

Abstract
Purpose
In a cohort of 8 families (11 patients) with autosomal recessive retinitis pigmentosa (arRP), we clinically characterized disease associated with mutations in CNGB1.
Methods
Visual function was determined by measuring the patients’ visual acuity, dark- and light-adapted perimetry, and by full field electroretinography. Retinal structure was evaluated with spectral-domain optical coherence tomography, fundus imaging, and autofluorescence imaging.
Results
Age of onset ranged from 4 to 49 years (mean [SD] 26 [17], median 27 years). The age at visit was 27 – 54 years, mean 37 (17). The range of visual acuity was logMAR −0.1 to 1.3 (Snellen 20/16 to 20/400) in the right eye and −0.1 to 0.9 (Snellen 20/16 to 20/160) in the left eye. Electrophysiological testing in 5 patients showed an absence of the rod response. Cone responses ranged from normal to severely reduced. The patients exhibited loss of rod vision more severe than cone vision. Funduscopic images showed widespread retinal degeneration with pigment clumping, optic disk pallor, arteriole attenuation, and a peri-foveal ring of hyper autofluorescence. Three families were tested for olfactory dysfunction and results indicated mild to complete anosmia in individuals with mutations in CNGB1.
Genetic analysis revealed 6 novel variants, c.2127C>G, p.Phe709Leu; c.1431C>A, p.Cys477*; c.2034G>A, p.Trp678*; c.2092T>C, p.Cys698Arg; and c.583+2T>C, c.2305-34G>A and 3 variants that have been previously described, c.2957A>T, p.Asn986Ile; c.2544dup, p.Leu849Alafs*3; and c.2492+1G>A.
Discussion
This is the first report for 6 novel CNGB1 variants associated with arRP. Two families had olfactory dysfunction in patients with arRP and family members who were heterozygous for a CNGB1 mutation. Additionally, findings demonstrated variable penetrance and expressivity of disease in these patients.
INTRODUCTION
Retinitis Pigmentosa (RP) is a group of inherited disorders involving progressive degeneration of the retina that leads to severe visual impairment. The disorder is characterized by retinal pigment epithelial (RPE) abnormalities visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically experience a decline and loss of night vision during adolescence, followed by decreased peripheral vision in young adulthood, and deterioration of the central vision in later life due to the progressive degeneration of rod and cone photoreceptors.(1)
To date 43 genes and loci (RETNET; https://sph.uth.edu/retnet/, May 2020) are associated with autosomal recessive RP (arRP), including genes encoding the cyclic nucleotide-gated (CNG) channel subunits, CNGA1 and CNGB1 expressed in rod photoreceptors. The CNG channels are nonselective cation channels in rod and cone photoreceptors and olfactory sensory neurons that translate changes of second-messenger cyclic guanosine monophosphate (cGMP) or cyclic adenosine monophosphate (cAMP) into voltage signals.(2, 3) The rod CNG channel is a heterotetramer formed by 3 α-subunits (encoded by gene CNGA1) and 1 β-subunit (encoded by CNGB1). (4, 5) Expression of the β-subunit, CNGB1 in the retina is restricted to the rod photoreceptors, and the observed cone dysfunction evident in patients is likely consequent to rod photoreceptor degeneration.(6) CNGB1 interacts with the C-terminus of the CNGA1 subunit and it is involved in the regulation of ion flow into the rod photoreceptor outer segment, in response to light-induced alteration of the levels of intracellular cGMP. Both CNGA1 and CNGB1 have been associated with arRP and similarly characterized by early onset nyctalopia, concentric loss of the visual field, macular involvement, and progressive visual acuity decline.(6–18) Mutations CNGB1 and CNGA2 but not CNGA1 have been linked to recessive olfactory dysfunction.(19–21)
Herein, we describe 11 individuals from 8 unrelated families affected by arRP due to mutations in CNGB1. Clinical and genetic examination of these families indicated a phenotype linked to arRP that revealed novel variants in CNGB1.
MATERIALS and METHODS
Patients were evaluated at the Retina Foundation of the Southwest in Dallas, TX, or the Dean McGee Eye Institute at the University of Oklahoma Health Sciences Center in Oklahoma City, OK. Informed consent was obtained prior to examinations. All procedures were approved by institutional ethics review boards and adhered to the Declaration of Helsinki.
Visual acuity was measured with a traditional Snellen chart or with the Electronic Visual Acuity Tester (Jaeb Center for Health Research, Tampa, FL, USA) and converted to a logarithm of minimal angle of resolution (logMAR). The eye with the lower acuity or the right eye if there was no difference between eyes, was dilated and dark-adapted. Full field electroretinography (ffERG) was performed on the dilated eye with the International Society of Electrophysiology of Vision standard protocol.(22) The ffERG was recorded using a Burian-Allen ERG electrode (Hansen Ophthalmic Development Laboratory). Fundus images of dilated eyes were acquired with the Optos® camera (Optos PLC, Dunfermline, UK) or with CF-60UD (Canon USA Inc, Lake Success, New York). Horizontal line scans were obtained with spectral-domain optical coherence tomography (SD-OCT; Spectralis HRA; Heidelberg Engineering, Heidelberg, Germany). Olfactory function was tested with the Smell Identification Test (SIT; Sensonics International, Haddon Heights, NJ). The SIT is a psychometric test that uses microencapsulation technology whereby olfactory function can be quantified.(23–26) Kinetic perimetry was performed on the un-dilated eye while the fellow eye was dark-adapting. Perimetry was performed using an Octopus900 (Haag-Streit AG, Switzerland). For the kinetic exam, the visual field was mapped with spot sizes V-4e, III-4e, and I-4e at a speed of 4°/sec. Visual field area was computer-calculated for each isopter.
To obtain genetic testing, whole blood or saliva was sent to Blueprint Genetics My Retina Tracker sponsored by Foundation Fighting Blindness using a retinal dystrophy panel or DNA extracted from whole blood was sent to Dr. Steve Daiger’s lab (LMDIED) using a 163 retinal gene–targeted NGS panel.(27–30)
RESULTS
Clinical examinations, imaging, and genetic analysis were used to characterize 11 patients from 8 unrelated families (Fig. 1) with arRP. The age of onset ranged from age 4 to 49 years (mean [SD] 26 [17], median 27 years; Table 1). The age at the reported visit was 27 – 54 years, mean 37 (17). Most patients remembered nyctalopia since childhood, but almost half (5) were not diagnosed with arRP until over the age of 30. Patient H1 noticed reduced visual field as the initial symptom and reported photophobia along with nyctalopia at her visit. The range of visual acuity was logMAR, −0.1 to 1.3 (Snellen 20/16 to 20/400) in right eyes and −0.1 to 0.9 (Snellen 20/16 to 20/160) in left eyes (Table 2). Electrophysiologic testing in 5 patients showed absence of a rod response and varying degrees of cone responses ranging from amplitudes within normal limits to those that were minimally detected (Table 2).
Figure 1.
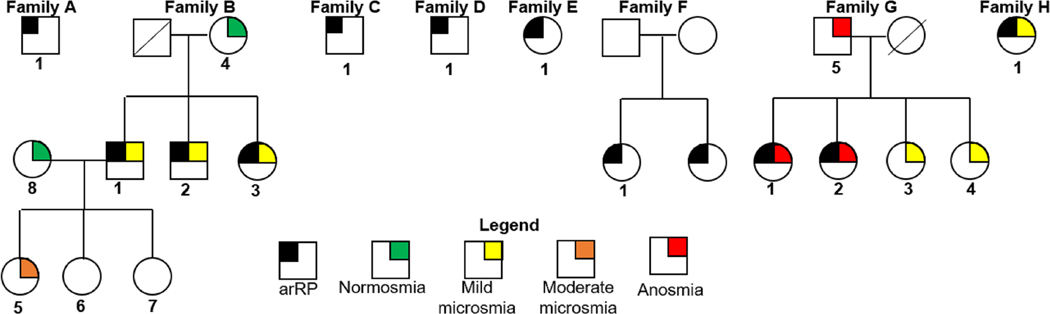
Pedigrees with diagnosis for 8 families with autosomal recessive retinitis pigmentosa (arRP). Numbered patients had genetic testing. Symbols with black were patients diagnosed with arRP. The results for patients who performed the smell identification test are in color.
Table 1.
Patient Demographics and mutations
| Family | Patient # | Gender | Age of diagnosis | Age at visit | Variant 1 | Variant 2 |
|---|---|---|---|---|---|---|
| A | 1 | Male | 49 | 49 | c.2957A>T, p.Asn986Ile28,29 | VUS c.2127C>G, p.Phe709Leu |
| B | 1 | Male | 5 | 30 | c.2957A>T, p.Asn986Ile28,29 | c.2957A>T, p.Asn986Ile28,29 |
| B | 3 | Female | 43 | 43 | c.2957A>T, p.Asn986Ile28,29 | c.2957A>T, p.Asn986Ile28,29 |
| B | 2 | Male | 17 | 27 | c.2957A>T, p.Asn986Ile28,29 | c.2957A>T, p.Asn986Ile28,29 |
| C | 1 | Male | 17 | 32 | c.2544dup, p.Leu849Alafs*328,30,31 | c.2957A>T, p.Asn986Ile28,29 |
| D | 1 | Male | 27 | 42 | c.2034G>A, p.Trp678* | c.2034G>A, p.Trp678* |
| E | 1 | Female | 4 | 54 | c.1431C>A, p.Cys477* | c.1431C>A, p.Cys477* |
| F | 1 | Female | 12 | 52 | c.2957A>T, p.Asn986Ile28,29 | c.2957A>T, p.Asn986Ile |
| G | 1 | Female | 34 | 53 | c.2492+1G>A, p.?19 | c.2092T>C, p.Cys698Arg |
| G | 2 | Female | 32 | 42 | c.2492+1G>A, p.?19 | c.2092T>C, p.Cys698Arg |
| H | 1 | Female | 49 | 52 | c.583+2T>C, p.? | VUS c.2305-34G>A, p.? |
Variant of uncertain significance, VUS
Table 2.
Visual function
| Patient # | BCVA (OD) | BCVA (OS) | Refraction (OD) | Refraction (OS) | ffERG | Area (deg2) kinetic (V4e, III4e, I4e) |
|---|---|---|---|---|---|---|
| A1 | 20/32 | 20/32 | −3.0 + 0 × 0 | −3.0 + 0 × 0 | not done | 2067, 168, 114 |
| B1 | 20/25 | 20/25 | none | none | rod ND; Mixed ND cone minimal amplitude | Not done |
| B3 | 20/16 | 20/16 | contacts | contacts | rod ND; Mixed decreased; normal cone | 12010,9716, 5934 |
| B2 | 20/20 | 20/25 | none | none | rod ND; Mixed and cone decreased | not done |
| C1 | 20/40 | 20/25 | none | none | rod ND; Mixed ND; cone minimal amplitude | 4419, 1489, 666 |
| D1 | 20/32 | 20/32 | 0.25+1.00×119 | 0.25+1.00×131 | not done | 1485, 354, 30 |
| E1 | 20/400 | 20/160 | none | none | not done | 77, 27, not done |
| F1 | 20/40 | 20/20 | none | none | rod ND; Mixed ND; cone minimal amplitude | 1200, 493, 251 |
| G1 | 20/40 | 20/40 | none | none | not done | 1245, 1372, not done |
| G2 | 20/32 | 20/60 | +4.00+0.75×015 | +1.25+0.75×117 | not done | 4085, 2683, 337 |
| H1 | 20/50 | 20/40 | −1.50+0.00×00 | −0.75+0.00×00 | not done | not done |
Right eye, OD; Left eye, OS; Full field electroretinography, ffERG; not detected, ND
Funduscopic images for 10 patients (all but B2, who did not have fundus photography) showed widespread retinal degeneration with pigment clumping, optic disk pallor, arteriole attenuation, and variable pigmentary changes in the macula (Figs. 3 & 4). The middle panel in Figure 3 shows the horizontal SD-OCT scan of the eye that performed the kinetic visual field. Patient A1, diagnosed at age 49 years, had central preservation of the inner segment ellipsoid (ISe) band of the photoreceptors revealed on SD-OCT (middle panel, blue arrows) consistent with 20/32 visual acuity (Table 2). RPE breakdown and pigment clumping (middle panel, yellow arrows) corresponded to constricted visual fields (approximately 5°) to spot sizes I4e and III4e (green and red isopters, respectively, right panel; Fig. 3A). Patient B3 was also diagnosed in her 40’s but had an ISe band that was preserved throughout the image which was presumed to contribute to the large visual field (Fig. 3B; Table 2). Patient C1 had cystoid macular edema, a full ISe band, temporally attenuated outer retina, and a complicated visual field showing preservation of the central 30° to spot sizes III4e and I4e (Fig. 3C). Vitreoretinal surface changes on SD-OCT suggested the formation of an epiretinal membrane (ERM) for patient C1. This patient also had a large wedge of vision in the temporal visual field. Patient D1 had a bull’s eye pattern of RPE breakdown on macular fundus imaging and foveal preservation of the ISe band on SD-OCT (Fig. 3D, left and middle panels, respectively). Kinetic perimetry measured approximately 20°, 10°, and 5° in the central field to spot sizes V4e, III4e, and I4e, respectively as well as temporal islands of vision measured with V4e (Fig. 3D, right panel). Patient E1 was diagnosed earliest (4 years) within this group and showed the most advanced disease severity with foveal ISe sparing and 5° of visual field (Fig. 3E). Peripapillary atrophy was noted in families 6–9 (Figs. 3E and 4B–4E, respectively).
Figure 3.
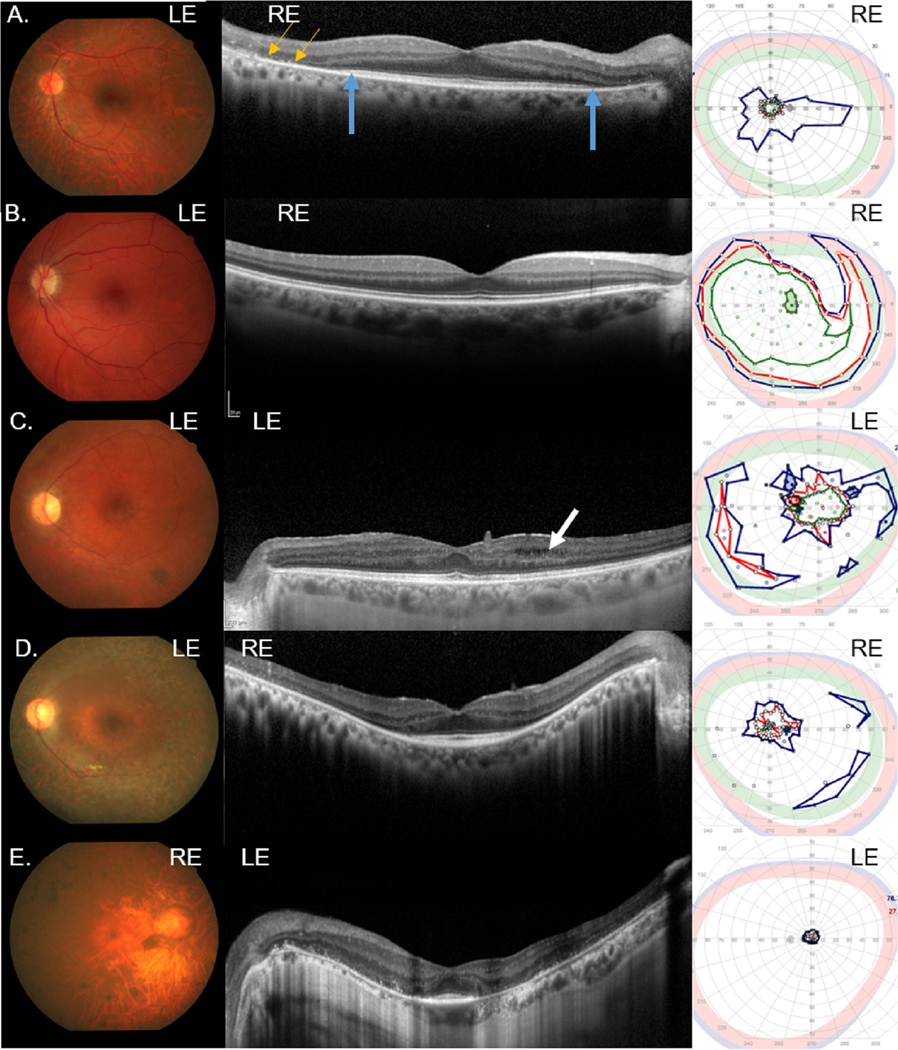
Color fundus images (left panel), SD-OCT horizontal line scans through the fovea (middle panel), and kinetic visual fields (right panel) for families A-E. (A) Patient A1; (B) Patient B3. (C) Patient C1 had cystoid macular edema (arrow) noted on SD-OCT. (D) Patient D1. (E) Patient E1. SD-OCT image on same eye as the kinetic visual field. Right eye, RE; Left eye, LE; blue isopter, spot size V4e; red isopter, spot size III4e; green isopter, spot size I4e.
Figure 4.
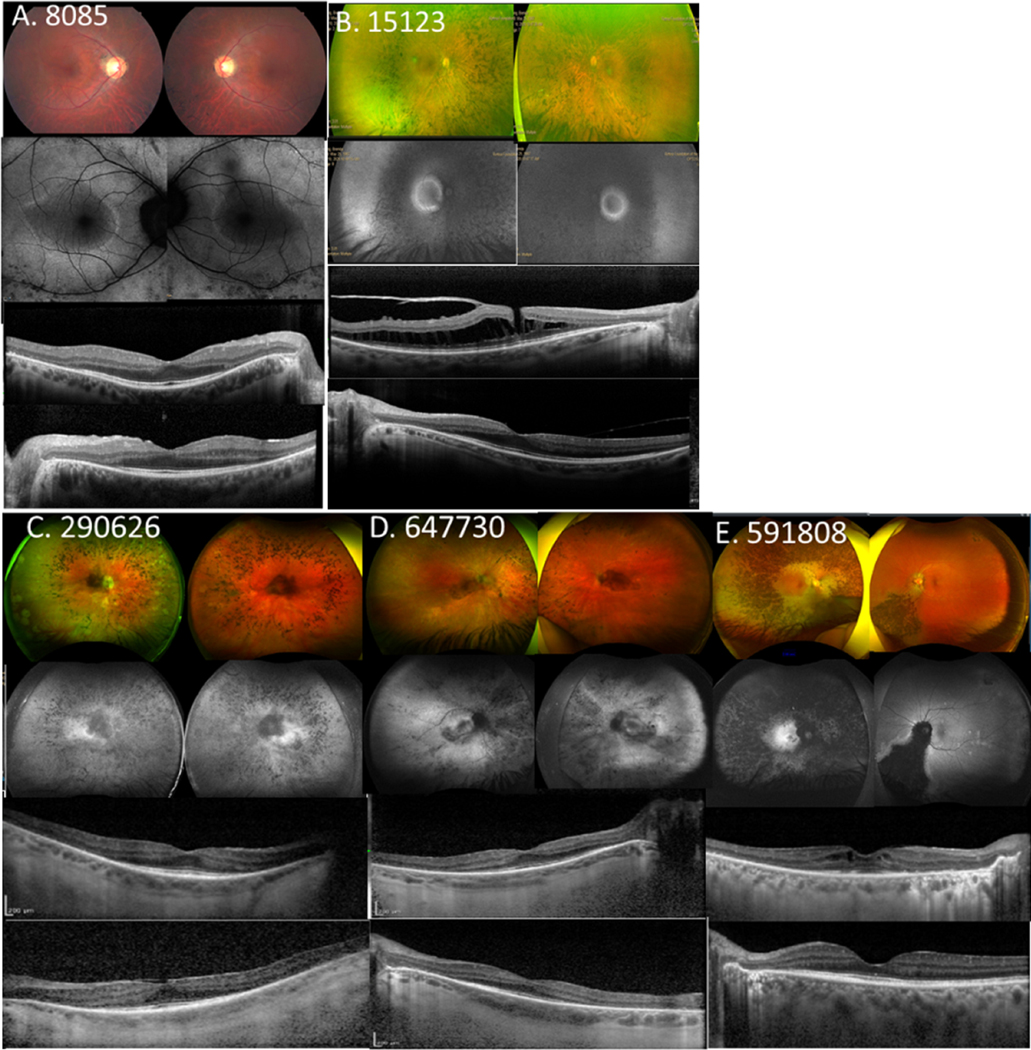
Spectrum of disease severity in patients with CNGB1-associated RP. Color fundus imaging (top row), fundus autofluorescence (2nd row) and right and left eye SD-OCT (bottom 2 rows) images, respectively for patient B1 (A) from family 2; patient F1 (B) from family 6; patients G1 (C) and G2 (D) from family 7; and (E) patient H1 from family 8, illustrating typical presentations of RP features (B–E) waxy pallor of the optic disc, severe attenuation of the retinal vasculature, RPE breakdown, and bone-spicule pigment clumping in the mid-periphery. (B and E) Cystoid macular edema (CME) and (A, B and C) epiretinal membrane. A macular hole is evident for patient F1 (B).
Five patients who had FAF imaging showed peri-macular rings of hyper-autofluorescence (AF) with a corresponding pattern of ISe attenuation on the SD-OCT (Fig. 4). The sub-foveal ISe band was preserved in all patients. Wide-field imaging revealed diffuse RPE breakdown with mid-peripheral pigment clumping and bone spicule formation (Fig. 4B–4E). A macular hole was detected in the right eye for patient F1 (Fig. 4B, 3rd row SD-OCT image). Patient H1 exhibited asymmetrical disease with severe chorioretinal degeneration, bone-spicule pigmentation, and bull’s eye pattern of AF in the right eye (Fig. 4E). However, the left macula looked relatively normal and degenerative changes were mainly confined to the infero-nasal retina.
Since CNGB1 has been associated with anosmia, we tested olfactory function in families B, G, and H. Those tested with the SIT have color indicators of the findings on their pedigrees in Figure 1. All 3 siblings in family B had arRP and mild microsmia (yellow; Fig. 1). Their mother, B4 and 3 children, B5-B7 were a heterozygous carriers of the CNGB1 mutation, c.2957A>T, p.Asn986Ile. B4 and B5 performed the SIT which revealed that B5 had moderate microsmia (orange) whereas B4 tested within the normal range (normosmia, green; Fig. 1). In family G, the father, G5 and all 4 sisters, G1-G4 tested below normosmia. The father, G5 and 2 normal-sighted sisters, G3 and G4 were heterozygous carriers of a CNGB1 mutation, c.2492+1G>A, p.?. The sisters with arRP, G1 and G2 and their father, G5 had anosmia (red). The 2 normal-sighted sisters, G3 and G4 had mild microsmia (yellow). In family H, patient 1 subjectively reported that her ability to taste was “not normal” and the SIT revealed that she had moderate microsmia (orange; Fig. 1).
GENETICS
We identified 9 variants in CNGB1 that were likely to explain the arRP from 8 independent families (Table 2). Six variants were novel, and 3 variants have been previously described.(19, 31–34) Two novel variants, c.2127C>G, p. Phe709Leu and c.2092T>C, p.Cys698Arg were nonsynonymous substitutions (missense) hat altered highly conserved amino acid residues (Table 3). These variants were found in the population (ALL) database, gnomAD Browser(35) with low frequencies that ranged from 0 – 0.0008% (Table 3), meaning that they were unlikely to be common benign variants. Two novel nonsense variants c.2034G>A, p.Trp678* and c.1431C>A, p.Cys477* introduced stop codons that were predicted to result in nonsense-mediated decay (NMD; Table 3). Finally, two variants, c.2305–34G>A and c.583+2T>C were predicted to affect mRNA splicing. Prediction modeling (Alamut® Visual 2.12, Interactive Biosoftware Inc.) revealed that c.2305-34G>A activated a cryptic splice site. The variant c.583+2T>C disrupted a donor splice site. The consequence of this change was not predictable, but would likely result in a skipping of the 9th exon in CNGB1. No other potentially disease-causing variants were identified in other genes known to be associated with RP.
Table 3.
Novel Variants
| Variant on Nucleotide Level | Consequence on Protein Level | Location in CNGB1 Polypeptide | gnomAD Browser MAF, ALL % | Conservation | Prediction |
|---|---|---|---|---|---|
| c.2127C>G | Phe709Leu | channel domain | 0.0024 | Highly; up to Fruitfly | Mutationtaster: disease causing |
| c.2034G>A | Trp678* | NA | 0 | Moderately; up to Tetradon | NMD |
| c.1431C>A | Cys477* | NA | 0 | Weakly; up to chimp | NMD |
| c.2092T>C | Cys698Arg | TM1 | 0.0008 | Highly; up to C. elegans | SIFT: Deleterious; PolyPhen-2: probably damaging; Mutationtaster: disease causing |
| c.2305–34G>A | Splice defect | NA | 0.0097 | NA | New acceptor site |
| c.583+2T>C | Splice defect | NA | 0.0012 | NA | Broken donor site |
Transmembrane domain, TM; nonsense mediated decay, NMD; not applicable, NA
DISCUSSION
CNG channels are necessary for normal phototransduction. In the dark, the rod cells have high levels of cGMP that act to keep open a proportion of the CNG channels, allowing an influx of cations that depolarizes the photoreceptor. Light stimulation of rhodopsin activates transducin, a G-protein, which in turn activates phosphodiesterase (PDE). The active PDE hydrolyses cGMP thus reducing its concentration, resulting in closure of CNG channels. This closure stops the influx of sodium ions and subsequently results in hyperpolarization of the rod photoreceptor.(3) CNGB1 is an important modulatory subunit for CNG formation and targeting to the rod outer segments. The mRNA of CNGB1 is alternatively spliced to create 3 distinct proteins in the retina that include the soluble glutamic acid rich proteins 1 and 2 (GARP1 and GARP2) and a full length CNGB1 protein.(36) The full-length CNGB1 protein in the retina called CNGB1a, consists of an N-terminal “GARP region” and a C-terminal “channel domain” (Fig. 2). Unique CNGB1 transcripts have been identified in the kidney, brain, testes, and olfactory epithelium(3) but, there is insufficient evidence that these tissues express functional CNG channels. In the brain, currents were recorded from hippocampal neurons but they were dissimilar to the currents flowing through the well-characterized CNG channels in the retina and olfactory neurons.(37) Further, the presence of the subunit polypeptides in the brain have not been clearly demonstrated. In spermatozoa, the CNG channel might be involved in chemotactic swimming behavior, capacitation, or acrosomal exocytosis. This has only been suggestive since there was no evidence for the presence of a receptor-type guanylyl cyclase (GC) that is needed to synthesize cGMP and there has been no identification of Ca2+-regulated GCs in the testes. For a full review see (3). Importantly, neither animal models, nor patient histories have identified CNGB1-related effects in organ systems other than the retina and olfactory neurons which means that functional CNGB channels are unlikely to be expressed in other tissues.
Figure 2.
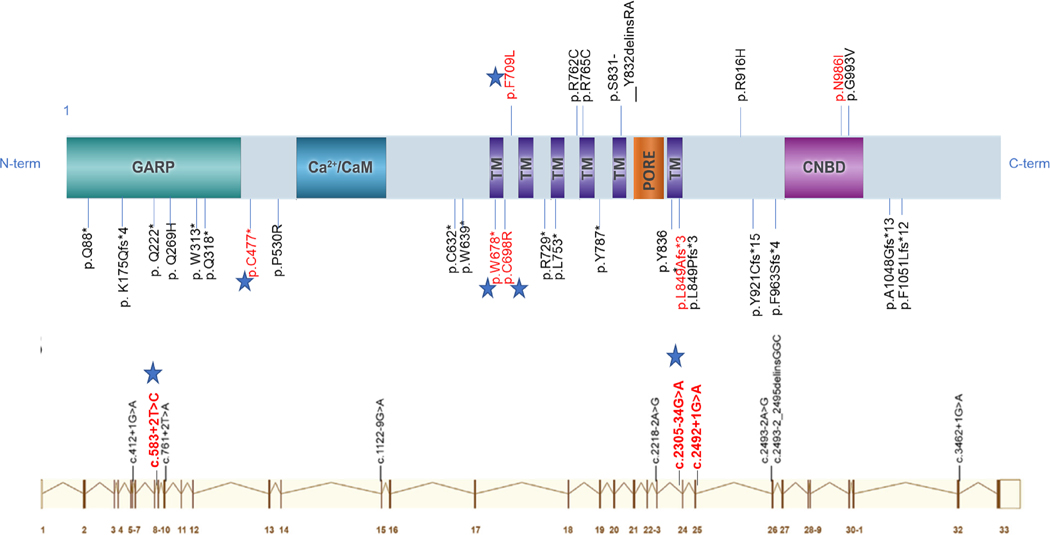
Schematic representation of the CNGB1 protein (top) from the canonical transcript NM_001297.4 (bottom). CNGB1 has six transmembrane (TM) domains, a pore loop domain between the fifth and sixth transmembrane domains, and intracellular N- and C-termini. CNGB1 has a large glutamic acid-rich protein (GARP) region in the N-terminal tail (Top panel). CNGB1 interacts with the C-terminus of the CNGA1 subunit through a region containing the calcium (Ca2+)/ calmodulin (CaM) binding site. Cyclic nucleotide binding domain, CNBD is located in the C-terminal tail. The previously reported disease-causing mutations are in black lettering. The previously reported splice-site mutations are aligned with the CNGB1 exons (Bottom panel). Red lettered are variants detected in our cohort. Stars represent novel variants (modified from mutagenetix website-database of mutations and phenotypes).
A recent report of ten patients with arRP due to CNGB1 mutations showed childhood-onset of night-blindness, peripheral visual field constriction earlier than the third decade of life, and subnormal cone ffERG responses in the absence of rod responses.(31) Here we show a wide range of ages for disease onset and variability in severity among patients. Patient A1, had constricted ISe band of the photoreceptors (Fig. 3A) that he likely did not notice the vision loss due to the slow progression over time since he was diagnosed in his 40’s. Patient B3 was also diagnosed in her 40’s but she had preservation of the ISe band and the largest visual field among all patients. Her 2 brothers had earlier onset, ages 5 and 17 compared to her onset, which was at age 43, revealing variable expressivity even within families for CNGB1-associated arRP.
Currently, there are 22 likely pathogenic and 34 pathogenic mutations listed in ClinVar for CNGB1. Of these variants, only 5 are missense, with the remaining presumed to be ‘null’ (10 nonsense; 11 frameshifts; 9 splice-site). Reported here are 3 missense and 6 null variants, including 1 frameshift, 2 nonsense, and 3 that alter splicing (Table 1).
The missense variant c.2957A>T, p.Asn986Ile is located in the cyclic nucleotide-binding domain (CNBD) which impairs cGMP binding resulting in a non-functional channel.(14, 38) The other 2 missense variants found in our patients, c.2127C>G, p.Phe709Leu and c.2092T>C, p. Cys698Arg, alter highly conserved amino acid residues within transmembrane domains (TM) close to TM1 (Figure 2). A recent study detected a strong physical interaction between an N-terminal region of CNGB1 (amino acids 677–764) and a C-terminal region of CNGA1. The results imply that a CNGA1/CNGB1 inter-subunit interaction regulates membrane expression which is required for surface targeting of intact channels.(39) Therefore, pathogenic variants in this region possibly prevent CNG channels from localizing to the surface of rod cells, resulting in abnormal of outer segment development, metabolic overload, dysfunction of retinal pigment epithelial cells, and sustained activation of the photoreceptor.(40)
Two nonsense variants c.2034G>A, p. Trp678* and c.1431C>A, p. Cys477* as well as the frameshift variant p.L849Afs*3 introduce stop codons in the mRNA whereas c.2492+1G>A, c.583+2T>C and c.2305-34G>A are predicted to upset splicing mechanisms. The null mutations likely trigger NMD and result in an absence of the full length CNGB1a subunit in the retina. In a CNGB1 mouse knockout model that lacks all retinal CNGB1 products, the GARPs have been shown to be necessary for outer segment disk development and the structural integrity of the rod outer segments.(18) Furthermore, in rods, the loss of CNGB1a induces down-regulation of several proteins of the phototransduction cascade and degradation of the CNGA1 subunit, thereby depleting the channel from the photoreceptors.
Olfactory sensory neurons convert odor stimuli into electrical signals. Olfactory signal transduction occurs in sensory neuron cilia. The olfactory CNG channels are open at physiological levels of cAMP. Binding of an odorant to surface receptors triggers a G protein-coupled cascade, which leads to an increase in the intracellular concentration of cAMP and the subsequent opening of CNG channels. Depolarization occurs upon the influx of sodium and calcium ions. The depolarization triggers action potentials that propagate along the olfactory neuronal axon to the olfactory bulb in the brain. Not only is CNGB1 is required for olfactory transduction, but it is also necessary for targeting the olfactory CNG channel to the cilia.(41)
The variant c.2492+1G>A has been associated with anosmia patients with arRP due to compound heterozygous mutations in CNGB1.(19) Here we tested extended family members who were carriers of a single CNGB1 mutation (c.2957A>T or c.2492+1G>A) with the SIT. The results for these individuals suggests that these mutations could be dominant with respect to olfactory dysfunction. Importantly, alternative splicing for CNGB1 creates distinct subunits, CNGB1a and CNGB1b where CNGB1a is rod specific while CNGB1b is olfactory specific.(3) The rod subunit, CNGB1a (1394 amino acids, aa) is the full length protein that includes the GARP domain whereas CNGB1b (856 aa) does not contain the GARP domain (Fig. 2). Therefore, mutations in CNGB1b (C-terminal half of CNGB1a) but not in N-terminus (538aa) of CNGB1a will likely cause olfactory dysfunction. Thus, patient E1, who had a homozygous mutation, c.1431C>A (p.Cys477*) is expected to be normosmic since the mutation would not be expressed in olfactory neurons. On the other hand, Issa et al(19) reported that a patient with a homozygous mutation, c.1312C>T (p.(Gln438*) had hyposmia that was borderline high. This is curious since the spliced transcript expressed in the nasal sensory neurons would translate a normal CNGB1a protein subunit.
Patient H1 in family 8 had asymmetrical RP and moderate anosmia. Two variants predicted to affect splicing were found upon genetic analysis, where the c.2305-34G>A variant would affect both CNGB1a and CNGB1b but c.583+2T>C would only be expressed in rod photoreceptors. Although variants that activate cryptic splice sites have not been associated with CNGB1, the resultant decrease in protein may regulated locally which could explain the asymmetric clinical presentation. We are currently testing whether this variant affects splicing and to what extent, if any, transcript or protein levels are disrupted. This would explain the range of olfactory dysfunction within families and supports a dose-dependent association with disease severity. All other families (excluding patient E1) in this series are expected to have some level of anosmia based on their genetic findings. Future visits for these families should include a SIT to quantify olfactory dysfunction and test whether variants in the CNGB1b subunit causes olfactory dysfunction in heterozygous carriers. The number of carriers tested here was too few to conclusively declare that CNGB1 is dominantly associated with olfactory dysfunction since other conditions or situations such as overuse of alcohol, lack of sleep, and cigarette smoke(42) may result in abnormal responses the SIT. All but sleep were queried and were not relative to the SIT results for these individuals that we tested. Further, COVID-19 infection has recently been associated with olfactory dysfunction. For a literature review, see (43). Although the individuals tested here were not likely exposed to COVID-19, we did not test for this virus. Therefore, we cannot rule out other factors that may contribute to olfactory dysfunction in this cohort.
This is the first report that the CNGB1 variants c.583+2T>C, c.2305-34G>A, c.2127C>G p.(Phe709Leu), c.2034G>A p.(Trp678*), c.1431C>A p.(Cys477*), and c.2092T>C, p.(Cys698Arg) were associated with autosomal recessive RP and that c.2957A>T and c.2492+1G>A may act as dominant variants for olfactory dysfunction. Furthermore, findings demonstrated variable penetrance and expressivity in these patients which could be induced from genetic, environmental, or metabolic factors that have yet to be determined. The expression, cellular localization, and the physical association of the channel subunits, CNGB1 and CNGA1 need to be evaluated to elucidate the mechanism of disease for the novel variants found in our patients.
ACKNOWLEDGMENTS
We would like to thank Dr. Xi-Qin Ding for thoughtful consideration and discussion of these findings. Research reported in this publication was supported by the Foundation Fighting Blindness (DGB) and the National Eye Institute of the National Institutes of Health under Award Numbers K99EY027460 (L.D.B.), R00EYE027460 (L.D.B.), US4GM104938 (L.D.B.), and EY09076 (D.G.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding information: K99EY027460 (LDB), R00EY027460 (LDB), US4GM104938 (LDB), and EY09076 (DGB)
Footnotes
Declaration of Interest Statement: The authors report no conflict of interest.
References:
- 1.Fahim AT, Daiger SP, Weleber RG. Nonsyndromic Retinitis Pigmentosa Overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. , editors. GeneReviews((R)). Seattle (WA)1993. [Google Scholar]
- 2.Biel M, Michalakis S. Function and dysfunction of CNG channels: insights from channelopathies and mouse models. Mol Neurobiol. 2007;35(3):266–77. [DOI] [PubMed] [Google Scholar]
- 3.Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82(3):769–824. [DOI] [PubMed] [Google Scholar]
- 4.Zheng J, Trudeau MC, Zagotta WN. Rod cyclic nucleotide-gated channels have a stoichiometry of three CNGA1 subunits and one CNGB1 subunit. Neuron. 2002;36(5):891–6. [DOI] [PubMed] [Google Scholar]
- 5.Peng C, Rich ED, Varnum MD. Subunit configuration of heteromeric cone cyclic nucleotide-gated channels. Neuron. 2004;42(3):401–10. [DOI] [PubMed] [Google Scholar]
- 6.Katagiri S, Hayashi T, Yoshitake K, Akahori M, Ikeo K, Gekka T, et al. Novel C8orf37 Mutations in Patients with Early-onset Retinal Dystrophy, Macular Atrophy, Cataracts, and High Myopia. Ophthalmic genetics. 2016;37(1):68–75. [DOI] [PubMed] [Google Scholar]
- 7.Comander J, Weigel-DiFranco C, Maher M, Place E, Wan A, Harper S, et al. The Genetic Basis of Pericentral Retinitis Pigmentosa-A Form of Mild Retinitis Pigmentosa. Genes (Basel). 2017;8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin X, Qu LH, Hou BK, Xu HW, Meng XH, Pang CP, et al. Novel compound heterozygous mutation in the CNGA1 gene underlie autosomal recessive retinitis pigmentosa in a Chinese family. Biosci Rep. 2016;36(1):e00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paloma E, Martinez-Mir A, Garcia-Sandoval B, Ayuso C, Vilageliu L, Gonzalez-Duarte R, et al. Novel homozygous mutation in the alpha subunit of the rod cGMP gated channel (CNGA1) in two Spanish sibs affected with autosomal recessive retinitis pigmentosa. Journal of medical genetics. 2002;39(10):E66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M, Gan D, Huang X, Xu G. Novel compound heterozygous mutations in CNGA1in a Chinese family affected with autosomal recessive retinitis pigmentosa by targeted sequencing. BMC ophthalmology. 2016;16:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiik AC, Ropstad EO, Ekesten B, Karlstam L, Wade CM, Lingaas F. Progressive retinal atrophy in Shetland sheepdog is associated with a mutation in the CNGA1 gene. Anim Genet. 2015;46(5):515–21. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Zulfiqar F, Riazuddin SA, Xiao X, Ahmad Z, Riazuddin S, et al. Autosomal recessive retinitis pigmentosa in a Pakistani family mapped to CNGA1 with identification of a novel mutation. Molecular vision. 2004;10:884–9. [PubMed] [Google Scholar]
- 13.Dryja TP, Li T. Molecular genetics of retinitis pigmentosa. Hum Mol Genet. 1995;4 Spec No:1739–43. [DOI] [PubMed] [Google Scholar]
- 14.Bareil C, Hamel CP, Delague V, Arnaud B, Demaille J, Claustres M. Segregation of a mutation in CNGB1 encoding the beta-subunit of the rod cGMP-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum Genet. 2001;108(4):328–34. [DOI] [PubMed] [Google Scholar]
- 15.Bocquet B, Marzouka NA, Hebrard M, Manes G, Senechal A, Meunier I, et al. Homozygosity mapping in autosomal recessive retinitis pigmentosa families detects novel mutations. Molecular vision. 2013;19:2487–500. [PMC free article] [PubMed] [Google Scholar]
- 16.Fradin M, Colin E, Hannouche-Bared D, Audo I, Sahel JA, Biskup S, et al. Run of homozygosity analysis reveals a novel nonsense variant of the CNGB1 gene involved in retinitis pigmentosa 45. Ophthalmic genetics. 2016;37(3):357–9. [DOI] [PubMed] [Google Scholar]
- 17.Kondo H, Qin M, Mizota A, Kondo M, Hayashi H, Hayashi K, et al. A homozygosity-based search for mutations in patients with autosomal recessive retinitis pigmentosa, using microsatellite markers. Invest Ophthalmol Vis Sci. 2004;45(12):4433–9. [DOI] [PubMed] [Google Scholar]
- 18.Winkler PA, Ekenstedt KJ, Occelli LM, Frattaroli AV, Bartoe JT, Venta PJ, et al. A large animal model for CNGB1 autosomal recessive retinitis pigmentosa. PloS one. 2013;8(8):e72229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charbel Issa P, Reuter P, Kuhlewein L, Birtel J, Gliem M, Tropitzsch A, et al. Olfactory Dysfunction in Patients With CNGB1-Associated Retinitis Pigmentosa. JAMA Ophthalmol. 2018;136(7):761–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karstensen HG, Mang Y, Fark T, Hummel T, Tommerup N. The first mutation in CNGA2 in two brothers with anosmia. Clinical genetics. 2015;88(3):293–6. [DOI] [PubMed] [Google Scholar]
- 21.Sailani MR, Jingga I, MirMazlomi SH, Bitarafan F, Bernstein JA, Snyder MP, et al. Isolated Congenital Anosmia and CNGA2 Mutation. Sci Rep. 2017;7(1):2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Documenta ophthalmologica Advances in ophthalmology. 2015;130(1):1–12. [DOI] [PubMed] [Google Scholar]
- 23.Doty RL, Shaman P, Dann M. Development of the University of Pennsylvania Smell Identification Test: a standardized microencapsulated test of olfactory function. Physiol Behav. 1984;32(3):489–502. [DOI] [PubMed] [Google Scholar]
- 24.Doty RL, Shaman P, Kimmelman CP, Dann MS. University of Pennsylvania Smell Identification Test: a rapid quantitative olfactory function test for the clinic. Laryngoscope. 1984;94(2 Pt 1):176–8. [DOI] [PubMed] [Google Scholar]
- 25.Doty RL. Influence of age and age-related diseases on olfactory function. Ann N Y Acad Sci. 1989;561:76–86. [DOI] [PubMed] [Google Scholar]
- 26.Doty RL, Frye RE, Agrawal U. Internal consistency reliability of the fractionated and whole University of Pennsylvania Smell Identification Test. Perception & psychophysics. 1989;45(5):381–4. [DOI] [PubMed] [Google Scholar]
- 27.Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clinical genetics. 2013;84(2):132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones KD, Wheaton DK, Bowne SJ, Sullivan LS, Birch DG, Chen R, et al. Next-generation sequencing to solve complex inherited retinal dystrophy: A case series of multiple genes contributing to disease in extended families. Molecular vision. 2017;23:470–81. [PMC free article] [PubMed] [Google Scholar]
- 29.Wang F, Wang H, Tuan HF, Nguyen DH, Sun V, Keser V, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014;133(3):331–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellingford JM, Barton S, Bhaskar S, O’Sullivan J, Williams SG, Lamb JA, et al. Molecular findings from 537 individuals with inherited retinal disease. Journal of medical genetics. 2016;53(11):761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hull S, Attanasio M, Arno G, Carss K, Robson AG, Thompson DA, et al. Clinical Characterization of CNGB1-Related Autosomal Recessive Retinitis Pigmentosa. JAMA Ophthalmol. 2017;135(2):137–44. [DOI] [PubMed] [Google Scholar]
- 32.Simpson DA, Clark GR, Alexander S, Silvestri G, Willoughby CE. Molecular diagnosis for heterogeneous genetic diseases with targeted high-throughput DNA sequencing applied to retinitis pigmentosa. Journal of medical genetics. 2011;48(3):145–51. [DOI] [PubMed] [Google Scholar]
- 33.Lingao MD, Ganesh A, Karthikeyan AS, Al Zuhaibi S, Al-Hosni A, Al Khayat A, et al. Macular cystoid spaces in patients with retinal dystrophy. Ophthalmic genetics. 2016;37(4):377–83. [DOI] [PubMed] [Google Scholar]
- 34.Ge Z, Bowles K, Goetz K, Scholl HP, Wang F, Wang X, et al. NGS-based Molecular diagnosis of 105 eyeGENE((R)) probands with Retinitis Pigmentosa. Sci Rep. 2015;5:18287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colville CA, Molday RS. Primary structure and expression of the human beta-subunit and related proteins of the rod photoreceptor cGMP-gated channel. The Journal of biological chemistry. 1996;271(51):32968–74. [DOI] [PubMed] [Google Scholar]
- 37.Leinders-Zufall T, Rosenboom H, Barnstable CJ, Shepherd GM, Zufall F. A calcium-permeable cGMP-activated cation conductance in hippocampal neurons. Neuroreport. 1995;6(13):1761–5. [DOI] [PubMed] [Google Scholar]
- 38.Michalakis S, Zong X, Becirovic E, Hammelmann V, Wein T, Wanner KT, et al. The glutamic acid-rich protein is a gating inhibitor of cyclic nucleotide-gated channels. J Neurosci. 2011;31(1):133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trudeau MC, Zagotta WN. An intersubunit interaction regulates trafficking of rod cyclic nucleotide-gated channels and is disrupted in an inherited form of blindness. Neuron. 2002;34(2):197–207. [DOI] [PubMed] [Google Scholar]
- 40.Pierce EA. Pathways to photoreceptor cell death in inherited retinal degenerations. BioEssays : news and reviews in molecular, cellular and developmental biology. 2001;23(7):605–18. [DOI] [PubMed] [Google Scholar]
- 41.Michalakis S, Reisert J, Geiger H, Wetzel C, Zong X, Bradley J, et al. Loss of CNGB1 protein leads to olfactory dysfunction and subciliary cyclic nucleotide-gated channel trapping. The Journal of biological chemistry. 2006;281(46):35156–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frye RE, Schwartz BS, Doty RL. Dose-related effects of cigarette smoking on olfactory function. JAMA. 1990;263(9):1233–6. [PubMed] [Google Scholar]
- 43.Meng X, Deng Y, Dai Z, Meng Z. COVID-19 and anosmia: A review based on up-to-date knowledge. Am J Otolaryngol. 2020;41(5):102581. [DOI] [PMC free article] [PubMed] [Google Scholar]