

Abstract
Introduction
This study investigates the metabolic activity of circulating monocytes and their impact on pro‐inflammatory responses in RA and explores whether this phenotype is already primed for inflammation before clinical manifestations of disease.
Methods
Blood was collected and CD14+ monocytes isolated from healthy control donors (HC), individuals at‐risk (IAR) and RA patients. Monocyte frequency in blood and synovial tissue was assessed by flow cytometry. Inflammatory responses and metabolic analysis ± specific inhibitors were quantified by RT‐PCR, Western blot, migration assays, Seahorse‐XFe‐technology, mitotracker assays and transmission electron microscopy. Transcriptomic analysis was performed on HC, IAR and RA synovial tissue.
Results
CD14+ monocytes from RA patients are hyper‐inflammatory following stimulation, with significantly higher expression of cytokines/chemokines than those from HC. LPS‐induced RA monocyte migratory capacity is consistent with increased monocyte frequency in RA synovial tissue. RA CD14+ monocytes show enhanced mitochondrial respiration, biogenesis and alterations in mitochondrial morphology. Furthermore, RA monocytes display increased levels of key glycolytic enzymes HIF1α, HK2 and PFKFB3 and demonstrate a reliance on glucose consumption, blockade of which abrogates pro‐inflammatory mediator responses. Blockade of STAT3 activation inhibits this forced glycolytic flux resulting in metabolic reprogramming and resolution of inflammation. Interestingly, this highly activated monocytic phenotype is evident in IAR of developing disease, in addition to an enhanced monocyte gene signature observed in synovial tissue from IAR.
Conclusion
RA CD14+ monocytes are metabolically re‐programmed for sustained induction of pro‐inflammatory responses, with STAT3 identified as a molecular regulator of metabolic dysfunction. This phenotype precedes clinical disease onset and may represent a potential pathway for therapeutic targeting early in disease.
Keywords: rheumatoid arthritis, monocytes, immunometabolism, STAT3, arthralgia
In this study, we demonstrated that rheumatoid arthritis CD14+ monocytes are metabolically re‐programmed for sustained induction of pro‐inflammatory responses, an effect that is mediated by STAT3 signalling and precedes clinical disease onset.
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease, characterised by synovial inflammation, and structural damage of cartilage and bone. 1 If not treated early and aggressively, RA can result in a loss of function, disability, reduced quality of life and increased mortality.2, 3, 4, 5 Recent research has focused on defining the different disease phases in RA, with an emphasis on those ‘at risk’ of developing disease. These individuals present with symptoms of arthralgia and positivity for circulatory autoantibodies (such as anti‐citrullinated protein antibodies [ACPA] or rheumatoid factor [RF]), yet have no clinical signs of inflammation. 6 It is now well established that autoantibodies can precede the onset of clinical symptoms by years in RA patients, 7 thus studying these ‘individuals at risk’ (IAR) may provide important clues in understanding the evolution of RA, and in identifying altered immune responses that may predict disease onset. 8 Although arthralgia along with autoantibody positivity greatly increases the risk of development of RA, reported conversion rates vary between 30 and 70%.9, 10 Furthermore, blood or tissue biomarkers have not yet been identified in this ‘at risk’ population. Studies have shown differences in circulatory lymphocyte subsets in arthralgia subjects during development of arthritis, with studies in synovial tissue limited, showing either no abnormalities or early T‐cell infiltration.11, 12 More recent studies have demonstrated an increase in immune cell gene signatures in IAR, suggesting pre‐existing autoimmune dysregulation.13, 14
Monocytes are crucial innate effector cells central to the initiation of inflammation in RA. Monocytes represent a phenotypically heterogeneous pool of immune cells capable of differentiating into macrophages, dendritic cells and osteoclasts subject to their microenvironmental demand. 15 In addition to their principal role as precursor cells, functionally, monocytes specialise in phagocytosis of foreign particles, secrete many pro‐inflammatory cytokines and chemokines such as TNFα, IL‐1β and CXCL10 and are involved in the production of reactive oxygen species (ROS).15, 16, 17 Peripheral blood monocytes may also act as cellular biomarkers for the assessment of RA disease activity and treatment response. Studies demonstrate that the proportion of circulating monocytes positively correlates with RA disease activity, while elevated levels of monocytes in treatment‐naïve RA patients are predictive of a reduced or poor clinical response to treatment with methotrexate (MTX).18, 19
During inflammation, monocytes have been shown to preferentially differentiate into inflammatory macrophages in response to local macrophage colony‐stimulating factor (M‐CSF), a process which is particularly enhanced in response to physiological stress and inflammatory cues.20, 21 Therefore, a key function of monocytes in RA, once recruited to the site of inflammation, is to differentiate into inflammatory macrophages which actively secrete key pro‐inflammatory cytokines including TNFα, IL‐1β and IL‐6, which in turn promote synovial fibroblast cell invasive mechanisms, resulting in pannus formation and cartilage destruction. In addition, it is now known that the bioenergetic status of macrophages determines their functionality, with studies suggesting that inflammatory macrophages have an enhanced glycolytic flux, a concomitant impairment in the Krebs cycle and utilise fatty acid synthesis (FAS) to support anabolic pathways needed for biosynthesis. 22 This is particularly interesting in the context of RA, whereby synovial CD68+ macrophages strongly correlate with metabolic activity, mitochondrial dysfunction, and oxidative stress in vivo.23, 24 CD68+ macrophages also positively correlate with the degree of clinical disease activity and are inversely related to in vivo synovial pO2 levels.23, 25, 26 RA macrophages also express high levels of the glycolytic enzyme α‐enolase, which is recognised by autoantibodies to induce cytokine production. 27 Furthermore, in animal models of arthritis, RA synovial fluid (SF) derived succinate induces IL‐1β release from macrophages, an effect mediated through GPR91. 28 Recent studies in coronary artery disease (CAD) patients demonstrate that CAD monocytes and macrophages share underpinning metabolic abnormalities to promote inflammation, characterised by increased glucose consumption, ATP production and cytokine secretion compared to their healthy counterparts. 29 Circulatory CAD monocytes were also pre‐programmed to become inflammatory macrophages, indicating that the initial immune insult occurs prior to monocyte differentiation.
This study focuses on assessing whether monocytes in RA and IAR are already primed in the circulation, and whether undifferentiated monocytes are pre‐programmed to differentiate into inflammatory macrophages. Specifically, we demonstrate that CD14+ RA monocytes are hyper‐inflammatory and hyper‐metabolic compared to healthy controls, with a signature indicative of an M1‐like pro‐inflammatory phenotype. This hyper‐inflammatory/metabolic phenotype is mediated by signal transducer and activator of transcription 3 (STAT3) signalling, as selective STAT3 inhibition significantly decreased M1‐like cytokines and metabolic mechanisms. In addition, despite their key role in RA development and pathogenesis, no studies to date have investigated the role of monocytes in IAR of developing RA. Here, we demonstrate that the pro‐inflammatory/metabolic phenotype is evident in CD14+ monocytes from arthralgia autoantibody‐positive subjects, demonstrating that monocyte activation may precede clinical manifestations of RA, and may represent a potential pathway for therapeutic targeting early in disease.
Results
Peripheral RA CD14+ monocytes are hyper‐inflammatory
To elucidate the role of peripheral blood monocytes in the pathogenesis of RA, we initially analysed the frequency of circulating monocytes in RA patients (88.9 ± 2.3) compared to HC (73.2 ± 2.6). RA patients displayed increased frequencies of CD14+ peripheral blood monocytes compared to healthy controls (Figure 1a, P < 0.05).
Figure 1.
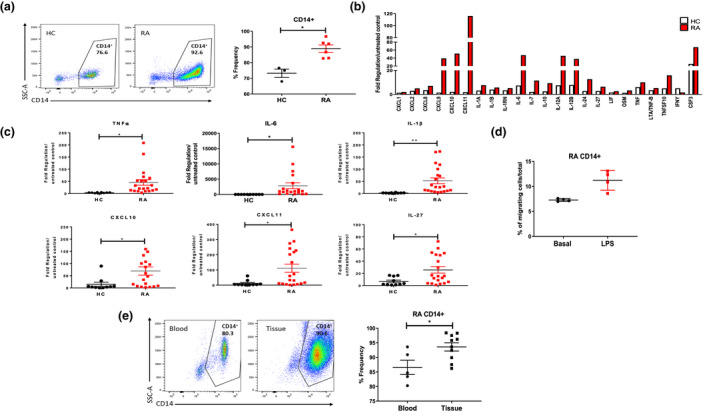
(a) Difference in expression of CD14+ cells in PBMC from HC and RA patients. Left, representative flow cytometry plots. Right, percentage frequency of CD14+ cells in healthy (n = 3) and RA (n = 6) blood. (b) Gene expression of RA vs HC monocytes stimulated with LPS with more than 1.5‐fold difference (n = 3). Data are expressed as upregulation compared to unstimulated control and normalised to housekeepers β‐actin, B2M, GAPDH, HPRT1 and RPLPO. (c) Gene expression between healthy (n = 14 or 15) and RA (n = 19 or 21) monocytes, represented as fold change in response to LPS stimulation. (d) CD14+ RA monocyte migrating cells in response to LPS stimulation (n = 4). (e) Percentage frequency of CD14+ cells in RA blood (n = 5) and RA synovial tissue (n = 10). Data are represented as mean ± SEM; Mann–Whitney U‐tests and t‐tests were used as appropriate. *P < 0.05, **P < 0.01.
In order to compare the expression of pro‐inflammatory genes by CD14+ monocytes isolated from the blood of RA and HC donors; an 84 cytokine/chemokine PCR array was performed. An increase in the expression of 34 cytokine/chemokines in response to LPS stimulation was demonstrated in RA and HC CD14+ monocytes (Supplementary figure 1). LPS stimulation resulted in a significant increase in 23 of these genes encoding key cytokines and chemokines in RA compared to HC CD14+ monocytes, suggesting a heightened immune response by RA monocytes (Figure 1b).
Selecting those genes with the greatest differences between RA and HC, and those of greater functional interest, 6 of these 23 genes were validated in a larger cohort. Dot plots representing these 6 genes; TNFα, IL‐6, IL‐1β, CXCL10, CXCL11 and IL‐27 demonstrate a significant increase in responsiveness to LPS stimulation in RA vs HC monocytes (Figure 1c, all P < 0.05). In addition to a hyper‐inflammatory response to LPS, RA CD14+ monocytes also demonstrated increased migratory capacity in response to LPS (Figure 1d). To support the concept of an increased migratory potential of RA CD14+ monocytes, we next compared the frequency of CD14+ monocytes in RA blood and synovial tissue cell suspensions to examine if monocyte frequency was increased at the site of inflammation in RA. Analysis of single‐cell synovial tissue suspensions demonstrated a significant increase in the frequency of infiltrating CD14+ monocytes in RA synovial tissue compared to peripheral blood monocytes (Figure 1e, P < 0.05).
Activated peripheral RA CD14+ monocytes are hyper‐metabolic
A greater inflammatory burden is known to be associated with a greater demand for ATP and higher metabolic activity, particularly in inflammatory M1 macrophages. Therefore, we next examined glucose utilisation and mitochondrial function of RA CD14+ monocytes using the Seahorse‐XFe96 analyzer. Figure 2a illustrates average bioenergetic profiles of oxidative phosphorylation (OCR) before and after injections of oligomycin, FCCP and antimycin A in basal (unstimulated) and LPS‐stimulated monocytes. Following stimulation, CD14+ monocytes display a significant decrease in baseline OCR and ATP synthesis compared to unstimulated basal control (Figure 2b, P < 0.05). When directly comparing ex vivo RA CD14+ monocytes to HC CD14+ monocytes, however, a significant increase in mitochondrial respiration (OCR) and ATP synthesis was observed in RA (P < 0.05), with no difference observed for maximal or spare respiratory capacity (Figure 2b). Similarly, LPS‐activated RA monocytes displayed significantly higher baseline OCR, maximal respiratory capacity and ATP synthesis compared to LPS‐activated healthy monocytes (Figure 2b, P < 0.05). Increased mitochondrial respiration, resulting in higher ATP production, demonstrates a state of heightened mitochondrial activity.
Figure 2.
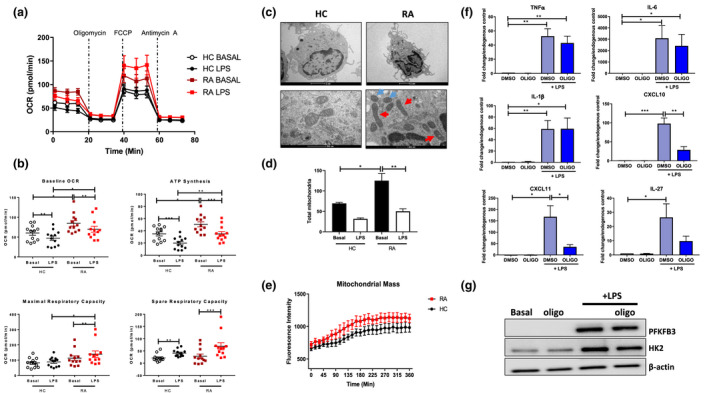
(a) Seahorse bioenergetics profile (average) OCR of healthy (n = 12) and RA (n = 12) monocytes before and after injections of oligomycin, FCCP and antimycin A in ex vivo resting and LPS‐stimulated (100 ng mL−1; 1 h) monocytes. (b) Baseline OCR, ATP synthesis, maximal respiratory capacity and spare respiratory capacity (mean ± SEM). (c) Representative TEM images of healthy control and RA monocytes in response to LPS stimulation. Regular‐ and irregular‐shaped mitochondria indicated by blue and red arrows, respectively. Scale bar represents 2 µm and 500 nm. (d) Bar graphs represent quantification of total mitochondria present from high‐powered images of 5 cells per sample in HC (n = 3) and RA (n = 3) CD14+ monocytes ± LPS. Data are represented as mean ± SEM, and the two‐way ANOVA with Tukey multiple comparisons test used, *P < 0.05, **P < 0.01. (e) Mitochondrial mass of HC (n = 4) and RA (n = 6) monocytes stained with MitoTracker Green in response to LPS stimulation. Kinetic mode readings performed every 15 min over a 6‐h time period. Data are expressed as mean ± SEM. (f) RA monocytes (n = 8) were stimulated with LPS (100 ng mL−1) for 3 h ± oligomycin. Gene expression of TNFα, IL‐6, IL‐1β, CXCL10, CXCL11 and IL‐27. Data are represented as mean ± SEM. (g) Representative Western blots showing PFKFB3, HK2 and β‐actin protein expression. The Wilcoxon signed rank test, a Mann–Whitney U‐test and a t‐test were used as appropriate. *P < 0.05, ** P < 0.01, ***P < 0.005.
Using ultrastructure analysis of mitochondrial morphology assessed by TEM, a mixture of regular‐ and irregular‐shaped mitochondria was observed in RA monocytes compared to HC (Figure 2c; blue and red arrows). We demonstrated a significant increase in the numbers of mitochondria in unstimulated RA monocyte compared to unstimulated HC monocytes (Figure 2d, P < 0.05). Upon stimulation, the total number of mitochondria decreased in both RA and HC monocytes compared to the resting state (Figure 2d), this is consistent with the concomitant decrease in baseline OCR and ATP synthesis observed in response to monocyte activation (Figure 2b, P < 0.05). RA CD14+ monocytes were also demonstrated to have increased mitochondrial mass compared to healthy controls, again suggesting mitochondrial dysfunction (Figure 2e). We also examined the number of elongated mitochondria (as a percentage of total mitochondria present), and while there was an increase in the number of elongated mitochondria in LPS‐stimulated RA monocytes compared to healthy monocytes, this was not statistically significant (Supplementary figure 1c).
To assess whether alterations in mitochondrial respiration contribute to the hyper‐inflammatory phenotype observed in RA CD14+ monocytes, oligomycin was utilised. Blockade of oxidative phosphorylation only partially inhibited the LPS‐induced inflammatory burden, with inhibition of CXCL10, CXCL11 (both P < 0.05) and IL‐27 (P = 0.07), but no change in TNFα, IL‐1β or IL‐6 (Figure 2f). Interestingly, oligomycin had no effect on protein expression of key glycolytic enzymes HK2 and PFKFB3 (Figure 2g and Supplementary figure 5a). The effect of oligomycin was also investigated in healthy control monocytes at both the gene and protein level (Supplementary figures 3a, 4a and b).
Activated peripheral RA CD14+ monocytes rely on glycolysis
Representative ECAR profiles are shown in Figure 3a. Average ECAR profiles indicate a clear increase in glycolytic capacity following LPS stimulation, an effect that is exacerbated when comparing RA CD14+ monocytes to HC (Figure 3a). Indeed, the significant induction in baseline ECAR and maximal glycolytic rate following LPS stimulation was significantly enhanced in RA monocytes compared to that of HC (Figure 3b, P < 0.05). This resulted in a shift in the ECAR/OCR ratio of RA and HC monocytes in favor of glycolysis in response to LPS stimulation (Figure 3b, P < 0.05). This hyper‐glycolytic phenotype was paralleled by overexpression of the master regulator of metabolism HIF1α (P < 0.05), in addition to PFKFB3 a key glycolytic enzyme (P < 0.05), with HK2 also increased although not significant (Figure 3c). In addition, the representative metabolic profile showing the changes in metabolic demands in RA and HA CD14+ monocytes following LPS stimulation demonstrates that both RA and HC monocytes switch their metabolism to a highly energetic state. This effect is, however, potentiated in RA monocytes compared to HC (Figure 3d).
Figure 3.
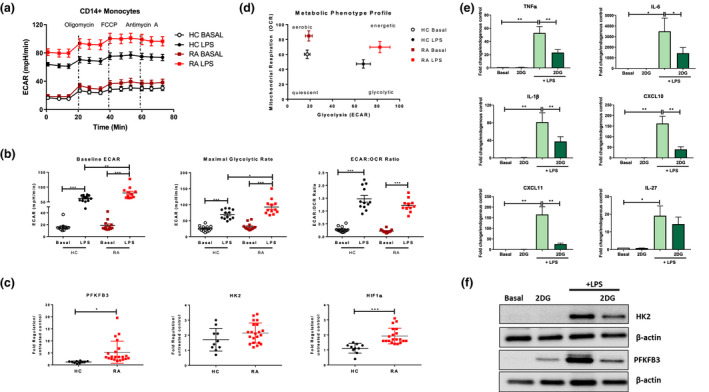
(a) Seahorse bioenergetics profile (average) demonstrating ECAR of HC (n = 12) and RA (n = 12) monocytes before and after injections of oligomycin, FCCP and antimycin A in ex vivo resting untreated and LPS‐stimulated (100 ng mL−1; 1 h) monocytes. (b) Baseline glycolysis, maximal glycolytic rate and ECAR:OCR ratio, expressed as mean ± SEM. (c) Dot plots represent gene expression of PFKFB3, HK2, HIF1α in HC (n = 10) and RA (n = 21) monocytes. Data are represented as fold change in response to LPS stimulation, mean ± SEM. RA CD14+ monocytes (n = 7) were stimulated with LPS (100 ng mL−1) for 3 h ± 2DG. (d) Overall metabolic profile of untreated and LPS‐stimulated HC (n = 12) and RA (n = 12) monocytes. (e) RA monocytes (n = 7) were stimulated with LPS (100 ng mL−1) for 3 h ± 2DG. Gene expression of RA monocytes TNFα, IL‐6, IL‐1β, CXCL10, CXCL11 and IL‐27, mean ± SEM. (f) Representative Western blots showing PFKFB3 and HK2 protein expression in CD14+ RA monocytes. The Wilcoxon signed rank test, a Mann–Whitney U‐test or a paired t‐test were used as appropriate. *P < 0.05, **P < 0.01, ***P < 0.005.
To examine whether glycolysis is fuelling the inflammatory phenotype of RA monocytes, we cultured monocytes in the presence or absence of 2DG. Blocking glycolysis significantly inhibited expression of all six pro‐inflammatory signature cytokines/chemokines; TNFα, IL‐6, IL‐1β, CXCL10, CXCL11 (all P < 0.05) and IL‐27 (P = 0.057) (Figure 3e). The effect of 2‐DG was also investigated in healthy control monocytes at both the gene and protein level (Supplementary figures 3b and 4c and d). This was paralleled by the inhibition of LPS‐induced protein expression of key glycolytic enzymes HK2 and PFKFB3 by 2DG treatment (Figure 3f and Supplementary figure 5b). Considering the partial effect of blocking mitochondrial respiration on LPS‐induced inflammatory mediators, these results would suggest that glycolysis and not oxidative phosphorylation regulates the RA CD14+ monocyte inflammatory phenotype.
Furthermore, analysis of unstimulated ex vivo monocytes also reveals striking metabolic differences. RA CD14+ unstimulated monocytes display significantly increased levels of oxidative phosphorylation with significantly elevated baseline OCR and ATP synthesis (Supplementary figure 2a). In addition, RA ex vivo monocytes display increased numbers of mitochondria compared to healthy control (Supplementary figure 2c and d, P < 0.05). Moreover, RA monocytes have slightly higher glycolytic capacities compared to HC basally (Supplementary figure 2b). Taken together, these data suggest that unstimulated basal RA monocytes are hyper‐metabolic, with a reliance on oxidative phosphorylation. Therefore, upon activation, this reliance on mitochondrial respiration in RA monocytes switches towards a predominantly glycolytic phenotype (Figure 3a–d). While baseline OCR and ATP levels decrease following LPS stimulation in RA monocytes, they are still significantly higher compared to that of HC, in addition to increases in maximal and spare respiratory capacity in RA monocytes compared to HC. Thus overall, RA CD14+ monocytes are hyper‐metabolic compared to basal and activated HC monocytes.
RA CD14+ phenotype is mediated through STAT3
The transcription factor STAT3 has previously been implicated in mediating invasive mechanisms in RA30, 31 and has recently been linked to regulation of metabolic activity, 32 therefore, we next assessed the role of STAT3 in mediating the hyper‐inflammatory and metabolic phenotype observed in RA CD14+ monocytes. Firstly, we demonstrate that STAT3 mRNA expression is significantly increased in LPS‐stimulated RA CD14+ monocytes compared to HC (Figure 4a, P < 0.05). In addition, pSTAT3 protein expression is increased in RA stimulated CD14+ monocytes compared to ex vivo unstimulated monocytes (Supplementary figure 6a and b). Furthermore, selective inhibition of STAT3 using STATTIC (Supplementary figure 6) resulted in almost complete inhibition of the signature inflammatory panel: TNFα (P < 0.05), IL‐6 (P < 0.05), IL‐1β (P < 0.05), CXCL10, CXCL11 and IL‐27 (P < 0.05) (Figure 4b). Additionally, STAT3 inhibition further decreased GLUT‐1, HK2 and PFKFB3 protein expression (Figure 4c and Supplementary figure 6c). To assess whether STAT3 mediates these effects through regulation of metabolic pathways, we assessed the effect of STATTIC on RA CD14+ bioenergetic profiles. Figure 4d demonstrates average energy profiles, showing that STAT3 inhibition results in a decrease in both oxidative phosphorylation and glycolysis. Interestingly, inhibition of glycolysis using 2DG also showed reciprocal inhibition of STAT3 expression, suggesting bi‐directional interplay or negative feedback, as STAT3 inhibition can reduce glycolysis; however, inhibiting glycolysis can also reduce STAT3 expression (Figure 4e, P < 0.05). Interestingly, no effect was observed in LPS‐induced STAT3 expression with inhibition of mitochondrial respiration with oligomycin (Supplementary figure 5c).
Figure 4.
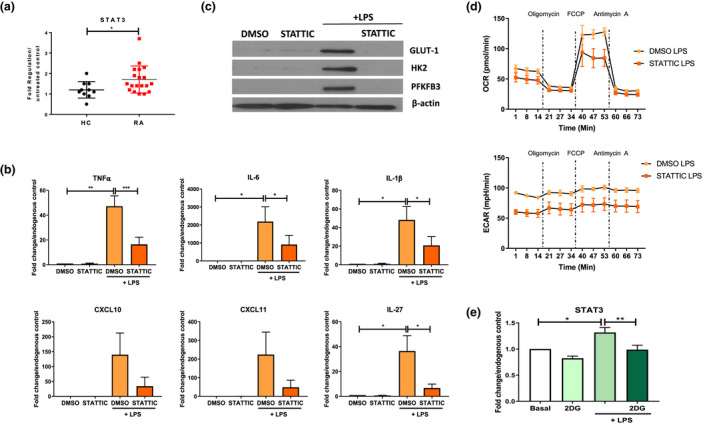
Gene expression of (a) STAT3 in HC (n = 10) and RA (n = 19) monocytes in response to LPS stimulation (100 ng mL−1; 3 h) and (b) TNFα, IL‐6, IL‐1β, IL‐27, CXCL10 and CXCL11 in response to LPS ± STATTIC in RA monocytes (n = 7–8). Data are represented as fold change in response to LPS stimulation, mean ± SEM, normalised to housekeeping control. (c) Representative Western blots demonstrating inhibition of LPS‐induced GLUT‐1, HK2 and PFKFB3 protein expression in RA CD14+ monocytes following incubation with STATTIC. (d) Seahorse bioenergetic profiles (average) demonstrating OCR (top) and ECAR (bottom) in LPS‐stimulated (100 ng mL−1; 1 h) ± STATTIC RA monocytes (n = 2). (e) STAT3 gene expression in response to LPS ± 2DG in RA CD14+ monocytes (n = 5). Data are represented as mean ± SEM. A Wilcoxon signed rank test, a Mann–Whitney U‐test or a paired t‐test was used as appropriate. *P < 0.05, **P < 0.01, ***P < 0.005.
CD14+ monocytes from ‘individuals at‐risk’ already display RA‐like phenotype
It is now appreciated that immunological events precede the onset of clinical manifestations33, 34 and so recent studies have focused on defining the different phases of RA disease development, with a particular emphasis on those ‘individuals at risk’ of RA 35 (Figure 5a). To assess whether the changes identified in established RA cells are already evident before disease onset, CD14+ monocytes from IAR were examined. Firstly, we identified a stepwise progression in expression of pro‐inflammatory mediators from HC to IAR to RA, with significant increases in TNFα (P < 0.05), IL‐6 (P < 0.05), IL‐1β (P < 0.05), and CXCL11 (P < 0.05) in LPS‐activated CD14+ IAR monocytes compared to HC (Figure 5b). Serum levels of IL‐6 expression in HC, IAR and RA serum samples were also analysed; however, no significant difference was observed (Supplementary figure 7b). In addition, the metabolic phenotype of IAR CD14+ monocytes is also altered. Figure 5c depicts representative OCR and ECAR) profiles of four IAR donors, showing a similar metabolic phenotype to RA CD14+ monocytes, with increases in both oxidative phosphorylation and glycolysis compared to healthy monocytes. The metabolic phenotype profile (Figure 5d) demonstrates similarities between RA and IAR CD14+ monocytes, compared to HC. Therefore, IAR CD14+ monocytes are distinctly different from HC, with a clearly heightened energetic state. In addition to increased inflammatory responsiveness, we also observed an increase in HIF1α (P < 0.05), PFKFB3 (P < 0.05) and HK2 in IAR CD14+ monocytes compared to HC (Figure 5e). This increase in PFKFB3 and HK2 in IAR and RA activated monocytes compared to HC monocytes was also similarly observed at the protein level (Figure 5f). Interestingly, expression levels of HIF1α, PFKFB3 and HK2 were similar between IAR and RA CD14+ monocytes (Figure 5e and f).
Figure 5.
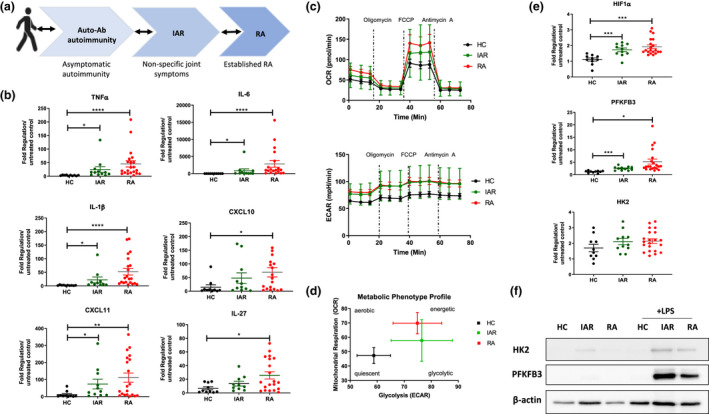
(a) Schematic representing the stages of RA. (b) Dot plots representing gene expression of TNFα, IL‐6, IL‐1β, IL‐27, CXCL10 and CXCL11 in healthy (n = 10), IAR (n = 10 or 12) and RA (n = 19 or 21) CD14+ monocytes. Data are represented as fold change in response to LPS stimulation, mean ± SEM, normalised to housekeeping control. (c) Average Seahorse bioenergetics profile demonstrating OCR (top) and ECAR (bottom) of healthy (n = 12), IAR (n = 4) and RA (n = 12) LPS‐stimulated (100 ng mL−1; 1 h) monocytes. (d) Overall metabolic profile of LPS‐stimulated HC, IAR and RA monocytes. (e) Expression of metabolic genes in healthy (n = 10), IAR (n = 11) and RA (n = 20 or 21) CD14+ monocytes in response to 100 ng mL−1; 3 h LPS stimulation. Data are expressed as fold change compared to unstimulated monocytes, mean ± SEM, normalised to housekeeping control using an unpaired t‐test and ANOVA. *P < 0.05, **P < 0.01, ***P < 0.005. (f) Representative Western blots demonstrating HK2 and PFKFB3 protein expression in HC, IAR and RA CD14+ monocytes following activation with LPS.
Finally, RNA‐seq analysis of synovial tissue from healthy donors, IAR and established RA patients was performed on a data set we previously described. 13 Transcriptomic analysis revealed enrichment of a monocyte gene signature pathway as assessed by Biocarta analysis in the synovium of IAR (P < 0.05) and RA (P < 0.05) compared to HC tissue (Figure 6a). In addition, STRING pathway analysis demonstrates known and predicted interactions of 11 genes represented in Figure 6b. Specifically, a significant stepwise increase in 6 of the 11 monocyte pathway genes from HC to IAR to RA was observed (Figure 6c). Significant increases were observed in gene expression of ITGA4, ITGAM, ITGB1, PECAM‐1, SELL and SELP in IAR synovial tissue compared to HC (Figure 6c, all P < 0.05). These data suggest that the altered phenotype of monocytes precedes clinical manifestations, and thus circulating macrophage precursors may have potential in identifying ‘individuals at risk’ who will go on to develop RA.
Figure 6.
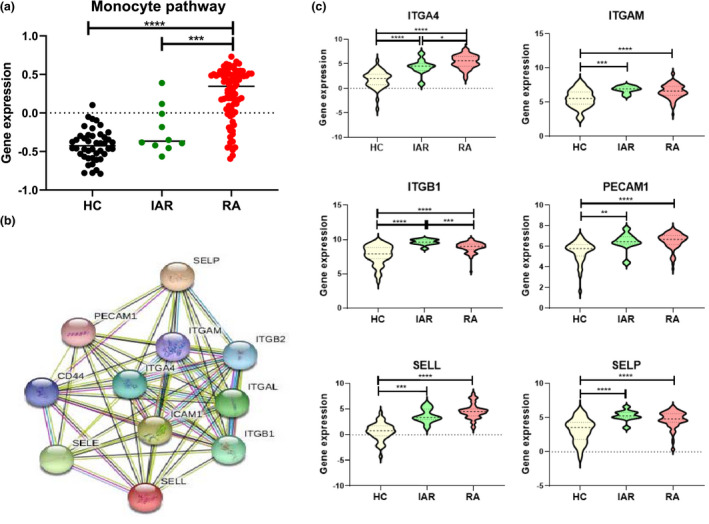
Following whole synovial tissue biopsy mRNA isolation, a high‐throughput mRNA‐seq coupled with a systems biology network computational approach was used to identify novel pathways as previously described. 13 (a) Biocarta analysis to examine gene interactive pathways, identified enrichment of specific monocyte genes in IAR (n = 10), established RA (RA) (n = 85) compared to normal healthy synovium (n = 44) synovial tissue, mean ± SEM. (b) STRING pathway analysis of specific monocyte pathway gene signature. (c) Violin plots depicting expression of monocyte‐associated genes in normal (HC) (n = 44), IAR (n = 10) and established RA (RA) (n = 85) synovial tissue, mean ± SEM. A Mann–Whitney U‐test was used. *P < 0.05, **P < 0.01, ***P < 0.005.
Discussion
Myeloid cells with a monocyte/macrophage phenotype are present in large numbers in the RA joint, significantly contributing to disease. In this study, we aimed to assess whether peripheral blood monocytes in RA are primed for inflammation prior to joint infiltration to become inflammatory macrophages. In this study, we demonstrate significantly more CD14+ monocytes in circulation in RA patients compared to HC. We report that CD14+ monocytes from RA patients are poised to produce pro‐inflammatory mediators upon stimulation, with significantly higher expression of IL‐1β, TNFα, IL‐6, IL‐27, CXCL10 and CXCL11 compared to healthy controls, which is indicative of an M1‐inflammatory phenotype. These hyper‐inflammatory cells also display elevated migratory capacity in response to LPS stimulation, paralleled by the observed increased frequency of infiltrating CD14+ monocytes in RA synovial tissue single‐cell analysis. Evaluation of the metabolic competence of CD14+ monocytes revealed a robust boost in both oxidative phosphorylation and glycolysis in RA compared to HC, coupled with altered mitochondrial morphology. In addition, we demonstrate a consistent upregulation of the glycolytic machinery, both at the gene and protein level, indicating a fundamental abnormality in the processing of glucose. This hyper‐inflammatory, hyper‐metabolic phenotype is mediated by STAT3, as selective STAT3 inhibition switches the inflammatory and metabolic phenotype of RA monocytes; significantly decreasing pro‐inflammatory cytokines, mitochondrial respiration and glycolytic mechanisms to promote resolution of inflammation. Finally, this distinct phenotype is evident in CD14+ monocytes from ‘individuals at risk’ of developing disease, demonstrating that monocyte activation may precede clinical manifestations of RA.
In this study, we report an inflammatory signature specific to circulating CD14+ monocytes in RA patients compared to healthy individuals. Gene expression analysis revealed that monocytes from RA patients are poised to produce high levels of the key pro‐inflammatory cytokines/chemokines, TNFα, IL‐6, IL‐1β, CXCL10, CXCL11 and IL‐27. Interestingly, a recent review on macrophage polarisation reported that these specific genes are overexpressed and are a hallmark of M1‐inflammatory macrophages. 36 This is in line with studies assessing monocyte/macrophage function in CAD, which suggest that monocyte precursors are primed for inflammation and subsequent differentiation into inflammatory macrophages. 29 Conversely, a recent study indicated that circulating monocytes from giant cell arteritis (GCA) patients display pro‐inflammatory signatures similar to healthy individuals, indicating that pre‐programming of monocytes is disease‐specific. 37 TLR4 stimulation is the model for activation used in this study, although other studies have demonstrated a role for other TLRs in RA myeloid cells. TLR2 and TLR9 have been shown to be overexpressed on monocyte subsets of active RA patients with enhanced pro‐inflammatory cytokine production in response to synthetic and viral TLR2 and TLR9 agonists compared to healthy. 38 Interestingly, previous studies have also shown that RA monocytes induce specific cytokines depending on exposure to different TLR agonists, thus in addition to the cytokine/chemokine responses demonstrated in response to TLR4 in this study, additional inflammatory responses may be observed for other stimulants.38, 39 Moreover, the direct role of certain TLRs on monocyte activation such as TLR9, is controversial, with certain studies suggesting that it is more of an indirect effect mediated by dendritic cells. 40
In RA, myeloid cell effector functions may be influenced by citrullinated proteins. ACPA positivity has been shown to influence the inflammatory status of monocyte‐derived macrophages, with ACPA from RA synovial fluid initiating a shift in peripheral blood monocytes towards a pro‐inflammatory macrophage phenotype in vitro. 41 Another study reported that ACPA promote IL‐1β release from macrophages via AKT/NFκB and subsequent activation of the NLRP3 inflammasome. 42 Additionally, studies have demonstrated that circulating RA monocytes tend to skew towards the CD14++/CD16+ ‘intermediate’ monocyte subset, which are primed to produce high levels of many pro‐inflammatory cytokines including TNFα, IL‐1β and IL‐6, and are thought to be the monocyte subset that predominantly differentiates into M1 inflammatory macrophages.18, 20, 43, 44, 45 Elevated levels of circulating monocytes positively correlate with disease activity and are predictive of a poor response to treatment in RA.18, 19 This underscores the potential role of monocytes as a promising biomarker in informing RA disease activity and treatment response.
We demonstrate that RA CD14+ monocytes are in a state of hyper‐metabolism, displaying heightened mitochondrial respiration coupled with increased mitochondrial number, along with a significant reliance on glycolytic mechanisms. Interestingly, mitochondrial respiration is significantly increased in ex vivo unstimulated RA monocytes compared to healthy with the reliance on glycolysis only becoming apparent following activation. While this is the first study to demonstrate the priming of circulatory monocytes in RA, similar mechanisms have been demonstrated in CAD. The hyper‐metabolic state, as represented by increased oxygen consumption, ATP production and cytokine secretion, in CAD monocytes is similar to that observed in pathogenic RA monocytes in this study. 29 Similarly, another study reports a remodelling of mitochondria‐associated membranes in RA and CAD macrophages to enhance mitochondrial activity, a phenomenon that is already present in circulating monocytes and dependent on inactivation of glycogen synthase kinase 3b (GSK3b). 46 Interestingly, functional abnormalities of circulating monocytes are not common to all autoimmune disorders, as a study comparing circulating monocytes from CAD and GCA demonstrated differential glucose requirements in GCA when compared to CAD. 37 Moreover, a reliance on glycolysis isn’t observed in all RA immune cells, indeed one study has demonstrated that naive T cells in RA in fact display defective glycolytic flux resulting in a hyper‐inflammatory phenotype. 47 Therefore, the effect of metabolic rewiring in RA appears to be cell type and context‐dependent.
The observed concomitant changes in inflammatory function and metabolic outputs are not surprising. A link between immune function and cellular metabolism has been extensively studied in recent years. As such, the metabolic plasticity of monocytes opens the possibility of bioenergetic interference as a therapeutic intervention for RA to re‐direct circulating monocytes away from inflammatory mechanisms. HIF1α deletion in myeloid cells of collagen‐induced arthritis (CIA) mice has been reported to result in decreased macrophage infiltration and joint swelling due to reduced macrophage mobility and invasive capacity, 48 while silencing of the amino acid transporter SLC7A5, results in a significant decrease of IL‐1β and glycolysis in RA monocytes and macrophages. 49 Furthermore, elevated glucose concentrations promote IL‐1β production from RA myeloid cells via NLRP3 inflammasome activation. 50
The distinct inflammatory and metabolic pre‐programming of monocytes may also involve altered epigenetic mechanisms. Studies indicate that monocytes are capable of building immunological memory via epigenetic imprinting.51, 52 Epigenomic profiling of ex vivo monocytes and differentiated macrophages demonstrates that differentiating monocytes undergo substantial epigenetic remodelling, implicating epigenetic mechanisms as a potential target for therapy. 52 Consequently, the epigenetic landscape is crucial in determining macrophage polarisation; HDAC3, a specific histone deacetylase, has been shown to support inflammatory macrophage polarisation while simultaneously blunting anti‐inflammatory macrophage activation. 53 Indeed, HDAC3‐deficient macrophages display an M2 pro‐resolution phenotype and are hyper‐responsive to IL‐4, thus pharmacological inhibition of HDAC3 may be of therapeutic benefit. 54 In addition, environmental factors such as smoking are associated with DNA methylation 55 which is interesting in the context of RA considering the strong association of smoking with disease pathogenesis. 56
In this study, we identify a key role for the transcription factor STAT3 in influencing RA monocyte inflammatory and bioenergetic mechanisms. This is in line with studies linking STAT3 to the activation of other cells involved in RA disease pathogenesis; phosphorylation of STAT3 mediates RA synovial fibroblast migration and invasive mechanisms. 31 Moreover, the STAT3‐LPS axis has been widely reported with TLR4 activation previously reported to induce STAT3 activation in other myeloid cells such as dendritic cells and macrophages.57, 58, 59 Studies have also shown that inhibition of STAT3 results in decreased LPS‐mediated cytokine expression in human lung carcinoma cells and THP‐1 human monocytic cells, 60 similar to the results observed in LPS‐activated RA monocytes in the presence of STATTIC in this study.
Indeed, activation of STAT3 has been shown to be associated with in vivo synovial hypoxia, while hypoxia‐induced STAT3 activation causes upregulation of HIF1α31, 61; thus, the observed activation of HIF1α in response to LPS stimulation in this study may indicate an indirect effect of LPS on STAT‐3 activation also. Inhibition of STAT3 suppresses disease severity in the CIA model of arthritis. 62 JAK‐STAT signalling is closely linked to altered cellular metabolism, with blockade of the key glycolytic enzyme PFKFB3 resulting in decreased phosphorylation of STAT3 in RA fibroblast‐like synovial cells, 63 while STAT3 regulation of glycolysis is achieved through HK2 in cancer cells. 64 Indeed, our group has also recently reported evidence that inhibition of JAK‐STAT signalling switches the metabolic and inflammatory profile of synovial cells away from pathogenic mechanisms and towards resolution of inflammation. 32 In addition, we demonstrate that inhibition of glycolysis with 2DG resulted in a reciprocal inhibition of STAT3 activation, indicating a negative feedback loop whereby inhibition of STAT3 blocks glycolysis, while inhibition of glycolysis reduces STAT3 expression. This is consistent with studies demonstrating bi‐directional interactions between STAT3 and key glycolytic enzymes HIF1α, PKM2 and HK2,29, 31, 61, 64 thus further supporting the concept of indirect effects of LPS on STAT3 activation.
We also demonstrate that circulating monocytes from IAR display phenotypic features of inflammatory RA monocytes prior to clinical manifestations. This is the first study to investigate peripheral blood monocytes in IAR, with their role pre‐disease largely unknown. One study has demonstrated that circulating monocytes are increased in early RA (< 1 year), and treatment with infliximab results in a decrease of this population after just two weeks. 65 ACPA purified from RA patients have been demonstrated to induce a pro‐inflammatory phenotype in healthy donor monocyte‐derived macrophages through IL‐1β and ATP release. 42 In addition, a pilot study of ACPA‐positive arthralgia patients observed increased uptake of C‐(R)‐PK11195, which specifically targets activated macrophages, in patients who went on to develop RA after two years, 66 suggesting macrophage activation may potentially identify subclinical arthritis. However, few studies have assessed the in vitro and ex vivo alterations occurring in IAR and data on immune dysfunction before disease onset are lacking. One study, however, identified a decrease in CD8+ T cells in patients with arthralgia who converted to RA 24 months after the blood analysis was completed. In addition, they demonstrated a difference in memory B cells in arthralgia subjects who did not go on to develop RA compared to those who did go on to develop RA within 12 months. 11
Finally, we have demonstrated increased expression of a specific monocyte gene signature in synovial tissue of IAR and RA compared to healthy controls. Specifically, integrins (ITAG4, ITGAM ITGB1), L‐selectin (SELL), P‐selectin (SELP) and PECAM are all enriched in IAR and RA synovial tissue compared to HC, all of which are involved in migration and adhesion of monocytes.67, 68, 69 The significance of this gene signature is supported by studies demonstrating that integrins are increased in the inflamed RA synovium to facilitate leukocyte trafficking and contribute to joint destruction,70, 71, 72, 73, 74 with others indicating enriched levels of selectins in the sera of active RA patients, correlating with markers of disease activity. 75 Further evidential support is provided by single‐cell analysis on monocytes, demonstrating enriched expression of these genes in distinct monocyte subsets. 67 Furthermore, we have previously demonstrated in the same data set an increase in the CD40–CD40L pathway signature in both RA and IAR, suggesting early inflammatory changes are already occurring in the synovium of IAR, even though there are no clinical signs of inflammation. 13 This study supports the concept that early changes in immune cells may serve to stratify arthralgia and IAR, into those that will or will not develop RA.
In conclusion, this study demonstrates the unique inflammatory and metabolic phenotype of RA monocytes, suggesting that peripheral CD14+ monocytes may be committed to become pro‐inflammatory macrophages. Mechanistically, we have identified a key role for STAT3 in regulating RA monocyte metabolism and pro‐inflammatory mechanisms. In addition, the observation of this phenotype in IAR indicates that these features may precede clinical manifestations of RA and represent a potential early target.
Methods
Patient Recruitment
Patients with established RA were recruited from the Rheumatology Department at St Vincent’s University Hospital, Dublin, Ireland. Patients fulfilling the ACR classification criteria 76 were included in this investigation. IAR presented with arthralgia, ACPA/RF positivity but had a normal CRP and no evidence of synovitis upon clinical assessment. Blood samples were collected from RA and IAR donors on the same day as clinical assessment. In total, 66 RA donors were collected for this study, of which 63% were female. The average age and disease duration were 57.8 and 12.2 years, respectively. 54% of RA donors were positive for ACPA antibodies, and 53% RF positive. Samples from a total of 14 IAR were collected in this study, of which 66.7 % were female with an average age of 50.8 years, 85.7% of which were ACPA positive, with 64% RF positive. Patients recruited for the RNA‐seq analysis were described previously. 13 All patients were required to give fully informed written consent, approved by the St Vincent’s Healthcare Group Medical Research and Ethics Committee. Healthy blood, used as a comparison, was obtained from anonymous healthy donors from St James’s Hospital and St Vincent’s University Hospital. Ethical approval was obtained by the School of Medicine Research Ethics Committee, Trinity College Dublin and St Vincent’s Healthcare Group Medical Research and Ethics Committee.
Peripheral CD14+ monocyte isolation and culture
Peripheral blood mononuclear cells (PBMC) were isolated from blood by density gradient centrifugation (Lymphoprep; Stemcell Technologies, Canada). A positive selection of CD14+ cells was performed by adding MACS superparamagnetic microbeads (Miltenyi‐Biotec, Germany) conjugated with monoclonal anti‐human CD14 antibodies to freshly prepared PBMC in MACS buffer (Miltenyi‐Biotec), according to the manufacturer’s instructions. Isolated PBMC were then magnetically sorted and labelled to yield a pure (≥95%) population of CD14+ monocytes. Freshly isolated monocytes were cultured in RPMI 1640 (Thermo Fisher Scientific, USA) supplemented with 10% foetal calf serum (FCS), HEPES (20 mM), penicillin–streptomycin (100 units mL−1 and 100 μg mL−1), amphotericin B (0.25 µg mL−1) (all Gibco‐BRL, UK) and 50 μg mL−1 of gentamycin (Sigma‐Aldrich, USA). CD14+ monocytes were analysed either ex vivo or following 3–24‐h stimulations with lipopolysaccharide (LPS; 100 ng mL−1) (Enzo Life Science, UK) as a model for monocyte activation consistent with previous studies.29, 49, 77, 78, 79, 80 For functional experiments, cells were treated with LPS in the presence or absence of the inhibitors; STATTIC (STAT3 inhibitor; 10 µM; VWR, USA), 2‐deoxy‐D‐glucose (2DG) (glycolytic inhibitor; 10 mM; Sigma‐Aldrich) and oligomycin (ATP‐synthase inhibitor [oxidative phosphorylation]; 2 µg mL−1; VWR). For all functional experiments, inhibitors were added 30 min prior to LPS stimulation.
Synovial tissue dissociation
Patients with active RA are continuously recruited from outpatient clinics at St Vincent’s University Hospital and The Adelaide and Meath Hospital, Tallaght. Ethical approval to conduct this study was granted by St Vincent’s Healthcare Group Medical Research and Ethics Committee and the Tallaght University Hospital/St James’ Hospital Joint Research Committee. All subjects gave fully informed written consent for use of biological samples and medical information, prior to inclusion in the study. Only patients with an actively inflamed knee joint and those that fulfilled the revised ACR/EULAR criteria were included. 76 Under local anaesthesia, arthroscopies of the inflamed knee were performed using Wolf 2.7 mm needle. RA synovial tissue biopsies were obtained from the site of inflammation under direct visualisation as previously described. 23 Healthy individuals undergoing arthroscopy during anterior cruciate ligament (ACL) reconstruction surgery were included in this study. Healthy subjects were defined as those who had no evidence of any form of arthritis on history or examination and had no cartilage damage or synovitis on knee arthroscopy.
Synovial tissue biopsies obtained at the time of arthroscopy were mechanically and enzymatically digested using the gentleMACS dissociator and a soft tumor dissociation kit (Miltenyi‐Biotec), according to the manufacturer’s instructions, to yield a single‐cell suspension of synovial tissue cells. Following arthroscopy, synovial tissue was sectioned into small pieces and added to an enzyme mix composed of 4.7 mL serum‐free and antibiotic‐free RPMI medium, 200 μL of enzyme H, 100 μL of enzyme R and 25 μL of enzyme A in a gentleMACS C tube (Miltenyi‐Biotec). Using the gentleMACS program; m_spleen_4, a mechanical stress was applied to the synovial tissue for 60 sec. Samples were then incubated at 37°C for 30 min under constant rotation using the MACSmix Tube Rotator (Miltenyi‐Biotec). The samples were subsequently exposed to a second mechanical agitation using the m_brain_03 gentle‐MACS program and incubated for a further 30 min at 37°C. A final mechanical agitation was applied for 30 sec using the gentleMACS h_tumor_03 program. The resulting cell suspension was passed through a 70μm cell strainer to remove any undigested clumps. Red blood cells were removed using Pharmlyse red blood cell lysis buffer (BD Biosciences, USA) before being washed and pelleted at 1400 RPM.
Gene expression analysis
For gene expression experiments, CD14+ monocytes were seeded at a density of 1 × 106 cells mL–1 and cultured in the presence or absence of LPS (100 ng mL−1) for 3 h. Total RNA was then isolated using a RNeasy Mini Kit (Qiagen, UK) according to the manufacturer’s instructions. To ensure the integrity of RNA was sufficient for PCR assays, RNA quality was assessed using a bioanalyzer (Agilent, USA). Samples with a 260:280 nm ratio of 1.8 and above, and an RNA integrity number (RIN) between 7 and 10 were used in subsequent experiments. 100 ng of total RNA was reverse transcribed to complementary DNA (cDNA) using a high capacity cDNA reverse transcription kit (Applied Biosystems, UK). PCR reaction mixtures contained 1 μL of cDNA, alongside specifically designed primer sequences (described in Supplementary table 1) in a dilution of RNase‐free water and SYBR Green Master Mix (Thermo Fisher Scientific). Relative quantification of gene expression was analysed with pre‐optimised conditions using the QuantStudio 5 (Thermo Fisher Scientific). Data were normalised to housekeeping genes RPLPO and HPRT1 (Supplementary table 1) and analysed using both the δCt (ex vivo analysis) and δδCt (fold‐regulation analysis) methods.
PCR Gene array
A preliminary human cytokine/chemokine RT2 Profiler™ PCR microarray (Qiagen, UK) was performed on monocytes to simultaneously quantify the expression of 84 cytokine/chemokine genes as per the manufacturer’s instructions. PCR was performed on a LightCycler 480 System (Roche Diagnostics, Switzerland). Relative changes in gene expression were determined using the GeneGlobe Data Analysis Centre (Web resource by Qiagen) using the 2−ΔΔCt method by normalising the data to four housekeeping genes; β‐actin, B2M, HPRT1 and RPLP0.
Migration assay
Migration was assessed in a 24‐trans‐well plate with a 5 µm pore size (Sigma‐Aldrich). Monocytes were pre‐treated with LPS (100 ng mL−1) for 24 h or left untreated, re‐suspended in serum‐free media and added to the top of the chamber at a concentration of 750 000 cells mL–1. 2% FBS in serum‐free media was added to the bottom of the chamber. Cells were incubated at 37°C for 1 h. Supernatants from top and bottom of the chamber were collected in separate tubes and centrifuged. The pellet was re‐suspended in 1% paraformaldehyde (PFA) for 10 min and washed with PBS. Cells were re‐suspended in PBS, and 100 µL of Precision Counting Beads (BioLegend, USA) was added directly before flow cytometry analysis. Samples were acquired using the Fortessa Flow Cytometer (Beckman Coulter, USA) and analysed using FlowJo software (Treestar Inc., USA). Absolute cell counts were calculated as per the manufacturer’s instructions. The frequency was calculated by the ratio of cells that migrated to the bottom of the chamber vs the total number of cells.
Flow cytometry
The percentage of CD14+ cells in PBMC or synovial tissue suspensions was analysed by flow cytometry. Cells were gated based on forward and side scatter and dead cells and doublets were removed. Live Dead Red (Molecular Probes, USA) was used to eliminate dead cells, according to gating strategy (Supplementary figure 1b). To eliminate non‐specific binding of mouse monoclonal antibodies to the Fc‐gamma receptor (FcγR), samples were blocked with a human FcγR‐binding inhibitor (eBioscience or BioLegend) prior to antibody staining. The following antibodies were used in combination to assess monocyte frequency: CD14 PE or PE‐DAZZLE (Clone M5E2), HLA‐DR Brilliant Violet 421 (Clone G46‐6), CD45 BV650 or FITC (Clone HI30), APC anti‐human Lineage Cocktail (CD3/19/20/56). Cells were identified as CD45+/LIN−/HLADR+/CD14+. Samples were acquired using the Fortessa Flow Cytometer and analysed using FlowJo software.
Protein isolation and western blotting
For protein analysis, CD14+ monocytes were seeded at a density of 1 × 106 cells/mL and cultured in the presence or absence of LPS (100 ng mL−1) for 24 h. Ice‐cold RIPA (Radio‐Immunoprecipitation Assay) buffer (Sigma‐Aldrich) containing 10 μg mL−1 phosphatase inhibitor cocktail and 10 μg mL−1 protease inhibitor cocktail (both Sigma‐Aldrich) was used to lyse cell suspensions. Total protein quantification and normalisation were performed using a BCA assay (Pierce Chemical Co., USA). Protein (1–2 μg) was resolved by SDS‐PAGE (5% stacking, 10% resolving), gels were then transferred onto PVDF membranes (Amersham Biosciences, UK) prior to 1 h blocking in wash buffer containing 3% BSA (0.1% Tween 20) with gentle agitation at room temperature. Membranes were incubated with mouse monoclonal anti‐HK2 (Novus Biologicals, USA), rabbit monoclonal anti‐PFKFB3 (Abcam, UK), rabbit polyclonal anti‐GLUT1 (Abcam), anti‐pSTAT3 and tSTAT3 (both Cell‐Signaling Technology, UK), and diluted in 3% BSA containing 0.1% Tween 20 at 4°C overnight with gentle agitation. β‐actin (1:5000, Sigma‐Aldrich) was used as a loading control. The signal was detected using SuperSignal® West Pico Chemiluminescent Substrate (Amersham Biosciences). Band densities were imaged using the ChemiDoc MP Imaging System (Bio‐Rad, USA). Densitometry analysis was carried out using ImageJ Software.
Measurement of Mitochondrial Mass
Freshly isolated monocytes were washed and incubated with 150 nM MitoTracker Green FM (Thermo Fisher Scientific) for 30 min at 37°C. Cells were washed in Hanks’ Balanced Salt Solution (HBSS; Sigma‐Aldrich) buffer and seeded at a density of 100 000 cells per well in a ViewPlate‐96 black (PerkinElmer, USA). Cells were stimulated with 50 µL of LPS solution (200 ng mL−1) or HBSS buffer. MitoTracker emission was read using the kinetic mode of the Spectra Max Gemini System with excitation and emission wavelengths of 490 nm and 516 nm, respectively. Readings were performed every 15 min for 6 h.
Cellular bioenergetic function analysis
To assess the metabolic function of CD14+ monocytes, the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), reflecting oxidative phosphorylation and glycolysis respectively, were measured using the Seahorse‐XFe96 analyzer (Agilent Technologies). CD14+ monocytes were seeded at a density of 3 × 105 cells per well in a 96‐well Seahorse‐XFe microplate (Agilent Technologies) and allowed to adhere for 1 h in RPMI. Following this, cells were then washed and further incubated with Seahorse assay medium (unbuffered DMEM supplemented with 10 mM glucose, pH‐7.4; Agilent Technologies) in the presence or absence of LPS (100 ng mL–1) for 1 h at 37°C in a non‐CO2 incubator, before being placed in the Seahorse‐XFe96 analyzer. Basal oxidative phosphorylation/glycolysis was calculated by the average of three baseline OCR/ECAR measurements, respectively. Specific metabolic inhibitors; oligomycin (2 µg mL−1), trifluorocarbonylcyanide phenylhydrazone (FCCP) (mitochondrial uncoupler) (5 μM) and antimycin A (complex‐III inhibitor) (2 μM) (all Agilent Technologies) were further injected to assess the metabolic capacity of CD14+ monocytes. The maximal respiratory capacity and maximal glycolytic capacity were calculated by averaging the three OCR or ECAR measurements following FCCP and oligomycin injection, respectively. The spare respiratory capacity was calculated by subtracting the baseline OCR from the maximal respiratory capacity OCR and ATP synthesis was calculated by subtracting the oligomycin OCR from the baseline OCR.
Transmission electron microscopy
To investigate the structure of mitochondria in CD14+ monocytes, transmission electron microscopy (TEM) was used. CD14+ monocytes were seeded at a density of 1 × 106 cells mL–1 and cultured in the presence or absence of LPS (100 ng mL−1) for 24 h. Following this, cells were fixed in glutaraldehyde (3% in 0.05 M potassium phosphate buffer, pH 6.8; Sigma‐Aldrich) for 1 h at room temperature. Samples were processed and analysed using a Jeol JEM2100 LaB6 (operated at 100 Kv). Digital images were obtained using an AMT XR80 capture system and ImageJ software. High‐powered images of 5 cells per sample were taken. The number of mitochondria was counted, and the average, per sample, was calculated. The number of elongated mitochondria was also counted and represented as a percentage of the total.
Cytokine measurements
Serum samples were collected from HC, IAR and RA donors and levels of IL‐6 were measured by enzyme‐linked immunosorbent assay (ELISA) according to the manufacturer’s conditions (R&D systems, UK).
RNA‐sequencing gene expression analysis
RNA‐seq analysis was performed on synovial tissue biopsies using the algorithm reported previously GEO database (GSE89408) 13 . Briefly, total RNA was extracted from synovial tissue biopsies and the quality was evaluated using an Agilent Bioanalyzer. Sequencing libraries were prepared using TruSeq Stranded Total RNA RiboZero protocol from Illumina. Libraries were pooled and sequenced with an Illumina HiSeq 2000 with paired‐end 100 bp flow cells. Raw read quality was evaluated using FastQC. Reads were trimmed for adaptors and sequence quality. The average number of clusters (post‐trimming) per sample was 8.9 × 107. Trimmed reads were aligned to human b37.3 reference genome using the STAR v2.4 aligner. Aligned reads were quantified using RSEM v1.2.14 algorithm with UCSC transcriptome model (accessed on 03/17/2014) that included lincRNAs from Ensembl v75. Aligned data were evaluated for quality using several metrics (e.g. mapping rate, coverage) and visually inspected for deviation from the population across multiple metrics and principal components analysis. Statistical testing of RNA‐seq data were performed in R with the ‘limma’ package. Counts were converted to log2 counts per million, quantile normalised and precision weighted. A linear model was fitted to each gene, and empirical Bayes moderated t‐statistics were used to assess differences in expression. The RNA‐seq data were deposited in GEO database (GSE89408). 13 Single sample Gene Set Enrichment Analysis (GSEA) was performed using the GSVA algorithm 81 as implemented in the R/Bioconductor package ‘GSVA’. Prior to the enrichment analysis, gene‐level counts were converted into counts per million and log2 transformed for each sample, and the enrichment analysis was performed on these transformed values. To examine pathway‐level enrichments, the Biocarta collection available in the canonical pathways (CP) subset of the MSigDb database 82 curated at the Broad Institute was used. Pathway analysis was also performed using STRING 11.0 software, a database of known and predicted protein interactions. 83
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7 software. The Wilcoxon signed rank test or Mann–Whitney U‐test was used for analysis of non‐parametric data. The Student’s t‐test was used for parametric data analysis. P‐values of less than 0.05 (*P < 0.05) were determined to be statistically significant.
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author Contribution
Trudy McGarry: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Writing‐original draft; Writing‐review & editing. Megan M Hanlon: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Writing‐original draft; Writing‐review & editing. Viviana Marzaioli: Conceptualization; Data curation; Formal analysis; Methodology; Writing‐original draft. Clare Cunningham: Data curation; Formal analysis; Investigation; Methodology; Writing‐original draft; Writing‐review & editing. Vinod Krishna: Data curation; Formal analysis; Writing‐original draft; Writing‐review & editing. Kieran Murray: Data curation; Methodology; Writing‐original draft; Writing‐review & editing. Conor Hurson: Data curation; Methodology; Writing‐original draft; Writing‐review & editing. Sunil Nagpal: Data curation; Formal analysis; Investigation; Methodology; Writing‐original draft; Writing‐review & editing. Douglas J Veale: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Supervision; Writing‐original draft; Writing‐review & editing. Ursula Fearon: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Writing‐original draft; Writing‐review & editing. Phil Gallagher: Patient recruitment; data analysis; writing – reviewing and editing of manuscript.
Supporting information
Acknowledgments
This study was funded by the Health Research Board Ireland, Irish Research Council, Centre for Arthritis and Rheumatic Diseases, and Arthritis Ireland.
References
- 1. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017; 389: 2328–2337. [DOI] [PubMed] [Google Scholar]
- 2. Emery P, McInnes IB, van Vollenhoven R, Kraan MC. Clinical identification and treatment of a rapidly progressing disease state in patients with rheumatoid arthritis. Rheumatol (Oxford) 2008; 47: 392–398. [DOI] [PubMed] [Google Scholar]
- 3. van der Helm‐van Mil AH, le Cessie S, van Dongen H, Breedveld FC, Toes RE, Huizinga TW. A prediction rule for disease outcome in patients with recent‐onset undifferentiated arthritis: How to guide individual treatment decisions. Arthritis Rheum 2007; 56: 433–440. [DOI] [PubMed] [Google Scholar]
- 4. Monti S, Montecucco C, Bugatti S, Caporali R. Rheumatoid arthritis treatment: the earlier the better to prevent joint damage. RMD Open 2015; 1(Suppl 1): e000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Combe B, Landewe R, Daien CI et al 2016 update of the EULAR recommendations for the management of early arthritis. Ann Rheum Dis 2017; 76: 948–959. [DOI] [PubMed] [Google Scholar]
- 6. Gerlag DM, Raza K, van Baarsen LG et al EULAR recommendations for terminology and research in individuals at risk of rheumatoid arthritis: Report from the Study Group for Risk Factors for Rheumatoid Arthritis. Ann Rheum Dis 2012; 71: 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nielen MM, van Schaardenburg D, Reesink HW et al Specific Autoantibodies Precede the Symptoms of Rheumatoid Arthritis: A Study of Serial Measurements in Blood Donors. Arthritis Rheum 2004; 50: 380–386. [DOI] [PubMed] [Google Scholar]
- 8. Falahee M, Finckh A, Raza K, Harrison M. Preferences of patients and at‐risk individuals for preventive approaches to rheumatoid arthritis. Clin Ther 2019; 41: 1346–1354. [DOI] [PubMed] [Google Scholar]
- 9. van de Sande MG, de Hair MJ, van der Leij C et al Different stages of rheumatoid arthritis: Features of the synovium in the preclinical phase. Ann Rheum Dis 2011; 70: 772–777. [DOI] [PubMed] [Google Scholar]
- 10. van de Stadt LA, van der Horst AR, de Koning MH et al The extent of the anti‐citrullinated protein antibody repertoire is associated with arthritis development in patients with seropositive arthralgia. Ann Rheum Dis 2011; 70: 128–133. [DOI] [PubMed] [Google Scholar]
- 11. Lübbers J, van Beers‐Tas MH, Vosslamber S et al Changes in peripheral blood lymphocyte subsets during arthritis development in arthralgia patients. Arthritis Res Ther 2016; 18: 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chalan P, Kroesen BJ, van der Geest KS et al Circulating CD4+CD161+ T lymphocytes are increased in seropositive arthralgia patients but decreased in patients with newly diagnosed rheumatoid arthritis. PLoS One 2013; 8: e79370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo Y, Walsh AM, Fearon U et al CD40L‐dependent pathway is active at various stages of rheumatoid arthritis disease progression. J Immunol 2017; 198: 4490–4501. [DOI] [PubMed] [Google Scholar]
- 14. Cole S, Walsh A, Yin X et al Integrative analysis reveals CD38 as a therapeutic target for plasma cell‐rich pre‐disease and established rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res Ther 2018; 20: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rana AK, Li Y, Dang Q, Yang F. Monocytes in rheumatoid arthritis: Circulating precursors of macrophages and osteoclasts and their heterogeneity and plasticity role in RA pathogenesis. Int Immunopharmacol 2018; 65: 348–359. [DOI] [PubMed] [Google Scholar]
- 16. Cros J, Cagnard N, Woollard K et al Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 2010; 33: 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoon BR, Yoo SJ, Choi Yh et al Functional phenotype of synovial monocytes modulating inflammatory T‐cell responses in rheumatoid arthritis (RA). PLoS One 2014; 9: e109775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsukamoto M, Seta N, Yoshimoto K, Suzuki K, Yamaoka K, Takeuchi T. CD14brightCD16+ intermediate monocytes are induced by interleukin‐10 and positively correlate with disease activity in rheumatoid arthritis. Arthritis Res Ther 2017; 19: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chara L, Sánchez‐Atrio A, Pérez A et al The number of circulating monocytes as biomarkers of the clinical response to methotrexate in untreated patients with rheumatoid arthritis. J Transl Med 2015; 13: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang J, Zhang L, Yu C, Yang XF, Wang H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomarker Res 2014; 2: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol 2016; 12: 472–485. [DOI] [PubMed] [Google Scholar]
- 22. Saha S, Shalova IN, Biswas SK. Metabolic regulation of macrophage phenotype and function. Immunol Rev 2017; 280: 102–111. [DOI] [PubMed] [Google Scholar]
- 23. Ng CT, Biniecka M, Kennedy A et al Synovial tissue hypoxia and inflammation in vivo. Ann Rheum Dis 2010; 69: 1389–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Biniecka M, Fox E, Gao W et al Hypoxia induces mitochondrial mutagenesis and dysfunction in inflammatory arthritis. Arthritis Rheum 2011; 63: 2172–2182. [DOI] [PubMed] [Google Scholar]
- 25. Kennedy A, Ng CT, Chang TC et al Tumor necrosis factor blocking therapy alters joint inflammation and hypoxia. Arthritis Rheum 2011; 63: 923–932. [DOI] [PubMed] [Google Scholar]
- 26. Harty LC, Biniecka M, O'Sullivan J et al Mitochondrial mutagenesis correlates with the local inflammatory environment in arthritis. Ann Rheum Dis 2012; 71: 582–588. [DOI] [PubMed] [Google Scholar]
- 27. Bae S, Kim H, Lee N et al α‐Enolase expressed on the surfaces of monocytes and macrophages induces robust synovial inflammation in rheumatoid arthritis. J Immunol 2012; 189: 365–372. [DOI] [PubMed] [Google Scholar]
- 28. Littlewood‐Evans A, Sarret S, Apfel V et al GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med 2016; 213: 1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shirai T, Nazarewicz RR, Wallis BB et al The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med 2016; 213: 337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ju JH, Heo YJ, Cho ML et al Modulation of STAT‐3 in rheumatoid synovial T cells suppresses Th17 differentiation and increases the proportion of treg cells. Arthritis Rheum 2012; 64: 3543–3552. [DOI] [PubMed] [Google Scholar]
- 31. Gao W, McCormick J, Connolly M, Balogh E, Veale DJ, Fearon U. Hypoxia and STAT3 signalling interactions regulate pro‐inflammatory pathways in rheumatoid arthritis. Ann Rheum Dis 2015; 74: 1275–1283. [DOI] [PubMed] [Google Scholar]
- 32. McGarry T, Orr C, Wade S et al JAK / STAT blockade alters synovial bioenergetics, mitochondrial function, and proinflammatory mediators in rheumatoid arthritis. Arthritis Rheumatol 2018; 70: 1959–1970. [DOI] [PubMed] [Google Scholar]
- 33. Quinn MA, Cox S. The evidence for early intervention. Rheum Dis Clin North Am 2005; 31: 575–589. [DOI] [PubMed] [Google Scholar]
- 34. van der Woude D, Rantapää‐Dahlqvist S, Ioan‐Facsinay A et al Epitope spreading of the anti‐citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann Rheum Dis 2010; 69: 1554–1561. [DOI] [PubMed] [Google Scholar]
- 35. Van Steenbergen HW, Da Silva JAP, Huizinga TWJ, Van Der Helm‐Van Mil AHM. Preventing progression from arthralgia to arthritis: Targeting the right patients. Nat Rev Rheumatol 2018; 14: 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shapouri‐Moghaddam A, Mohammadian S, Vazini H et al Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 2018; 233: 6425–6440. [DOI] [PubMed] [Google Scholar]
- 37. Watanabe R, Hilhorst M, Zhang H et al Glucose metabolism controls disease‐specific signatures of macrophage effector functions. JCI insight 2018; 3: e123047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lacerte P, Brunet A, Egarnes B, Duchêne B, Brown JP, Gosselin J. Overexpression of TLR2 and TLR9 on monocyte subsets of active rheumatoid arthritis patients contributes to enhance responsiveness to TLR agonists. Arthritis Res Ther 2016; 18: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tolusso B, Fabris M, Di Poi E, Assaloni R, Tomietto P, Ferraccioli GF. Response of mononuclear cells to lipopolysaccharide and CpG oligonucleotide stimulation: Possible additive effect in rheumatoid inflammation. Ann Rheum Dis 2003; 62: 284–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blackwell SE, Krieg AM. CpG‐A‐Induced Monocyte IFN‐γ‐Inducible Protein‐10 Production Is Regulated by Plasmacytoid Dendritic Cell‐Derived IFN‐α. J Immunol 2003; 170: 4061–4068. [DOI] [PubMed] [Google Scholar]
- 41. Zhu W, Li X, Fang S et al Anti‐citrullinated protein antibodies induce macrophage subset disequilibrium in RA patients. Inflammation 2015; 38: 2067–2075. [DOI] [PubMed] [Google Scholar]
- 42. Dong X, Zheng Z, Lin P et al ACPAs promote IL‐1β production in rheumatoid arthritis by activating the NLRP3 inflammasome. Cell Mol Immunol 2019; 17: 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Klimek E, Mikołajczyk T, Sulicka J et al Blood monocyte subsets and selected cardiovascular risk markers in rheumatoid arthritis of short duration in relation to disease activity. Biomed Res Int 2014; 2014: 736853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U. The CD14brightCD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis Rheum 2012; 64: 671–677. [DOI] [PubMed] [Google Scholar]
- 45. Weldon AJ, Moldovan I, Cabling MG et al Surface April is elevated on myeloid cells and is associated with disease activity in patients with rheumatoid arthritis. J Rheumatol 2015; 42: 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zeisbrich M, Yanes RE, Zhang H et al Hypermetabolic macrophages in rheumatoid arthritis and coronary artery disease due to glycogen synthase kinase 3b inactivation. Ann Rheum Dis 2018; 77: 1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang Z, Shen Y, Oishi H et al Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med 2016; 8: 331ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cramer T, Yamanishi Y, Clausen BE et al HIF‐1α is essential for myeloid cell‐mediated inflammation. Cell 2003; 112: 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yoon BR, Oh YJ, Kang SW, Lee EB, Lee WW. Role of SLC7A5 in metabolic reprogramming of human monocyte/macrophage immune responses. Front Immunol 2018; 9: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ruscitti P, Cipriani P, Di Benedetto P et al Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)‐1β via the nucleotide‐binding domain and leucine‐rich repeat containing family pyrin 3(NLRP3)‐inflammasome activation: a possible implication for therapeutic decision in these patients. Clin Exp Immunol 2015; 182: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Neele AE, Van den Bossche J, Hoeksema MA, de Winther MP. Epigenetic pathways in macrophages emerge as novel targets in atherosclerosis. Eur J Pharmacol 2015; 763: 79–89. [DOI] [PubMed] [Google Scholar]
- 52. Saeed S, Quintin J, Kerstens HH et al Epigenetic programming of monocyte‐to‐macrophage differentiation and trained innate immunity. Science 2014; 345: 1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hoeksema MA, Gijbels MJ, Van den Bossche J et al Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med 2014; 6: 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mullican SE, Gaddis CA, Alenghat T et al Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev 2011; 25: 2480–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reynolds LM, Wan M, Ding J et al DNA Methylation of the Aryl Hydrocarbon Receptor Repressor Associations with Cigarette Smoking and Subclinical Atherosclerosis. Circ Cardiovasc Genet 2015; 8: 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sugiyama D, Nishimura K, Tamaki K et al Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta‐analysis of observational studies. Ann Rheum Dis 2010; 69: 70–81. [DOI] [PubMed] [Google Scholar]
- 57. Kortylewski M, Kujawski M, Herrmann A et al Toll‐like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist‐based immunotherapy. Cancer Res 2009; 69: 2497–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chaves de Souza JA, Nogueira AV, Chaves de Souza PP et al SOCS3 expression correlates with severity of inflammation, expression of proinflammatory cytokines, and activation of STAT3 and p38 MAPK in LPS‐induced inflammation in vivo . Mediators Inflamm 2013; 2013: 650812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fu XQ, Liu B, Wang YP et al Activation of STAT3 is a key event in TLR4 signaling‐mediated melanoma progression. Cell Death Dis 2010; 11: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu X, Yin S, Chen Y et al LPS‐induced proinflammatory cytokine expression in human airway epithelial cells and macrophages via NF‐κB, STAT3 or AP‐1 activation. Mol Med Rep 2018; 17: 5484–5491. [DOI] [PubMed] [Google Scholar]
- 61. Jung JE, Lee HG, Cho IH et al STAT3 is a potential modulator of HIF‐1‐mediated VEGF expression in human renal carcinoma cells. FASEB J 2005; 19: 1296–1298. [DOI] [PubMed] [Google Scholar]
- 62. Mori T, Miyamoto T, Yoshida H et al IL‐1β and TNFα‐initiated IL‐6‐STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int Immunol 2011; 23: 701–712. [DOI] [PubMed] [Google Scholar]
- 63. Biniecka M, Canavan M, McGarry T et al Dysregulated bioenergetics: a key regulator of joint inflammation. Ann Rheum Dis 2016; 75: 2192–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li M, Jin R, Wang W et al STAT3 regulates glycolysis via targeting hexokinase 2 in hepatocellular carcinoma cells. Oncotarget 2017; 8: 24777–24784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Coulthard LR, Geiler J, Mathews RJ et al Differential effects of infliximab on absolute circulating blood leucocyte counts of innate immune cells in early and late rheumatoid arthritis patients. Clin Exp Immunol 2012; 170: 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gent YY, Voskuyl AE, Kloet RW et al Macrophage positron emission tomography imaging as a biomarker for preclinical rheumatoid arthritis: Findings of a prospective pilot study. Arthritis Rheum 2012; 64: 62–66. [DOI] [PubMed] [Google Scholar]
- 67. Gren ST, Rasmussen TB, Janciauskiene S, Håkansson K, Gerwien JG, Grip O. A single‐cell gene‐expression profile reveals inter‐cellular heterogeneity within human monocyte subsets. PLoS One 2015; 10: e0144351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res 2015; 107: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wong KL, Tai JJ, Wong WC et al Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011; 118: e16–e31. [DOI] [PubMed] [Google Scholar]
- 70. Grisar J, Hahn P, Brosch S, Peterlik M, Smolen JS, Pietschmann P. Phenotypic characteristics of human monocytes undergoing transendothelial migration. Arthritis Res 2001; 3: 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Taylor PC, Chu CQ, Plater‐Zyberk C, Maini RN. Transfer of type II collagen‐induced arthritis from DBA/1 to severe combined immunodeficiency mice can be prevented by blockade of Mac‐1. Immunology 1996; 88: 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cush JJ, Lipsky PE. Phenotypic analysis of synovial tissue and peripheral blood lymphocytes isolated from patients with rheumatoid arthritis. Arthritis Rheum 1988; 31: 1230–1238. [DOI] [PubMed] [Google Scholar]
- 73. Takahashi H, Söderström K, Nilsson E, Kiessling R, Patarroyo M. Integrins and other adhesion molecules on lymphocytes from synovial fluid and peripheral blood of rheumatoid arthritis patients. Eur J Immunol 1992; 22: 2879–2885. [DOI] [PubMed] [Google Scholar]
- 74. Schittenhelm L, Hilkens CM, Morrison VL. β2 integrins as regulators of dendritic cell, monocyte, and macrophage function. Front Immunol 2017; 8: 1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ateş A, Kinikli G, Turgay M, Duman M. Serum‐Soluble Selectin Levels in Patients with Rheumatoid Arthritis and Systemic Sclerosis. Scand J Immunol 2004; 59: 315–320. [DOI] [PubMed] [Google Scholar]
- 76. Aletaha D, Neogi T, Silman AJ et al 2010 rheumatoid arthritis classification criteria: an American college of rheumatology/European league against rheumatism collaborative initiative. Arthritis Rheum 2010; 62: 2569–2581. [DOI] [PubMed] [Google Scholar]
- 77. Rodgers LC, Cole J, Rattigan KM et al The rheumatoid synovial environment alters fatty acid metabolism in human monocytes and enhances CCL20 secretion. Rheumatol (Oxford) 2020; 59: 869–878. [DOI] [PubMed] [Google Scholar]
- 78. Zhang L, Yao Y, Tian J et al Alterations and abnormal expression of A20 in peripheral monocyte subtypes in patients with rheumatoid arthritis. Clin Rheumatol 2020; 40: 341–348. [DOI] [PubMed] [Google Scholar]
- 79. Zasłona Z, Pålsson‐McDermott EM, Menon D et al The Induction of Pro‐IL‐1β by Lipopolysaccharide Requires Endogenous Prostaglandin E2 Production. J Immunol 2017; 198: 3558–3564. [DOI] [PubMed] [Google Scholar]
- 80. Evans HG, Gullick NJ, Kelly S et al In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proc Natl Acad Sci USA 2009; 106: 6232–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hänzelmann S, Castelo R, Guinney J. GSVA: Gene set variation analysis for microarray and RNA‐Seq data. BMC Bioinformatics 2013; 14: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinforma 2011; 27: 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Szklarczyk D, Gable AL, Lyon D et al STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res 2018; 47: 607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials