

Abstract
While the significance of acquired genetic abnormalities in the initiation of hepatocellular carcinoma (HCC) has been established, the role of epigenetic modification remains unknown. Here we identified the pivotal role of histone methyltransferase G9a in the DNA damage-triggered initiation of HCC. Using liver-specific G9a-deficient (G9aΔHep) mice, we revealed that loss of G9a significantly attenuated liver tumor initiation caused by diethylnitrosamine (DEN). In addition, pharmacological inhibition of G9a attenuated the DEN-induced initiation of HCC. After treatment with DEN, while the induction of γH2AX and p53 were comparable in the G9aΔHep and wild-type livers, more apoptotic hepatocytes were detected in the G9aΔHep liver. Transcriptome analysis identified Bcl-G, a pro-apoptotic Bcl-2 family member, to be markedly upregulated in the G9aΔHep liver. In human cultured hepatoma cells, a G9a inhibitor, UNC0638, upregulated BCL-G expression and enhanced the apoptotic response after treatment with hydrogen peroxide or irradiation, suggesting an essential role of the G9a-Bcl-G axis in DNA damage response in hepatocytes. The proposed mechanism was that DNA damage stimuli recruited G9a to the p53-responsive element of the Bcl-G gene, resulting in the impaired enrichment of p53 to the region and the attenuation of Bcl-G expression. G9a deletion allowed the recruitment of p53 and upregulated Bcl-G expression. These results demonstrate that G9a allows DNA-damaged hepatocytes to escape p53-induced apoptosis by silencing Bcl-G, which may contribute to the tumor initiation. Therefore, G9a inhibition can be a novel preventive strategy for HCC.
Subject terms: Liver cancer, Apoptosis, Gene silencing, Liver cancer, Experimental models of disease
Introduction
Hepatocellular carcinoma (HCC) accounts for the majority of primary liver cancers and is the fourth most common cause of cancer-related deaths worldwide1. HCC occurs due to a variety of risk factors, including viral hepatitis, alcoholic and nonalcoholic fatty liver disease, carcinogen exposure, and metabolic liver diseases. Despite recent advances in the pathophysiology and treatment of HCC, the overall prognosis remains poor. A better understanding of the mechanisms of hepatocarcinogenesis will provide novel and efficacious targets for treatment of HCC.
HCC develops through a complex multistep process in which various genomic abnormalities are acquired. In addition to genetic alterations, accumulating evidence has highlighted the key role of epigenetic dysregulation in liver cancer pathogenesis2. Consistently, recent genome analysis has revealed a high number of genetic disturbances in the genes related to epigenetics in HCC3,4. Global DNA hypomethylation and promoter hypermethylation of specific tumor suppressor genes are well-characterized epigenetic changes in human carcinogenesis5. In addition to DNA methylation, aberrant expression of histone modifiers has also been implicated in the tumor development. Histone methylation critically determines the chromosomal structure as well as accessibility to transcription factors.6 Disturbances in histone methylation associate with HCC development7–9. Therefore, modulating the enzymatic activities of histone modifiers might be a potential therapeutic strategy to prevent HCC.
G9a (also known as euchromatic histone-lysine N-methyltransferase 2, EHMT2) is a SET domain-containing protein that catalyzes specifically the di-methylation of histone 3 lysine 9 (H3K9me2), which is a prominent epigenetic marker of transcriptional repression10. G9a plays important roles in diverse cellular processes, such as proliferation, differentiation, senescence, and autophagy11. The dysregulation of G9a and aberrant levels of H3K9me2 are involved in different types of human cancers12–15. As for HCC, G9a promotes tumor progression by silencing tumor suppressor genes or enhancing epithelial-mesenchymal transition16,17, and its inhibition reduces tumor aggressiveness18,19. In contrast, how G9a contributes to the development or initiation of HCC has not yet been investigated in vivo.
In chronic hepatic injury caused by viral infection or excessive fat accumulation, DNA damage from produced reactive oxygen species is a critical pathogenic factor for liver carcinogenesis20. Given the role of G9a for DNA damage repair and cell survival21, we hypothesized that G9a might be involved in DNA damage-induced liver cancer initiation. In this study, we demonstrate that liver-specific G9a-deficient (G9aΔHep) mice suppress HCC development triggered by diethylnitrosamine (DEN) and the pivotal role of G9a in DNA damaged hepatocytes.
Results
G9a is frequently upregulated in human HCC
First, we analyzed the expression levels of histone modifiers from 373 HCC cases identified in The Cancer Genome Atlas (TCGA) dataset. From the 82 identified histone modifiers, many histone methyltransferases were upregulated; consistent with previous papers, SETDB1 was the most significantly upregulated9,22 (Figs. 1A, S1). G9a was also identified as one of the most upregulated genes, with high expression levels (Z-score > 2) in 16.6% (62/373) of the cases (Fig. 1A). Expression levels of G9a were not significantly different among etiologies but rather tended to increase according to the pathological malignancy grade of HCC (Fig. 1B). G9a expression was also higher in many HCC cell lines than in normal hepatocytes (Fig. 1C). In addition, in an original cohort consisting of 40 pairs of HCC and adjacent NT livers in our institution, we successfully validated the upregulation of G9a in HCC compared with NT livers in almost half of the cases (Fig. 1D). In the TCGA cohort, copy number gain of G9a gene was detected in 4.29% (16/373) of the cases, and it was positively correlated with higher G9a mRNA expression levels (Fig. 1E). These data confirm the hypothesis that accumulated genomic alterations in advanced HCC are linked to G9a upregulation as well as SETDB116,22, and highlight the pathological significance of G9a in HCC. Of note, given the enzymatic roles of G9a and SETDB1 related to H3K9 methylation, the above findings suggest the molecular significance of H3K9 methylation in HCC pathogenesis. In contrast, however, the involvement of G9a and its H3K9 methylation activity in hepatocarcinogenesis in vivo remains unclear. Thus, we next examined the significance of G9a in liver tumor development using liver-specific G9a-deficient (G9aΔHep) mice.
Fig. 1. G9a is highly expressed in human HCC.

A Relative expression levels of histone-lysine methyltransferase (KMT) and demethylase (KDM) in hepatocellular carcinoma using The Cancer Genome Atlas (TCGA) data set. B G9a expression is comparable among etiologies (nBnC, n = 180; HBV, n = 97; HCV, n = 49; p = 0.79, one-way ANOVA) and increases according to the pTNM stage in the TCGA cohort (Stage I, n = 171; II, n = 85; III, n = 85; IV, n = 5; p < 0.01, one-way ANOVA). The top and bottom boxes represent the first and third quartiles, respectively. The thick line inside each box represents the median. Whiskers represent 1.5 interquartile ranges from the hinges. Open circles represent outliers. C G9a is upregulated in many HCC cell lines. Data are mean ± SEM from three independent experiments. **p < 0.01, Student’s t test. D G9a expression levels in 40 pairs of HCC (T) and surrounding non-tumorous (NT) liver obtained in our institution. G9a expression level is more than double in the tumor in 19 of 40 cases (47.5%). E G9a gene copy number is positively correlated with mRNA expression levels in the TCGA data set (p < 0.01, one-way ANOVA).
Loss of G9a attenuates liver tumorigenesis in a carcinogen-induced HCC model
G9aΔHep mice were established by crossing G9aflox/flox mice with albumin-Cre transgenic mice. To confirm efficient deletion of G9a in hepatocytes, the expression levels of G9a protein were measured in whole liver and isolated primary hepatocytes. Deletion of G9a was confirmed in G9aΔHep mouse hepatocyte, while residual G9a expression was detected in the whole liver lysate of G9aΔHep mice, probably from nonhepatocyte fractions (Fig. 2A). Immunohistochemistry revealed that H3K9me2 levels in G9aΔHep mouse hepatocytes nuclei decreased significantly, suggesting the critical role of G9a in the H3K9me2 modification (Fig. 2B). G9aΔHep mice lineage was consistent with Mendel’s laws of inheritance, and there were no significant issues with their growth. As for the liver, there were no differences in liver weight or histology between G9aΔHep and wild-type (WT) mice (Fig. 2B).
Fig. 2. G9a inhibition attenuates DEN-induced hepatocarcinogenesis.

A G9a deletion is confirmed using immunoblotting of liver lysates and isolated primary hepatocytes. B Representative macro- and micro-images of the liver and liver/body weight (BW) ratio of 30-week-old mice. There is no obvious difference between wild-type (WT) and G9aΔHep livers, but H3K9me2 levels are markedly attenuated in the G9aΔHep hepatocytes. Scale bars, 50 µm; each, n = 7; NS not significant, Student’s t test. C WT and G9aΔHep mice are subjected to diethylnitrosamine (DEN) (25 mg/kg i.p.) at day 15 postpartum, followed by 22 injections of carbon tetrachloride (CCl4) (0.5. mL/kg i.p.) per week and euthanized 6.5 months after DEN administration. Liver tumor development is significantly attenuated in G9aΔHep mice (WT, n = 16; G9a+/−, n = 15; G9aΔHep, n = 13; **p < 0.01, NS not significant, Student’s t test). D Immunoblots using liver lysates after five sequential days of UNC0642 injection reveal that G9a inhibitor successfully reduces H3K9me2 levels in the liver. E UNC0642 treatment significantly attenuates DEN-induced liver tumor development (DEN/MOCK, n = 7; DEN/UNC0642, n = 7; DEN + CCl4/MOCK, n = 11; DEN + CCl4/UNC0642, n = 8; *p < 0.05, **p < 0.01, NS not significant, Student’s t test).
To address the role of G9a in liver carcinogenesis, we injected a combination of DEN and hepatotoxin carbontetrachloride (CCl4) solution into WT and G9aΔHep mice. The chemical carcinogenic model mimics chronic inflammation and fibrosis of the human liver, and the developed tumor has the characteristics of human HCC23. In the inflammation-related liver-tumorigenic setting, G9aΔHep mice displayed a profound reduction in tumor number and size (Fig. 2C). G9aΔHep mice did not show significant changes in the pathology, proliferation, and apoptosis of the barely formed tumors (Fig. S2A), which leads us to suggest that G9a is critical in the initial stage of liver tumorigenesis. Supportive of this notion, decreased liver tumor initiation in G9aΔHep mice was also observed under a purely genotoxic carcinogenic protocol without inflammation, namely, simple DEN administration without CCl4 injection (Fig. S2B).
To confirm the tumor-initiating role for G9a, we investigated the effects of pharmacological inhibition of G9a in the above in vivo settings. Treatment with the G9a inhibitor UNC0642 effectively reduced H3K9me2 levels in liver cells (Fig. 2D). Notably, treatment with UNC0642 resulted in significant attenuation of DEN-induced liver tumor development, regardless of subsequent CCl4 administration (Fig. 2E). In contrast, the size of largest tumors was not efficiently affected by the administration of UNC0642 for 10 days, suggesting that the inhibition of G9a may predominantly affect the tumor initiation. These findings support our hypothesis that the enzymatic function of G9a is involved in the liver tumor initiation following DEN administration.
G9a deletion induced apoptosis in DNA-damaged hepatocytes
DEN, a DNA-damaging carcinogen, is metabolized and activated to form DNA adducts mainly in Cyp2E1-expressing pericentral hepatocytes24. DNA damage caused by DEN is a significant factor for subsequent HCC development25. To assess the implication of G9a in DNA damage-triggered tumor initiation, both WT and G9aΔHep mice were injected with a higher dose of DEN and were euthanized 48 h post-injection. Expression of the G9a protein was predominantly observed around the central vein in WT hepatocytes after DEN administration (Fig. 3A), suggesting that G9a is upregulated in DNA-damaged hepatocytes. Although the levels of DNA damage, detected by γH2AX staining and p53 induction, were identical in WT and G9aΔHep mice livers, higher numbers of cleaved-Caspase 3-positive hepatocytes were observed around the central vein in G9aΔHep mice than in WT mice (Fig. 3A). The serum levels of alanine aminotransferase (ALT) after DEN injection were markedly higher in G9aΔHep mice than in WT mice (Fig. 3B). To exclude the possibility that the chemical activity of DEN was enhanced by G9a deletion, we examined the expression of Cyp2e1. As reported previously, Cyp2e1 was downregulated after DEN treatment in WT mice liver26. As we did not observe any significant difference in Cyp2e1 levels between WT and G9aΔHep liver, we suggest that G9a deletion affects the cellular response against DEN-induced DNA damage, but not the chemical activity of DEN, in pericentral hepatocytes (Fig. 3C).
Fig. 3. G9a protects hepatocytes against apoptosis after a high dose of DEN.

A Immunohistochemical analysis of the mice liver 48 h after administering a high dose of diethylnitrosamine (DEN) (100 mg/kg i.p.). G9a is upregulated after DEN administration, and cleaved-Caspase 3 induction occurs only in the G9aΔHep liver, whereas γH2AX and p53 levels are comparable between wild-type (WT) and G9aΔHep livers. Bcl-G expression is elevated in the pericentral hepatocytes of G9aΔHep mice. Scale bars, 50 µm. The number of positive hepatocytes is shown in the graph below (**p < 0.01, NS not significant, Student’s t test). B Serum ALT levels are significantly elevated in G9aΔHep mice 48 h after administering a high dose of DEN. Data are represented as mean ± SEM (WT/DEN0h, n = 4; G9aΔHep/DEN0h, n = 3; WT/DEN48h, n = 5; G9aΔHep/DEN48h, n = 3). C Cyp2e1 expression, as assessed by qRT-PCR, is comparable between WT and G9aΔHep livers (each, n = 3; NS not significant, Student’s t test). D Top 10 upregulated genes in the non-tumorous area of G9aΔHep liver are listed. E qRT-PCR analysis validates Bcl-G upregulation in the G9aΔHep liver (*p < 0.05, Student’s t test). F Immunoblots reveal increased Bcl-G, cleaved-PARP, and cleaved-Caspase3 protein levels in the G9aΔHep liver 48 h after high-dose DEN administration. Right graph shows relative protein level of Bcl-G (each, n = 3; **p < 0.01, NS not significant, Student’s t test). G Relative expression levels of G9a and BCL-G in human non-tumorous (NT) liver obtained in our institution. BCL-G expression is negatively correlated with G9a expression. (rho = −0.37, p = 0.02, Spearman’s rank correlation coefficient).
Given the epigenetic function of G9a as a transcriptional repressor, we hypothesized that G9a might regulate the expression of genes which determine cellular responses under DNA damage. To identify such genes regulated by G9a, comprehensive gene expression profiles of NT areas were analyzed in WT and G9aΔHep livers. Based on the function of G9a as a transcriptional repressor, we focused on the genes upregulated in the G9aΔHep liver. The top ten genes with the greatest fold inductions in G9aΔHep liver are shown in Fig. 3D. Among them, Bcl2-like 14 (also known as Bcl-G), a pro-apoptotic Bcl-2 family member, was significantly upregulated in the G9aΔHep liver. Quantitative real-time PCR confirmed that Bcl-G mRNA expression levels increased significantly in the NT areas of G9aΔHep liver (Fig. 3E). Immunoblotting revealed that Bcl-G protein, together with cleaved-PARP and cleaved-Caspase 3, accumulate after DEN administration in G9aΔHep liver but not in WT liver (Fig. 3F). In addition, immunohistochemistry showed that Bcl-G expression is markedly elevated in pericentral hepatocytes of G9aΔHep mice (Fig. 3A). These findings suggest that G9a-dependent transcriptional regulation of Bcl-G gene suppresses apoptosis under DNA damage in murine liver. Furthermore, to study the relationships in the expression of G9a and Bcl-G gene, we measured the levels of G9a and BCL-G mRNA in human NT liver samples. Supporting the findings in murine liver, the expression levels of BCL-G were negatively correlated to that of G9a in human NT liver (Fig. 3G).
G9a inhibits DNA damage-induced apoptosis by regulating BCL-G expression
We hypothesized that the regulation of Bcl-G expression by G9a might be a gatekeeper of apoptosis in DNA-damaged hepatocytes. To prove this hypothesis, we first examined the induction of apoptosis after BCL-G overexpression in a normal human hepatocyte cell line. BCL-G overexpression induced the cleavage of PARP and Caspase3, suggesting the pro-apoptotic role of BCL-G in human hepatocytes (Fig. 4A).
Fig. 4. BCL-G is involved in apoptosis during the DNA damage response in hepatocytes.

A BCL-G overexpression induces apoptotic changes in normal human hepatocytes. Representative immunoblot images and relative expression levels of the proteins are shown (each, n = 3; *p < 0.05, Student’s t test). B, D The G9a inhibitor UNC0638 (5 µM) promotes apoptosis in hepatocytes after treatment with hydrogen peroxide (H2O2, 2 mM) or UVB irradiation (300 J/m2) with an increase in BCL-G expression. Bcl-G knockdown (KD) suppresses the excessive induction of apoptosis caused by a G9a inhibitor. Right graphs show quantification of relative protein levels. C, E Representative dot-plot diagrams showing the percentage of the hepatocyte lines stained with Annexin V-FITC and propidium iodide (PI) after 24 h of treatment with H2O2 or UVB irradiation. The results of three independent experiments are shown in the right graph (**p < 0.01, NS not significant, Student’s t test).
Next, we examined if the G9a-BCL-G axis is involved in apoptosis caused by DNA damage in the hepatocytes. For this purpose, the hepatocyte lines were treated with hydrogen peroxide (H2O2, 2 mM) or UVB irradiation (300 J/m2), which are well-known DNA damage inducers. As expected, both H2O2 administration and UVB irradiation increased total p53 and phosphorylated p53 levels, the active form often induced by DNA damage27, followed by the upregulation of cleaved-PARP and cleaved-Caspase3 (Fig. 4B, D). Treatment with 5 µM of a G9a inhibitor UNC0638 significantly attenuated the global levels of H3K9me2 in the hepatocyte lines (Fig. S3A), without affecting cell cycle status (Fig. S3B). In order to examine the role of G9a in DNA-damaged hepatocyte, the hepatocyte lines were treated with 5 µM of UNC0638 for 24 h before treatment with DNA damage inducers. In contrast to the slight increase of BCL-G after the introduction of DNA damage inducers in the control hepatocytes, the G9a inhibitor UNC0638 strikingly enhanced the expression of BCL-G (Fig. 4B, D). UNC0638 also induced PARP or Caspase3 cleavage while it did not always affect p53 activation, and it was suppressed in BCL-G knockdown cells (Fig. 4B, D). The enhancement of apoptotic changes by G9a inhibitor and their attenuation by BCL-G knockdown were also confirmed by flow cytometric analysis (Fig. 4C, E). These findings indicate that BCL-G, induced by G9a inhibition, is involved in the induction of apoptosis in DNA-damaged hepatocytes. The global levels of H3K9me2 increased after both treatments, and decreased, at least in part, by UNC0638, indicating that G9a contributes to DNA damage-induced H3K9me2 modification in hepatocytes. The residual H3K9me2 marks imply the involvement of other histone modifiers for H3K9me2.
G9a regulates Bcl-G expression by mediating p53 recruitment to its response element
Bcl-G is a p53 direct target gene.28 Notably, UNC0638 altered BCL-G expression without affecting p53 activity in DNA-damaged hepatocytes (Fig. 4B, C). In addition, although p53 activation was comparable between WT and G9aΔHep mice livers after administering a higher dose of DEN (Fig. 3A), Bcl-G protein and gene expression levels were significantly higher in the liver of G9aΔHep mice (Figs. 3G and 5A). These findings suggest that G9a regulates Bcl-G expression without affecting p53 status in DNA-damaged hepatocytes. Histone modifiers regulate gene expression by mediating the recruitment of transcriptional machinery to their target genes.29–31 Therefore, to determine the mechanism through which G9a regulates the expression of Bcl-G, we first examined the binding of G9a to the p53 response element (RE) on the Bcl-G gene using the chromatin immunoprecipitation (ChIP) assay in mice livers. G9a was significantly enriched in the p53 RE of the Bcl-G gene in the WT liver after administration of a high dose of DEN, while it did not bind to the p53 RE in non-treated WT liver cells (Fig. 5B). H3K9me2 levels at the p53 RE were elevated in the WT liver after DEN administration, but it remained unchanged in the G9aΔHep liver (Fig. 5C). Importantly, while p53 binding to the RE of the Bcl-G gene was not detected in the WT liver, G9a deletion increased significantly the recruitment of p53 to the RE of the Bcl-G gene after DEN administration (Fig. 5D). Thus, G9a might regulate Bcl-G expression by interfering with the p53 recruitment in an H3K9 methylase-dependent manner.
Fig. 5. G9a inhibits p53 recruitment to its response element on Bcl-G in an H3K9 methylase-dependent manner.

A, E Relative expression levels of Bcl-G and p21 are determined by qRT-PCR in WT and G9aΔHep mice livers before and after a high dose of diethylnitrosamine (DEN) administration (each, n = 3; **p < 0.01, NS not significant, Student’s t test). B–D, F–H Chromatin immunoprecipitation analyzes G9a, p53, and H3K9me2 binding at the indicated loci of Bcl-G and p21. The recruitment of G9a and increased H3K9me2 levels are shown in the Bcl-G gene (B, C), but not in the P21 gene (F, G) after a high dose of DEN. G9a ablation, combined with DEN treatment, allows p53 recruitment onto the Bcl-G locus via a reduction in H3K9me2 levels (D). In contrast, p53 is recruited onto the p21 locus after DEN treatment, independent of G9a status (H). Transcriptional start site (TSS), p53 response element (RE), and amplicons (10 kb upstream or downstream regions of TSS) are shown in the above diagrams. Results are expressed as the mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, Student’s t test. I Diagram showing the regulation of p53 target gene expression by G9a. G9a inhibits p53 binding to RE of Bcl-G gene in an H3K9me2-dependent manner and suppresses Bcl-G expression. G9a deletion enables p53 recruitment to Bcl-G gene RE and upregulates Bcl-G expression.
Next, we examined whether the regulation of p53 recruitment by G9a is a common phenomenon for p53 target genes. We focused on p21, one of the major target genes of p53, because it is well known that DEN induces p53-dependent p21 upregulation.32 High doses of DEN significantly upregulated the expression of p21, and importantly, the upregulation was independent of G9a status (Fig. 5E). The ChIP assay revealed that G9a did not bound to the p53 RE of the p21 gene after DEN administration (Fig. 5F). Furthermore, the levels of H3k9me2 were not elevated following DEN administration (Fig. 5G), and p53 was enriched in the p53 RE of the p21 gene in both WT and G9aΔHep liver (Fig. 5H). These findings indicate that G9a is not involved in the regulation of p21 expression. Thus, G9a individually regulates p53 target genes expression through selective control of p53 recruitment (Fig. 5I).
Discussion
Accumulating evidence suggests that epigenetic dysregulation plays an important role in human carcinogenesis.5 Histone modifiers play crucial roles in regulating oncogenes and tumor suppressor genes in various cancer types, including HCC. The establishment of HCC is a complex process which involves the accumulation of multiple gene mutations. However, it is unclear how mutated hepatocytes escape p53-dependent genome surveillance and progress to HCC. In this study, we reveal the role of histone methyltransferase G9a in hepatocarcinogenesis in vivo. In a pro-carcinogen DEN-induced murine HCC model, G9a prevented DNA-damaged hepatocytes from undergoing apoptosis by modulating p53 transactivation (Fig. 6). This is a novel finding of the role of epigenetic regulation to liver cancer initiation.
Fig. 6. Graphical scheme.
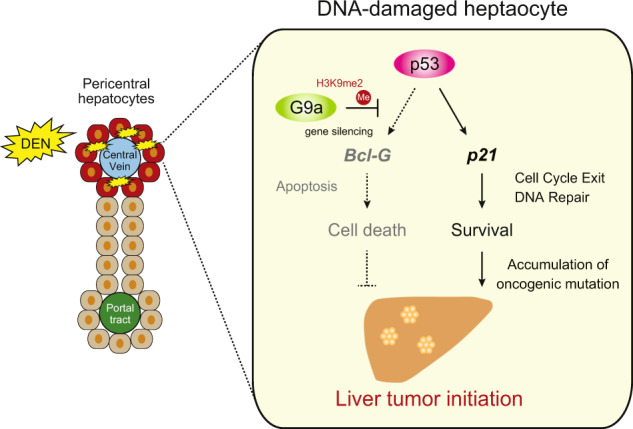
Schematic representation of the G9a-p53-Bcl-G circuit that controls liver tumor initiation from DNA-damaged hepatocytes. DEN-exposed pericentral hepatocytes undergo DNA damage and mutagenesis. G9a allows DNA-damaged hepatocytes to escape p53-induced apoptosis, the potent genome surveillance checkpoint, via Bcl-G silencing, which results in the fixation of mutations and promotion of future HCC development. In summary, G9a determines whether hepatocytes undergo apoptosis or survive during DNA damage response via the regulation of p53 transactivation.
We showed, by analyzing a TCGA data set, that several histone methyltransferases, including SETDB1 and G9a, are overexpressed in HCC. Interestingly, the loci of both genes, chromosome 6p21 of G9a and chromosome 1q21 of SETDB1, are amplified in human HCC22,33, suggesting that upregulation of G9a may be due to copy number amplification, as in the case of SETDB1. H3K9 methyltransferases including G9a, GLp, SETDB1, and Suv39h1, functionally cooperate as components of the transcriptional megacomplex34. Since they are important in liver carcinogenesis8,9,16, we suggest that the coordinated activity of H3K9 methyltransferase may play a role in liver tumor initiation. Indeed, we observed a residual increase in H3K9 levels regardless of G9a inhibition in DNA-damaged human hepatocytes, suggesting that other H3K9 methyltransferases may be involved.
Surviving DNA damage, such as that induced by reactive oxygen species or radiation exposure, involves the cells; ability to adapt to DNA damage. However, these cellular adaptations may enhance the viability of premalignant cells and their transformation to tumor cells. One group reported that G9a is required for DNA damage repair and allowed cancer cells to survive under DNA damage21. Another group demonstrated that G9a is recruited to DNA damage sites with other repressive chromatin proteins such as enhancer of zeste homolog 2 (EZH2) and DNA methyltransferase (DNMT) and maintained transcriptional silencing of tumor suppressor genes in colorectal cancer35. These reports indicate the important roles for G9a in transcriptional repression and tumor initiation under DNA damage. Consistent with this notion, G9a ablation or pharmacological inhibition resulted in enhanced apoptosis in DNA-damaged hepatocytes and attenuated liver tumor initiation. Our data emphasize the pro-tumorigenic role of G9a by suppressing DNA damage-induced apoptosis in mammalian cells, including hepatocytes.
Transcriptional analysis revealed that G9a silenced Bcl-G expression, a pro-apoptotic member of the Bcl-2 family. In humans, there are two BCL-G isoforms, BCL-GS and BCL-GL, generated by alternative splicing36. Both isoforms are related to the induction of apoptosis, with BCL-GL being widely expressed in adult human tissues, whereas BCL-GS is found only in the testicles. In mice, only one form of Bcl-G exists, which is homologous to human BCL-GL and is expressed in similar tissues37. Studies on breast and colorectal cancers suggest that BCL-G has a tumor-suppressive function38,39. Bcl-G expression in HCC was less than half of that in the surrounding NT liver in our cohort (data not shown). Moreover, we found that overexpression of BCL-G in hepatoma cell lines in vitro induces apoptotic changes, suggesting its role as a tumor suppressor in hepatocarcinogenesis. Taken together, our study demonstrates that G9a allows cell survival and future malignant transformation by silencing the expression of tumor-suppressing Bcl-G in DNA-damaged hepatocytes.
Recent papers have reported the functional link of G9a with p53 activity. G9a specifically methylates p53 at lysine 373, resulting in its inactivation40. Loss of G9a delayed malignant transition in progenitors of a murine chemical mutagen-induced squamous tumor through p53 overactivation15. These reports suggest that G9a promotes tumorigenesis by impairing p53 transactivation. Moreover, G9a and HDAC1 epigenetically repress p53 target genes by maintaining H3K9 methylation and histone deacetylation of the p53 target genes promoter41. Consistently, our ChIP experiments revealed that G9a regulates p53 recruitment onto its target pro-apoptotic gene, Bcl-G, in an H3K9-methylase dependent manner. In contrast, the regulation was not in the case of p21 promoter. It remains unclear how the specificity of G9a binding to p53 target genes is determined. Since histone modifiers regulate gene expression through interaction with chromatin remodeling complexes29,42, a chromatin remodeling mechanism or chromatin conformational changes may be involved in the processes. Further studies are needed to elucidate this point.
Finally, G9a inhibitors could provide additional effects when combined with other agents such as DNMT inhibitors19,43 or immune checkpoint inhibitors43,44, which are expected to be effective against HCC45. Our results might point to a new therapeutic strategy of combinatory therapies, including G9a inhibitors, against HCC.
Materials and methods
Human clinical samples
Surgically resected HCC samples were obtained from patients who underwent hepatectomy at the University of Tokyo between November 2013 and October 2014. These procedures were approved by the Ethical Committee for Clinical Research of our institution and written informed consent was obtained from each patient. The clinical diagnosis of all samples as HCC was confirmed by the Department of Pathology at the University of Tokyo Hospital.
The cancer genome atlas (TCGA) dataset
373 HCC samples with both mutation and mRNA data were obtained from TCGA via cBioportal (http://www.cbioportal.org/). Among them, putative copy-number alterations data calculated by GISTIC was available in 360 cases.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from frozen clinical liver specimens, HCC cell lines, and mouse livers, using NucleoSpin RNAII (Takara, Shiga, Japan) or ISOGEN-II (Nippon Gene, Tokyo, Japan). The purified RNA was reverse transcribed using the ImProm_II Reverse Transcription System (Promega, Madison, WI, USA) and amplified by reverse transcription PCR. The qRT-PCR analysis was performed using a PCR mixture containing a complementary DNA sample, forward and reverse primers, and THUNDERBIRD SYBR qPCR Mix (Toyobo, Osaka, Japan), using the StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. Values were internally normalized against β-actin mRNA expression. The primer sequences are listed in Supplementary Table 1.
Cell lines and treatment
The human cell lines Huh1, Huh6, Huh7, HLE, HLF, JHH1, JHH4, and JHH7 were obtained from the Japanese Collection of Research Bioresouces (JCRB; Osaka, Japan). HepG2 and PLC/PRF/5 were obtained from RIKEN Cell Bank (RCB; Tsukuba, Japan). Hep3B and SkHep1 were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA). Human normal hepatocyte was obtained from Applied Cell Biology Research Institute (ACBRI; Kirkland, WA, USA). JHH1, JHH6, and JHH7 were maintained in William’s E Medium (Sigma, St Louis, MO, USA) containing 10% heat-inactivated fetal bovine serum (FBS). Other cell cultures were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Sigma) containing 10% heat-inactivated FBS. All the cells were incubated at 37 °C and 5% CO2.
Animal experiments
Floxed G9a (G9aflox/flox) mice were generated as described previously10,46 and Alb-Cre mice were purchased from The Jackson Laboratory47. Mice were maintained in a temperature- and light-controlled facility, and permitted ad libitum regular chow diet and autoclaved water. All mice were backcrossed with the C57BL/6 strain and the male progeny were analyzed. All the experiments were performed in accordance with protocols approved by the Animal Ethics Committee of the University of Tokyo.
HCC was induced as previously described23. 15-day-old WT and G9aΔHep mice were injected intraperitoneally (i.p.) with DEN (Sigma, St. Louis, MO, USA) (25 mg/kg) alone or in combination with 22 weekly injections of CCl4 (Wako, Osaka, Japan) (0.5 ml/kg i.p., dissolved in corn oil). To evaluate acute effects of G9a in damaged hepatocytes, 8-week-old WT and G9aΔHep mice were treated with DEN (100 mg/kg i.p.) and sacrificed 48 h after DEN administration. In vivo G9a inhibitor treatment was performed as previously described48. WT mice were injected i.p. with G9a inhibitor UNC0642 (5 mg/kg) for 10 days before and after DEN administration. Randomization or blinding of animal experiments were not possible.
Primary hepatocyte isolation
Primary hepatocytes were isolated from mouse livers as previously described49. Briefly, 40 μg/ml Liberase (Roche, Grenzacherstrasse, Basel, Switzerland) was perfused through the liver. Digested liver was passed through a 40 μm cell strainer, and centrifuged several times at 50 g for 1 min to remove nonparenchymal cells.
Immunoblotting
Whole-liver protein homogenates or cell lysates were prepared. Immunoblot was performed as previously described29. The primary antibodies used were against β-actin (Sigma, A5441), G9a (R&D Systems, Minneapolis, MN, USA, PP-A8320A-00), H3K9me2 (Abcam, Cambridge, UK, ab1220), Histone H3 (Abcam, ab1791), Bcl-G (Thermo Fisher Scientific, Waltham, MA, USA, PA5-20029), Cleaved-PARP (Cell Signaling, Danvers, MA, USA, 9541: human, 9544: mouse), Cleaved-Caspase 3 (Cell Signaling, 9661), Flag (Sigma, F1804), γH2AX (Abcam, ab2893), p53 (Cell Signaling, 2524), Phospho-p53 (Ser 15) (Cell signaling, 9284). Immunoblot signals were analyzed using Image J software and relative protein expression levels were internally normalized against β-actin expression levels.
Histology and immunohistochemistry
Mouse livers were fixed with 4% paraformaldehyde and embedded in paraffin. Tissue sections were hematoxylin and eosin (H&E) stained for pathological evaluation. Immunohistochemistry was performed using Histofine Mousestain Kit (Nichirei Bioscience, Tokyo, Japan). The slides were subjected to heat mediated antigen retrieval using 10 mM citrate buffer (pH 6.0) and incubated overnight at 4 °C with the indicated primary antibodies. The antibodies used were against H3K9me2 (Abcam, ab1220), Ki67 (Abcam, ab15580), Cleaved-Caspase3 (Cell Signaling, 9661), G9a (R&D Systems, PP-A8320A-00), γH2AX (Abcam, ab2893), p53 (Leica Biosystems, Wetzlar, Germany, P53-CM5P-L), Bcl-G (Thermo Fisher Scientific, PA5-20029).
Serum alanine aminotransferase (ALT) measurement
Serum samples for ALT measurement were collected after a 16 h starvation (SRL, Tokyo, Japan).
Microarray analysis
GeneChip Mouse Genome 430 2.0 Arrays were performed according to the manufacturer’s protocol (Affymetrix, Santa Clara, CA, USA). Total RNA was extracted from non-tumorous (NT) liver of 30-week-old WT and G9aΔHep mice treated with DEN followed by 22 repeated CCl4 administration (each, n = 2). Probe design files and microarray data have been submitted to the National Center for Biotechnology Information Gene Expression Omnibus database under accession number GSE147061.
Plasmids, short hairpin RNA (shRNA), and transfection
The human BCL-G expression plasmid, pCAGGS human flag-BCL-G, was kindly provided by Dr. Toyomasa Katagiri (Division of Genome Medicine, Institute of Advanced Medical Sciences, Tokushima University, Japan)38 The lentiviral-based knockdown plasmid expressing the specific shRNA of BCL-G was purchased from Open Biosystems (Huntsville, AL, USA). The cells were transfected using the Effectene Transfection Reagent (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
Flow cytometry
For cell cycle distribution analysis, the hepatocyte lines were collected after 24 h of treatment with a G9a inhibitor UNC0638 or mock. The cells were fixed in 1 mL ethanol (70 %) at 4 °C for 30 min and were resuspended in phosphate buffered saline containing 50 μg/mL propidium iodide (PI) and incubated at room temperature for 30 min before analysis.
For apoptosis detection assay, the hepatocyte lines were collected 24 h after treatment with ultraviolet B (UVB) irradiation (300 J/m2) by a UVB lamp (UVP, Upland, CA, USA) or hydrogen peroxide (2 mM, lasting 24 h). The cells were stained with 5 μL of FITC Annexin V (BD Biosciences, Franklin Lakes, NJ, USA) and PI (50 μg/mL) for 15 min in the dark. PI negative and Annexin V positive cells were considered early apoptotic; PI and Annexin V positive cells were considered to be in late apoptosis.
Data analysis and acquisition were performed using the Guava® EasyCyte™ Plus Flow Cytometry System and the Guava Express Pro Software (Guava Technologies, Hayward, CA, USA).
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation were performed as previously described29,50. Fifty milligrams of mouse livers were crosslinked in 1% formaldehyde/Phosphate-buffered saline for 15 min at room temperature, which was quenched by 0.125 M glycine for 5 min. The tissue was homogenized, resuspended in cell lysis buffer (10 mM Hepes pH 7.9, 0.5% NP-40, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT), and incubated at 4 °C for 4 h. The nuclei were resuspended in nuclear lysis buffer (20 mM Hepes pH7.9, 25% Glycerol, 0.5% NP-40, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM ethylenediaminetetraacetic acid), and sonicated for 15 cycles (30 s ON/30 s OFF) using Bioruptor UCD-250 (Cosmo Bio, Carlsbad, CA, USA) to yield DNA fragments approximately of 500 bp. The soluble chromatin lysate was then immunoprecipitated using antibodies against G9a (Cell Signaling, 3306), p53 (Cell Signaling, 2524), and H3K9me2 (Abcam, ab1220) or control IgG (rabbit: Cell Signaling, 2729, mouse: Santa Cruz, sc-2025).
The ChIPed DNA was purified using QIA quick PCR purification kit (Qiagen). Results from each immunoprecipitation were presented as input percentage. The primer sequences for the target regions are available in Supplementary Table 2.
Statistical analyses
All statistical analyses were performed using R software (version 3.6.1; R Development Core Team, Vienna, Austria). All the results are expressed as mean ± standard error of the mean (SEM) of at least three independent experiments. Statistical significance was determined by the two-tailed Student’s t test and Fisher’s exact probability test. p values < 0.05 were considered as statistically significant.
Supplementary information
Acknowledgements
We thank Dr. Toyomasa Katagiri for providing the BCL-G expression vector and Mitsuko Tsubouchi and Sayaka Ito for their technical assistance. We would like to thank Editage (www.editage.com) for English language editing.
Author contributions
Conceptualization: T.N., K.T. Data acquisition: T.N., H.K., H.F., K.Y., Y.K., H.N., Y.T., H.I. Data analysis and interpretation: T.N., K.T. Material support: T.I., T.I., K.H., M.T., Y.S. Drafting of the manuscript: T.N. Critical review of the manuscript: K.T. Study supervision: K.K.
Ethics
Ethical approval was obtained from the Ethical Committee for Clinical Research of our institution and written informed consent was obtained from each patient. All the animal experiments were performed in accordance with protocols approved by the Animal Ethics Committee of the University of Tokyo.
Funding
This research was supported by Grants-in-Aid for Scientific Research (18K15741) from Japan Society for the Promotion of Science and the Research Program on Hepatitis from Japan Agency for Medical Research and Development (AMED) under Grant Number JP17fk0210304, JP18fk0210040, JP19fk0210040, and JP20fk0210040.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by M. Agostini
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information The online version contains supplementary material available at (10.1038/s41419-020-03381-1).
References
- 1.Villanueva A. Hepatocellular carcinoma. N. Engl. J. Med. 2019;380:1450–1462. doi: 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 2.Mann DA. Epigenetics in liver disease. Hepatology. 2014;60:1418–1425. doi: 10.1002/hep.27131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Totoki Y, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- 4.Fujimoto A, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016;48:500–509. doi: 10.1038/ng.3547. [DOI] [PubMed] [Google Scholar]
- 5.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 7.Au SL, et al. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology. 2012;56:622–631. doi: 10.1002/hep.25679. [DOI] [PubMed] [Google Scholar]
- 8.Fan DN, et al. Histone lysine methyltransferase, suppressor of variegation 3-9 homolog 1, promotes hepatocellular carcinoma progression and is negatively regulated by microRNA-125b. Hepatology. 2013;57:637–647. doi: 10.1002/hep.26083. [DOI] [PubMed] [Google Scholar]
- 9.Fei Q, et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat. Commun. 2015;6:8651. doi: 10.1038/ncomms9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tachibana M, et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–1791. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding J, et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013;18:896–907. doi: 10.1016/j.cmet.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cha ST, et al. G9a/RelB regulates self-renewal and function of colon-cancer-initiating cells by silencing Let-7b and activating the K-RAS/beta-catenin pathway. Nat. Cell Biol. 2016;18:993–1005. doi: 10.1038/ncb3395. [DOI] [PubMed] [Google Scholar]
- 14.Tu WB, et al. MYC Interacts with the G9a Histone Methyltransferase to Drive Transcriptional Repression and Tumorigenesis. Cancer Cell. 2018;34:579–595. e8. doi: 10.1016/j.ccell.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Avgustinova A, et al. Loss of G9a preserves mutation patterns but increases chromatin accessibility, genomic instability and aggressiveness in skin tumours. Nat. Cell Biol. 2018;20:1400–1409. doi: 10.1038/s41556-018-0233-x. [DOI] [PubMed] [Google Scholar]
- 16.Wei L, et al. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J. Hepatol. 2017;67:758–769. doi: 10.1016/j.jhep.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 17.Hu Y, et al. G9a and histone deacetylases are crucial for Snail2-mediated E-cadherin repression and metastasis in hepatocellular carcinoma. Cancer Sci. 2019;110:3442–3452. doi: 10.1111/cas.14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yokoyama M, et al. Histone lysine methyltransferase G9a is a novel epigenetic target for the treatment of hepatocellular carcinoma. Oncotarget. 2017;8:21315–21326. doi: 10.18632/oncotarget.15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barcena-Varela M, et al. Dual Targeting of Histone Methyltransferase G9a and DNA-Methyltransferase 1 for the Treatment of Experimental Hepatocellular Carcinoma. Hepatology. 2019;69:587–603. doi: 10.1002/hep.30168. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Li Z, Ye Y, Xie L, Li W. Oxidative Stress and Liver Cancer: Etiology and Therapeutic Targets. Oxid. Med Cell Longev. 2016;2016:7891574. doi: 10.1155/2016/7891574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Q, et al. G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proc. Natl Acad. Sci. USA. 2017;114:E6054–E6063. doi: 10.1073/pnas.1700694114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong CM, et al. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology. 2016;63:474–487. doi: 10.1002/hep.28304. [DOI] [PubMed] [Google Scholar]
- 23.Dapito DH, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang JS, Wanibuchi H, Morimura K, Gonzalez FJ, Fukushima S. Role of CYP2E1 in diethylnitrosamine-induced hepatocarcinogenesis in vivo. Cancer Res. 2007;67:11141–11146. doi: 10.1158/0008-5472.CAN-07-1369. [DOI] [PubMed] [Google Scholar]
- 25.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 26.Bergmann J, et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology. 2017;65:89–103. doi: 10.1002/hep.28874. [DOI] [PubMed] [Google Scholar]
- 27.Siliciano JD, et al. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11:3471–3481. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miled C, Pontoglio M, Garbay S, Yaniv M, Weitzman JB. A genomic map of p53 binding sites identifies novel p53 targets involved in an apoptotic network. Cancer Res. 2005;65:5096–5104. doi: 10.1158/0008-5472.CAN-04-4232. [DOI] [PubMed] [Google Scholar]
- 29.Nakatsuka T, et al. Impact of histone demethylase KDM3A-dependent AP-1 transactivity on hepatotumorigenesis induced by PI3K activation. Oncogene. 2017;36:6262–6271. doi: 10.1038/onc.2017.222. [DOI] [PubMed] [Google Scholar]
- 30.Tateishi K, Okada Y, Kallin EM, Zhang Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature. 2009;458:757–761. doi: 10.1038/nature07777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamoto S, et al. Histone demethylase KDM4C regulates sphere formation by mediating the cross talk between Wnt and Notch pathways in colonic cancer cells. Carcinogenesis. 2013;34:2380–2388. doi: 10.1093/carcin/bgt174. [DOI] [PubMed] [Google Scholar]
- 32.Nakagawa H, et al. Apoptosis signal-regulating kinase 1 inhibits hepatocarcinogenesis by controlling the tumor-suppressing function of stress-activated mitogen-activated protein kinase. Hepatology. 2011;54:185–195. doi: 10.1002/hep.24357. [DOI] [PubMed] [Google Scholar]
- 33.Moinzadeh P, Breuhahn K, Stutzer H, Schirmacher P. Chromosome alterations in human hepatocellular carcinomas correlate with aetiology and histological grade-results of an explorative CGH meta-analysis. Br. J. Cancer. 2005;92:935–941. doi: 10.1038/sj.bjc.6602448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fritsch L, et al. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol. Cell. 2010;37:46–56. doi: 10.1016/j.molcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 35.Xia L, et al. CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell. 2017;31:653–668. e7. doi: 10.1016/j.ccell.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo B, Godzik A, Reed JC. Bcl-G, a novel pro-apoptotic member of the Bcl-2 family. J. Biol. Chem. 2001;276:2780–2785. doi: 10.1074/jbc.M005889200. [DOI] [PubMed] [Google Scholar]
- 37.Giam M, et al. Detection of Bcl-2 family member Bcl-G in mouse tissues using new monoclonal antibodies. Cell Death Dis. 2012;3:e378. doi: 10.1038/cddis.2012.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin ML, Park JH, Nishidate T, Nakamura Y, Katagiri T. Involvement of maternal embryonic leucine zipper kinase (MELK) in mammary carcinogenesis through interaction with Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer Res. 2007;9:R17. doi: 10.1186/bcr1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen PM, et al. Loss of Bcl-G, a Bcl-2 family member, augments the development of inflammation-associated colorectal cancer. Cell Death Differ. 2019;27:742–757. doi: 10.1038/s41418-019-0383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang J, et al. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 2010;285:9636–9641. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heo K, et al. Cell-penetrating H4 tail peptides potentiate p53-mediated transactivation via inhibition of G9a and HDAC1. Oncogene. 2013;32:2510–2520. doi: 10.1038/onc.2012.273. [DOI] [PubMed] [Google Scholar]
- 42.Abe Y, et al. JMJD1A is a signal-sensing scaffold that regulates acute chromatin dynamics via SWI/SNF association for thermogenesis. Nat. Commun. 2015;6:7052. doi: 10.1038/ncomms8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segovia C, et al. Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat. Med. 2019;25:1073–1081. doi: 10.1038/s41591-019-0499-y. [DOI] [PubMed] [Google Scholar]
- 44.Urrutia G, et al. Combined targeting of G9a and checkpoint kinase 1 synergistically inhibits pancreatic cancer cell growth by replication fork collapse. Mol. Cancer Res. 2020;18:448–462. doi: 10.1158/1541-7786.MCR-19-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng AL, Hsu C, Chan SL, Choo SP, Kudo M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 2020;72:307–319. doi: 10.1016/j.jhep.2019.09.025. [DOI] [PubMed] [Google Scholar]
- 46.Tachibana M, Nozaki M, Takeda N, Shinkai Y. Functional dynamics of H3K9 methylation during meiotic prophase progression. EMBO J. 2007;26:3346–3359. doi: 10.1038/sj.emboj.7601767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26:149–150. doi: 10.1002/(SICI)1526-968X(200002)26:2<149::AID-GENE16>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 48.Kim Y, et al. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat. Med. 2017;23:213–222. doi: 10.1038/nm.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakagawa H, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. 2014;26:331–343. doi: 10.1016/j.ccr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamamoto K, et al. Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget. 2016;7:61469–61484. doi: 10.18632/oncotarget.11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.