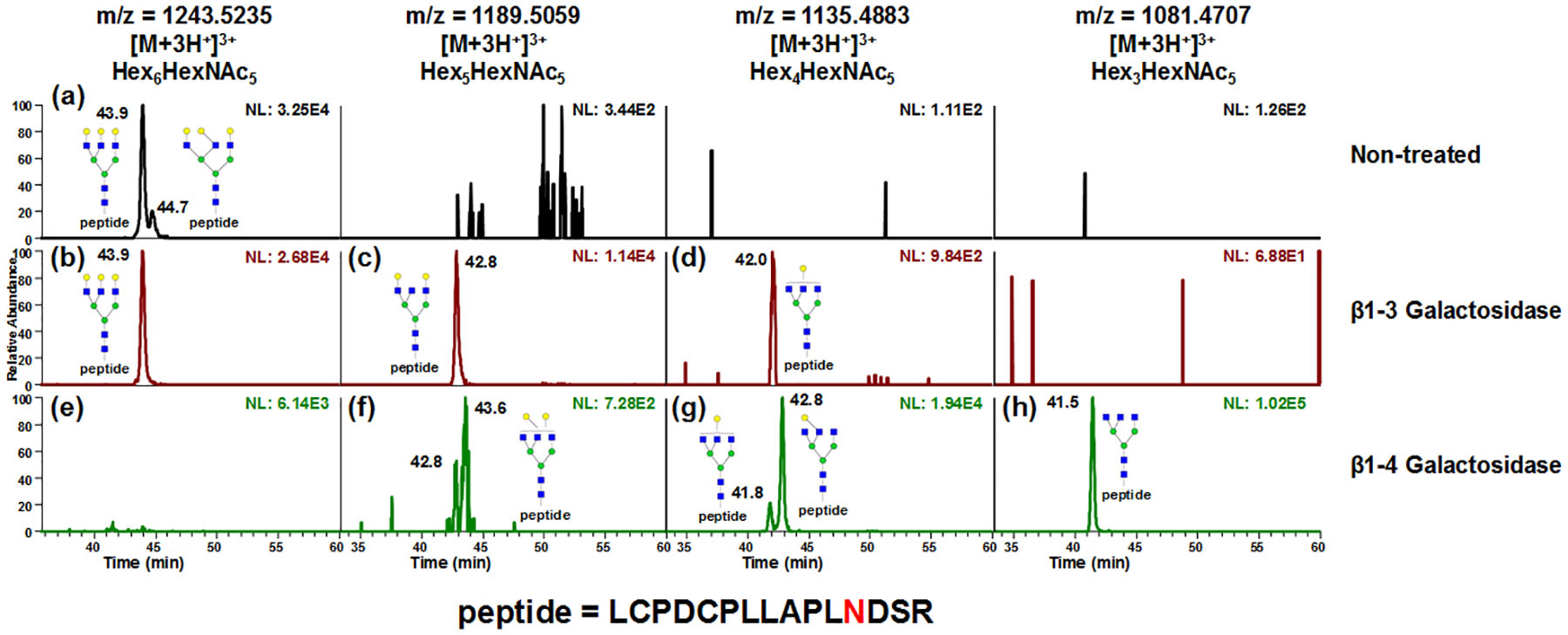
EICs of A3G3, A3G2, A3G1 and A3 glycopeptides in non-treated (top traces), β1–3 galactosidase treated (middle traces) and β1–4 galactosidase treated asialofetuin glycopeptide samples (bottom traces). LC flow rate was decreased to 0.05 ml/min to improve chromatographic behavior. Comparing between A3G3 in non-treated (a) and β1–3 galactosidase treated samples (b), the 43.9 mins peak remain non-reactive to β1–3 galactosidase, indicating that the 43.9 min peak has all galactose β1–4 linked. The corresponding digestion product of 44.7 min peak in (a) can be identified in (c) with a minor amount of overly digested product observed in (d). Meanwhile, with β1–4 galactosidase treatment, both peaks in (a) were found to be digested (e). Despite minor amount of under digested product (f and g), the β1–3 linked A3G1 can be identified in (g). The linkage difference in different enzymatic digestion product resulted in retention time discrepancy between 42.0 min (d) and 42.8 min (g). The major product of digestion was A3 (h), which also proved that the assignment of isomeric structure in (a) was correct.
Not only was it able to separate the linkage isomers prompted by galactose; partial separation of branch isomers caused by galactose positions were also observed in tryptic digested murine IgG1. As shown in Fig 5a–c, A2F1, A2G1F1 and A2G2F1 were observed. After β1–4 galactosidase digestion, the doublets in Fig 5b produced singlet A2F1 (Fig 5d) that was the same as the A2F1 originally in the sample, indicating that the isomeric separation was only driven by galactose linkages. Since both peaks were truncated by the β1–4Gal, the isomeric separation was caused by galactose positions on different arms. The same conclusion can be draw from β1–3,4 galactosidase (Fig 5 bottom traces). The structures were assigned based on the elution order observed by Altmann et al.56 However, due to the lack of enzyme that can branch specifically cleave the monosaccharide residues and the instrumentation to perform MSn study of A2G1F1 glycopeptides, the galactose linkages to the arms were not further confirmed. Another example of positional isomer separation was the glycopeptide with high mannose glycans digested from RNase B. Man5 through Man9 from tryptic glycopeptides were analyzed as shown in Fig 6, in which isomeric separation was observed for Man6, Man7 and Man8 (Fig 6b–6d). The isomeric structures associated with each peak were annotated based on the retention order of the isoforms from works by Altmann et al57 and Lu et al.58 Similar isomeric separation was reported by Lu et al. under room temperature, however, a 2D -LC was employed where PGC is the second dimension of separation.58 Quantification of RNase B glycoform distribution by PGC-LC-MS was compared to previous 1H NMR results59 (Fig 6f and Table S2). In both cases, Man5 was the most abundant structure detected while Man9 had Fthe least abundance. Though similar distributions were observed, Man5 was found to be less abundant by present method than the NMR study, possibly due to the sample differences.
Figure 5.
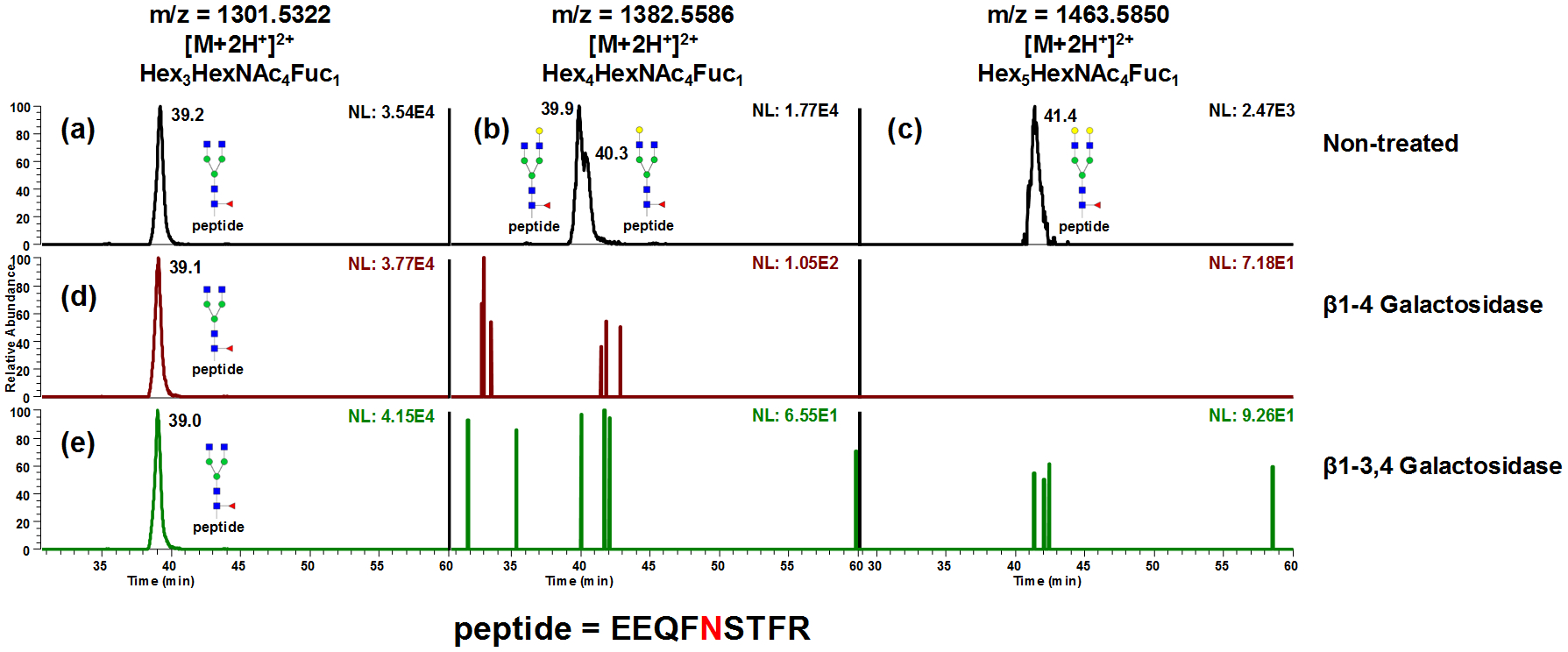
Separation of tryptic digested murine IgG1 glycopeptides. A2F1, A2G1F1 and A2G2F1 were observed (a-c). After β1–4 galactosidase digestion, the doublets in (b) produced singlet A2F1 (d) that is the same to the A2F1 originally in the sample, indicating that the isomeric separation was only driven by galactose linkages. Since both peaks were truncated by the β1–4 galactosidase, the isomeric separation was caused by galactose positions on different arms. The same conclusion can be draw from β1–3,4 galactosidase digestion (e). The structures of A2G1F1 isomers were assigned based on the elution order observed by Altmann et al.56
Figure 6.
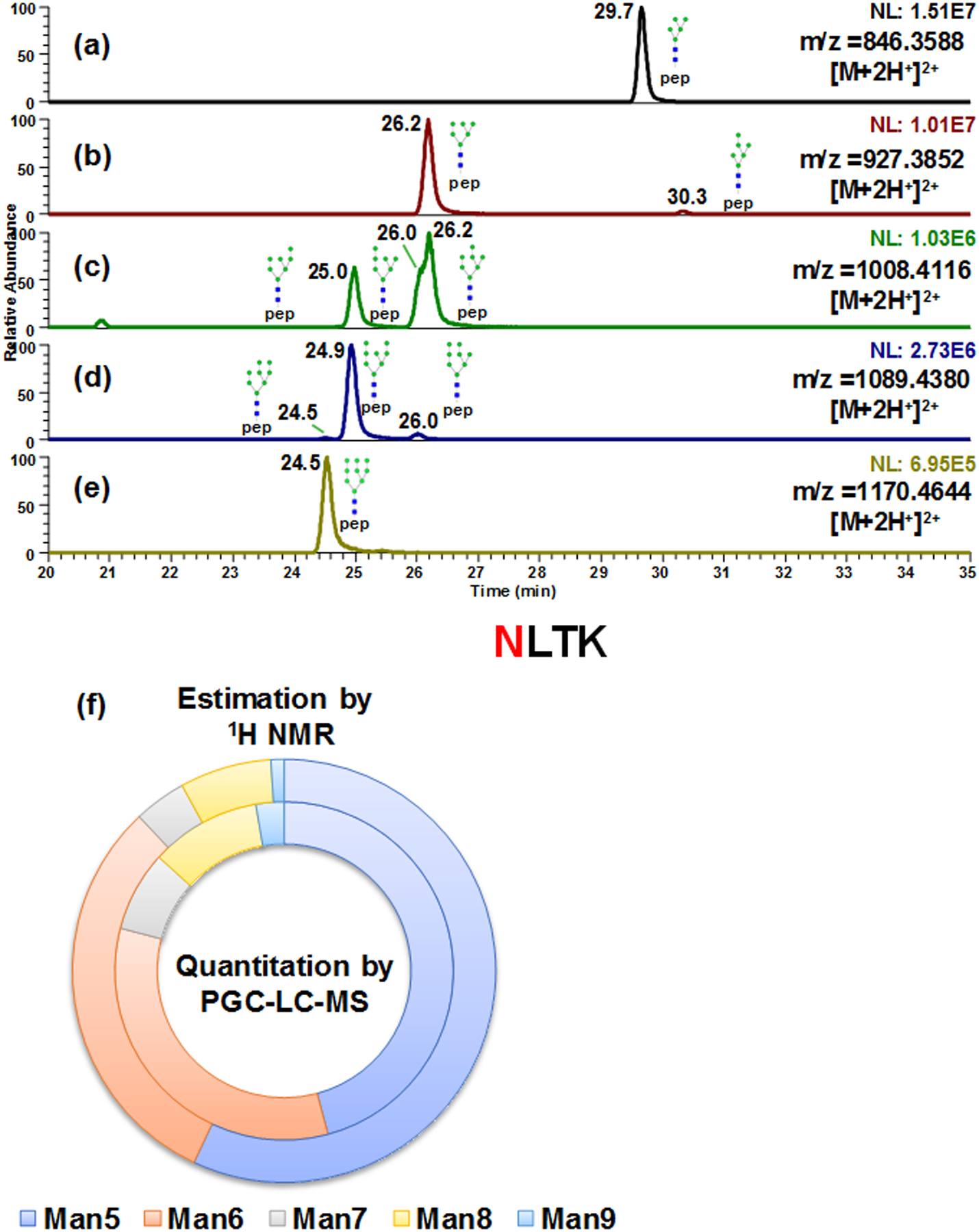
Isomeric separation of high mannose glycopeptides (a-e) derived from bovine Ribonuclease B. Isomeric structures for Man6, Man7 and Man8 were separated (b-d). Quantitative results by present method (inner ring) was compared to previous 1H NMR study59 (outer ring) (f).
Isomeric separation of complex glycopeptides derived from AGP
Elution profiles of glycopeptides composed of different peptide moieties, antennary types, degrees of sialylation and fucosylation were shown in Fig 7 and Fig S8. The isomeric separation was achieved comprehensively over all detected AGP glycopeptides. The PGC-LC-MS analysis permitted the identification of 62 glycopeptide isoforms derived from all 5 glycosylation sites originated from trypsin and chymotrypsin digested AGP. The retention times and glycoforms for each glycopeptide isomers were summarized in Table S3. A neuraminidase digestion experiment was also performed on the AGP sample. Fig S9a depicts the EIC of tetra-sialylated glycopeptide on 103Asn. After α2–3 neuraminidase treatment, only the first peak at 24.9 min remained, suggesting that the sialic acids in the first eluted peak were all α2–6 linked (Fig S9b). Since no peaks were detected after α2–3,6,8,9 neuraminidase digestion (Fig S9c), the peaks in Fig S9a were designated as true peaks for glycopeptides. This is also in consensus with the results in bovine fetuin sample as aforementioned. However, no other isomeric structures were assigned due to the complexity of the sample and lack of the enzyme that is specific to α2–6 linked sialic acids.
Figure 7.
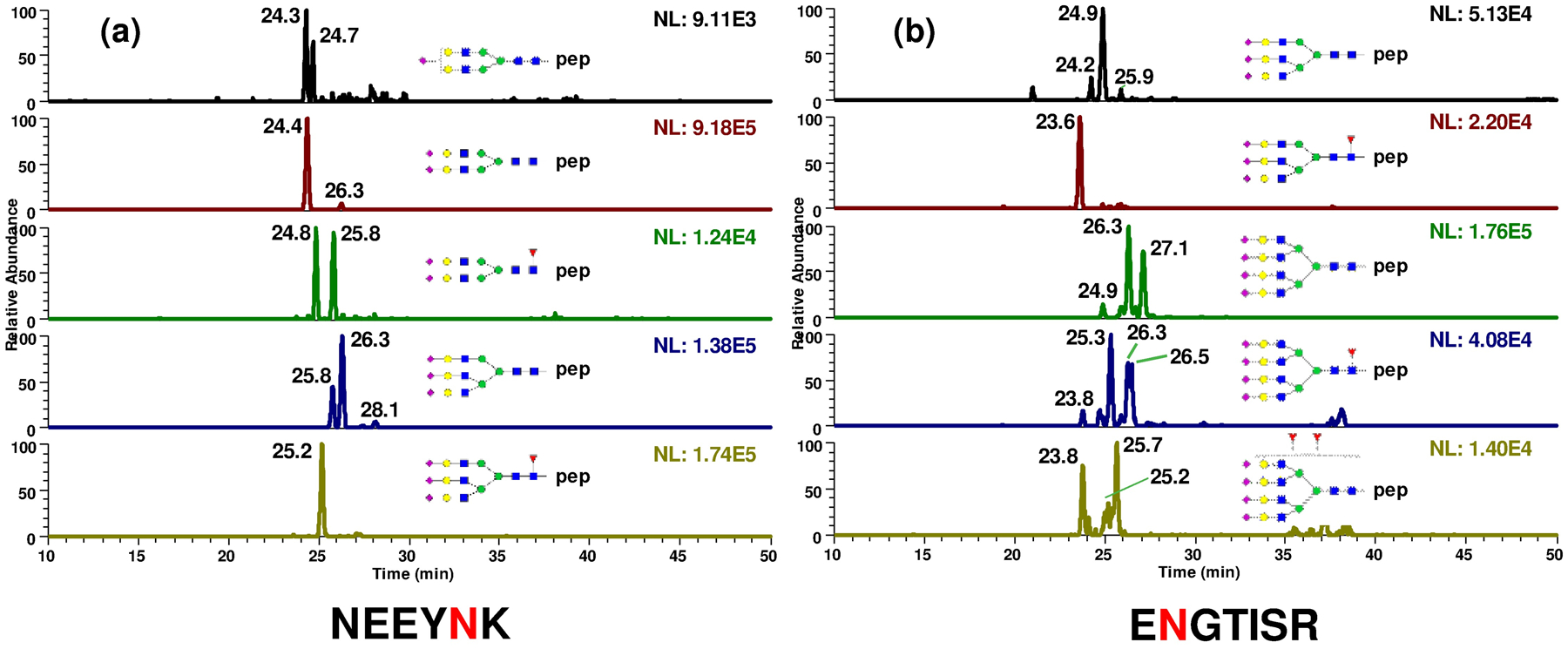
Glycopeptides composed of different peptide moieties, antennary types, degrees of sialylation derived from α1-acid glycoprotein can be achieved on PGC. The glycoform distribution as well as their isoform distribution of all 5 glycosylation sites were characterized in the same run. Glycopeptide structures identified from glycosylation sites 56Asn (a) and 103Asn (b) were shown here, 33Asn, 72Asn and 93Asn were depicted in Fig S8.
In addition, the glycosylation mirco-heterogeneity of AGP presented different profiles corresponding to different sites. For example, none of the tetra-antennary type glycans were found to be attached to 56Asn (Fig 7a), while 10 glycopeptides isoforms with tetra-antennary type glycans were detected to be attached to 103Asn (Fig 7b). This distinction suggested the glycan occupancy on each site of AGP was expected to differ from the overall released glycan profiles. Considering that previous reports have shown alterations in not only compositions but also isomeric distributions of released glycans correlating to diseases,60,61 the capability of site specific isomeric analysis is expected to prompt more understanding of the biological significance of AGP.
However, due to the great complexities of biological samples such as blood serum or tissues, it is noteworthy to point out that the site-specific glycopeptide isoform profiles could be too complicated to interpret and illustrate. This method is suggested to be used for targeted analysis of a single glycoprotein with purification methods such as immunoprecipitation62 and size-exclusion chromatography.63 It is also recommended to perform a PNGase F digestion, which gives the overall N-glycosylation profiles, prior to the separation to prevent the false assignment of the isomers.
Conclusion
The first extensive characterization of isomeric separation of glycopeptides using PGC column is conducted in this study. As the key factors in PGC separation, both pH and temperature were optimized to achieve the efficient separation. Reproducibility testing showed the method had less than 0.3 min retention time differences in different days. The high temperature PGC-LC-MS method was applied to a variety of standard glycoproteins possessing high mannose, sialylated, neutral and fucosylated glycans including both linkage isomers and positional isomers. This method allowed for a micro-heterogeneity study of isomeric separation of glycoproteins in interest with a combination of site-specific enzymatic digestions. Structure elucidation was performed using exoglycosidase digestion strategies. Furthermore, the isomeric separation of glycopeptide derived from AGP allows for more detailed analyses of AGP glycosylation, which will be advantageous to future biomarker research. In conclusion, conducting PGC-LC-MS in basic conditions and at high temperature is a useful and practical method for glycoprotein characterization.
Supplementary Material
Table S1. Retention times and theirs RSDs of tryptic digested fetuin glycopeptide isomers on LCPDCPLLAPL156NDSR over 15 runs
Table S2. Comparison of quantitative results of glycopeptides derived from RNaseB between PGC-LC-MS and 1H NMR study by Fu et al.59
Table S3. Summary of the retention times and glycoforms for each glycopeptide isomers derived from AGP glycopeptides. AGP was digested with trypsin and chymotrypsin. See additional information provided in the method section of the paper.
Figure S1. EICs of peptides (a-d) and glycopeptides (e-f) in tryptic digested fetuin sample under acidic conditions. The highly sialylated structures were not eluted from the column.
Figure S2. EICs of peptides (a-d) and glycopeptides (e-f) in tryptic digested fetuin sample under basic conditions. Glycopeptides were isomerically separated with stronger retention and uniformed charge states. Deglycosylated peptide backbone were not resolved indicating the separation was prompted by glycan isoforms (g).
Figure S3. Intensity comparison of tryptic digest of bovine fetuin under (a) 100% water, 0.1% formic acid (pH 2.8) and (b) 100% water, 25mM ammonium acetate and 1.25% ammonium hydroxide (pH 9.9) in direct infusion mode. Each trace represented an average of 100 scans. No significant decrease in intensities were observed.
Figure S4. Full MS spectrum of A2G2S2 glycopeptide derived from bovine fetuin. Both ammoniated and sodiated adducts were detected due to the additives in mobile phases.
Figure S5. EICs of 156Asn glycopeptide PGC-LC-MS with 15 injections. The first 10 runs were made back to back and runs 11–15 were made on the second day of experiment. The retention time between each run was less than 1 min, demonstrating the reproducibility of the separation method.
Figure S6. MS/MS spectra of A2G2S2 glycopeptide isomers with (a) both sialic acids with α2–6 linkage and (b) one sialic acid with α2–3 linkage and the other with α2–6 linkage. No significant differences were observed between the two spectra.
Figure S7. EICs of A3G3 glycopeptides from (a) 156Asn, (b) 176Asn and (c) 99Asn sites in desialylated bovine fetuin. Partial separation of galactose linkage isomers can be obtained (a), while no isomeric separation was observed on the other two glycosylation sites.
Figure S8. EICs of glycopeptides identified from glycosylation sites 33Asn, 72Asn and 93Asn in AGP sample sequentially digested by trypsin and chymotrypsin.
Figure S9. The results from neuraminidase digestion experiment of AGP glycopeptide ENGTISR with A4G4S4. Only the glycopeptide eluted at 24.9 min in (a) remained after α2–3 neuraminidase treatment, indicating that the peak is fully occupied with α2–6 sialic acids. The other two glycopeptides (26.3 min and 27.1 min peaks in a) possessed at least one α2–3 linked sialic acid, since both peaks disappeared after the α2–3 enzyme digestion (b). All the three peaks (24.9 min, 26.3 min and 27.1 min) are associated with sialylated glycopeptides because none was detected after α2–3,6,8,9 neuraminidase digestion (c). However, no further identification was able to achieve due to the complexity of the sample and lack of enzymes specific to α2–6 linked sialic acids.
Acknowledgement
NIH 1R01GM130091-03 and 1U01CA225753-02.
Footnotes
Conflict of interest statement
The authors declare no conflict of interest.
References
- (1).Apweiler R; Hermjakob H; Sharon N Biochim Biophys Acta 1999, 1473, 4–8. [DOI] [PubMed] [Google Scholar]
- (2).Helenius A; Aebi M Science 2001, 291, 2364–2369. [DOI] [PubMed] [Google Scholar]
- (3).Varki A Glycobiology 1993, 3, 97–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).O’Connor SE; Imperiali B Chem Biol 1996, 3, 803–812. [DOI] [PubMed] [Google Scholar]
- (5).Dube DH; Bertozzi CR Nature reviews. Drug discovery 2005, 4, 477–488. [DOI] [PubMed] [Google Scholar]
- (6).Campbell BJ; Yu LG; Rhodes JM Glycoconjugate journal 2001, 18, 851–858. [DOI] [PubMed] [Google Scholar]
- (7).Song E; Zhu R; Hammoud ZT; Mechref Y J Proteome Res 2014, 13, 4808–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Smith KD; Pollacchi A; Field M; Watson J Biomedical chromatography : BMC 2002, 16, 261–266. [DOI] [PubMed] [Google Scholar]
- (9).Elliott MA; Elliott HG; Gallagher K; McGuire J; Field M; Smith KD Journal of chromatography. B, Biomedical sciences and applications 1997, 688, 229–237. [DOI] [PubMed] [Google Scholar]
- (10).Botella-Lopez A; Burgaya F; Gavin R; Garcia-Ayllon MS; Gomez-Tortosa E; Pena-Casanova J; Urena JM; Del Rio JA; Blesa R; Soriano E; Saez-Valero J Proceedings of the National Academy of Sciences of the United States of America 2006, 103, 5573–5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang JZ; Grundke-Iqbal I; Iqbal K Nature medicine 1996, 2, 871–875. [DOI] [PubMed] [Google Scholar]
- (12).Abou-Abbass H; Abou-El-Hassan H; Bahmad H; Zibara K; Zebian A; Youssef R; Ismail J; Zhu R; Zhou S; Dong X; Nasser M; Bahmad M; Darwish H; Mechref Y; Kobeissy F Electrophoresis 2016, 37, 1549–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Abou-Abbass H; Bahmad H; Abou-El-Hassan H; Zhu R; Zhou S; Dong X; Hamade E; Mallah K; Zebian A; Ramadan N; Mondello S; Fares J; Comair Y; Atweh S; Darwish H; Zibara K; Mechref Y; Kobeissy F Electrophoresis 2016, 37, 1562–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mechref Y; Hu Y; Garcia A; Hussein A Electrophoresis 2012, 33, 1755–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tsai TH; Song E; Zhu R; Di Poto C; Wang M; Luo Y; Varghese RS; Tadesse MG; Ziada DH; Desai CS; Shetty K; Mechref Y; Ressom HW Proteomics 2015, 15, 2369–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zacharias LG; Hartmann AK; Song E; Zhao J; Zhu R; Mirzaei P; Mechref Y J Proteome Res 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dwek RA Chem Rev 1996, 96, 683–720. [DOI] [PubMed] [Google Scholar]
- (18).Palmisano G; Larsen MR; Packer NH; Thaysen-Andersen M RSC Advances 2013, 3, 22706–22726. [Google Scholar]
- (19).Ruhaak LR; Xu G; Li Q; Goonatilleke E; Lebrilla CB Chem Rev 2018, 118, 7886–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yu A; Zhao J; Peng W; Banazadeh A; Williamson SD; Goli M; Huang Y; Mechref Y Electrophoresis 2018, 39, 3104–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ahn YH; Kim JY; Yoo JS Mass spectrometry reviews 2015, 34, 148–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Thaysen-Andersen M; Packer NH Biochim Biophys Acta 2014, 1844, 1437–1452. [DOI] [PubMed] [Google Scholar]
- (23).Chovanec M; Plzak J; Betka J; Brabec J; Kodet R; Smetana K Jr. Oncology reports 2004, 12, 297–301. [PubMed] [Google Scholar]
- (24).Holikova Z; Hrdlickova-Cela E; Plzak J; Smetana K Jr.; Betka J; Dvorankova B; Esner M; Wasano K; Andre S; Kaltner H; Motlik J; Hercogova J; Kodet R; Gabius HJ APMIS : acta pathologica, microbiologica, et immunologica Scandinavica 2002, 110, 845–856. [DOI] [PubMed] [Google Scholar]
- (25).Mondal G; Chatterjee U; Chawla YK; Chatterjee BP Glycoconjugate journal 2011, 28, 1–9. [DOI] [PubMed] [Google Scholar]
- (26).Halim A; Nilsson J; Ruetschi U; Hesse C; Larson G Molecular & cellular proteomics : MCP 2012, 11, M111 013649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Anonsen JH; Vik A; Egge-Jacobsen W; Koomey M J Proteome Res 2012, 11, 5781–5793. [DOI] [PubMed] [Google Scholar]
- (28).Yin X; Bern M; Xing Q; Ho J; Viner R; Mayr M Molecular & cellular proteomics : MCP 2013, 12, 956–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Halim A; Ruetschi U; Larson G; Nilsson J J Proteome Res 2013, 12, 573–584. [DOI] [PubMed] [Google Scholar]
- (30).Ji ES; Lee HK; Park GW; Kim KH; Kim JY; Yoo JS J Chromatogr B Analyt Technol Biomed Life Sci 2019, 1110–1111, 101–107. [DOI] [PubMed] [Google Scholar]
- (31).West C; Elfakir C; Lafosse M J Chromatogr A 2010, 1217, 3201–3216. [DOI] [PubMed] [Google Scholar]
- (32).Wuhrer M; Deelder AM; Hokke CH J Chromatogr B Analyt Technol Biomed Life Sci 2005, 825, 124–133. [DOI] [PubMed] [Google Scholar]
- (33).Thaysen-Andersen M; Wilkinson BL; Payne RJ; Packer NH Electrophoresis 2011, 32, 3536–3545. [DOI] [PubMed] [Google Scholar]
- (34).Alley WR Jr.; Mechref Y; Novotny MV Rapid Commun Mass Spectrom 2009, 23, 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).An HJ; Peavy TR; Hedrick JL; Lebrilla CB Anal Chem 2003, 75, 5628–5637. [DOI] [PubMed] [Google Scholar]
- (36).Froehlich JW; Barboza M; Chu C; Lerno LA Jr.; Clowers BH; Zivkovic AM; German JB; Lebrilla CB Anal Chem 2011, 83, 5541–5547. [DOI] [PubMed] [Google Scholar]
- (37).Hua S; Hu CY; Kim BJ; Totten SM; Oh MJ; Yun N; Nwosu CC; Yoo JS; Lebrilla CB; An HJ J Proteome Res 2013, 12, 4414–4423. [DOI] [PubMed] [Google Scholar]
- (38).Stavenhagen K; Plomp R; Wuhrer M Anal Chem 2015, 87, 11691–11699. [DOI] [PubMed] [Google Scholar]
- (39).Dodds ED; Seipert RR; Clowers BH; German JB; Lebrilla CB J Proteome Res 2009, 8, 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Giansanti P; Tsiatsiani L; Low TY; Heck AJ Nature protocols 2016, 11, 993–1006. [DOI] [PubMed] [Google Scholar]
- (41).Pabst M; Bondili JS; Stadlmann J; Mach L; Altmann F Anal Chem 2007, 79, 5051–5057. [DOI] [PubMed] [Google Scholar]
- (42).Dolan JW Journal of Chromatography A 2002, 965, 195–205. [DOI] [PubMed] [Google Scholar]
- (43).Zhou S; Hu Y; Mechref Y Electrophoresis 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhou S; Dong X; Veillon L; Huang Y; Mechref Y Analytical and bioanalytical chemistry 2017, 409, 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Huang Y; Zhou S; Zhu J; Lubman DM; Mechref Y Electrophoresis 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dong X; Zhou S; Mechref Y Electrophoresis 2016, 37, 1532–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hennion MC; Coquart V; Guenu S; Sella C Journal of Chromatography A 1995, 712, 287–301. [Google Scholar]
- (48).Pereira L J Liq Chromatogr R T 2008, 31, 1687–1731. [Google Scholar]
- (49).Mirza UA; Chait BT Anal Chem 1994, 66, 2898–2904. [DOI] [PubMed] [Google Scholar]
- (50).Blades AT; Ikonomou MG; Kebarle P Analytical Chemistry 1991, 63, 2109–2114. [Google Scholar]
- (51).Van Berkel GJ; Kertesz V Anal Chem 2007, 79, 5510–5520. [DOI] [PubMed] [Google Scholar]
- (52).Kruve A; Kaupmees K Journal of the American Society for Mass Spectrometry 2017, 28, 887–894. [DOI] [PubMed] [Google Scholar]
- (53).Pabst M; Altmann F Anal Chem 2008, 80, 7534–7542. [DOI] [PubMed] [Google Scholar]
- (54).Zhou S; Huang Y; Dong X; Peng W; Veillon L; Kitagawa DAS; Aquino AJA; Mechref Y Anal Chem 2017, 89, 6590–6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Green ED; Adelt G; Baenziger JU; Wilson S; Van Halbeek H J Biol Chem 1988, 263, 18253–18268. [PubMed] [Google Scholar]
- (56).Stadlmann J; Pabst M; Kolarich D; Kunert R; Altmann F Proteomics 2008, 8, 2858–2871. [DOI] [PubMed] [Google Scholar]
- (57).Pabst M; Grass J; Toegel S; Liebminger E; Strasser R; Altmann F Glycobiology 2012, 22, 389–399. [DOI] [PubMed] [Google Scholar]
- (58).Lu J; Fu DM; Yu L; Cao CY; Zou LJ; Liang XM Anal Lett 2017, 50, 315–324. [Google Scholar]
- (59).Fu D; Chen L; O’Neill RA Carbohydr Res 1994, 261, 173–186. [DOI] [PubMed] [Google Scholar]
- (60).Balmana M; Gimenez E; Puerta A; Llop E; Figueras J; Fort E; Sanz-Nebot V; de Bolos C; Rizzi A; Barrabes S; de Frutos M; Peracaula R J Proteomics 2016, 132, 144–154. [DOI] [PubMed] [Google Scholar]
- (61).Gimenez E; Balmana M; Figueras J; Fort E; de Bolos C; Sanz-Nebot V; Peracaula R; Rizzi A Anal Chim Acta 2015, 866, 59–68. [DOI] [PubMed] [Google Scholar]
- (62).Zhu J; Lin Z; Wu J; Yin H; Dai J; Feng Z; Marrero J; Lubman DM J Proteome Res 2014, 13, 2986–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Graslund S; Nordlund P; Weigelt J; Hallberg BM; Bray J; Gileadi O; Knapp S; Oppermann U; Arrowsmith C; Hui R; Ming J; dhe-Paganon S; Park HW; Savchenko A; Yee A; Edwards A; Vincentelli R; Cambillau C; Kim R; Kim SH, et al. Nature methods 2008, 5, 135–146.18235434 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Retention times and theirs RSDs of tryptic digested fetuin glycopeptide isomers on LCPDCPLLAPL156NDSR over 15 runs
Table S2. Comparison of quantitative results of glycopeptides derived from RNaseB between PGC-LC-MS and 1H NMR study by Fu et al.59
Table S3. Summary of the retention times and glycoforms for each glycopeptide isomers derived from AGP glycopeptides. AGP was digested with trypsin and chymotrypsin. See additional information provided in the method section of the paper.
Figure S1. EICs of peptides (a-d) and glycopeptides (e-f) in tryptic digested fetuin sample under acidic conditions. The highly sialylated structures were not eluted from the column.
Figure S2. EICs of peptides (a-d) and glycopeptides (e-f) in tryptic digested fetuin sample under basic conditions. Glycopeptides were isomerically separated with stronger retention and uniformed charge states. Deglycosylated peptide backbone were not resolved indicating the separation was prompted by glycan isoforms (g).
Figure S3. Intensity comparison of tryptic digest of bovine fetuin under (a) 100% water, 0.1% formic acid (pH 2.8) and (b) 100% water, 25mM ammonium acetate and 1.25% ammonium hydroxide (pH 9.9) in direct infusion mode. Each trace represented an average of 100 scans. No significant decrease in intensities were observed.
Figure S4. Full MS spectrum of A2G2S2 glycopeptide derived from bovine fetuin. Both ammoniated and sodiated adducts were detected due to the additives in mobile phases.
Figure S5. EICs of 156Asn glycopeptide PGC-LC-MS with 15 injections. The first 10 runs were made back to back and runs 11–15 were made on the second day of experiment. The retention time between each run was less than 1 min, demonstrating the reproducibility of the separation method.
Figure S6. MS/MS spectra of A2G2S2 glycopeptide isomers with (a) both sialic acids with α2–6 linkage and (b) one sialic acid with α2–3 linkage and the other with α2–6 linkage. No significant differences were observed between the two spectra.
Figure S7. EICs of A3G3 glycopeptides from (a) 156Asn, (b) 176Asn and (c) 99Asn sites in desialylated bovine fetuin. Partial separation of galactose linkage isomers can be obtained (a), while no isomeric separation was observed on the other two glycosylation sites.
Figure S8. EICs of glycopeptides identified from glycosylation sites 33Asn, 72Asn and 93Asn in AGP sample sequentially digested by trypsin and chymotrypsin.
Figure S9. The results from neuraminidase digestion experiment of AGP glycopeptide ENGTISR with A4G4S4. Only the glycopeptide eluted at 24.9 min in (a) remained after α2–3 neuraminidase treatment, indicating that the peak is fully occupied with α2–6 sialic acids. The other two glycopeptides (26.3 min and 27.1 min peaks in a) possessed at least one α2–3 linked sialic acid, since both peaks disappeared after the α2–3 enzyme digestion (b). All the three peaks (24.9 min, 26.3 min and 27.1 min) are associated with sialylated glycopeptides because none was detected after α2–3,6,8,9 neuraminidase digestion (c). However, no further identification was able to achieve due to the complexity of the sample and lack of enzymes specific to α2–6 linked sialic acids.