

Abstract
Cereblon (CRBN), the substrate receptor of an E3 ubiquitin ligase complex, is a target of thalidomide and thalidomide-derived immunomodulatory drugs (IMiDs). The binding of these IMiDs to CRBN alters the substrate specificity of the ligase, thereby mediating multiple effects that are exploited in cancer therapy. However, to date, it is not clear which other possible targets might be involved in the efficacy of IMiDs. One especially prominent effect of a number of thalidomide analogs is their ability to inhibit angiogenesis, which is typically enhanced in fluorinated analogs. So far, the involvement of CRBN in antiangiogenic effects is under debate. Here, starting from a systematic set of thalidomide analogs and employing a quantitative in vitro CRBN-binding assay, we study the correlation of fluorination, CRBN binding and antiangiogenic effects. We clearly identify fluorination to correlate both with CRBN binding affinity and with antiangiogenic effects, but do not find a correlation between the latter two phenomena, indicating that the main target for the antiangiogenic effects of thalidomide analogs still remains to be identified.
Keywords: IMiDs, CRBN, cancer therapy, microscale thermophoresis, MST
Graphical Abstract
Introduction
Thalidomide and its derivatives constitute the class of immunomodulatory drugs (IMiDs). These have proven to be effective in diverse clinical settings, most prominently in cancer therapy [1,2]. A major factor for the anticancer effects are the antiangiogenic properties of various thalidomide analogs, but potentially also of hydrolysis products and physiological thalidomide metabolites [3]. Thalidomide consists of a glutarimide and a phthalimide ring, which were both subject of a plethora of variations and substitutions in various studies focusing on the design and biological evaluation of analogs [4–9]. Previous studies have demonstrated that fluorinated thalidomide derivatives exhibit improved antiangiogenic efficiency [10–12]; however, a defined mechanism of action is still elusive [13,14].
About a decade ago, the protein cereblon (CRBN) was identified as a target of thalidomide [15]; CRBN is the substrate recognition module of an E3 ubiquitin ligase, which can be targeted towards different substrate proteins via the binding of thalidomide derivatives [16–19]. The structural basis for the binding of thalidomide analogs to CRBN is well understood [20–23] and exploited in the development of degraders targeting CRBN [24–26] as well as applied in a variety of PROTACs for the selective degradation of a multitude of protein targets [27–30]. However, the relevance of CRBN for the antiangiogenic properties of thalidomide derivatives is currently under debate [31].
Recently, we have characterized the CRBN binding mode of α-(2-carboxybenzamido)glutarimide (CBG), a major thalidomide metabolite resulting from partial hydrolysis of the phthalimide ring. This hydrolysis leads to the formation of an amide group, which we found is forming additional interactions with CRBN [32]. Interestingly, this metabolite is similar in structure to a set of tetrafluorinated, benzamide-based thalidomide analogs with promising antiangiogenic properties that we had characterized previously [13]. We therefore sought to test for correlations between CRBN binding, tetrafluorination, and the antiangiogenic properties for a set of compounds that are based on thalidomide and its CBG metabolite.
Materials and Methods
Chemistry
Thalidomide was purchased from Tocris (Bristol, UK). Gu991, Gu993 and Gu1032 were prepared as described [33]. General chemical methods and materials were as described elsewhere [34].
2-(2,6-Dioxo-3-piperidyl)-4,5,6,7-tetrafluoro-isoindoline-1,3-dione (Gu3041) [35].
To a round bottom flask was added tetrafluorophthalic anhydride (1.65 g, 7.5 mmol) and 3-aminopiperidine-2,6-dione hydrochloride (0.82 g, 5.0 mmol). A solution of sodium acetate (0.5 g, 6.0 mmol) in glacial acetic acid (20 mL) was added and the colorless solution was refluxed for 3 h. After cooling, the purple suspension was filtered, and H2O (50 mL) was added to the filtrate. The colorless solid formed was collected, washed with H2O (3 × 5 mL) and petroleum ether (3 × 5 mL) and it was further dried in vacuo to give the title product as a colorless solid. Yield (1.13 g, 68%); mp 238 – 240 °C; 1H NMR (500 MHz, DMSO-d6) δ 2.01 – 2.11 (m, 1H), 2.41 – 2.65 (m, 2H), 2.82 – 2.93 (m, 1H), 5.18 (dd, J = 5.4, 13.0 Hz, 1H), 11.15 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 21.78, 30.95, 49.69, 113.52 (d, 3J (C, F) = 8.3 Hz), 142.72 (m, 1J (C, F) = 266 Hz), 145.14 (m, 1J (C, F) = 264 Hz); 161.98, 169.31, 172.68; LC-MS (ESI) 99% purity, m/z [M + NH4]+ calcd for C13H6F4N2O4, 348.06; found, 348.1; HRMS m/z [M – H]− calcd for C13H6F4N2O4, 329.0191; found, 329.0201.
N-(2,6-Dioxo-3-piperidyl)benzamide (Gu3408).
3-Aminopiperidine-2,6-dione hydrochloride (0.25 g, 1.5 mmol) was suspended in dry CH2Cl2 (15 mL), and it was cooled to 0 °C. Subsequently, Et3N (0.30 g, 0.42 mL, 3.0 mmol) and benzoyl chloride (0.21 g, 172 μL, 1.5 mmol) were added. After stirring the mixture for 18 h at rt, it was quenched by the addition of half-saturated NH4Cl solution (50 mL), and it was extracted with 10% MeOH in EtOAc (2 × 50 mL). The combined organic layers were washed with H2O (50 mL) and brine (50 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (gradient of petroleum ether/EtOAc 1:2 to EtOAc) to give a colourless solid. Yield (0.21 g, 59%); mp 219 – 221 °C (lit. 221 – 223 °C [36]); Rf = 0.50 (EtOAc); 1H NMR (600 MHz, DMSO-d6) δ 1.86 – 2.04 (m, 1H), 2.04 – 2.26 (m, 1H), 2.50 – 2.67 (m, 1H), 2.68 – 2.91 (m, 1H), 4.78 (ddd, J = 5.3, 8.3, 12.2 Hz, 1H), 7.48 (t, J = 7.6 Hz, 2H), 7.52 – 7.62 (m, 1H), 7.79 – 7.94 (m, 2H), 8.74 (d, J = 8.3 Hz, 1H), 10.84 (br s, 1H); 13C NMR (151 MHz, DMSO-d6) δ 24.36, 31.15, 49.69, 127.46, 128.51, 131.63, 134.08, 166.27, 172.37, 173.18; LC-MS (ESI) 99% purity, m/z [M + H]+ calcd for C12H12N2O3, 233.09; found, 232.9.
N-(2,6-Dioxo-3-piperidyl)-2,3,4,5-tetrafluoro-benzamide (Gu3364).
This compound was synthesized by analogy with compound Gu3408, but using 2,3,4,5-tetrafluorobenzoyl chloride. The crude product was purified by column chromatography (petroleum ether/EtOAc 1:2), followed by recrystallization from n-hexanes/EtOAc to give a colorless solid. Yield (0.13 g, 29%); mp 208 – 210 °C; Rf = 0.52 (petroleum ether/EtOAc 1:2); 1H NMR (500 MHz, DMSO-d6) δ 1.97 – 2.06 (m, 1H), 2.03 – 2.14 (m, 1H), 2.50 – 2.58 (m, 1H), 2.72 – 2.83 (m, 1H), 4.71 – 4.79 (m, 1H), 7.55 – 7.64 (m, 1H), 8.82 (d, J = 8.0 Hz, 1H), 10.86 (br s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 24.02, 30.99, 50.05, 111.88 (d, J = 20.6 Hz), 120.05, 137.82 – 148.70 (m), 161.06, 171.61, 173.02; LC-MS (ESI) 99% purity, m/z [M + H]+ calcd for C12H8F4N2O3, 305.05; found, 305.0.
N-(1-Isopropyl-5-methyl-2,4,6-trioxo-hexahydropyrimidin-5-yl)benzamide (Gu3407).
This compound was synthesized by analogy with compound Gu3408, but using 5-amino-1-isopropyl-5-methyl-hexahydropyrimidine-2,4,6-trione hydrochloride (0.35 g) [33]. The crude product was purified by column chromatography (petroleum ether/EtOAc 1:2) to give a colorless solid. Yield (0.38 g, 84%); mp > 250 °C; 1H NMR (600 MHz, DMSO-d6) δ 1.34 (t, J = 6.7 Hz, 6H), 1.63 (s, 3H), 4.78 – 4.88 (m, 1H), 7.49 (t, J = 7.7 Hz, 2H), 7.58 (t, J = 7.4 Hz, 1H), 7.88 – 7.92 (m, 2H), 9.49 (s, 1H), 11.51 (s, 1H); 13C NMR (151 MHz, DMSO-d6) δ 19.10, 19.87, 22.56, 45.89, 60.07, 127.95, 128.53, 132.09, 132.29, 149.89, 166.78, 170.18, 171.03; LC-MS (ESI) 99% purity, m/z [M + H]+ calcd for C15H17N3O4, 304.13; found, 303.8; HRMS m/z [M – H]− calcd for C15H17N3O4, 304.1292; found, 304.1310.
Cereblon binding assay
Affinity measurements were performed using TBD (residues 319–425 of human CRBN) in a competitive assay based on microscale thermophoresis (MST), following the thermophoretic behavior of the reporter ligand BODIPY-uracil [40]. Dilution series of all compounds were generated in DMSO and subsequently diluted 1:100 in water to yield a final constant concentration of 0.5 % (v/v) DMSO. All experiments were performed as described previously [40], using a NanoTemper Monolith NT.115 with a Nano BLUE detector, MO.Control v1.6, MST power medium, temperature 25 °C, excitation power 20 %, on-time 20s. Data were analyzed using PRISM 8, and IC50 values converted to Ki values as described previously [40].
Cell culture
Human Umbilical Vein Endothelial Cells (HUVECs) were purchased from Lonza (Walkersville, MD), cultured in EGM-plus media (Lonza, Walkersville, MD) and only low passage cells (before passage 10) were used. To split, the cells were detached using TryplE Express (ThermoFisher Scientific, Waltham, MA), spun at 1280 rpm, and resuspended in EGM-plus media. Cells were cultured in 5% CO2 and 95% air at 37 °C. HUVECs were authenticated by Lonza and included testing against mycoplasma, bacteria, yeast, and fungi.
Endothelial cell tube formation assay (Lattice)
The in vitro angiogenesis assay kit was purchased from EMD Millipore (Darmstadt, Germany). Briefly, ECMatrix (50μL/well) was plated to a 96-well plate and left to set for 30 minutes. HUVECs were plated atop the gel (35,000 cells/well) and treated with the vehicle control (0.5% DMSO), 30 μM CPS49 (positive control) or test compounds at a range of concentrations. Wells were imaged after 18 hours of incubation. Tubule formation was quantified using ImageJ. Experiments were independently repeated thrice (with n=3–6 for each experiment). Statistical analysis was conducted using GraphPad Prism software (Version 7, GraphPad Software, La Jolla, CA). Statistical significance was assessed using two-tailed Students t-tests and error bars represent mean ± standard error of the mean.
Rat aortic ring assay of angiogenesis (RAR)
The antiangiogenic effects of the test compounds were evaluated in the rat aortic ring angiogenesis model as previously described [13,14]. Briefly, 24-well tissue culture plates were covered with 250 μL of Matrigel (BD Biosciences) and allowed to set for 1 hour at room temperature. Six- to eight-week old male Sprague Dawley rats were euthanized and the descending aortas were dissected. Following excision of fibroadipose tissue, the aortic sections were cut into 1 mm cross-sections, placed on Matrigel-coated wells, and layered with additional Matrigel (250 μl). These were then allowed to set, after which the cross-sectional rings were covered with endothelial cell growth media (EGM-II, Lonza, Walkersville, MD) and incubated under 5 % CO2 at 37 ° C overnight. EGM-II consists of endothelial cell basal medium (EBM-II) and endothelial cell growth factors. The next day, media was replaced with EBM-II containing either the vehicle control (0.5% DMSO), known angiogenesis inhibitors as the positive controls (30 μM carboxyamidotriazole (CAI) or 50 μM TNP-470), or the test compounds at a range of concentrations. Rings were incubated for 4 days and then imaged on day 5 using an EVOS scope. This was independently replicated three times using aortas from 3–4 different rats. The area of angiogenic sprouting, reported in square pixels, was quantified using Adobe Photoshop. Data was presented as percent growth based on the negative control (vehicle), which was normalized to 100% growth. Statistical analysis was conducted using GraphPad Prism software (Version 7, GraphPad Software, La Jolla, CA). Statistical significance was assessed using two-tailed Students t-tests and error bars represent mean ± standard error of the mean.
Results and Discussion
Design of compounds
For a systematic analysis, we employed a set of compounds based on 2-phthalimidinoglutarimide (thalidomide, scaffold A) and 2-benzamidoglutarimide (Gu3408, scaffold B) (Figure 1). The latter represents a simplified form of CBG, which should be able to bind to CRBN via its glutarimide moiety [37]. In the variants Gu991 and Gu3407, which have a modified glutarimide moiety reminiscent of antiangiogenic compounds that we had characterized previously [13], CRBN binding should be abolished [20]. For all four compounds, we designed the counterparts Gu3041, Gu3364, Gu993 and Gu1032, which are tetrafluorinated in their phthalimide or benzoyl portion. This final set of eight compounds should allow us to study potential correlations between tetrafluorination, CRBN binding, and antiangiogenic properties of thalidomide analogs.
Figure 1.

Top left: Markush structures of thalidomide analogs based on scaffolds A and B. Top right: Dose response curves for Thalidomide, Gu3041, Gu3408 and 3364 as assayed by competitive MST experiments to human TBD. Bottom: Affinities to hTBD and IC50 values in the tube formation and RAR assay.
Assessment of cereblon binding
We have recently described an in-vitro CRBN binding assay based on microscale thermophoresis (MST) [40]. This assay follows the behavior of a fluorescent reporter ligand in a thermal gradient, which is largely different for the free reporter and the reporter bound to CRBN. In a titration experiment, the out-competition of the reporter by increasing concentrations of a test compound can be followed, which allows for the determination of affinity constants. Here, we used this assay to determine the affinities of the test compounds to the thalidomide binding domain (TBD) of human CRBN.
We had previously determined the affinity of thalidomide to the TBD, resulting in a Ki of 8.6 μM [40], and now derived a Ki of 63 μM for the CBG-based Gu3408. Interestingly, their tetrafluorinated counterparts Gu3041 and Gu3364 generally displayed improved affinities (6.1 and 30 μM, respectively); the compounds with modified glutarimide moieties (Gu991, Gu993, Gu1032 and Gu3407) did not show any binding independent of fluorination (Figure 1).
Endothelial cell tube formation assay
We investigated the antiangiogenic activity of the thalidomide analogs on endothelial cell tube formation. Human Umbilical Vein Endothelial Cells (HUVECs) can form hollow tube-like structures when cultured upon biological gels, such as ECMatrix. The formation of the tubules can then be used as a simple in vitro measurement of angiogenesis, with the extent of inhibition corresponding to the antiangiogenic effects of the compounds.
We have previously optimized the lattice assay for screening the compound series and selected 10 μM as the initial screening dose to determine which compounds exhibit potent antiangiogenic activity prior to further concentration-range testing. All compounds were tested at this initial 10 μM dose along with the positive control CPS49 and thalidomide. Thalidomide at 100 μM did not demonstrate potent antiangiogenic activity, consistent with previous results which tested the concentration range of 12.5–100 μM and failed to significantly block tube formation [38,39]. The two compounds with tetrafluorinated phthalimide rings, Gu3041 and Gu993, were found to be the most potent and efficacious analogs. Compared with the vehicle control (0.5% DMSO), significant inhibitions of 83% and 88% were observed at 10 μM for Gu3041 and Gu993, respectively, comparable to CPS49 (89%). Gu991, Gu3408, Gu3364, Gu3407, and Gu1032 demonstrate no inhibition of tube formation at this initial screening dose. Compounds Gu991, Gu3408, Gu3364, Gu3407, and Gu1032 were further tested in the concentration range of 10–100 μM and found to also exhibit no significant inhibition of tube formation at any concentrations. Gu3041 and Gu993 were subsequently tested in the concentration range of 0.1–10 μM and reduced tube formation in a concentration-dependent manner. Results and representative tube images are shown in Figure 2.
Figure 2.
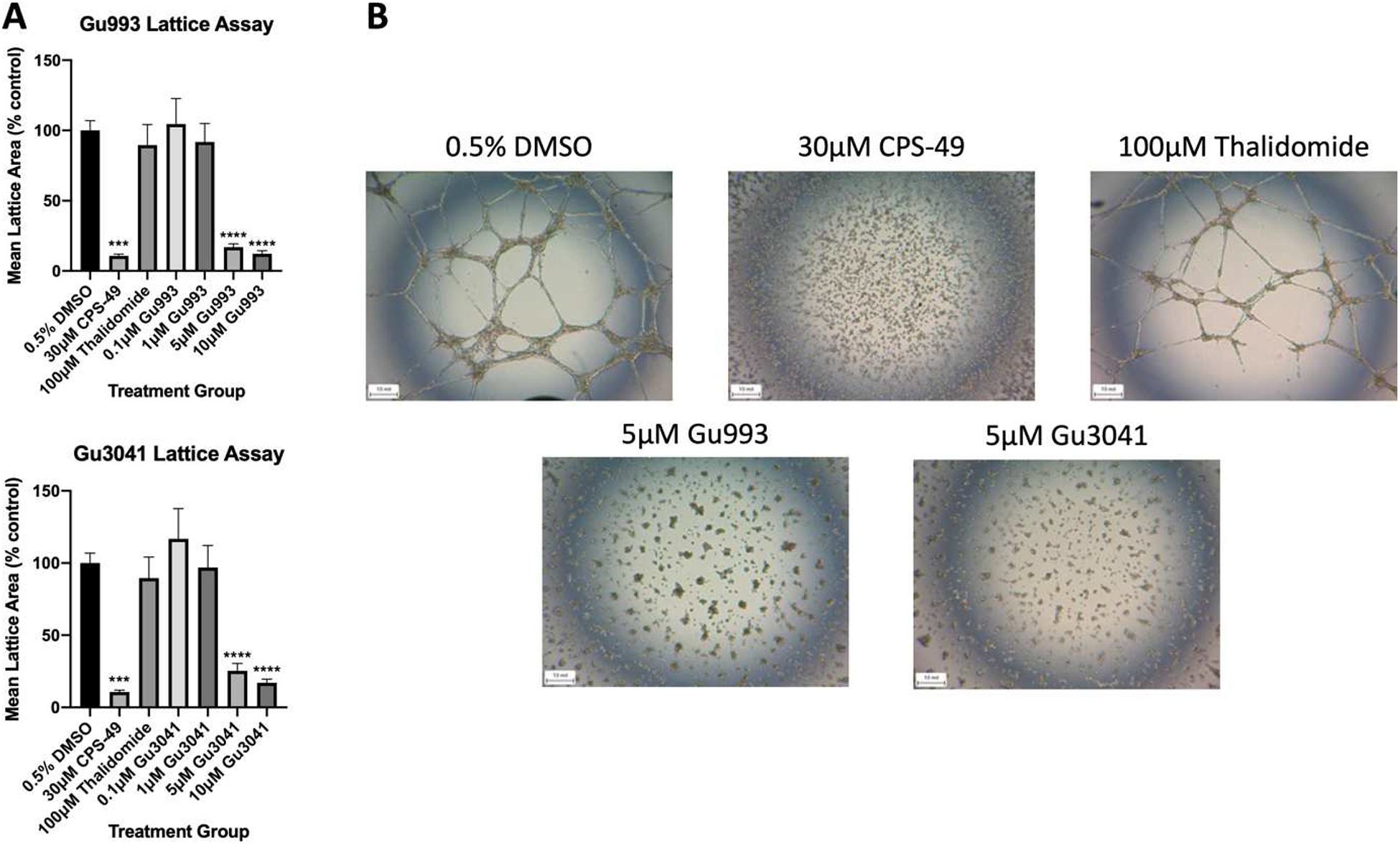
Screening conducted in the endothelial tube formation angiogenesis assay. Vehicle control was 0.5% DMSO, CPS49 (30 mM) was used as a positive control, and Thalidomide (100 mM) was used as a comparator. (A) Graph represents the mean area of lattice formation relative to vehicle control. Gu993 and Gu3041 both inhibited tubule formation in HUVECs in a dose-dependent manner after 18 hours (***p=0.0002, ****p<0.0001). (B) Representative images of the tube formation assay. Results shown are representative of at least 3 independent experiments with at least 3 replicates per experiment.
Rat aortic ring (RAR) assay
All compounds were tested in the RAR model at the initial 50 μM screening dose (previously optimized for this series of compounds) and active compounds that demonstrated >50% inhibition were further tested in the concentration range of 5–100 μM. Microvessel outgrowth was not significantly blocked by thalidomide at 100 μM, consistent with previous results which tested the concentration range of 12.5–100 μM [14]. Positive controls TNP-470 (92%) or CAI (95%) markedly suppressed angiogenesis. Gu991 and Gu3364 demonstrated no significant inhibition of microvessel outgrowth at the initial screening dose, all other compounds reduced microvessel outgrowth in a concentration-dependent manner. Results and representative images of aortic rings are illustrated in Figure 3.
Figure 3.
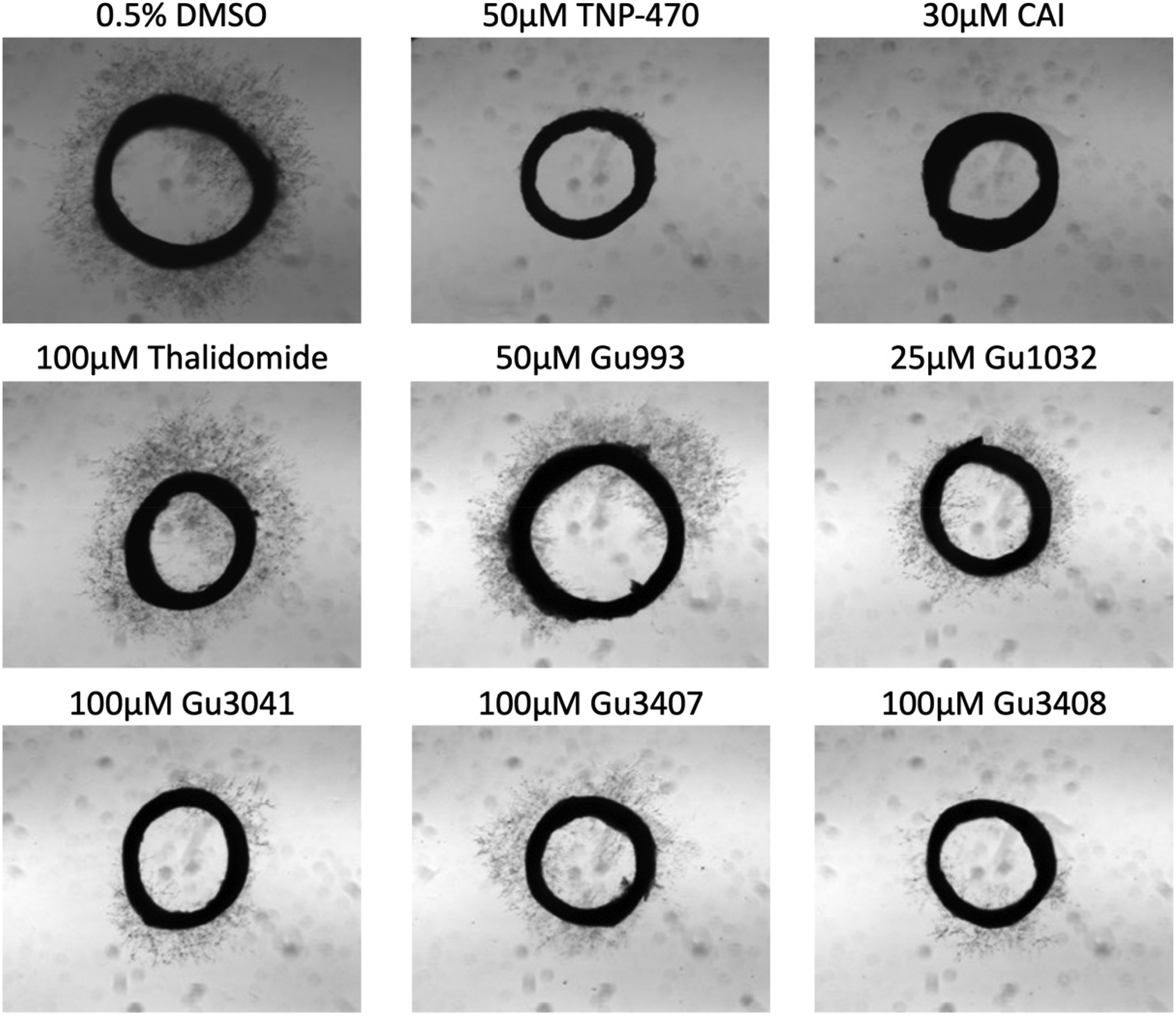
Ex vivo screening was conducted in the rat aorta ring (RAR) angiogenesis assay. Vehicle control was 0.5% DMSO, TNP-470 (50 mM) and CAI (30 mM) were used as positive controls, and Thalidomide (100 mM) was used as a comparator. Representative images of microvessel outgrowth from ring explants taken after 4 days of incubation and treatment with or without compounds.
Conclusions
Our experiments reveal different correlations between structural features and the activity of the compounds. With respect to CRBN binding, as expected, binding was only observed for the four compounds with an unmodified glutarimide moiety. Here, the affinities for the 2-phthalimidinoglutarimide scaffold (A) were generally higher than for the 2-benzamidoglutarimide scaffold (B), while fluorination yielded an increased affinity for both scaffolds.
These structure-activity relationships were not fully paralleled in the antiangiogenesis assays. Generally, the tube formation assay revealed clear correlations. Here, only fluorinated compounds based on the 2-phthalimidinoglutarimide scaffold showed significant efficacy, irrespective of modifications on the glutarimide moiety, while nonfluorinated compounds or compounds based on the 2-benzamidoglutarimide scaffold (B) did not. However, the RAR assay showed more ambiguous results. While this assay generally exhibits a higher complexity due to the involvement of multiple cell types, it reflects most key steps of angiogenesis in not pre-selected explants, closely mimicking a real-life microenvironment during substance testing. Here, tetrafluorination led to increased angiogenesis inhibition for test compounds of both scaffolds, with the exception of Gu3364. Notably, the CBG-inspired and tetrafluorinated compound Gu1032 showed comparable efficacy to the positive control TNP-470.
Taken together, these results describe controversary relationships. On one hand, both for the inhibition of tube formation and CRBN binding, tetrafluorination seems necessary or strongly preferred, and the 2-phthalimidinoglutarimide scaffold (A) appears more advantageous than the 2-benzamidoglutarimide scaffold (B). However, the tube formation assay seemed to be insensitive to modifications of the glutarimide moiety that abolish CRBN binding. Here, the efficacy of the CRBN binder Gu3041 was almost identical to that of the non-binder Gu993. These findings highlight that only a subset of immunomodulatory drugs possess antiangiogenic properties and suggest that these properties are not mediated via CRBN. While it seems conceivable that CRBN might play an auxiliary role in this process, the main target for the antiangiogenic properties of thalidomide analogs remains to be identified.
Highlights.
fluorination of thalidomide analogs enhances binding to cereblon
fluorination increases antiangiogenic properties of thalidomide analogs
affinity to cereblon does not correlate with antiangiogenic properties
Acknowledgements
This work was supported by institutional funds of the Max Planck Society and by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (ZIA SC006538).
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Bartlett JB, Dredge K, Dalgleish AG, The evolution of thalidomide and its IMiD derivatives as anticancer agents, Nat Rev Cancer 4 (2004) 314–322. 10.1038/nrc1323. [DOI] [PubMed] [Google Scholar]
- [2].Li SR, Gill N, Lentzsch S, Recent advances of IMiDs in cancer therapy, Curr Opin Oncol 22 (2010) 579–585. 10.1097/CCO.0b013e32833d752c. [DOI] [PubMed] [Google Scholar]
- [3].Price DK, Ando Y, Kruger EA, Weiss M, Figg WD, 5’-OH-thalidomide, a metabolite of thalidomide, inhibits angiogenesis, Ther Drug Monit 24 (2002) 104–110. 10.1097/00007691-200202000-00017. [DOI] [PubMed] [Google Scholar]
- [4].Gutschow M, Hecker T, Thiele A, Hauschildt S, Eger K, Aza analogues of thalidomide: Synthesis and evaluation as inhibitors of tumor necrosis factor-alpha production in vitro, Bioorgan Med Chem 9 (2001) 1059–1065. Doi 10.1016/S0968-0896(00)00323-0. [DOI] [PubMed] [Google Scholar]
- [5].Luzzio FA, Mayorov AV, Ng SS, Kruger EA, Figg WD, Thalidomide metabolites and analogues. 3. Synthesis and antiangiogenic activity of the teratogenic and TNFalpha-modulatory thalidomide analogue 2-(2,6-dioxopiperidine-3-yl)phthalimidine, J Med Chem 46 (2003) 3793–3799. 10.1021/jm020079d. [DOI] [PubMed] [Google Scholar]
- [6].Tokunaga E, Akiyama H, Soloshonok VA, Inoue Y, Hara H, Shibata N, Biological evaluation of both enantiomers of fluoro-thalidomide using human myeloma cell line H929 and others, Plos One 12 (2017). ARTN e0182152 10.1371/journal.pone.0182152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Iacopetta D, Carocci A, Sinicropi MS, Catalano A, Lentini G, Ceramella J, Curcio R, Caroleo MC, Old Drug Scaffold, New Activity: Thalidomide-Correlated Compounds Exert Different Effects on Breast Cancer Cell Growth and Progression, Chemmedchem 12 (2017) 381–389. 10.1002/cmdc.201600629. [DOI] [PubMed] [Google Scholar]
- [8].Cieslak M, Kazmierczak-Baranska J, Krolewska-Golinska K, Napiorkowska M, Stukan I, Wojda U, Nawrot B, New Thalidomide-Resembling Dicarboximides Target ABC50 Protein and Show Antileukemic and Immunomodulatory Activities, Biomolecules 9 (2019). ARTN 446 10.3390/biom9090446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xiao DH, Wang YJ, Wang HL, Zhou YB, Li J, Lu W, Jin JY, Design and synthesis of new lenalidomide analogs via Suzuki cross-coupling reaction, Arch Pharm 353 (2020). ARTN e1900376 10.1002/ardp.201900376. [DOI] [PubMed] [Google Scholar]
- [10].Capitosti SM, Hansen TP, Brown ML, Thalidomide analogues demonstrate dual inhibition of both angiogenesis and prostate cancer, Bioorgan Med Chem 12 (2004) 327–336. 10.1016/j.bmc.2003.11.007. [DOI] [PubMed] [Google Scholar]
- [11].Lepper ER, Ng SSW, Gutschow M, Weiss M, Hauschildt S, Hecker TK, Luzzio FA, Eger K, Figg WD, Comparative molecular field analysis and comparative molecular similarity indices analysis of thalidomide analogues as angiogenesis inhibitors, Journal of Medicinal Chemistry 47 (2004) 2219–2227. 10.1021/jm0304820. [DOI] [PubMed] [Google Scholar]
- [12].Steinebach C, Ambrozak A, Dosa S, Beedie SL, Strope JD, Schnakenburg G, Figg WD, Gutschow M, Synthesis, Structural Characterization, and Antiangiogenic Activity of Polyfluorinated Benzamides, Chemmedchem 13 (2018) 2080–2089. 10.1002/cmdc.201800263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Beedie SL, Peer CJ, Pisle S, Gardner ER, Mahony C, Barnett S, Ambrozak A, Gutschow M, Chau CH, Vargesson N, Figg WD, Anticancer Properties of a Novel Class of Tetrafluorinated Thalidomide Analogues, Mol Cancer Ther 14 (2015) 2228–2237. 10.1158/1535-7163.MCT-15-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ng SS, Gutschow M, Weiss M, Hauschildt S, Teubert U, Hecker TK, Luzzio FA, Kruger EA, Eger K, Figg WD, Antiangiogenic activity of N-substituted and tetrafluorinated thalidomide analogues, Cancer Res 63 (2003) 3189–3194. [PubMed] [Google Scholar]
- [15].Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H, Identification of a primary target of thalidomide teratogenicity, Science 327 (2010) 1345–1350. 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- [16].Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, Tichkule RB, Schebesta M, Forrester WC, Schirle M, Hassiepen U, Ottl J, Hild M, Beckwith RE, Harper JW, Jenkins JL, Thoma NH, Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide, Nature 512 (2014) 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, Svinkina T, Schneider RK, McConkey M, Jaras M, Griffiths E, Wetzler M, Bullinger L, Cathers BE, Carr SA, Chopra R, Ebert BL, Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS, Nature 523 (2015) 183–188. 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, Ciarlo C, Hartman E, Munshi N, Schenone M, Schreiber SL, Carr SA, Ebert BL, Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells, Science 343 (2014) 301–305. 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Petzold G, Fischer ES, Thoma NH, Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase, Nature 532 (2016) 127–130. 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- [20].Boichenko I, Bar K, Deiss S, Heim C, Albrecht R, Lupas AN, Hernandez Alvarez B, Hartmann MD, Chemical Ligand Space of Cereblon, ACS Omega 3 (2018) 11163–11171. 10.1021/acsomega.8b00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chamberlain PP, Lopez-Girona A, Miller K, Carmel G, Pagarigan B, Chie-Leon B, Rychak E, Corral LG, Ren YJ, Wang M, Riley M, Delker SL, Ito T, Ando H, Mori T, Hirano Y, Handa H, Hakoshima T, Daniel TO, Cathers BE, Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs, Nat Struct Mol Biol 21 (2014) 803–809. 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- [22].Hartmann MD, Boichenko I, Coles M, Lupas AN, Hernandez Alvarez B, Structural dynamics of the cereblon ligand binding domain, Plos One 10 (2015) e0128342 10.1371/journal.pone.0128342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hartmann MD, Boichenko I, Coles M, Zanini F, Lupas AN, Hernandez Alvarez B, Thalidomide mimics uridine binding to an aromatic cage in cereblon, J Struct Biol 188 (2014) 225–232. 10.1016/j.jsb.2014.10.010. [DOI] [PubMed] [Google Scholar]
- [24].Steinebach C, Lindner S, Udeshi ND, Mani DC, Kehm H, Kopff S, Carr SA, Gutschow M, Kronke J, Homo-PROTACs for the Chemical Knockdown of Cereblon, ACS Chem Biol 13 (2018) 2771–2782. 10.1021/acschembio.8b00693. [DOI] [PubMed] [Google Scholar]
- [25].Girardini M, Maniaci C, Hughes SJ, Testa A, Ciulli A, Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs, Bioorg Med Chem 27 (2019) 2466–2479. 10.1016/j.bmc.2019.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Steinebach C, Kehm H, Lindner S, Vu LP, Kopff S, Marmol AL, Weiler C, Wagner KG, Reichenzeller M, Kronke J, Gutschow M, PROTAC-mediated crosstalk between E3 ligases, Chem Commun 55 (2019) 1821–1824. 10.1039/c8cc09541h. [DOI] [PubMed] [Google Scholar]
- [27].Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J, Crews CM, Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL, Angew Chem Int Ed Engl 55 (2016) 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chopra R, Sadok A, Collins I, A critical evaluation of the approaches to targeted protein degradation for drug discovery, Drug Discov Today Technol 31 (2019) 5–13. 10.1016/j.ddtec.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nalawansha DA, Crews CM, PROTACs: An Emerging Therapeutic Modality in Precision Medicine, Cell Chem Biol 27 (2020) 998–1014. 10.1016/j.chembiol.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Maniaci C, Ciulli A, Bifunctional chemical probes inducing protein-protein interactions, Curr Opin Chem Biol 52 (2019) 145–156. 10.1016/j.cbpa.2019.07.003. [DOI] [PubMed] [Google Scholar]
- [31].Beedie SL, Huang PA, Harris EM, Strope JD, Mahony C, Chau CH, Vargesson N, Figg WD, Role of cereblon in angiogenesis and in mediating the antiangiogenic activity of immunomodulatory drugs, FASEB J (2020). 10.1096/fj.201903060RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Heim C, Pliatsika D, Mousavizadeh F, Bar K, Hernandez Alvarez B, Giannis A, Hartmann MD, De-Novo Design of Cereblon (CRBN) Effectors Guided by Natural Hydrolysis Products of Thalidomide Derivatives, J Med Chem 62 (2019) 6615–6629. 10.1021/acs.jmedchem.9b00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ambrozak A, Steinebach C, Gardner ER, Beedie SL, Schnakenburg G, Figg WD, Gutschow M, Synthesis and Antiangiogenic Properties of Tetrafluorophthalimido and Tetrafluorobenzamido Barbituric Acids, Chemmedchem 11 (2016) 2621–2629. 10.1002/cmdc.201600496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Steinebach C, Ng YLD, Sosic I, Lee CS, Chen SR, Lindner S, Vu LP, Bricelj A, Haschemi R, Monschke M, Steinwarz E, Wagner KG, Bendas G, Luo J, Gutschow M, Kronke J, Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders, Chem Sci 11 (2020) 3474–3486. 10.1039/d0sc00167h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Burslem GM, Ottis P, Jaime-Figueroa S, Morgan A, Cromm PM, Toure M, Crews CM, Efficient Synthesis of Immunomodulatory Drug Analogues Enables Exploration of Structure-Degradation Relationships, Chemmedchem 13 (2018) 1508–1512. 10.1002/cmdc.201800271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Abou-Zeid L, El-Mowafy AM, El-Kerdawy MM, Hamza H, Abdel-Hamid ME, Synthesis, spectroscopic characterization, stability assessment and DNA-binding of new 2,6-piperidinedione derivatives, Farmaco 56 (2001) 763–770. 10.1016/s0014-827x(01)01131-4. [DOI] [PubMed] [Google Scholar]
- [37].Boichenko I, Deiss S, Bar K, Hartmann MD, Hernandez Alvarez B, A FRET-Based Assay for the Identification and Characterization of Cereblon Ligands, J Med Chem 59 (2016) 770–774. 10.1021/acs.jmedchem.5b01735. [DOI] [PubMed] [Google Scholar]
- [38].El-Aarag BY, Kasai T, Zahran MA, Zakhary NI, Shigehiro T, Sekhar SC, Agwa HS, Mizutani A, Murakami H, Kakuta H, Seno M, In vitro anti-proliferative and anti-angiogenic activities of thalidomide dithiocarbamate analogs, Int Immunopharmacol 21 (2014) 283–292. 10.1016/j.intimp.2014.05.007. [DOI] [PubMed] [Google Scholar]
- [39].Lu L, Payvandi F, Wu L, Zhang LH, Hariri RJ, Man HW, Chen RS, Muller GW, Hughes CC, Stirling DI, Schafer PH, Bartlett JB, The anti-cancer drug lenalidomide inhibits angiogenesis and metastasis via multiple inhibitory effects on endothelial cell function in normoxic and hypoxic conditions, Microvasc Res 77 (2009) 78–86. 10.1016/j.mvr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- [40].Maiwald S, Heim C, Hernandez Alvarez B, Hartmann MD Sweet and blind spots in E3 ligase ligand space revealed by a thermophoresis-based assay ACS Med. Chem. Lett (2020), 10.1021/acsmedchemlett.0c00440 In press [DOI] [PMC free article] [PubMed] [Google Scholar]