

Abstract
The trichophycin family of compounds are chlorinated polyketides first discovered from environmental collections of a bloom-forming Trichodesmium sp. cyanobacterium. In an effort to fully capture the chemical space of this group of metabolites, the utilization of MS/MS-based molecular networking of a Trichodesmium thiebautii extract revealed a metabolome replete with halogenated compounds. Subsequent MS-guided isolation resulted in the characterization of isotrichophycin C and trichophycins G-I (1–4). These new metabolites had intriguing structural variations from those trichophycins previously characterized, which allowed for a comparative study to examine structural features that are associated with toxicity to murine neuroblastoma cells. Additionally, we propose the absolute configuration of the previously characterized trichophycin A (5). Overall, the metabolome of the Trichodesmium bloom is hallmarked by an unprecedented amount of chlorinated molecules, many of which remain to be structurally characterized.
Graphical Abstract
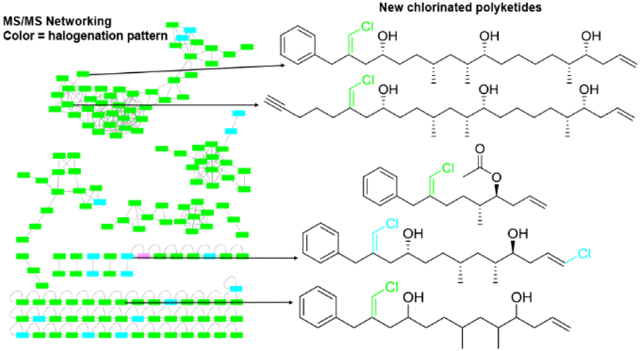
Studies on the ecological role of members of the cyanobacterial genus Trichodesmium have been predominantly centered on the capacity for nitrogen fixation in this group,1,2 and their substantial role in productivity and biogeochemistry in the oceans.3,4 The specialized metabolite composition of these cyanobacteria has been less well studied. However, recent efforts from our group and others have shown that environmental collections of Trichodesmium sp. contain many specialized metabolites, especially chlorinated polyketides and hybrid polyketide-peptide metabolites.5–9 The trichophycins are a new group of chlorinated polyketides, with six described members, that have shown intriguing structural differences, and some members have shown moderately potent cytotoxicity against neuronal cells lines.7,8 These metabolites are related by structure to the trichotoxins,10 and a stereoisomer of trichophycin B has recently been characterized from a marine sponge and given the name smenolactone C.11 As this group of molecules continues to grow in number, we are interested in determining the full suite of trichophycin analogs, and determining their biological activities for their potential for use in therapeutically-relevant areas. For instance, trichophycin B showed sub-micromolar anti-proliferative activity against the MCF7 breast cancer cell line.11 There are many examples of families of molecules isolated from cyanobacteria comprised of dozens of individual members. One such example, the malyngamides, are a group of well-known cyanobacterial metabolites predominantly isolated from Moorea producens with over 30 members.12 These metabolites have shown diverse biological activities such as cytotoxicity, inhibition of quorum sensing, and anti-inflammatory activity.13,14 Trichodesmium blooms are noteworthy for the diversity of organisms present during the course of a bloom event, such as diatoms, dinoflagellates, and heterotrophic bacteria.15–17 Extracts and chromatography fractions from environmental collections of bloom material are very complex with respect to metabolite composition making a comprehensive characterization of all metabolites present challenging.9
MS/MS-based molecular networking provides an approach to aid in chemical space assessments of extracts, and recent workflows have been developed to target geometric isomers and chlorinated metabolites.11 In the current report, we utilized MS/MS-based molecular networking to capture the halogenated chemical space present in Trichodesmium extracts. Further isolation efforts resulted in the structure characterization of four new trichophycin compounds. These new metabolites (1–4) add new chemical diversity to the trichophycin family (Figure S1) and cytotoxicity assays with members of this group identified trichophycin G as a moderately potent cytotoxin, and additionally identified structural features of the trichophycins that are associated with cytotoxicity.
RESULTS AND DISCUSSION
The metabolites in the current report, and the previously characterized trichophycins were isolated from the lipophilic extracts of an environmentally collected cyanobacterial sample, which was identified as a Trichodesmium species according to morphological characters. Phylogenetic analysis supported identification as a strain of Trichodesmium thiebautii (Figure S35). As trichophycins are hallmarked by a chlorovinylidene functionality, MS/MS-based molecular networking of the original Trichodesmium extract was focused on analyzing only compounds that contained at least one chlorine atom. Halogenation patterns of individual compounds were color-coded in the networking analysis, and it was clear that there were over 100 metabolites possessing at least a single chlorine in their chemical structure (Figure 1). We focused isolation efforts on molecules that HRMS analysis indicated did not contain any nitrogen atoms and contained at least one chlorine atom. Using this mass spectrometry-guided isolation approach, four new metabolites in the trichophycin family were isolated and characterized (1-4).
Figure 1.
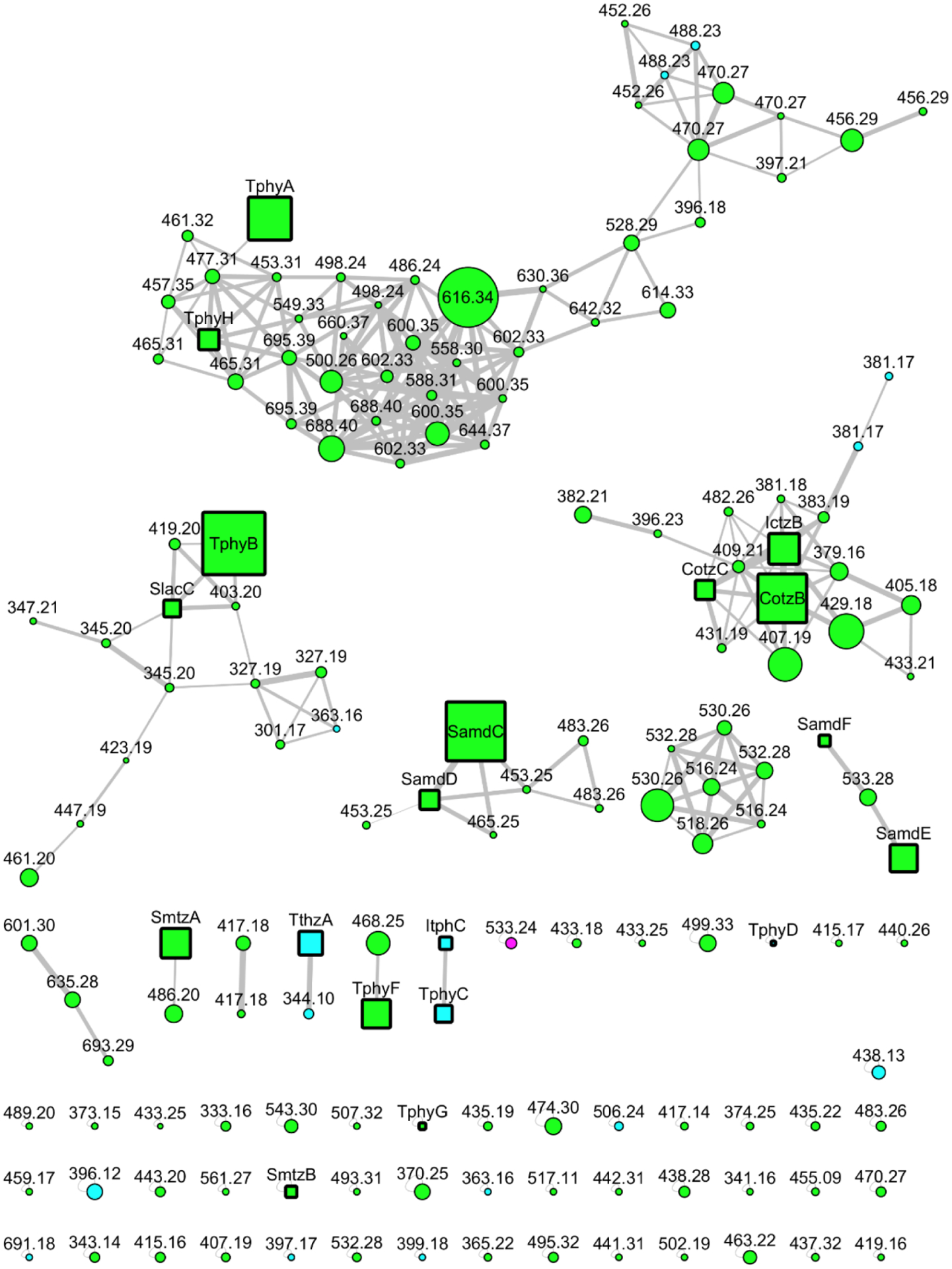
Molecular network of the Trichodesmium bloom extract. Only halogenated molecules are shown in the network. Color-coding of halogenation pattern is as follows: green = Cl, cyan = 2 × Cl, and pink = ClBr. Node size is proportional to LC-MS peak area. Previously reported compounds and newly reported molecules (1–4) are shown as square nodes and abbreviated by name (Cotz, conulothiazole; Ictz, isoconulothiazole; Samd, smenamide; Tphy, trichophycin; Itph, isotrichophycin; Slac, smenolactone; Smtz, smenothiazole). Circular nodes are currently uncharacterized metabolites.
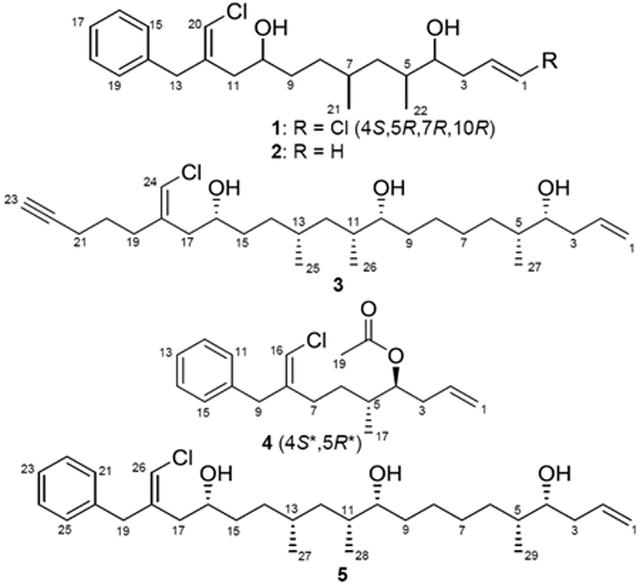
HRESIMS analysis of 1 gave an [M+H]+ of m/z 399.1855, suggesting an identical molecular formula to that of the previously characterized trichophycin C, C22H32Cl2O2.8 Comparison of the 1H NMR (Figure S2) and 13C NMR spectra of 1 indicated that the metabolite was nearly identical in structure to trichophycin C. Additionally, this metabolite was clustered with trichophycin C in the molecular network (Figure 1, TphyC-ItphC cluster). NMR signals in 1 from C-9 to C-13 showed the greatest divergence in chemical shift from those of the previously characterized metabolite. NOE correlations between H-20 (δH 6.00) and H-15 (δH 7.17) and H2-13 (δH 3.46) supported a Z configuration of the chlorovinylidene group at C-12, unlike that of the C-12 chlorovinylidene of trichophycin C, which has an E configuration. The C-1–C-2 olefin in 1 was determined to be E from the large vicinal 1H-1H coupling constant (13.2 Hz). Our initial speculation based on NMR data was that 1 was simply a cis-trans isomer of trichophycin C (Table 1). Examination of the 13C NMR chemical shifts at the C-4, C-5 and C-7 stereogenic centers showed no deviation in shifts from those of trichophycin C. However, examination of the specific rotation of 1 (−9.5) showed the opposite sign to that of trichophycin C (+26.8). Examination of 1H chemical shifts of diastereomeric bis-MTPA esters of 1 indicated that the absolute configurations of C-10 and C-4 were R and S, respectively (Figure S31), identical to that of trichophycin C. ECD analysis showed nearly superimposable spectra (Figure S11). The large difference in the chemical shifts between the diastereotopic protons attached to C-6 in 1 supported a syn relationship between the two methyl groups (H-6a, δH 1.38; H-6b, δH 0.98),18 and the 5.3 Hz coupling constant measured for H-4 and H-5 was identical to that of trichophycin C and supported an anti relationship between these two substituents.8 Careful inspection of the spectroscopic data, strongly supported 1 as a cis-trans isomer of trichophycin C, garnering 1 the name isotrichophycin C.
Table 1.
NMR Data for Isotrichophycin C (1).
| pos | δC, type | δH (J in Hz) | HMBC | COSY |
|---|---|---|---|---|
| 1 | 119.2, CH | 6.05, d (13.2) | 2, 3 | 2 |
| 2 | 130.7, CH | 5.95, m | 1, 3, 4 | 1, 3a, 3b |
| 3a | 35.0, CH2 | 2.24, dddd (14.4, 7.1, 3.3, 1.5) | 1, 2, 4, 5 | 2, 3b, 4 |
| 3b | 2.13, dddd (14.4, 9.2, 8.6, 1.2) | 1, 2, 4, 5 | 2, 3a, 4 | |
| 4 | 74.8, CH | 3.46, ddd (8.7, 5.3, 3.4) | 2, 3, 5, 6, 22 | 3a, 3b, 5 |
| 5 | 35.8, CH | 1.66, m | 3, 4, 6, 7, 22 | 4, 6a, 6b, 22 |
| 6a | 39.8, CH2 | 1.38, ddd (13.4, 8.5, 4.6) | 4, 5, 7, 8, 21, 22 | 5, 6b |
| 6b | 0.98, ddd (13.6, 9.1, 5.5) | 4, 5, 7, 8, 21, 22 | 5, 6a | |
| 7 | 30.1, CH | 1.51, ovlpa | 6, 8, 9, 21 | 8a, 8b, 21 |
| 8a | 31.7, CH2 | 1.37, ovlp | 6, 7, 9, 10, 21 | 7, 8b |
| 8b | 1.17, m | 6, 7, 9, 10, 21 | 7, 8a | |
| 9a | 34.7, CH2 | 1.50, ovlp | 7, 8, 10, 11 | 8b, 9b |
| 9b | 1.41, m | 7, 8, 10, 11 | 8b | |
| 10 | 70.5, CH | 3.78, m | 8, 9, 11, 12 | 9a, 9b, 11a, 11b |
| 11a | 38.2, CH2 | 2.39, dd (13.5, 8.8) | 9, 10, 12, 13, 20 | 10, 11b |
| 11b | 2.28, dd (13.5, 4.2) | 9, 10, 12, 13, 20 | 10, 11a | |
| 12 | 139.5, C | |||
| 13 | 42.0, CH2 | 3.46, ovlp | 11, 12, 14, 20 | |
| 14 | 138.0, C | |||
| 15 | 129.0, CH | 7.17, d (7.6) | 13, 17 | 16 |
| 16 | 128.6, CH | 7.30, t (7.6) | 14 | 17 |
| 17 | 126.7, CH | 7.24, t (7.6) | 15/19 | |
| 18 | 128.6, CH | 7.30, t (7.6) | 14 | |
| 19 | 129.0, CH | 7.17, d (7.6) | 13, 17 | 18 |
| 20 | 116.2, CH | 6.00, s | 11, 12, 13 | |
| 21 | 20.7, CH3 | 0.90, d (6.6) | 6, 7, 8 | 7 |
| 22 | 15.7, CH3 | 0.89, d (6.8) | 4, 5, 6 | 5 |
overlapping signals
Having prepared bis-MTPA esters of trichophycin C previously and bis-MTPA esters of isotrichophycin C in the current report, we attempted to generate tris-MTPA esters of the previously reported trichophycin A (5). The absolute and relative configurations of the metabolite were not addressed in the initial publication.7 Examination of 1H chemical shifts of diastereomeric tris-MTPA esters of trichophycin A indicated that the absolute configurations of C-16, C-10, and C-4 were all R (Figure S32). The large difference in chemical shift between H-12a (δH 1.38) and H-12b (δH 1.00) strongly supported a syn 1,3 methyl system.18 The absolute configurations of C-11 and C-5 were relayed from C-10 and C-4, respectively by virtue of J-coupling analysis. The coupling constants between H-4 and H-5 and between H-10 and H-11 in trichophycin A were both 4.0 Hz (determined following interpretation of DQF-COSY). This was very similar to the coupling constant between H-5 and H-6 of the previously reported trichophycin B (3.9 Hz).8 A syn relationship was determined between these substituents in trichophycin B by means of J-coupling analysis and DFT calculations.8 An anti relationship was determined for H-5 and H-6 in smenolactone C (a stereoisomer of trichophycin B) and the JH5–H6 was determined to be 5.8 Hz.11 The relationship between H-4 and H-5 in trichophycin C and isotrichophycin C was determined to be 5.3 Hz in each metabolite and anti relationships were assigned. This was strongly supported by DFT calculations (M06–2X/6–31G(d,p) level of theory), which showed that theoretically calculated J-coupling constants between H-4 and H-5 were 4.2 Hz for 4R5S (syn) and 6.3 Hz for 4R5R (anti) for trichophycin C.8 In the analysis of trichophycin A configurations, J-coupling analysis strongly supported syn relationships between H-4 and H-5 and H-10 and H-11, which established an absolute configuration of 4R5R10R11R13R16R for this compound (5).
HRESIMS analysis of 2 gave an [M+Na]+ of m/z 387.2071, suggesting a molecular formula of C22H33ClO2. Inspection of the HRESIMS data and the intensity ratio in the isotopic pattern of 2 indicated that this molecule contained a single chlorine atom (Figure S14). The 1H NMR spectra of 1 and 2 were very similar (cf. Tables 1 and S1). However in 2, resonances at δH 5.17 and 5.14 (H-1a and H-1b, respectively) were correlated to H-2 (δH 5.84) following interpretation of the COSY spectrum, and showed that 2 possessed a terminal alkene instead of the terminal vinyl chloride of 1. We did not isolate enough of 2 for further derivative formation experiments and only propose the planar structure at this time. We named this molecule trichophycin G, adding it to the series of trichophycins.
HRESIMS analysis of 3 gave an [M+Na]+ of m/z 477.3107, suggesting a molecular formula of C27H47ClO3 and a requirement of four degrees of unsaturation. Comparison of the 1H NMR spectrum of 3 and that of the previously reported trichophycin A indicated that these metabolites were very similar, except that the 1H NMR spectrum of 3 showed no resonances consistent with the presence of a benzene ring (Figure S15). Additionally, the M+H+ of this metabolite (m/z 455, TphyH) clustered with trichophycin A (TphyA) in the molecular network (Figure 1). Examining the 1H NMR spectrum, a resonance in 3 (δH 1.98, H-23) was not present in the 1H NMR spectrum of trichophycin A (Table S2, Figure S16). H-23 showed a correlation to H2-21 following interpretation of the COSY spectrum. COSY analysis showed that H2-21 was correlated to H2-20 (δH 1.68), which itself was correlated to the deshielded methylene H2-19 (δH 2.28). H2-19 was correlated to C-18 (δC 139.0) following interpretation of the HMBC spectrum of 3. Analysis of HMBC data showed correlations between H2-20, H2-21 and C-22 (δC 83.5), which firmly established an alkyne group and the planar structure of 3, which was named trichophycin H. The configuration of the chlorovinylidene in 3 was assigned following analysis of 13C NMR shift values of an adjacent methylene group in Z-configured and E-configured chlorovinylidenes in the trichophycins and other metabolites.7,8,11,19 In Z-configured molecules, the methylene carbon adjacent to the chlorovinylidene (C-17 in 3) is shielded compared to the same carbon in E-configured molecules (Table S4). The chemical shift of C-17 in 3 (δC 38.2) is consistent with a Z-configured chlorovinylidene. The absolute configuration of 3 is proposed to be identical to that of trichophycin A following analysis of tris-MTPA esters of 3 (4R,10R,16R). Additionally, the coupling constants between H-4 and H-5 and between H-10 and H-11 were identical to trichophycin A, and the large difference in the chemical shifts of the diastereotopic protons attached to C-12 (Δ = 0.39 ppm) supported this configuration assignment (cf. Table S2 and Figure S33).
HRESIMS analysis of 4 gave an [M+Na]+ of m/z 343.1454, suggesting a molecular formula of C19H25ClO2 and a requirement of seven degrees of unsaturation. Examination of NMR data showed the presence of the monosubstituted benzene ring, deshielded methylene and chlorovinylidene (C-8 to C-16) consistent with the trichophycin family (Table S3). NOE correlations between H-16 (δH 5.84) and H2-9 (δH 3.36) supported a Z configuration of the vinyl chloride group. Moderately deshielded methylene protons (H2-7, δH 2.14) showed an HMBC correlation to C-8 (δC 141.9) and a COSY correlation to H2-6 (H-6a, δH 1.50; H-6b, δH 1.17). The H-6 methylene group was correlated by COSY to the H-5 methine (δH 1.67), which itself was correlated by COSY to H3-17 (δH 0.90) and the H-4 oxymethine (δH 4.80). The H2-3 methylene group showed COSY correlations to the oxymethine and H-2 (δH 5.71). H-2 was correlated by COSY to H2-1 (H-1a, δH 5.07; H-1b, δH 5.03), and established a terminal alkene group in 4. The H-4 oxymethine in 4 was considerably deshielded (δH 4.80) compared to other trichophycins. An HMBC correlation from a deshielded methyl group (H3-19, δH 2.02) to a carbonyl (C-18, δC 170.7) supported the presence of an acetyl group and satisfied the remaining degree of unsaturation. Compound 4 was named trichophycin I. We were not able to determine the absolute configuration of 4 due to limited quantities isolated. However, the coupling constant of 5.9 Hz between H-4 and H-5 strongly supported an anti relative configuration with respect to these stereocenters.
Isotrichophycin C (1) did show a lower EC50 value (EC50: 13 ± 1 μM) against neuro-2A cells than trichophycin C (EC50: 24 ± 4 μM).8 However, neither metabolite is considered cytotoxic. Trichophycin G (2) showed modest cytotoxicity (EC50: 8.4 ± 3.0 μM) and was close to the potency of trichophycin A (EC50: 6.5 ± 1.4 μM).7 Only trichophycin A and isotrichophycin C are cytotoxic. However, there are some interesting insights when examining the cell cytotoxicity data of a panel of trichophycins as we now have data for 12 out of the 15 trichophycin/trichotoxin metabolites including tricholactone, which does not contain a chlorovinylidene group and is not toxic. (Table 2). Trichophycin A and 1 both have a terminal alkene while most of the trichophycins have a terminal vinyl chloride or lactone. Although, trichotoxins A and B both contain a terminal alkene and are not active. It may be that longer polyketide chains or the presence of more hydroxyl groups increase potency. In an intriguing result, the ability to donate hydrogen bonds may play a role, as acetylated versions of isotrichophycin C and trichophycin A lost potency. This also may be due to added steric bulk of the acetyl groups. While we have provided some initial observations, a comprehensive structure-activity relationship study will need to be carried out to explore these relationships further.
Table 2.
Cytotoxicity comparison of trichophycins, trichotoxins, and tricholactone against neuro-2a cells.
| Compound Name | Structure | EC50 [mean ± SE (μM)] |
|---|---|---|
| Trichophycin A | 6.5 ± 1.47 | |
| Trichophycin G (2) | 8.4 ± 3.0 | |
| Trichophycin H (3) | 12 ± 1 | |
| Isotrichophycin C (1) | 13 ± 1 | |
| Trichophycin F | 14 ± 28 | |
| Trichophycin B | 15 ± 28 | |
| Trichophycin C | 24 ± 48 | |
| Trichophycin D | 40 ± 48 | |
| Trichophycin E | >1008 | |
| Trichotoxin A | >5010 | |
| Trichotoxin B | >5010 | |
| Trichophycin A tri Ac | >100 | |
| Isotrichophycin C di Ac | >100 | |
| Tricholactone | 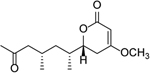 |
>100 |
The molecular network generated in this study showed an abundance of halogenated molecules, especially chlorinated metabolites, and displayed several compounds that have been previously characterized from Trichodesmium (Figure 1). Additionally, we have detected known metabolites characterized from a marine sponge that are present in the bloom material, such as smenothiazole B,20 conulothiazole B,21 and smenamide F.22 Thus far, all of the metabolites we have isolated from Trichodesmium that are chlorinated contain the Cl atom as part of a chlorovinylidene functional group. Furthermore, we will be able to use the network as a target guide for additional isolations, and for comparisons with future collections of Trichodesmium. The network had certain advantages for isolating new trichophycins in this current report, especially isotrichophycin C and trichophycin H, which clustered with related metabolites that had previously been characterized. However, due to a lack of informative MS/MS fragments, or possibly low abundance, certain trichophycins were observed as single nodes in the network (trichophycin G), and some were not observed in the network at all (trichophycin I). These metabolites were isolated as a result of polarity and serendipity, eluting near other compounds of interest during chromatographic procedures. The current work was an improvement in chemical space coverage from previous efforts to examine the Trichodesmium metabolome,9 and its similarity to that of marine sponges,11 and should be useful in longitudinal studies of bloom metabolite composition.
In the current report, we have isolated and characterized four new members of the trichophycin compound family. Additionally, we have shown that MS/MS-based profiling and analysis techniques continue to be useful in describing the chemical space of a sample and providing potential targets for isolation and evaluation. We have provided some initial insights about how structure affects cytotoxicity in this compound class, and we remain interested in testing these molecules against other biological endpoints. Future work will determine if the trichophycins and other metabolites are found repeatedly from colonies of Trichodesmium collected over time (we have completed a 2019 collecting trip of Trichodesmium colonies in the Gulf of Mexico), and we will continue to search for potential therapeutic lead molecules from this rich source of new chemistry. If Trichodesmium thiebautii is unequivocally shown to be the true producer of these network metabolites, the genetic architecture responsible for the abundance of these halogenated compounds will be of extreme interest.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations were measured using a Jasco P-2000 polarimeter. UV spectra were measured using a Beckman Coulter DU-800 spectrophotometer. ECD spectra were recorded using a Jasco J-1100 CD spectrometer, and IR spectra were recorded using a Thermo Scientific Nicolet 380 FT-IR spectrometer. NMR spectra were collected using both a Bruker 800 MHz NMR instrument equipped with a cryoprobe and a Varian 500 MHz instrument. The chemical shifts reported were referenced to the residual solvent peaks of CDCl3 (δH 7.26 and δC 77.2). HRESIMS analysis was performed using an AB SCIEX TripleTOF 4600 mass spectrometer with Analyst TF software. LC-HRESIMS experiments for molecular networking were performed using a Thermo LTQ Orbitrap XL high-resolution ESI mass spectrometer coupled to Thermo U3000 HPLC system, equipped with a solvent reservoir, in-line degasser, binary pump and refrigerated autosampler. Low resolution LC-MS was performed using a Thermo Fisher Scientific ISQ mass spectrometer with an electrospray ionization (ESI) source. Semi-preparative HPLC was carried out using a Dionex UltiMate 3000 HPLC system equipped with a micro vacuum degasser, an autosampler, and a diode-array detector.
Collection of Biological Material.
Biomass from a localized bloom of Trichodesmium sp. was collected at the water’s surface as previously described.10 The individual filaments of the bloom were identified as Trichodesmium following the examination of morphological characteristics using light microscopy. Biomass was frozen for further chemical analysis.
DNA Extraction, Amplification, Sequencing, and Phylogenetic Analysis.
DNA was extracted from cyanobacterial filaments stored in RNAlater using the Qiagen DNeasy Plant Mini Kit following the manufacturer’s specifications. A Mini-Beadbeater-96 (BioSpec Products Inc.) was used for 2 min to facilitate cell lysis using 0.5 mm and 0.1 mm beads. The integrity and purity of the extracted DNA was measured using a NanoDrop 2000c (Thermo Scientific). Lineage-specific primers were designed using IDT primer quest tool to amplify the SSU rRNA gene. The primer set used was: Forward 5’- GTA GCG GTG AAA TGC GTA GA −3’ and Reverse 5’- CTC CCT TTC GGG TTA GAG TAA TG −3’. The PCR reaction components consisted of 3 μL of DNA (36 ng), 7 μL of PCR grade H2O (ThermoFisher), 1.25 μL of forward and reverse primers (10 μM), and 12.5 μL of 2X Platinum SuperFi PCR Master Mix (ThermoFisher) totaling 25 μL. The PCR reaction was performed using an Eppendorf Mastercycler X50a using the following method. Denaturation occurred for 30 seconds at 98 °C followed by 33 cycles of 10 seconds at 98 °C, 30 seconds at 47 °C, 30 seconds at 72 °C, and a final elongation for 5 min at 72 °C. PCR product purification was performed using a QIAquick PCR purification Kit following the manufacturers specifications. Sanger sequencing of the PCR product was carried out using the same primers used for amplification. Partial 16S rRNA sequences from relevant cyanobacterial strains were collected from GenBank, aligned using ClustalW, and trimmed. A phylogenetic tree was generated using the maximum likelihood method and Tamura-Nei model in MEGA X.23 The final tree used for phylogenetic inference was created from 1000 bootstrap replicates. The partial 16S sequence has been deposited in GenBank under the accession number MT478931.
Data-dependent LC-HRMS/MS Analysis.
The CH2Cl2:MeOH extract of Trichodesmium was dissolved in MeOH (10 mg/mL) for LC-HRMS/MS analysis. A 5-μm Kinetex C18 column (50 × 2.10 mm) was used and a method with a flow rate of 200 μL·min−1 and a mobile phase of 0.1% formic acid in H2O (eluent A) and CH3CN (eluent B). The gradient program was as follows: 45% B for 1 min, 30%→80% B over 30 min, 80%→100% B over 1 min, 100% B for 9 min.
Acquisition-time selection of halogenated compounds was used to avoid that abundant, but uninteresting non-halogenated compound are selected for fragmentation. This was achieved using the data-dependent acquisition mode of the Orbitrap spectrometer software. Based on known metabolites from Trichodesmium, the halogenation patterns Cl, Cl2, BrCl, and BrCl2 were considered of interest, and a separate run was performed for each pattern. Fragmentation was triggered by an M+2 isotope peak with intensity 34±10% to select Cl ions, an M+2 isotope peak with intensity 60±10% to select Cl2 ions, an M+4 isotope peak with intensity 32±10% to select BrCl ions, and an M+4 isotope with intensity 72±10% to select BrCl2 ions. For each full scan, the 5 most intense ions with the required isotopic ratio were subjected to fragmentation. The spectrometer software did not allow data-dependent acquisition to be triggered by an accurate mass difference. Therefore, some non-halogenated ions were also selected when they showed the required M+2 or M+4 isotope peak ratios because of other co-eluting compounds. These were filtered out by the subsequent data processing.
MZmine Processing and Molecular Networking.
Processing of LC-HRMS/MS data was performed using MZmine 2.51.24 The LC-MS .raw files generated by the Orbitrap mass spectrometer are supported by MZmine and were used with no conversion. After standard preliminary data treatment (data crop between 18–37 min and m/z 70–700, mass detection, ADAP chromatogram building, smoothing, and deconvolution using the local minimum search algorithm), the Adduct Search module was used to identify Na+, NH4+, and K+ adducts and 13C isotope peaks (mass_difference = 1.0033) and annotate them for subsequent filtering. The four .raw files from the four runs selecting different halogenation patterns (see above) were aligned using the Join Aligner module, and peaks annotated as adducts were filtered out using the Feature List Rows Filter module. Known compounds were identified using the Custom Database Search module and a list of mass and retention times in our standard chromatographic conditions.
Automatic identification of isotope patterns is not implemented in MZmine, but could be achieved using the Adduct Search module repeatedly with the following parameters (in this order): for BrCl2, mass_difference = −3.995, Max relative adduct peak height < 160%; for BrCl, mass_difference = −3.995, Max relative adduct peak height < 400%; for Cl2, mass_difference = −1.997, Max relative adduct peak height < 180%; for Cl, mass_difference = −1.997, Max relative adduct peak height < 400% (because negative mass differences are used, the M peak is seen by the software as an “adduct” of the M+2 or M+4 peaks). Therefore, those M peaks which showed M+2 or M+4 isotope peaks with the required mass and intensity were annotated as BrCl2, BrCl, Cl2, or Cl. After this, the peaks with no annotation and the peaks with no associated MS2 data were filtered out using the Feature List Rows Filter module. Finally, very minor compounds (peak area < 2.0E+7) were also filtered out, and MS data were exported to a MGF file, while quantitative data, along with the annotated isotope pattern and identification, were exported to a CSV file.
The molecular network was then generated using the GNPS platform.25 Although we were undoubtedly performing Feature Based Molecular Networking (FBMN),26 we used the Metabolomics workflow because the FBMN workflow on GNPS was not able to propagate our isotope pattern annotations. The following parameters were used: parent mass tolerance 0.02 Da, MS/MS fragment ion tolerance 0.02 Da, the cosine score > 0.6, matched peaks > 6, and maximum neighbor number (topK) = 12. Quantitative data and annotations in the CSV file were mapped to the relevant nodes of the network (https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=416bebdcdeb1468cb39610db0aab871f) using Cytoscape 3.7.2,27 which was also used for network visualization and analysis.
Isolation of 1–4.
The frozen biomass was thawed and repeatedly extracted with 2:1 CH2Cl2:MeOH, and the organic extract was subjected to vacuum liquid chromatography as previously described resulting in nine VLC fractions (A-I).7 Fraction C (80% hexanes in EtOAc, 144.2 mg) was subjected to SPE fractionation using a 2 g C18 SPE column eluting with 100% MeOH to generate an HPLC pre-fraction C-1. The same sample preparation procedure was used for fraction D (60% hexanes in EtOAc, 293.4 mg) resulting in the D-1 HPLC pre-fraction (53.3 mg). Fraction I (100% MeOH, 2,090.0 mg) was separated over a 10 g C18 SPE eluting with 50% CH3CN in H2O, 100% CH3CN, 100% MeOH, and 100% EtOAC. The 100% CH3CN fraction (I-2, 273.3 mg) was used for further purifications. Fraction D-1 did not contain many metabolites (determined by analytical HPLC-DAD analysis) and was subjected to semi-preparative RP-HPLC using a YMC 5 μm ODS column (250 × 10 mm) with an mobile phase of 75% CH3CN in H2O with 0.05% formic acid added, and a flow rate of 3 mL/min. Isotrichophycin C (1) (5.0 mg, tR: 13.65 min) was isolated from this fraction. Fraction C-1 was also subjected to RP-HPLC using the same YMC column and flow rate with a mobile phase of 65% CH3CN in H2O with 0.05% formic acid added. Trichophycin I (4) (2 mg, tR: 20.25 min) was isolated from this fraction. Fraction I-2 was much more complex with respect to metabolite composition and was subjected to RP-HPLC using the YMC column, a 3 mL/min flow rate, and a gradient HPLC method and time-based collection (5 min increments). The mobile phase consisted of H2O (eluent A) and CH3CN (eluent B) each amended with 0.05% formic acid. The gradient program was as follows: 50% B for 5 min, 50%→100% B over 25 min, 100% B over 5 min, and a return to initial conditions for 10 min. Time-based collection of peaks from 20–25 min was designated as fraction E and solvent was evaporated under reduced pressure leaving a clear oily residue (57.9 mg). The residue was further processed using a Kinetex 5 μm C18 column (250 × 10 mm); mobile phase: 75% CH3CN in H2O with 0.05% formic acid added to each solvent, flow rate 3 mL/min, and 2.2 mg and 4.7 mg of impure fractions were collected (tR, 10–11 min for fraction E-3, and 11–12 min for fraction E-4). Fraction E-3 was subjected to a final purification using the same Kinetex column and flow rate with a mobile phase of 65% CH3CN in H2O with 0.05% formic acid added to each solvent and 2 was isolated (1.4 mg, tR: 23.75 min). Fraction E-4 was also subjected to a final purification using the Kinetex 5 μm C18 column (250 × 10 mm), a flow rate of 3 mL/min and a mobile phase of 75% CH3CN in H2O with 0.05% formic acid added to each solvent. 1.1 mg of 3 was isolated (tR: 9.30 min).
Isotrichophycin C (1):
colorless oil; [α]23D −9.5 (c 0.20, MeOH); UV (MeOH) λmax (log ε) 208 (4.2) nm; ECD (c 1.0 mM, CH3CN) λmax (Δε) 228 (0.68) nm; IR (ZnSe) νmax 3400 (br), 2917, 2849, 1743, 1733, 1183 cm−1; 1H NMR (800 MHz, CDCl3), and 13C NMR (200 MHz, CDCl3), Table 1; HRESIMS m/z 399.1855 [M+H]+ (calcd for C22H33Cl2O2, 399.1858).
Trichophycin G (2):
colorless oil; [α]23D −21 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 203 (3.6); 1H NMR (500 MHz, CDCl3) Table S1; HRESIMS m/z 387.2071 [M+Na]+ (calcd for C22H33ClO2Na, 387.2067).
Trichophycin H (3):
colorless oil; [α]23D +23 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 201 (3.6) nm; IR (ZnSe) νmax 3420 (br), 2920, 2313, 1646 cm−1; 1H NMR (500 MHz, CDCl3), and 13C NMR (125 MHz, CDCl3), Table S2; HRESIMS m/z 477.3107 [M+Na]+ (calcd for C27H47ClO3Na, 477.3111).
Trichophycin I (4)
colorless oil; α22D −6.2 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 202 (3.8) nm; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), Table S4; HRESIMS m/z 343.1454 [M+Na]+ (calcd for C19H25ClO2Na, 343.1441).
Preparation of 1 and Trichophycin A Peracetylation Products.
In separate reaction vials, 1 mg of 1 and a sample of impure trichophycin A were stirred for 24 h in a 1:1 mixture of pyridine and acetic anhydride. The reaction mixtures were dried under a stream of N2, and the residues were subjected to RP-HPLC using a Kinetex 5 μm C18 column (250 × 10 mm), a flow rate of 3 mL/min, and a mobile phase of 95% CH3CN in H2O with 0.05% formic acid added to each solvent. Isotrichophycin C diacetate (1.0 mg, tR: 7.25 min) and trichophycin A triacetate (1.5 mg, tR: 7.65 min) were isolated.
Isotrichophycin C diacetate:
1H NMR (500 MHz, CDCl3) δ 7.31 (2H, t, J = 7.4 Hz, H-16, H-18), 7.23 (1H, m, H-17), 7.17 (2H, d, J = 7.4 Hz, H-15, H-19), 6.01 (1H, d, J = 13.4 Hz, H-1), 5.93 (1H, s, H-20), 5.84 (1H, m, H-2), 5.09 (1H, m, H-10), 4.78 (1H, dt, J = 7.6, 5.1 Hz, H-4), 3.43 (2H, d, J = 3.7 Hz, H-13), 2.54 (1H, dd, J = 13.7, 8.5 Hz, H-11a), 2.28 (1H, m, H-11b), 2.27 (2H, m, H-3), 2.05 (6H, s, 2-Ac groups), 1.79 (1H, m, H-5), 1.61 (2H, m, H-9), 1.45 (1H, m, H-7), 1.31 (1H, m, H-8a), 1.25 (1H, m, H-6a), 1.05 (1H, m, H-8b), 0.97 (1H, m, H-6b), 0.89 (3H, d, J = 6.7 Hz, H-21), 0.87 (3H, d, J = 6.8 Hz, H-22); HRESIMS m/z 505.1889 [M+Na]+ (calcd for C26H36Cl2O4, 505.1888).
Trichophycin A triacetate:
1H NMR (500 MHz, CDCl3) δ 7.30 (2H, d, J = 7.6 Hz, H-22, H-24), 7.23 (1H, m, H-23), 7.17 (2H, d, J = 7.6 Hz, H-21, H-25), 5.91 (1H, s, H-26), 5.73 (1H, m, H-2), 5.09 (1H, m, H-16), 5.05 (1H, m, H-1a), 5.02 (1H, m, H-1b); 4.88 (1H, m, H-4), 4.81 (1H, m, H-10), 3.43 (2H, d, J = 4.2 Hz, H-19), 2.53 (1H, dd, J = 13.7, 8.5 Hz, H-17a), 2.29 (2H, m, H-3), 2.27 (1H, dd, J = 13.7, 4.6 Hz, H-17b), 2.03–2.04 (9H, ovlp, 3-Ac groups), 1.70 (1H, m, H-11), 1.65 (1H, m, H-5), 1.58 (1H, m, H-15a), 1.48 (1H, ovlp, H-13), 1.46 (1H, ovlp, H-15b), 1.46 (2H, ovlp, H-9), 1.31 (1H, m, H-7a), 1.26 (1H, ovlp, H-7b), 1.26 (2H, ovlp, H-14), 1.25 (1H, ovlp, H-12a), 1.25 (1H, ovlp, H-6a), 1.23 (2H, ovlp, H-8), 1.04 (1H, m, H-6b), 0.92 (1H, m, H-12b), 0.90 (3H, d, J = 6.8 Hz, H-29), 0.86 (3H, ovlp, H-28), 0.85 (3H, ovlp, H-27); HRESIMS m/z 627.3437 [M+Na]+ (calcd for C35H53ClO6Na, 627.3428).
Preparation and Analysis of MTPA Esters.
The procedure was carried out according to Hoye et al. with minor modification.28 1.5 mg of isotrichophycin C (1) was dissolved in dry CDCl3 and separated into two equal portions in 4 mL vials. Dry pyridine (10 μL) and (S)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (15 μL) were added to the first vial. The vial was capped and the reaction mixture was stirred for 24 h. The identical procedure was repeated with an equal amount of 1 and (R)-(−)-a-methoxy-a-(trifluoromethyl)phenylacetyl chloride. After 24 h, the contents of the vials were immediately transferred to NMR tubes for 1H NMR analysis to determine the progress of the esterification by examining the presence of deshielded oxymethine signals at 5.29 and 4.99 ppm. Following NMR analysis, each sample was separated between CH2Cl2 and H2O, and the CH2Cl2 layers were dried under a stream of N2. The residues were subjected to RP-HPLC using a Kinetex 5 μm C18 column (250 × 10 mm) with a mobile phase of 100% CH3CN and a flow rate of 3 mL/min and both bis-MTPA esters were isolated (tR, 7.80 min). The same procedure was performed using 1.5 mg of trichophycin A for each reaction and identical reaction procedures were followed. The tris-MTPA esters were isolated using the identical HPLC method (tR,12.0). The procedure was also carried out with 0.6 mg of 3 separated into two equal portion of 0.3 mg. However, we replaced CH2Cl2 with CDCl3 and analyzed the reaction products directly via 1H NMR, COSY, and TOCSY (Figure S33).
Isotrichophycin C bis-MTPA:
S-ester (partial) 1H NMR (500 MHz, CDCl3) δ 7.28 (2H, t, J = 7.4 Hz, H-16, H-18), 7.23 (1H, m, H-17), 7.11 (2H, d, J = 7.4 Hz, H-15, H-19), 5.87 (1H, s, H-20), 5.91 (1H, s, H-1), 5.71 (1H, m, H-2), 5.29 (1H, m, H-10), 5.02 (1H, m, H-4), 3.26 (1H, d, m, H-13), 2.40 (2H, m, H-11), 2.27 (2H, m, H-3), 1.93 (1H, m, H-5), 1.64 (1H, m, H-9), 1.55 (2H, m, H-8), 1.47 (1H, m, H-7), 1.25 (1H, m, H-6), 0.90 (3H, d, J = 6.6 Hz, H-21), 0.88 (3H, d, J = 6.6 Hz, H-22); HRESIMS m/z 853.2498 [M+Na]+ (calcd for C42H46Cl2F6O6Na, 853.2473); R-ester (partial) 1H NMR (500 MHz, CDCl3) δ 7.28 (2H, d, J = 7.4 Hz, H-16, H-18), 7.23 (1H, m, H-17), 7.13 (2H, d, J = 7.4 Hz, H-15, H-19), 5.96 (1H, s, H-20), 5.99 (1H, s, H-1), 5.81 (1H, m, H-2), 5.29 (1H, m, H-10), 4.99 (1H, m, H-4), 3.36 (1H, m, H-13), 2.54 (2H, m, H-11), 2.28 (2H, m, H-3), 1.83 (1H, m, H-5), 1.56 (1H, m, H-9), 1.47 (1H, m, H-8), 1.38 (1H, m, H-7), 1.13 (1H, m, H-6), 0.79 (3H, d, J = 6.6 Hz, H-21), 0.75 (3H, d, J = 6.8 Hz, H-22); HRESIMS m/z 853.2464 [M+Na]+ (calcd for C42H46Cl2F6O6Na, 853.2473).
Trichophycin A tris-MTPA:
S-ester (partial) 1H NMR (500 MHz, CDCl3) δ 7.29 (2H, d, J = 7.4 Hz, H-22, H-24), 7.23 (1H, m, H-23), 7.10 (2H, d, J = 7.4 Hz, H-21, H-25) 5.85 (1H, s, H-26), 5.73 (1H, m, H-2), 5.28 (1H, m, H-16), 5.10 (1H, m, H-4), 5.11 (1H, m, H-1a), 5.08 (1H, m, H-1b) 5.00 (1H, m, H-10), 3.24 (2H, d, J = 3.9 Hz), 2.41 (2H, m, H-3), 2.34 (2H, m, H-17), 1.72 (1H, m, H-11), 1.67 (2H, ovlp, H-15), 1.67 (1H, m, H-5), 1.63 (2H, m, H-9), 1.52 (2H, ovlp, H-14), 1.51 (2H, ovlp, H-8), 1.40 (1H, m, H-13), 1.23 (2H, m, H-6), 1.19 (2H, m, H-7), 1.14 (2H, m, H-12), 0.85 (3H, d, J = 6.8, H-29), 0.81 (3H, d, J = 6.8 Hz, H-28), 0.78 (3H, d, J = 6.6 Hz, H-27); HRESIMS m/z 1149.4317 [M+Na]+ (calcd for C59H68ClF9O9Na, 1149.4306); R-ester (partial) 1H NMR (500 MHz, CDCl3) δ 7.28, (2H, d, J = 7.4 Hz, H-22, H-24), 7.23 (1H, m, H-23), 7.13 (2H, d, J = 7.4 Hz, H-21, H-25), 5.95 (1H, s, H-26), 5.65 (1H, m, H-2), 5.29 (1H, m, H-16), 5.09 (1H, m, H-4), 5.03 (1H, m, H-1a), 5.00 (1H, m, H-1b), 5.00 (1H, m, H-10), 3.37 (2H, s, H-19), 2.53 (2H, dd, J = 14.0, 7.9, H-17), 2.35 (2H, m, H-3), 1.71 (1H, m, H-11), 1.70 (1H, m, H-5), 1.59 (2H, m, H-9), 1.53 (2H, m, H-15), 1.47 (2H, m, H-14), 1.45 (2H, m, H-8), 1.39 (1H, m, H-13), 1.27 (2H, m, H-6), 1.16 (2H, ovlp, H-12), 1.15 (2H, ovlp, H-7), 0.89 (3H, d, J = 6.7 Hz, H-29), 0.83 (3H, d, J = 6.7 Hz, H-28), 0.74 (3H, d, J = 6.7 Hz, H-27); HRESIMS m/z 1149.4316 [M+Na]+ (calcd for C59H68ClF9O9Na, 1149.4306).
Biological assays.
Cytotoxicity assays using murine neuroblastoma cells (neuro-2A) were conducted as previously described.7 Four technical replicates were prepared for each concentration of 1–3 tested, and each assay was performed in triplicate to determine EC50 values. Doxorubicin was used as a positive control (EC50: 127 μM). Four technical replicates were prepared for each concentration of isotrichophycin C diacetate and trichophycin A triacetate and viability values were determined. EC50 curves were generated and statistical procedures were performed using GraphPad Prism software.
Supplementary Material
Figure 2.
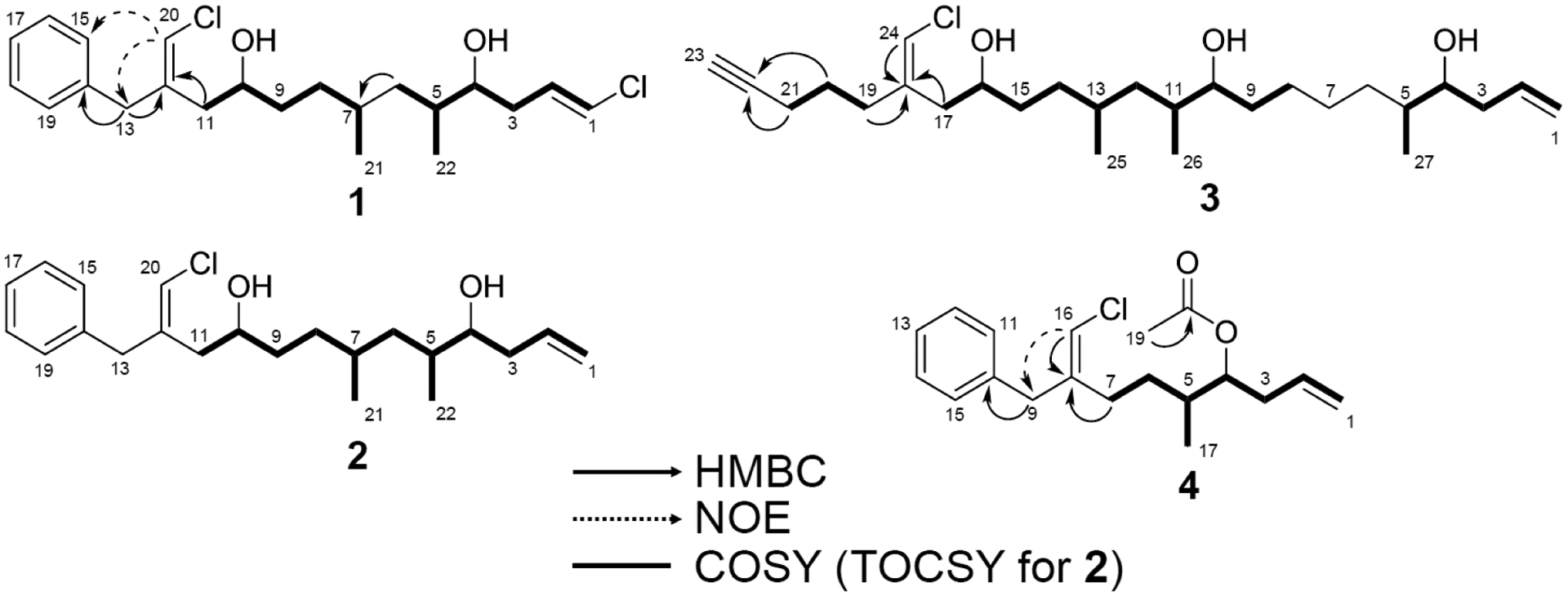
Key 2D NMR correlations for 1–4.
ACKNOWEDGEMENTS
The acquisition of high resolution mass spec, polarimetry, ECD, IR and UV-vis data in this publication was made possible by the use of spectrometric and spectroscopic equipment and services available through the RIINBRE Centralized Research Core Facility, which is supported by the Institutional Development Award (IDeA) Network for Biomedical Research Excellence from the National Institute of General Medical Sciences of the National Institutes of Health under Grant P20GM103430. Certain NMR experiments (500 MHz) were conducted at a research facility at the University of Rhode Island supported in part by the National Science Foundation EPSCoR Cooperative Agreement #EPS-1004057. This work was supported in part by an American Society of Pharmacognosy Starter Grant awarded to M. Bertin.
Footnotes
Supporting Information. The supporting information is available free of charge on the ACS publications website and includes: NMR data tables for 2–4, NMR and MS data for compounds 1–4 and derivatives of 1, 3, and trichophycin A. ECD data of 1 and trichophycin C. EC50 curves of 1–3 and acetylated derivatives, and phylogenetic analysis.
The authors declare no financial conflicts of interest.
REFERENCES
- (1).Davis CS; McGillicuddy DJ Transatlantic abundance of the N2-fixing colonial cyanobacterium Trichodesmium. Science 2006, 312, 1517–1520. DOI: 10.1126/science.1123570. [DOI] [PubMed] [Google Scholar]
- (2).Bergman B; Sandh G; Lin S; Larsson J; Carpenter EJ Trichodesmium – a widespread marine cyanobacterium with unusual nitrogen fixation properties. FEMS Microbiol. Rev 2013, 37, 286–302. DOI: 10.1111/j.1574-6976.2012.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Karl D; Michaels A; Bergman B; Capone D; Carpenter E; Letelier R; Lipschultz F; Paerl H; Sigman D; Stal L Dinitrogen fixation in the world’s oceans. Biogeochemistry 2002, 57/58, 47–98. DOI: 10.1023/A:1015798105851. [DOI] [Google Scholar]
- (4).Carpenter EJ; Capone DG Nitrogen fixation in the marine environment In: Capone DG; Bronk DA; Mulholland MR; Carpenter EJ (eds) Nitrogen in the marine environment, 2nd edition Academic Press/Elsevier, San Diego CA: 2008, pp. 141–198. [Google Scholar]
- (5).Schock TB; Huncik K; Beauchesne KR; Villareal TA; Moeller PDR Identification of trichotoxin, a novel chlorinated compound associated with the bloom forming cyanobacterium, Trichodesmium thiebautii. Environ. Sci. Technol 2011, 45, 7503–7509. DOI: 10.1021/es201034r. [DOI] [PubMed] [Google Scholar]
- (6).Malloy KL; Suyama TL; Engene N; Debonsi H; Cao Z; Matainaho T; Spadafora C; Murray TF; Gerwick WH Credneramides A and B: neuromodulatory phenethylamine and isopentylamine derivatives of a vinyl-chloride-containing fatty acid from cf. Trichodesmium sp. nov. J. Nat. Prod 2012, 75, 60–66. DOI: 10.1021/np200611f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bertin MJ; Wahome PG; Zimba PV; He H; Moeller PDR Trichophycin A, a cytotoxic linear polyketide isolated from a Trichodesmium thiebautii bloom. Mar. Drugs 2017, 15, DOI: 10.3390/md15010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bertin MJ; Saurí J; Liu Y; Via CW; Roduit AF; Williamson RT. Trichophycins B-F, chlorovinylidene-containing polyketides isolated from a cyanobacterial bloom. J. Org. Chem 2018, 83, 13256–13266. DOI: 10.1021/acs.joc.8b02070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Via CW; Glukhov E; Costa S; Zimba PV; Moeller PDR; Gerwick WH; Bertin MJ The metabolome of a cyanobacterial bloom visualized by MS/MS-based molecular networking reveals new neurotoxic smenamide analogs (C, D, and E). Front. Chem 2018, 6, DOI: 10.3389/fchem.2018.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bertin MJ; Zimba PV; He H; Moeller PDR Structure revision of trichotoxin, a chlorinated polyketide isolated from a Trichodesmium thiebautii bloom. Tetrahedron Lett. 2016, 57, 5864–5867. DOI: 10.1016/j.tetlet.2016.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Teta R; Dela Salla G; Esposito G; Via CW; Mazzoccoli C; Piccoli C; Bertin MJ; Costantino V; Mangoni A A joint molecular networking study of a Smenospongia sponge and a cyanobacterial bloom revealed new antiproliferative chlorinated polyketides. Org. Chem. Front 2019, 6, 1762–1774. DOI: 10.1039/C9QO00074G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gerwick L; Boudreau P; Choi H; Mascuch S; Villa FA; Balunas MJ; Malloy KL; Teasdale ME; Rowley DC; Gerwick WH Interkingdom signaling by structurally related cyanobacterial and algal secondary metabolites. Phytochem. Rev 2013, 12, 459–465. [Google Scholar]
- (13).Kwan JC; Teplitski M; Gunasekera SP; Paul VJ; Luesch H Isolation and biological evaluation of 8-epi-malyngamide C from the Floridian marine cyanobacterium Lyngbya majuscula. J. Nat. Prod 2010, 73, 463–466. DOI: 10.1021/np900614n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Villa FA; Lieske K; Gerwick L Selective MyD88-dependent pathway inhibition by the cyanobacterial natural product malyngamide F acetate. Eur. J. Pharmacol 2010, 629, 140–146. DOI: 10.1016/j.ejphar.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sheridan CC; Steinberg DK; Kling GW The microbial and metazoan community associated with colonies of Trichodesmium spp.: a quantitative survey. J. Plank. Res 2002, 24, 913–922. DOI: 10.1093/plankt/24.9.913. [DOI] [Google Scholar]
- (16).Rouco M; Haley ST; Dyhrman ST Microbial diversity within the Trichodesmium holobiont. Environ. Microbiol 2016, 18, 5151–5160. DOI: 10.1111/1462-2920. [DOI] [PubMed] [Google Scholar]
- (17).Frischkorn KR; Rouco M; Van Mooy BAS; Dyhrman ST Epibionts dominate metabolic functional potential of Trichodesmium colonies from oligotrophic ocean. ISME J. 2017, 11, 2090–2101. DOI: 10.1038/ismej.2017.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Schmidt Y; Lehr K; Colas L; Breit B Assignment of relative configuration of desoxypropionates by 1H NMR spectroscopy: method development, proof of principle by asymmetric total synthesis of xylarinic acid A and applications. Chem. Eur. J 2012, 18, 7071–7081. DOI: 10.1002/chem.201103988. [DOI] [PubMed] [Google Scholar]
- (19).Han B; Reinscheid UM; Gerwick WH; Gross H The structure elucidation of isomalyngamide K from the marine cyanobacterium Lyngbya majuscula by experimental and DFT computational methods. J. Mol. Struct 2011, 989, 109–113. DOI: 10.1016/j.molstruc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Esposito G; Teta R; Miceli R; Ceccarelli LS; Della Sala G; Camerlingo R; Irollo E; Mangoni A; Pirozzi G; Costantino V Isolation and assessment of the in vitro anti-tumor activity of smenothiazole A and B, chlorinated thiazole-containing peptide/polyketides from the Caribbean sponge, Smenospongia aurea. Mar. Drugs 2015, 13, 444–459. DOI: 10.3390/md13010444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Esposito G; Della Sala G; Teta R; Caso A; Bourguet-Kondracki M-L-; Pawlik JR, Mangoni A; Costantino V Chlorinated thiazole-containing polyketide-peptides from the caribbean sponge Smenospongia conulosa: structure elucidation on microgram scale. Eur. J. Org. Chem 2016, 2016, 2871–2875. DOI: 10.1002/ejoc.201600370. [DOI] [Google Scholar]
- (22).Caso A; Esposito G; Della Sala G; Pawlik JR; Teta R; Mangoni A; Costantino V Fast detection of two smenamide family members using molecular networking. Mar. Drugs 2019, 17, E618 DOI: 10.3390/md17110618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kumar S; Stecher G; Li M; Knyaz C; Tamura K MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol 2018, 35, 1547–1549. DOI: 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pluskal T; Castillo S; Villar-Briones A; Orešič M MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinformatics 2010, 11, 395 DOI: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Global Natural Products Social Molecular Networking: http://gnps.ucsd.edu//
- (26).Nothias LF; Petras D; Schmid R; Dührkop K; Rainer J; Sarvepalli A; Protsyuk I; Ernst M; Tsugawa H; Fleischauer M; Aicheler F; Aksenov A; Alka O; Allard P-M; Barsch A; Cachet X; Caraballo M; Da Silva RR; Dang T; Garg N; Gauglitz JM; Gurevich A; Isaac G; Jarmusch AK; Kameník Z; Kang K. Bin; Kessler N; Koester I; Korf A; Gouellec A. Le; Ludwig M; Christian MH; McCall L-I; McSayles J; Meyer SW; Mohimani H; Morsy M; Moyne O; Neumann S; Neuweger H; Nguyen NH; Nothias-Esposito M; Paolini J; Phelan VV; Pluskal T; Quinn RA; Rogers S; Shrestha B; Tripathi A; van der Hooft JJJ; Vargas F; Weldon KC; Witting M; Yang H; Zhang Z; Zubeil F; Kohlbacher O; Böcker S; Alexandrov T; Bandeira N; Wang M; Dorrestein PC Feature-Based Molecular Networking in the GNPS Analysis Environment. bioRxiv 2019, 812404 DOI: 10.1101/812404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shannon P; Markiel A; Ozier O; Baliga NS; Wang JT; Ramage D; Amin N; Schwikowski B; Ideker T Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504 DOI: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hoye TR; Jeffrey CS; Shao F Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc 2007, 2, 2451–2458. DOI: 10.1038/nprot.2007.354 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.