

Abstract
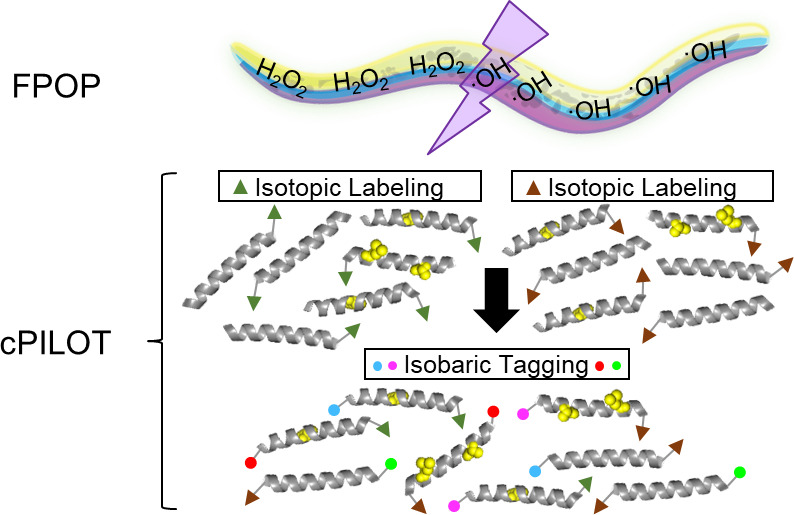
In vivo fast photochemical oxidation of proteins (IV-FPOP) is a hydroxyl radical protein footprinting method used to study protein structure and protein–protein interactions. Oxidatively modified proteins by IV-FPOP are analyzed by mass spectrometry (MS), and the extent of oxidation is quantified by label-free MS. Peptide oxidation changes yield useful information about protein structure, due to changes in solvent accessibility. However, the sample size necessary for animal studies requires increased sample preparation and instrument time. Here, we report the combined application of IV-FPOP and the enhanced multiplexing strategy combined precursor isotopic labeling and isobaric tagging (cPILOT) for higher-throughput analysis of oxidative modifications in C. elegans. Key differences in the performance of label-free MS and cPILOT were identified. The addition of oxygen (+16) was the most abundant modification identified among all known possible FPOP modifications. This study presents IV-FPOP coupled with enhanced multiplexing strategies such as cPILOT to increase throughput of studies seeking to examine oxidative protein modifications.
Proteins are multifaceted macromolecules found in living organisms that function as catalysts for numerous biochemical reactions and that regulate biological processes such as cell signaling, regulation, and structure. Structural understanding of these interactions has been predominantly studied by X-ray crystallography,1 nuclear magnetic resonance (NMR),2 cryogenic electron microscopy,3 and most recently mass spectrometry (MS).4 MS-based methods have the advantage over other techniques of identifying and quantifying macromolecules from complex mixtures with high sensitivity and low detection limits (i.e., atto- to zeptomole).
In recent years, protein footprinting by MS has become increasingly used to study protein conformation and protein–ligand interactions.4 These methods monitor changes in protein solvent accessible surface area (SASA) using reversible or irreversible chemical labels, which can be detected and quantified by MS. One covalent labeling technique, hydroxyl radical (OH) protein footprinting (HRPF),5 irreversibly labels the side chains of solvent accessible amino acids using OH. Although there are multiple ways to generate OH labels (e.g., Fenton chemistry,6 water radiolysis,7 and electrochemistry8), fast photochemical oxidation of proteins (FPOP) generates OH on the submillisecond time scale.9 FPOP utilizes an excimer laser at 248 nm to photolyze hydrogen peroxide (H2O2) to generate OH.9,10In vitro applications of this technique have identified protein–ligand11 and protein–protein interaction sites12 and regions of conformational change.13
FPOP is a versatile method and has been used to study complex protein biological systems in-cell (IC-FPOP).14,15 FPOP is particularly suited for in-cell protein studies because the irreversible OH label allows for postlabeling sample handling procedures, including protein extraction, precipitation, purification, and digestion, prior to liquid chromatography (LC) coupled to tandem MS analysis. Recently, Espino and Jones extended the use of FPOP for in vivo structural analysis in Caenorhabditis elegans (C. elegans).16 The roundworm is suitable for in vivo FPOP (IV-FPOP) studies because of its facile uptake of H2O2 and its transparency to the laser light.17 Due to its conserved genome with higher-ordered species, C. elegans has been used to study molecular and developmental biology pathways and even host–pathogen response.18,19C. elegans is an excellent animal model also to study biology in the context of human diseases.
IV-FPOP of C. elegans for various applications leads to substantial analysis time and costs. For example, most protein footprinting experiments require control conditions (e.g., no laser/H2O2 and no H2O2) to subtract background oxidation signals. In addition, protein footprinting studies typically require technical replication and experimental conditions, such as ligand-free against ligand-bound or normal versus diseased. As protein footprinting methods are further developed to study complex protein systems in-cell and animals, the number of controls, experimental conditions, plus the number of biological and workflow replicates can result in as many as 24 samples for a given biological question. Recently, IC- and IV-FPOP have been effectively demonstrated using label-free MS. For complex mixtures, it is desirable to reduce analysis time and variation introduced from post-labeling procedures. Here, we coupled IV-FPOP to an enhanced multiplexing strategy termed combined precursor isotopic labeling and isobaric tagging (cPILOT).20 cPILOT increases the number of samples that can be analyzed simultaneously by combining isotopic labels (i.e., dimethylation) with isobaric chemical reagents (i.e., tandem mass tags (TMT), isobaric tag for relative and absolute quantitation (iTRAQ), or N,N-dimethyl leucine (DiLeu)).20−22 For example, the N-terminus of peptides is labeled by light or heavy dimethylation (C2H6 or 13C22H6, respectively), while lysine residues are labeled by TMT 6-16 plex reagents. Recent advancements in isobaric reagents with DiLeu23 have increased the capabilities of cPILOT to 24 samples.24
Here, we describe the analysis of the C. elegans proteome modified by IV-FPOP using cPILOT enhanced multiplexing. Briefly, after IV-FPOP, proteins are extracted and digested, and peptides are labeled by cPILOT. Labeled peptides are then analyzed using strong cation exchange (SCX) fractionation LC-MS, MS/MS, and MS3 (Figure 1). A comparison of the evaluation of oxidatively modified proteins and peptides by label-free quantification and cPILOT shows the latter’s capability to reduce analysis time while also maintaining the large sample sizes required for IV-FPOP. In addition, the coupling of FPOP with cPILOT led to a higher reproducibility in both the identification and quantification of oxidatively modified proteins across biological replicates. Oxidatively modified proteins were further analyzed to evaluate site-specific modifications and coverage with label-free and cPILOT approaches.
Figure 1.

Experimental workflow for IV-FPOP cPILOT. C. elegans (N = 20,000) is grown to its fourth larva stage (L4). Three conditions (control, control oxidation, and FPOP) have three biological replicates. Worms in the presence of hydrogen peroxide are flown through a 250 μm i.d. fused silica and irradiated using a KrF excimer at 248 nm wavelength. Each biological replicate has two workflow replicates, resulting in 18 samples. Following oxidative labeling, samples are lysed and digested, and peptides are further labeled by cPILOT. Specifically, peptides (50 μg) are labeled by either light- or heavy dimethylation at the N-terminus and isobarically tagged by TMT 10-plex at lysine residues. Finally, peptides are analyzed using LC-MS, MS/MS, and MS3 on an Orbitrap Fusion Lumos, and the extent of modification of each protein of interest for every condition is calculated.
Experimental Procedures
IV-FPOP
Covalent labeling studies were performed as previously described with slight modifications.16 Each sample was prepared with approximatively 20,000 worms. Worms were kept separated from hydrogen peroxide and mixed together prior to IV-FPOP labeling using two 5 mL syringes (SGE Analytical Science). Each syringe was connected to a fused silica capillary (Polymicro Technologies), 250 μm inner diameter (i.d.), and advanced by a syringe pump (KD Scientific, Legato Model 101) at a final flow rate of 363.26 μL*min–1. Worms were mixed in the syringe using a VP710 tumble stirrer (V&P Scientific) with six stir discs (VP722fF, V&P Scientific) to prevent settling. The final peroxide concentration was 200 mM. The KrF excimer laser (GAM Laser Inc.) at a wavelength of 248 nm was set to 50 Hz pulse frequency, laser energy of 145 ± 1.01 mJ, pulse width of 2.46 mm, and zero exclusion volume. Immediately after labeling, worms were collected in a 15 mL conical tube containing the cell permeable quench solution [20 mM N′-dimethylthiourea (DMTU) and 20 mM N-tert-butyl-α-phenylnitrone (PBN)], in order to eliminate excess hydrogen peroxide and OH radicals, respectively. Methionine sulfoxide reductase was inhibited by adding 1% DMSO to the final sample. All irradiated samples were labeled in technical duplicates and biological triplicates with an equal number of controls (peroxide and no laser irradiation, no peroxide, and no laser irradiation).
Labeling by cPILOT
Peptides (50 μg) generated from trypsin digestion were dissolved in 1% acetic acid (0.25 μg*μL–1). Formaldehyde/deuterated formaldehyde (Sigma-Aldrich) (8 μL) and sodium cyanoborohydride/-deuteride (Sigma-Aldrich) (8 μL) were added to label peptides with either light (−C2H6) or heavy (−13C22H6) dimethylation, respectively. Peptides were reacted for 10 min (room temperature) with shaking. The reactions were quenched with 1% ammonia (16 μL, 5 min). Dimethylated peptides were reacidified with 5% FA, and light and heavy pairs were pooled (Supplemental Figure S1 and Table S1), desalted, and dried down by centrifugal evaporation. Desalted dimethylated peptides were dissolved in 100 mM triethylammonium bicarbonate buffer, and TMT6-plex reagents were prepared according to the manufacturer’s protocol. TMT6 -plex reagents were added to peptides and reacted at room temperature for 1 h with shaking. Peptides labeled by cPILOT were quenched with 5% (w/v) hydroxylamine hydrochloride for 15 min at room temperature and reacidified with FA. Peptides were pooled into a single sample, dried down to remove ACN, desalted, and dried down again by centrifugal evaporation. Peptides were then separated by SCX fractionation.
Offline Strong Cation Exchange (SCX) Fractionation
The pooled sample containing peptides labeled by cPILOT was fractionated by SCX according to the manufacturer’s protocol (Protea Biosciences). Briefly, peptides (500 μg) were dissolved in buffer A and loaded onto a preactivated spin column. Peptides were eluted from the spin column in eight intervals (room temperature, 6 min, 4000 × g) with increasing ammonium formate solutions (20, 40, 60, 80, 100, 150, 250, and 500 mM). Fractionated peptides were dried down by centrifugal evaporation and dissolved in 0.1% FA in water (v/v).
Liquid Chromatography and Mass Spectrometry Analyses
Label-Free MS Analysis
Online desalting and reversed-phase (RP) chromatography was performed with an Acquity UPLC M-Class System (Waters). Mobile phases A and B were 0.1% FA in water (v/v) and 0.1% FA in ACN (v/v), respectively. Peptides (∼50 μg) were loaded onto a commercial (Waters) trapping column (180 μm × 20 mm) containing C18 (5 μm, 100 Å) at 15 μL*min–1 in 0.1% FA in water (v/v) for 10 min. After desalting, samples were loaded onto an analytical column (75 μm i.d. × 20 cm). Peptides were separated on a RP analytical column packed in-house with C18 (Aqua, 5 μm, 125 Å, Phenomenex). The gradient was as follows: 0–1 min, 3% B; 2–90 min 10–45% B; 100–105 min 100% B; 106–120 min 3% B. Data-dependent acquisition parameters were performed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher) as follows: the MS survey scan in the Orbitrap (OT, 375–1500 m/z) was 60,000 resolution; the most intense peaks within 4 s (Top Speed) were isolated (1.2 m/z) and fragmented with high-energy collisional dissociation (HCD) in the OT (15,000 resolution) with a normalized collision energy (NCE) of 32%, AGC target of 5.0 × 105, dynamic exclusion of 60 s, ppm mass tolerance of 10, a maximum IT of 35 ms, and an intensity threshold of 5.0 × 104.
FPOP-cPILOT MS Analysis
Online desalting and RP chromatography was performed with a nano-UHPLC system equipped with an autosampler (Dionex, ThermoFisher Scientific). Mobile phases A and B were 0.1% FA in water (v/v) and 0.1% FA in ACN (v/v), respectively. Peptides (250 ng) were loaded onto a commercial (Thermo Fisher Scientific) trapping column (75 μm i.d. × 2 cm) containing C18 (XBridge BEH, 3 μm, 100 Å) at 2 μL*min–1 in 0.1% FA in water for 10 min. After desalting, each fraction was loaded onto an analytical column (100 μm i.d. × 23 cm), packed in-house with C18 (2.5 μm, 150 Å, Waters). The gradient was as follows: 0–10 min, 10% mobile phase B; 10–30 min, 10–15% B; 30–75 min, 15–30% B; 75–88 min, 30–60% B; 88–92 min, 60–90% B; 92–99 min, 90% B; 99–100 min, 90–10% B; 100–120 min, 10% B. Data-dependent acquisition parameters were performed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher) as follows: the MS survey scan in the Orbitrap (375–1500 m/z) was 120,000 resolution; the most intense peaks within 3 s (Top Speed) were isolated (2 m/z) and fragmented with CID in the ion trap with an NCE of 35%, AGC of 1 × 104, dynamic exclusion of 20 s, ppm mass tolerance of 10, and maximum IT of 100 ms. Directly after each MS/MS scan, the ten most intense fragment ions (over varying m/z ranges) were selected for an additional fragmentation (MS3) event by HCD and analyzed in the OT (scan range: 100–400 m/z, isolation width: 2 m/z, AGC: 5 × 104, NCE: 55%, resolution: 60,000, maximum IT: 118 ms). Other parameters such as precursor selection range, precursor ion exclusion, and isobaric tag loss exclusion were set as default. The targeted mass difference node was employed. Mass differences of 8.0444 Da (heavy dimethylation (DM) – light DM)) and 7.0381 Da (dimethyl 7-light DM) were listed, and the partner intensity range relative to the most intense precursor was set to 70–100%. A subsequent scan was performed on both ions in the pair and matching charge states for ions in the pair.
Data Analysis
RAW files were analyzed with Proteome Discoverer v. 2.2 software (Thermo Scientific). Spectra were searched against the Uniprot C. elegans database (07/17/2018, 26,794 sequences) to obtain sequence information. Parameters applied to SEQUEST HT were as follows: one maximum trypsin missed cleavage, precursor mass tolerance of 15 ppm, fragment mass tolerance of 1 Da; static modifications were either light (+28.031 Da) or heavy (+36.028 or +35.070 Da) dimethyl on peptide N-termini and carbamidomethyl (+57.021 Da) of cysteine residues; dynamic modifications were TMT six-plex (+229.163 Da) of lysine residues. Additionally, all known hydroxyl radical side-chain modifications25,26 were searched as dynamic modifications. Raw data was searched twice to account for light or heavy dimethylation modifications. Percolator database searching was employed to generate medium (p < 0.05) confidence peptide lists. Peptides with high or medium confidence were used to identify and quantify proteins. Filters applied for peptides were as follows: PSMs (peptide to spectral match) > 1 across biological cohorts, peptide confidence level of medium, peptide rank of 1, peptide deviation of 10 ppm, and S/N ≥ 10. Reporter ion (m/z 126–131) intensities had the following parameters: most confident centroid and reporter ion mass tolerance of 30 ppm. Furthermore, reporter ion values were normalized using internal reference scaling.27 The extent of oxidation per peptide was calculated as previously described28 using the following equation
where reporter ion abundance (RIA) modified is the abundance intensity of the peptide with the hydroxyl radical modification, and RIA total is the total abundance intensity of the same peptide with and without the hydroxyl radical modification.
Results and Discussion
C. elegans is a suitable model organism to study biologically relevant interactions as it grows quickly, has a short lifespan, is transparent, and has an evolutionarily conserved innate immune response. Due to noted transparency at the wavelength required for FPOP (248 nm), C. elegans is readily labeled by IV-FPOP. IV-FPOP results in irreversible oxidative modifications that allow for sample homogenization, digestion, and cleanup prior to LC-MS analysis.
Espino and Jones’ extension of FPOP for in vivo HRPF covalent labeling in C. elegans allows for the study of protein interactions and protein structural changes in native environments.16 Herein, wild-type C. elegans (L4 stage) had three conditions: 1) laser irradiated (KrF excimer, 248 nm) in the presence of H2O2 (sample), 2) exposed to H2O2 in the absence of laser irradiation (control oxidation), and 3) not exposed to H2O2 (control). Three biological and two technical replicates generated a total of 18 samples for analysis. Oxidatively modified peptides and proteins modified by IV-FPOP have been quantified by label-free MS, traditionally. However, individually processing 18 samples can be laborious and time-consuming; therefore, cPILOT was employed as a multiplexing strategy to increase sample throughput. Oxidatively modified proteins were digested, and resulting peptides were labeled by cPILOT (Figure 1). Labeled peptides were separated by offline SCX and online RP chromatography and analyzed by MS, MS/MS, and MS3. This analysis resulted in ∼180,000 PSMs corresponding to 2,682 proteins. Protein groups identified from light (2,334) and heavy (2,193) dimethylated peptides (Supplemental Table S2) were similar, with 1,844 proteins (79–84%) overlapping between these groups. TMT labeling efficiencies of both light and heavy dimethylated peptides were ∼98% (Supplemental Table S2), and among identified proteins, over 65% were quantified across all 18 reporter ion channels.
Biological Replicate Reproducibility Increases among Modified Proteins Using cPILOT
More than 700 oxidatively modified proteins were identified by both IV-FPOP label-free MS and IV-FPOP cPILOT quantification. A comparison of label-free quantification versus cPILOT demonstrated that label-free MS identified more oxidatively modified proteins across all biological replicates; however, cPILOT had higher reproducibility among technical and biological replicates (Figure 2). Additionally, SCX fractionation contributed to the increased proteome depth of the cPILOT approach. The cPILOT analysis requires MS3 on the Fusion Lumos which increases instrument duty cycle and lowers the number of proteins identified.21
Figure 2.

Protein oxidation quantification across biological and technical replicates. (a) IV-FPOP representative biological replicate of oxidatively modified proteins identified by label-free MS across two technical (tech) replicates: one (yellow, N = 157) and two (orange, N = 143). (b) IV-FPOP oxidatively modified proteins identified by cPILOT across technical replicates one (purple, N = 565) and two (green, N = 564). Venn diagrams of oxidatively modified proteins by IV-FPOP across three biological replicates (BR) identified by (c) label-free MS (N = 830) and (d) cPILOT (N = 703). (e) Venn diagram of common oxidatively modified proteins among label-free (red, N = 48) and cPILOT (blue, N = 429).
Among three biological replicates, 90, 67, and 111 (43, 12, and 40%) oxidatively modified proteins overlapped between label-free MS technical replicates (Figure 2a, Supplemental Figure S2a-b), respectively. Upon implementing cPILOT, the overlap between technical replicates increased to 563, 431, and 416 (99, 75, and 64%) oxidatively modified proteins, respectively (Figure 2b, Supplemental Figure S2a-b). When comparing oxidatively modified proteins identified by label-free MS and cPILOT for the same biological replicate as Figure 2a-b, 678 oxidatively modified proteins were identified across both methods (Supplemental Figure S2c). Though most identifications were unique to cPILOT, 98 proteins were in common between both approaches; this trend was also true for the other biological replicates. Among the proteins that overlapped, the number of oxidatively modified peptides observed and the number of oxidatively modified residues per peptide are greater with cPILOT in 79% of these proteins (Supplemental Table S3).
Across three biological replicates, 830 oxidatively modified proteins were identified by label-free MS (Figure 2c), with 177 proteins being present in at least two biological replicates. Upon implementing cPILOT to multiplex samples, 703 oxidatively modified proteins (Figure 2d) were identified with 662 proteins being present in at least two biological replicates. Interestingly, the number of proteins shared across at least two biological replicates increased approximatively 4-fold with cPILOT. This is an advantage when compared to label-free quantitation which has shown large variations in protein oxidation due to differences in protein abundance.16,29
In addition, quantifying 703 proteins by cPILOT is promising as quantitative protein information can be gleaned in future experiments that study other conditions, such as age, disease-state, or drug treatments. Since the IV-FPOP reaction and the time of laser irradiation occur very quickly (<30 s), significant changes in peptide oxidation are mostly due to rapid natural occurring structural differences.
Most importantly, the number of common oxidatively modified proteins increased approximatively 9-fold from 48 to 429 proteins from using label-free to cPILOT quantitative strategies, respectively (Figure 2c–d). This greatly increases the potential number of proteins that can be compared in future analyses. Among all biological replicates and between both quantification strategies, 36 proteins (Figure 2e) were identified. When assessing the coefficient of variation (CV) of 36 unique proteins common across both approaches, cPILOT had a 2-fold less CV, on average, in comparison to label free (data not shown). However, for most of these proteins, single or few peptides were oxidatively modified and used to calculate the CV across biological replicates. Greater numbers of peptides would generally give more measurements, resulting in lower CVs.
Multiplexing by cPILOT Increases Biological Reproducibility in IV-FPOP Oxidatively Modified Peptides
The extent of oxidation of the tubulin beta chain protein was evaluated as previously described.28,30 Eleven oxidatively modified peptides for tubulin beta chain were identified by label-free MS (Figure 3a), with no peptides being present in all three biological replicates. In contrast, six oxidatively modified peptides were identified with cPILOT; four of which were observed in all biological replicates (Figure 3b).
Figure 3.

Protein oxidation for tubulin beta chain. Calculated ln(PF) for tubulin beta chain oxidatively modified peptides identified by (a) label-free MS (N = 11) and (b) cPILOT (N = 6) across three biological replicates. SASA calculated values using the Homo sapiens tubulin beta chain cryo-EM structure (PDB: 5N5N(31)) are displayed on top of each bar. (c) Oxidatively modified peptides identified by label-free MS only (red), cPILOT only (blue), and both methods only (yellow) mapped on the cryo-EM structure of the human tubulin beta chain. CID-MS/MS spectra of the tubulin beta chain peptide 325–336 showing b- and y-ions for (d) light and (e) heavy dimethylation plus a +16 FPOP modification for residue M330 and isobaric-tag. HCD-MS3 spectra generated from the 10 most intense fragment ions (SPS-10) of the (f) light and (g) heavy CID-MS/MS ion.
Oxidation coverage was higher for tubulin beta chain from label-free MS (33%) in comparison to cPILOT (19%); however, peptides quantified in cPILOT were more reproducible in their detection across biological replicates. Oxidatively modified peptides identified by label-free MS (red), cPILOT (blue), or both methods (yellow) are mapped on the human tubulin beta chain cryo-EM structure (PDB: 5N5N(31)) (Figure 3c). Both approaches identified ∼30% of the protein sequence, with contributions from each method.
An example, MS/MS spectrum of peptide 325EVDEQMLSVQNK336, shows a +16 modification at M330 for both light (Figure 3d) and heavy (Figure 3e) labeled peptides. Reporter ion intensities were present for light (Figure 3f) and heavy (Figure 3g) labeled peptides in all IV-FPOP conditions (sample, control oxidation, control) and biological replicates. Similarly, for the actin protein, multiple oxidatively modified peptides were identified in both label-free MS and cPILOT (Supplemental Figure S3). Notably, 12 oxidatively modified peptides were identified using label-free MS (Supplemental Figure 3a), while three peptides were identified with cPILOT (Supplemental Figure S3b). MS/MS spectra of peptide 41HQGVMVGMGQK51 displayed differences in fragmentation patterns between HCD-MS/MS (label-free MS) (Supplemental Figure S3c) and CID-MS/MS (cPILOT) (Supplemental Figure S3d). HCD-MS/MS yielded a more disperse range of fragments and the identification of oxidatively modified residues M41, V46, and M48, whereas CID-MS/MS resulted in less fragments and the sole identification of M48. The identification of oxidative modifications by CID has been shown to be limited in peptides containing Met and His.32,33 Additionally, this suggests HCD-MS/MS and HCD-MS3 could improve identifications of cPILOT for IV-FPOP oxidatively modified proteins.
Peptides identified by FPOP-cPILOT were fragmented by CID, resulting in the identification of residues with oxidative modifications. Conversely, the label-free MS analysis used HCD fragmentation, which herein resulted in a wider range of oxidative modifications. For the cPILOT acquisition, it was more suitable to not use HCD fragmentation for peptide identification, as the time required to obtain HCD data for both peptide identification (MS/MS) and quantification (MS3) would greatly increase the duty cycle. This would have resulted in a reduction of the number of peptides identified. In addition, the use of HCD-MS/MS may have reduced the accuracy of reporter ion intensities obtained.34,35 Using CID fragmentation is beneficial in this application of IV-FPOP, as an additional fragmentation cycle is being employed to obtain quantitative information.
Owing to the fragmentation differences between label-free MS and cPILOT data generated herein, known OH amino acid modifications25 for all oxidatively modified peptides (N = 1005, 703 proteins) were compared. These modifications are found on most amino acids in which MS/MS spectra provide the location of these modifications. Peptides identified from label-free MS (Figure 4a, top) and cPILOT (Figure 4b, top) were mostly oxidatively modified by the addition of an oxygen atom, leading to a +16 modification. This modification occurred at M330 in the peptide 325EVDEQMLSVQNK336. This is promising, as the major modification (+16) was identified in both quantification strategies. Many other OH modifications were present in the label-free MS data, in comparison to that from cPILOT, with the exception of +32 modifications, which were higher in cPILOT data. Less abundant OH modifications to His (+5 and −10 Da mass shifts) residues were observed with cPILOT. We note that the use of different MS/MS fragmentation methods for label-free and cPILOT could contribute to the overall differences in fragmentation patterns and intensities of fragment peaks as shown (Figure 4.)
Figure 4.
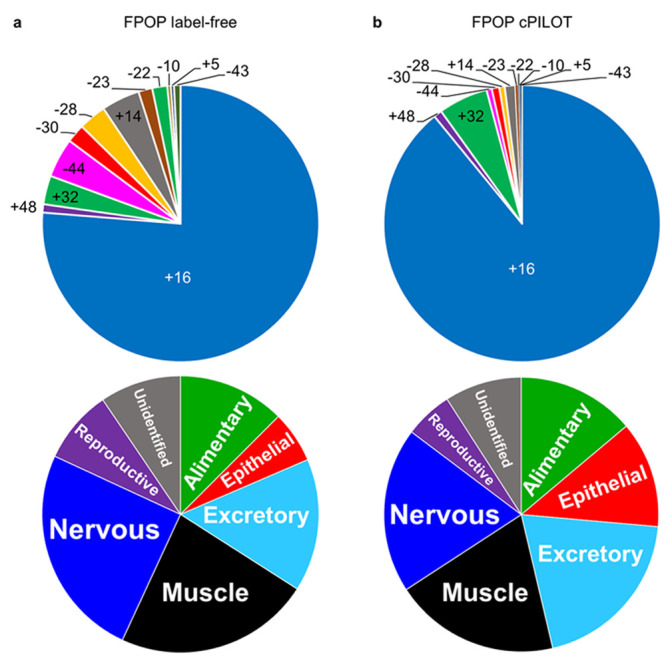
Oxidative mass shift modifications and body systems identified by IV-FPOP. (a) Oxidative mass shift modifications observed by HCD-MS/MS fragmentation (top) and body systems (bottom) identified by IV-FPOP label-free MS. (b) Oxidative mass shift modifications observed by CID-MS/MS fragmentation (top) and body systems (bottom) identified IV-FPOP-cPILOT.
Next, oxidatively modified proteins were matched to their primary gene and localized to relevant systems using the online Gene Search tool by The Genome BC C. elegans Gene Expression Consortium.36 The percentage of proteins identified in both label-free MS (Figure 4a, bottom) and cPILOT (Figure 4b, bottom) were very similar across nervous, muscle, epithelial, and reproductive body systems. This demonstrates that cPILOT probes oxidation in similar body systems as label-free; however, it extends the number of proteins observed in each of these bodily systems, providing more insight into the biological implications of readily accessible oxidatively modified proteins. Additionally, the epithelial system is the most outer-part system of C. elegans and has 13% of oxidatively modified proteins, which is intuitive given it is the first contact for the laser to target proteins. However, the FPOP strategy is able to probe deeper into the organism and study oxidation as demonstrated from reproductive, nervous, and muscle systems also being observed.
Some factors that should be considered when using enhanced multiplexing include the additional steps added to the sample workflow, data acquisition, and processing. Multiplexing strategies require a chemical labeling step. For cPILOT, two chemical labeling steps are necessary for dimethylation and TMT. Next, offline fractionation is performed to ensure proteome depth. Critical to the detection of multiple samples and quantitatively for cPILOT is the inclusion of HCD-MS3 to achieve accurate reporter ion information.34 The MS3 increases the instrument duty cycle; however, the overall gradient times are similar to the label-free approach. Generally, the time spent for label-free for this study from sample preparation to MS acquisition is 20 h, and for cPILOT it is 22 h. The additional time for cPILOT is not attributed to the multiplexing but more so to the incorporation of fractionation, which arguably could have improved the coverage in the label-free approach also but would have been over 12 days longer than that from cPILOT for 18 samples.
The MS time between both approaches was kept at 120 min. Once raw data is acquired, files are searched to obtain peptide and protein identifications/quantifications. Searching label-free versus cPILOT quantified data entails including all possible FPOP oxidative modifications, along with the specified quantification strategy. The time required to search 18 files as opposed to two files (one file per dimethylation modification) will also increase the overall time necessary to complete label-free experiments. In terms of quantification, ideally, resulting data would contain quantitative information from all channels; however, reporter ion intensities may be missing. To decrease the number of channels with missing reporter ion intensities, SPS has been employed. The collection of multiple fragment ions increases the intensities of reporter ions, thus increasing the probability of having quantitative information in all channels. As seen in the recent work of King and Robinson, using SPS-MS3, in addition to the targeted mass difference strategy, increased both the percentage of light and heavy dimethylation pair detections and peptide quantifications.37 Previous analyses21 have shown that using Lys-C results in longer peptides, thus reducing ionization and detection. Hence, trypsin was employed, resulting in ∼80% of K-terminated peptides (Supplemental Table S2) available for TMT labeling and subsequent detection. In short, more peptides were detected and quantified.
Overall, the complexity of this data set must be considered. Quantification information is obtained when 1. both light and heavy dimethylation pairs are detected and 2. reporter ion intensities are above the searching threshold. The experimental design may result in missing channels as peptides not oxidatively modified by FPOP or control oxidation would not be present. This may contribute to missing channels within the data set.
Conclusions
Here, we demonstrated the power of sample multiplexing for quantification of IV-FPOP modifications by using cPILOT.20,21 Peptide oxidation levels were quantified across three biological replicates, thus increasing sample reproducibility among technical and biological replicates. Moreover, within a single experiment control, sample and background conditions that typically are performed in independent label-free MS analyses were probed simultaneously with cPILOT. Enhanced multiplexing has been demonstrated as a valuable technique to reduce sample preparation time while increasing sample throughput for IV-FPOP. Despite trade-offs with throughput and protein depth, the quantification, multiplexing, and reproducibility of cPILOT informed of robust changes to protein oxidation for C. elegans.
Acknowledgments
The authors acknowledge the Vanderbilt University Start-up funds and NIH, NIGMS R01 GM117191 to R.A.S.R., University of Maryland, Baltimore start-up funds and the NIH, NIGMS R01 GM 127595 to L.M.J., and S-Stem Fellowship (University of Pittsburgh) and T32-AG058524 (Vanderbilt University) to C.D.K.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c00174.
Additional experimental methods; Supplemental Figure S1, schematic of peptides labeled by cPILOT; Supplemental Figure S2, oxidatively modified proteins across technical and biological replicates; and Supplemental Figure S3, protein oxidation for actin (PDF)
Supplemental Table S1, FPOP-cPILOT labeling scheme; Supplemental Table S2, protein and peptide identification/quantification results of FPOP-cPILOT experiment; and Supplemental Table S3, biological replicate 1 common IV-FPOP oxidatively modified peptides and residues (XLSX)
Author Contributions
§ L.M.J. and R.A.S.R. devised the project. J.A.E. performed IV-FPOP experiments, tissue homogenization, protein extraction, protein digestion, label-free MS data interpretation, and FPOP oxidation quantification. C.D.K. performed cPILOT labeling and cPILOT MS data interpretation. All authors contributed to writing. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. J.A.E. and C.D.K. contributed equally. This article was written in partial fulfillment of J.A.E. Ph.D. thesis. J.A.E. and C.D.K. share authorship.
The authors declare no competing financial interest.
Notes
Additional supporting research data, including LC-MS/MS and MS3, for this article are available from the corresponding authors upon request.
Supplementary Material
References
- Shi Y. A glimpse of structural biology through X-ray crystallography. Cell 2014, 159 (5), 995–1014. 10.1016/j.cell.2014.10.051. [DOI] [PubMed] [Google Scholar]
- Huang C.; Kalodimos C. G. Structures of Large Protein Complexes Determined by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Biophys. 2017, 46, 317–336. 10.1146/annurev-biophys-070816-033701. [DOI] [PubMed] [Google Scholar]
- Adrian M.; Dubochet J.; Lepault J.; McDowall A. W. Cryo-electron microscopy of viruses. Nature 1984, 308 (5954), 32–6. 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- Kaur U.; Johnson D. T.; Chea E. E.; Deredge D. J.; Espino J. A.; Jones L. M. Evolution of Structural Biology through the Lens of Mass Spectrometry. Anal. Chem. 2019, 91 (1), 142–155. 10.1021/acs.analchem.8b05014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chea E. E.; Jones L. M. Analyzing the structure of macromolecules in their native cellular environment using hydroxyl radical footprinting. Analyst 2018, 143 (4), 798–807. 10.1039/C7AN01323J. [DOI] [PubMed] [Google Scholar]
- Leser M.; Chapman J. R.; Khine M.; Pegan J.; Law M.; Makkaoui M. E.; Ueberheide B. M.; Brenowitz M. Chemical Generation of Hydroxyl Radical for Oxidative ’Footprinting’. Protein Pept. Lett. 2019, 26 (1), 61–69. 10.2174/0929866526666181212164812. [DOI] [PubMed] [Google Scholar]
- Chance M. R.; Sclavi B.; Woodson S. A.; Brenowitz M. Examining the conformational dynamics of macromolecules with time-resolved synchrotron X-ray ’footprinting’. Structure 1997, 5 (7), 865–9. 10.1016/S0969-2126(97)00241-4. [DOI] [PubMed] [Google Scholar]
- Monroe E. B.; Heien M. L. Electrochemical generation of hydroxyl radicals for examining protein structure. Anal. Chem. 2013, 85 (13), 6185–9. 10.1021/ac400107c. [DOI] [PubMed] [Google Scholar]
- Hambly D. M.; Gross M. L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 2005, 16 (12), 2057–63. 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Johnson D. T.; Di Stefano L. H.; Jones L. M. Fast photochemical oxidation of proteins(FPOP): A powerful mass spectrometry based structural proteomics tool. J. Biol. Chem. 2019, 294, 11969. 10.1074/jbc.REV119.006218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Moniz H.; Wang S.; Ramiah A.; Zhang F.; Moremen K. W.; Linhardt R. J.; Sharp J. S. High structural resolution hydroxyl radical protein footprinting reveals an extended Robo1-heparin binding interface. J. Biol. Chem. 2015, 290 (17), 10729–40. 10.1074/jbc.M115.648410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. M.; B. Sperry J.; A. Carroll J.; Gross M. L. Fast Photochemical Oxidation of Proteins for Epitope Mapping. Anal. Chem. 2011, 83 (20), 7657–7661. 10.1021/ac2007366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselar J. G.; Janmey P. A.; Almo S. C.; Chance M. R. Structural analysis of gelsolin using synchrotron protein footprinting. Mol. Cell. Proteomics 2003, 2 (10), 1120–32. 10.1074/mcp.M300068-MCP200. [DOI] [PubMed] [Google Scholar]
- Espino J. A.; Mali V. S.; Jones L. M. In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells. Anal. Chem. 2015, 87 (15), 7971–8. 10.1021/acs.analchem.5b01888. [DOI] [PubMed] [Google Scholar]
- Rinas A.; Mali V. S.; Espino J. A.; Jones L. M. Development of a Microflow System for In-Cell Footprinting Coupled with Mass Spectrometry. Anal. Chem. 2016, 88 (20), 10052–10058. 10.1021/acs.analchem.6b02357. [DOI] [PubMed] [Google Scholar]
- Espino J. A.; Jones L. M. Illuminating Biological Interactions with in Vivo Protein Footprinting. Anal. Chem. 2019, 91 (10), 6577–6584. 10.1021/acs.analchem.9b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C. I.; Calkins J.; Hartman P. S.; Rupert C. S. UV photobiology of the nematode Caenorhabditis elegans: action spectra, absence of photoreactivation and effects of caffeine. Photochem. Photobiol. 1987, 46 (4), 483–8. 10.1111/j.1751-1097.1987.tb04799.x. [DOI] [PubMed] [Google Scholar]
- King C. D.; Singh D.; Holden K.; Govan A. B.; Keith S. A.; Ghazi A.; Robinson R. A. S. Proteomic identification of virulence-related factors in young and aging C. elegans infected with Pseudomonas aeruginosa. J. Proteomics 2018, 181, 92–103. 10.1016/j.jprot.2018.04.006. [DOI] [PubMed] [Google Scholar]
- Nussbaum-Krammer C. I.; Morimoto R. I. Caenorhabditis elegans as a model system for studying non-cell-autonomous mechanisms in protein-misfolding diseases. Dis. Models & Mech. 2014, 7 (1), 31–39. 10.1242/dmm.013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson R. A.; Evans A. R. Enhanced sample multiplexing for nitrotyrosine-modified proteins using combined precursor isotopic labeling and isobaric tagging. Anal. Chem. 2012, 84 (11), 4677–86. 10.1021/ac202000v. [DOI] [PubMed] [Google Scholar]
- Evans A. R.; Robinson R. A. Global combined precursor isotopic labeling and isobaric tagging (cPILOT) approach with selective MS(3) acquisition. Proteomics 2013, 13 (22), 3267–72. 10.1002/pmic.201300198. [DOI] [PubMed] [Google Scholar]
- Frost D. C.; Greer T.; Li L. High-resolution enabled 12-plex DiLeu isobaric tags for quantitative proteomics. Anal. Chem. 2015, 87 (3), 1646–54. 10.1021/ac503276z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G. C.; Huttlin E. L.; Haas W.; Ting L.; Jedrychowski M. P.; Rogers J. C.; Kuhn K.; Pike I.; Grothe R. A.; Blethrow J. D.; Gygi S. P. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012, 84 (17), 7469–78. 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost D. C.; Rust C. J.; Robinson R. A. S.; Li L. Increased N,N-Dimethyl Leucine Isobaric Tag Multiplexing by a Combined Precursor Isotopic Labeling and Isobaric Tagging Approach. Anal. Chem. 2018, 90 (18), 10664–10669. 10.1021/acs.analchem.8b01301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G.; Chance M. R. Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem. Rev. 2007, 107 (8), 3514–3543. 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- Gau B. C.; Chen H.; Zhang Y.; Gross M. L. Sulfate radical anion as a new reagent for fast photochemical oxidation of proteins. Anal. Chem. 2010, 82 (18), 7821–7. 10.1021/ac101760y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plubell D. L.; Wilmarth P. A.; Zhao Y.; Fenton A. M.; Minnier J.; Reddy A. P.; Klimek J.; Yang X.; David L. L.; Pamir N. Extended Multiplexing of Tandem Mass Tags (TMT) Labeling Reveals Age and High Fat Diet Specific Proteome Changes in Mouse Epididymal Adipose Tissue. Mol. Cell. Proteomics 2017, 16 (5), 873–890. 10.1074/mcp.M116.065524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinas A.; Espino J. A.; Jones L. M. An efficient quantitation strategy for hydroxyl radical-mediated protein footprinting using Proteome Discoverer. Anal. Bioanal. Chem. 2016, 408 (11), 3021–31. 10.1007/s00216-016-9369-3. [DOI] [PubMed] [Google Scholar]
- Johnson D. T.; Punshon-Smith B.; Espino J. A.; Gershenson A.; Jones L. M. Implementing In-Cell Fast Photochemical Oxidation of Proteins in a Platform Incubator with a Movable XY Stage. Anal. Chem. 2020, 92 (2), 1691–1696. 10.1021/acs.analchem.9b04933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Ravikumar K. M.; Chance M. R.; Yang S. Quantitative mapping of protein structure by hydroxyl radical footprinting-mediated structural mass spectrometry: a protection factor analysis. Biophys. J. 2015, 108 (1), 107–15. 10.1016/j.bpj.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemu A.; Atherton J.; Spector J. O.; Moores C. A.; Roll-Mecak A. Tubulin isoform composition tunes microtubule dynamics. Mol. Biol. Cell 2017, 28 (25), 3564–3572. 10.1091/mbc.e17-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgewater J. D.; Srikanth R.; Lim J.; Vachet R. W. The effect of histidine oxidation on the dissociation patterns of peptide ions. J. Am. Soc. Mass Spectrom. 2007, 18 (3), 553–62. 10.1016/j.jasms.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth R.; Wilson J.; Bridgewater J. D.; Numbers J. R.; Lim J.; Olbris M. R.; Kettani A.; Vachet R. W. Improved sequencing of oxidized cysteine and methionine containing peptides using electron transfer dissociation. J. Am. Soc. Mass Spectrom. 2007, 18 (8), 1499–506. 10.1016/j.jasms.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Ting L.; Rad R.; Gygi S. P.; Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8 (11), 937–40. 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G. C.; Nusinow D. P.; Jedrychowski M. P.; Wuhr M.; Huttlin E. L.; Erickson B. K.; Rad R.; Haas W.; Gygi S. P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86 (14), 7150–8. 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt-Newbury R.; Viveiros R.; Johnsen R.; Mah A.; Anastas D.; Fang L.; Halfnight E.; Lee D.; Lin J.; Lorch A.; McKay S.; Okada H. M.; Pan J.; Schulz A. K.; Tu D.; Wong K.; Zhao Z.; Alexeyenko A.; Burglin T.; Sonnhammer E.; Schnabel R.; Jones S. J.; Marra M. A.; Baillie D. L.; Moerman D. G. High-Throughput In Vivo Analysis of Gene Expression in Caenorhabditis elegans. PLoS Biol. 2007, 5 (9), e237. 10.1371/journal.pbio.0050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C. D.; Robinson R. A. S. Evaluating Combined Precursor Isotopic Labeling and Isobaric Tagging Performance on Orbitraps To Study the Peripheral Proteome of Alzheimer’s Disease. Anal. Chem. 2020, 92 (4), 2911–2916. 10.1021/acs.analchem.9b01974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.