

Key Points
Question
Are there differential associations between genetic liability to smoking and atherosclerotic cardiovascular disease (ASCVD) outcomes (coronary artery disease, peripheral artery disease, and ischemic stroke)?
Findings
In this mendelian randomization study including summary data for more than 1 million individuals, genetic liability to smoking was associated with increased risk of ASCVD, with the largest association with peripheral artery disease, independent from other cardiovascular risk factors.
Meaning
Findings of this study indicate that genetic liability to smoking has a strong, independent effect on ASCVD but is most strongly associated with peripheral artery disease; further studies of the differential effects of other ASCVD risk factors may improve risk stratification and treatment.
Abstract
Importance
Smoking is associated with atherosclerotic cardiovascular disease, but the relative contribution to each subtype (coronary artery disease [CAD], peripheral artery disease [PAD], and large-artery stroke) remains less well understood.
Objective
To determine the association between genetic liability to smoking and risk of CAD, PAD, and large-artery stroke.
Design, Setting, and Participants
Mendelian randomization study using summary statistics from genome-wide associations of smoking (UK Biobank; up to 462 690 individuals), CAD (Coronary Artery Disease Genome Wide Replication and Meta-analysis plus the Coronary Artery Disease Genetics Consortium; up to 60 801 cases, 123 504 controls), PAD (VA Million Veteran Program; up to 24 009 cases, 150 983 controls), and large-artery stroke (MEGASTROKE; up to 4373 cases, 406 111 controls). This study was conducted using summary statistic data from large, previously described cohorts. Review of those publications does not reveal the total recruitment dates for those cohorts. Data analyses were conducted from August 2019 to June 2020.
Exposures
Genetic liability to smoking (as proxied by genetic variants associated with lifetime smoking index).
Main Outcomes and Measures
Risk (odds ratios [ORs]) of CAD, PAD, and large-artery stroke.
Results
Genetic liability to smoking was associated with increased risk of PAD (OR, 2.13; 95% CI, 1.78-2.56; P = 3.6 × 10−16), CAD (OR, 1.48; 95% CI, 1.25-1.75; P = 4.4 × 10−6), and stroke (OR, 1.40; 95% CI, 1.02-1.92; P = .04). Genetic liability to smoking was associated with greater risk of PAD than risk of large-artery stroke (ratio of ORs, 1.52; 95% CI, 1.05-2.19; P = .02) or CAD (ratio of ORs, 1.44; 95% CI, 1.12-1.84; P = .004). The association between genetic liability to smoking and atherosclerotic cardiovascular diseases remained independent from the effects of smoking on traditional cardiovascular risk factors.
Conclusions and Relevance
In this mendelian randomization analysis of data from large studies of atherosclerotic cardiovascular diseases, genetic liability to smoking was a strong risk factor for CAD, PAD, and stroke, although the estimated association was strongest between smoking and PAD. The association between smoking and atherosclerotic cardiovascular disease was independent of traditional cardiovascular risk factors.
This mendelian randomization study evaluates summary statistics from genome-wide associations involving more than 1 million individuals to assess whether there are differential associations between genetic liability to smoking and risk of coronary artery disease, peripheral artery disease, and large-artery stroke.
Introduction
Atherosclerotic cardiovascular disease (ASCVD) can affect numerous vascular beds throughout the body, with clinical manifestations including coronary artery disease (CAD), stroke, and peripheral artery disease (PAD). Smoking tobacco is consistently among the leading risk factors for ASCVD; however, the relative contribution of smoking to the individual ASCVD outcomes remains less well studied. Observational studies have examined these ASCVD outcomes together, with a recent study by Ding et al finding the strongest association between smoking and incident PAD compared with CAD or stroke.1,2,3,4 Observational study designs may be limited, however, by modest overall sample size, measurement error, and risk of residual confounding.5
A number of studies during the last several decades have identified detrimental effects of smoking on traditional cardiovascular risk factors, including blood pressure, lipids, and diabetes.6,7 Smoking also has independent effects on inflammation, endothelial function, and platelet aggregation.6 Despite the clear observational links between smoking and atherosclerosis, whether the effect of smoking on ASCVD is primarily mediated through correlated alterations of traditional cardiovascular risk factors, or operates via independent mechanisms is less clear. Because the detrimental effects of smoking may persist for decades,4 clarifying the basis of the smoking-atherosclerosis relationship could enable more targeted risk-reduction strategies among both current and former smokers and identify novel treatment strategies for those at highest risk of ASCVD.
Recently, large genome-wide association studies (GWASs) of smoking, coronary artery disease, stroke, and peripheral artery disease have identified genetic loci associated with each of these conditions.8,9,10,11 The mendelian randomization (MR) framework leverages the natural randomization of genetic variation at conception to mitigate risks of confounding that limit other observational methods. Genetic variants are randomly allocated across the population at meiosis and conception, mimicking randomization in a clinical trial. Under certain assumptions, the MR framework mitigates risks from environmental confounding and reverse causality that may affect other observational methods.12 This approach may allow for more precise quantification of potential differences between exposure-outcome pairs.12 The method has further been extended to consider exposures jointly, a form of mediation analysis that enables the estimation of the direct effect of each exposure on an outcome of interest.13 In the present analysis, we studied the association between genetic liability to smoking (defined by genetic variants associated with measures of smoking) and ASCVD and cardiometabolic outcomes.
Here, leveraging population-scale human genetics data from GWASs, we used the MR framework with genetic variants as instrumental variables to (1) estimate the total effect sizes for associations between smoking and risk of PAD, CAD, and stroke, the primary manifestations of ASCVD; (2) validate the association between smoking and traditional cardiovascular and inflammatory risk factors for ASCVD; and (3) assess the extent to which traditional cardiovascular and inflammatory risk factors affect the relationship between smoking and ASCVD outcomes.
Methods
Smoking Genetic Instrument Selection
Genetic variants were used as instrumental variables for 2 different measures of smoking. These genetic exposures used as proxies for smoking are referred to herein as “genetic liability to smoking.” The 2 measures of smoking used throughout the study are lifetime smoking index11 and smoking initiation.14 The primary measure of smoking was lifetime smoking index, a previously validated continuous measure that accounts for self-reported smoking status, age at initiation, age at cessation, number of cigarettes smoked per day, and a simulated half-life constant that captures the decreasing effect of smoking on health outcomes following a given exposure (Table).11 A genetic instrument for lifetime smoking index was constructed from summary statistics of a GWAS of UK Biobank participants (UK Biobank; N = 462 690) using independent (r2 = 0.001; distance, 10 000 kilobases; European-ancestry participants of the 1000 genomes project) genome-wide significant variants (P < 5 × 10−8) as the exposure, as previously described.11 Each SD increase in the lifetime smoking index instrument corresponds to an individual smoking 20 cigarettes daily for 15 years and stopping 17 years ago, or smoking 60 cigarettes daily for 13 years and stopping 22 years ago. The smoking index instrument was previously validated using an independent set of participants and with positive-control outcomes of coronary artery disease and lung cancer.11 To further validate the instrument, we performed 2-sample MR using lifetime smoking index as the exposure, and smoking initiation, age of smoking initiation, smoking cessation, and cigarettes per day,14 as reported by GWAS and Sequencing Consortium of Alcohol and Nicotine, as outcomes. To evaluate the strength of the smoking index instrument, we calculated the F statistic.15 In sensitivity analyses, genetic liability to smoking was assessed using smoking initiation as the exposure, rather than lifetime smoking index. This study is reported according to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline for observational studies.16 This study used only publicly available, deidentified summary statistics from previously published works, making it exempt from institutional review board review according to the University of Pennsylvania regulations.
Table. Overview of Genetic Data Setsa.
| Trait | Cohort | Year | PMID | No. | ||
|---|---|---|---|---|---|---|
| Sample size | Cases | Controls | ||||
| Smoking (lifetime smoking index) | UKB | 2019 | 31689377 | 462 690 | ||
| Coronary artery disease | CARDIoGRAMplusC4D | 2015 | 26343387 | 184 305 | 60 801 | 123 504 |
| Peripheral artery disease | MVP | 2019 | 31285632 | 174 992 | 24 009 | 150 983 |
| Large-artery stroke | MEGASTROKE | 2018 | 29531354 | 150 765 | 4373 | 406 111 |
| Overweight | GIANT | 2013 | 23563607 | 158 855 | 93 015 | 65 840 |
| Diabetes | DIAGRAM+GERA+UKB | 2018 | 30054458 | 655 666 | 61 714 | 1178 |
| Hyperlipidemia | UKB | 2018 | 29846171 | 462 933 | 56 753 | 406 180 |
| Chronic kidney disease | CKDGen | 2015 | 26831199 | 117 165 | 12 385 | 104 780 |
| Hypertension | UKB | 2018 | 29846171 | 462 933 | 2095 | 460 838 |
| Body mass index | GIANT | 2015 | 25673413 | 339 224 | NA | NA |
| C-reactive protein | INTERVAL | 2018 | 29875488 | 3301 | NA | NA |
| Fasting glucose | MAGIC | 2012 | 22581228 | 58 074 | NA | NA |
| Fasting insulin | MAGIC | 2012 | 22581228 | 51 750 | NA | NA |
| HbA1C | MAGIC | 2010 | 20858683 | 46 368 | NA | NA |
| HDL cholesterol | GLGC | 2013 | 24097068 | 187 167 | NA | NA |
| Interleukin 6 receptor | INTERVAL | 2018 | 28369058 | 3394 | NA | NA |
| Interleukin 1β | INTERVAL | 2018 | 29875488 | 3301 | NA | NA |
| Interleukin 6 | INTERVAL | 2018 | 29875488 | 3301 | NA | NA |
| LDL cholesterol | GLGC | 2013 | 24097068 | 173 082 | NA | NA |
| Serum creatinine (eGFRcrea) | CKDGen | 2015 | 26831199 | 133 814 | NA | NA |
| Systolic blood pressure | UKB | 2018 | 29846171 | 436 419 | NA | NA |
| Total cholesterol | GLGC | 2013 | 24097068 | 187 365 | NA | NA |
| Triglycerides | GLGC | 2013 | 24097068 | 177 861 | NA | NA |
| Waist circumference | GIANT | 2015 | 25673412 | 232 101 | NA | NA |
| Waist to hip ratio | GIANT | 2015 | 25673412 | 224 459 | NA | NA |
Abbreviations: CARDIoGRAMplusC4D, Coronary Artery Disease Genome Wide Replication and Meta-analysis plus the Coronary Artery Disease Genetics Consortium; CKDGen, Chronic Kidney Disease Genetics Consortium; DIAGRAM, Diabetes Genetics Replication and Meta-analysis consortium; eGFRcrea, estimated glomerular filtration rate (creatinine); GERA, Genetic Epidemiology Research on Aging; GIANT, Genetic Investigation of Anthropometric Traits consortium; GLGC, Global Lipid Genetics Consortium; HbA1C, hemoglobin A1C; HDL, high-density lipoprotein; LDL, low-density lipoprotein; MAGIC, Meta-Analyses of Glucose and Insulin-Related Traits Consortium; MVP, Million Veteran Program; NA, not applicable; PMID, PubMed identification number; UKB, UK Biobank.
Overview of genetic data sets used in the mendelian randomization analyses. The number of cases and controls is reported for case-control studies, with total sample size reported for all studies.
ASCVD Outcome Selection
The ASCVD outcomes were obtained from large GWASs of CAD, PAD, and large-artery stroke (Table). The CAD effects were obtained from the Coronary Artery Disease Genome Wide Replication and Meta-analysis plus the Coronary Artery Disease Genetics Consortium 1000 Genomes GWAS, which included 60 801 CAD cases and 123 504 controls of primarily European ancestry (77%) across 48 studies, including a combination of incident and prevalent disease.8 The PAD effects were obtained from the Million Veterans Program GWAS, which included 24 009 prevalent PAD cases and 150 983 controls of European ancestry enrolled at 63 Veterans Affairs (VA) Medical Centers across the United States.9 The large-artery stroke effects were obtained from the MEGASTROKE consortium GWAS, which included 4373 cases and 406 111 controls of European ancestry enrolled across 10 studies, including a combination of incident and prevalent cases.10
Cardiometabolic Risk Factor Selection
The effects of genetic variants on cardiometabolic risk factors were obtained from publicly available summary statistics from GWASs of continuous traits (total cholesterol level, low-density lipoprotein cholesterol level, high-density lipoprotein cholesterol level, level of triglycerides, body mass index, waist to hip ratio, fasting glucose level, fasting insulin level, systolic blood pressure, estimated glomerular filtration rate, circulating C-reactive protein level, circulating interleukin 1B level, circulating interleukin 6 level, and circulating interleukin 6R level), and binary traits (type 2 diabetes, hypertension, hyperlipidemia, chronic kidney disease, and overweight) identified using the MR-Base platform (Table).17,18,19,20,21,22,23,24 These risk factors were selected as common markers of cardiometabolic risk, inclusion in risk calculators, and genetic, observational, and randomized clinical trial evidence of association with coronary artery disease.25,26,27,28,29
Mendelian Randomization
In the primary analysis, the total effect of lifetime smoking index on ASCVD outcomes (CAD, PAD, and stroke) was estimated using random-effects inverse-variance–weighted MR within the TwoSampleMR package in R (Figure 1).5,30 In sensitivity analyses, fixed-effects inverse-variance–weighted, maximum likelihood, weighted-median, penalized weighted-median, and MR pleiotropy residual sum and outlier methods were performed to account for potential violations of the instrumental variable assumptions, presence of horizontal pleiotropy, heterogeneity, and error in the instrument-exposure associations.5,31,32 The latter method (1) tests for the presence of horizontal pleiotropy, (2) removes pleiotropic genetic variants, and (3) tests for differences in estimates before and after outlier removal.33 Diagnostic leave-one-out, single single-nucleotide variant, and funnel-plot analyses were performed to visually assess for outliers and bias. The Egger bias intercept test was used to quantitatively detect evidence of horizontal pleiotropy. In sensitivity analysis, a genetic instrument for smoking initiation was used as the exposure.14 Because 2-sample MR of binary exposures provides effect estimates per 1-unit change in the exposure, the results of the effect of the smoking initiation exposure on ASCVD outcomes are expressed as odds of the outcome per 2.72-fold (1 log odds unit) increase in the odds of ever smoking.34 Differences in the effect of smoking on ASCVD outcomes in each vascular bed were estimated using the ratio of the odds ratios (ORs), based on a null hypothesis that the ORs for the associations between smoking and each ASCVD outcome are not different.35
Figure 1. Mendelian Randomization Analysis Overview.

Overview of mendelian randomization analyses and major assumptions. Solid lines represent the direct pathway in which genetic variants serve as instruments for a risk factor of interest, and the effect on a disease outcome is measured. Dashed red lines represent pathways that potentially violate mendelian randomization assumptions. For atherosclerotic cardiovascular disease outcomes, the number of cases and controls are listed. For cardiometabolic outcomes, the total sample size is listed. See Table for additional cohort information. HDL indicates high-density lipoprotein; LDL, low-density lipoprotein; and SNV, single-nucleotide variant.
The effect of lifetime smoking on cardiometabolic risk factors was estimated using random-effects inverse-variance–weighted 2-sample MR (Figure 1). Because the genetic exposure for lifetime smoking index was derived from UK Biobank participants, and MR estimates derived from studies with a high proportion of overlapping samples may be biased, studies of cardiometabolic outcomes that included non–UK Biobank participants were preferred when available.36 The genetic exposure for smoking initiation, which included both UK Biobank participants and participants from several other studies, was used in sensitivity analyses to further minimize bias from participant overlap. The MR Steiger test of directionality was performed to validate direction of association between smoking (as the exposure) and cardiometabolic risk factors (as the outcomes).37
To determine whether any effect of smoking on ASCVD may be attenuated by effects of smoking on traditional cardiovascular risk factors, multivariable MR was performed. This method can be used to jointly estimate the direct effect of multiple exposure on an outcome, and account for potential violations of MR assumptions (Figure 1).38,39 Independent (r2 = 0.001; distance, 10 000 kilobases) genome-wide significant (P < 5 × 10−8) variants associated with traditional cardiovascular and inflammatory risk factors (low-density lipoprotein cholesterol level, high-density lipoprotein cholesterol level, level of triglycerides, body mass index, type 2 diabetes, systolic blood pressure, and circulating interleukin 6 levels) were identified using the MR-Base platform. The direct effect of lifetime smoking index was then estimated in models accounting for each traditional risk factor alone and in a model considering all risk factors simultaneously.
Statistical Analysis
For the primary analysis of smoking on ASCVD outcomes, the statistical significance threshold was set at a 2-sided P < .05. The relative associations between lifetime smoking index and the primary ASCVD outcomes were compared using the ratio of the ORs.35 For the secondary analysis of smoking on cardiometabolic risk factors, correction for multiple comparisons was made using the Bonferroni method (P < .05 / 21 = .002). Cardiometabolic traits with nominal associations (.002 ≤P < .05) were considered suggestive. In the multivariable analysis considering the joint effects of smoking and risk factors on ASCVD outcomes, the statistical significance threshold was set at a 2-sided P < .05. All analyses were performed using R version 3.6.2 (R Foundation for Statistical Computing). Review of the publications used in the present analysis does not reveal the total recruitment dates for the included cohorts. Data analyses were conducted from August 2019 to June 2020.
Results
Selection of Genetic Variants as Proxies for Smoking
Of the 126 independent single-nucleotide variants associated with lifetime smoking index, 116 were available in the summary statistics for stroke, 107 for CAD, and 105 for PAD (eTable 1 in the Supplement). For the lifetime smoking index genetic instrument, the F statistic was greater than 10 (range, 25-163; mean, 41), suggesting low risk of weak-instrument bias. To validate the genetic instrument for lifetime smoking index, we performed MR using smoking traits from the GWAS and Sequencing Consortium of Alcohol and Nicotine consortium as outcomes. The lifetime smoking index was significantly associated with increased smoking initiation (OR, 1.66; 95% CI, 1.59-1.73; P < .001), smoking cessation (OR, 1.43; 95% CI, 1.37-1.5; P < .001), increased amount of smoking (cigarettes per day) (β = 0.514; 95% CI, 0.4-0.63; P < .001), and decreased age of smoking initiation (β = −0.38; 95% CI, −0.44 to −0.32) (eFigure 1 in the Supplement).
Association of Smoking With Risk of ASCVD Outcomes
In inverse-variance–weighted MR analysis, each 1 SD increase in genetic liability to lifetime smoking index was associated with increased risk of PAD (OR, 2.13; 95% CI, 1.78-2.56; P = 3.6 × 10−16), CAD (OR, 1.48; 95% CI, 1.25-1.75; P = 4.4 × 10−6), and stroke (OR, 1.40; 95% CI, 1.02-1.92; P = .04) (Figure 2). The Egger bias intercept test identified horizontal pleiotropy for the smoking-CAD pathway (intercept, 0.01; P = .046) (eTable 2 in the Supplement) although the findings remained robust in sensitivity analyses using MR methods that account for the possibility of horizontal pleiotropy (Figure 2B; eFigures 2, 3, 4, and 5 in the Supplement), and when using an alternative genetic instrument for smoking (smoking initiation). Genetic liability to smoking initiation was associated with increased risk of PAD (OR, 2.36; 95% CI, 1.81-3.09; P < .001), CAD (OR, 1.76; 95% CI, 1.39-2.23; P < .001), and stroke (OR, 2.02; 95% CI, 1.33-3.06; P < .001) (eFigure 6 in the Supplement).
Figure 2. Total Effect Sizes for Associations Between Smoking and Risk of Peripheral Artery Disease (PAD), Coronary Artery Disease (CAD), and Stroke.
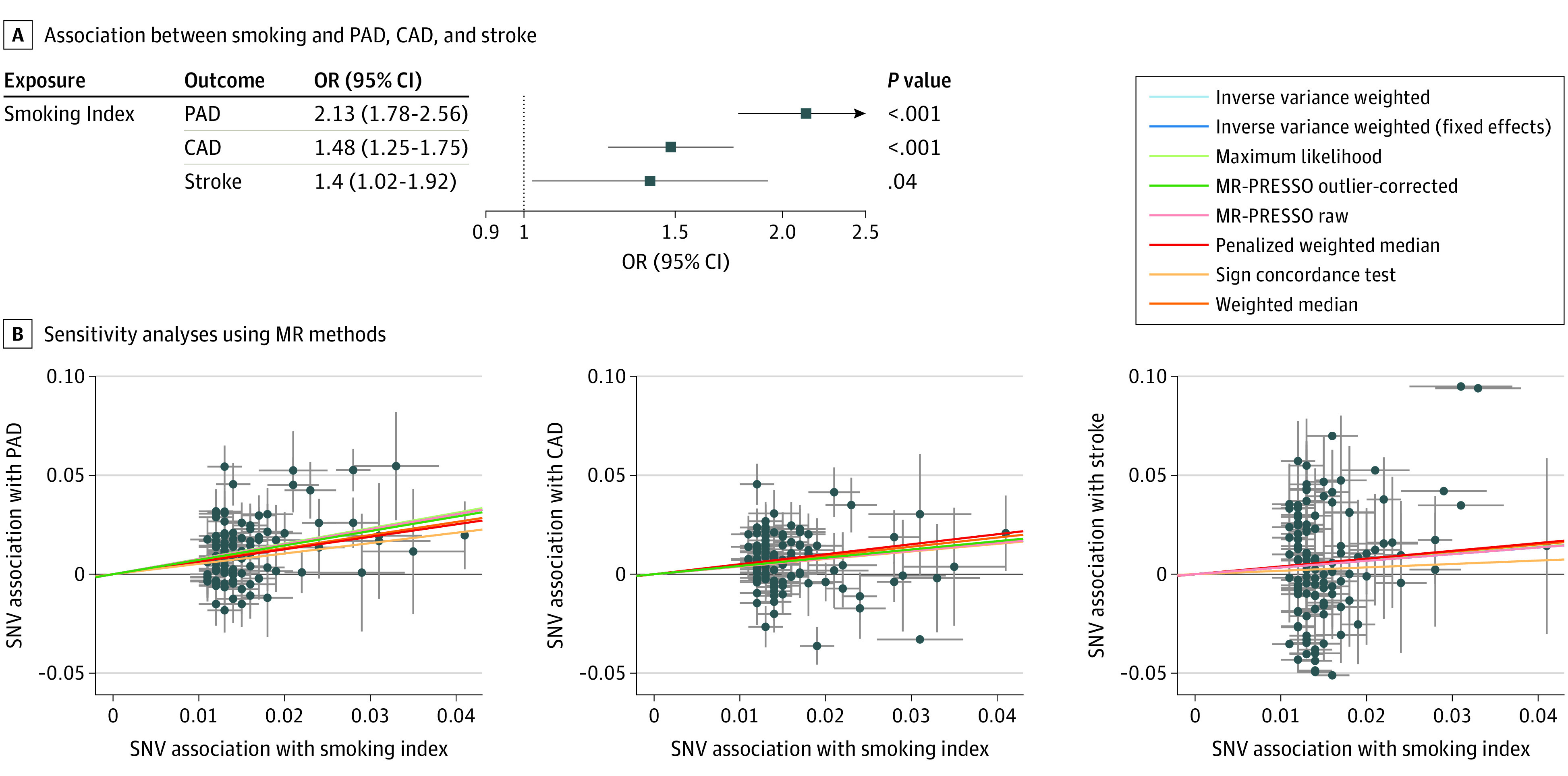
A, In inverse-variance–weighted mendelian randomization, each 1 SD increase in genetic liability to smoking is associated with significantly increased risk of PAD, CAD, and large-artery stroke. Smoking is most strongly associated with increased risk of PAD compared with large-artery stroke (P = .04) and CAD (P < .001). Odds ratios (ORs) are expressed per 1 SD increase in lifetime smoking index. B, Scatter plots demonstrating the effect of each smoking-associated genetic variant on each atherosclerotic cardiovascular disease outcome on the log-odds scale. Colored lines represent results of each mendelian randomization sensitivity analysis, making different assumptions about horizontal pleiotropy, heterogeneity, and error in the instrument-exposure associations. SNV represents single-nucleotide variant.
We next determined whether the estimated effect sizes for the association of smoking differed between the ASCVD endpoints. The primary inverse-variance weighted point estimate for the association of smoking with PAD was significantly greater than the estimates for large-artery stroke (ratio of ORs, 1.52; 95% CI, 1.05-2.19; P = .02) or CAD (ratio of ORs, 1.44; 95% CI, 1.12-1.84; P = .004).
Association of Smoking With Risk of Traditional Cardiometabolic Risk Factors
We next considered the effect of increasing genetic liability to lifetime smoking on other cardiometabolic traits that are known ASCVD risk factors. Increasing genetic liability to lifetime smoking was significantly associated (P < .001) with increased risk of type 2 diabetes (OR, 1.89; 95% CI, 1.53-2.33), hypertension (OR, 1.05; 95% CI, 1.04-1.07), waist circumference (β = 0.33; 95% CI, 0.17-0.49), body mass index (β = 0.35; 95% CI, 0.14-0.57), and waist to hip ratio (β = 0.23; 95% CI, 0.12-0.33), with a suggestive (P < .05) increase in the risk of hyperlipidemia (OR, 1.00; 95% CI, 1.00-1.01) and risk of being overweight (OR, 1.43; 95% CI, 1.07-1.91) (Figure 3). In a sensitivity analysis considering an alternative genetic instrument for smoking, the results were similar (eFigure 7 in the Supplement). The MR Steiger test confirmed the directionality of these significant and suggestive findings (P < .001) (eTable 3 in the Supplement).
Figure 3. Total Effect Sizes for Associations Between Smoking and Cardiometabolic Risk Factors for Atherosclerotic Cardiovascular Disease.
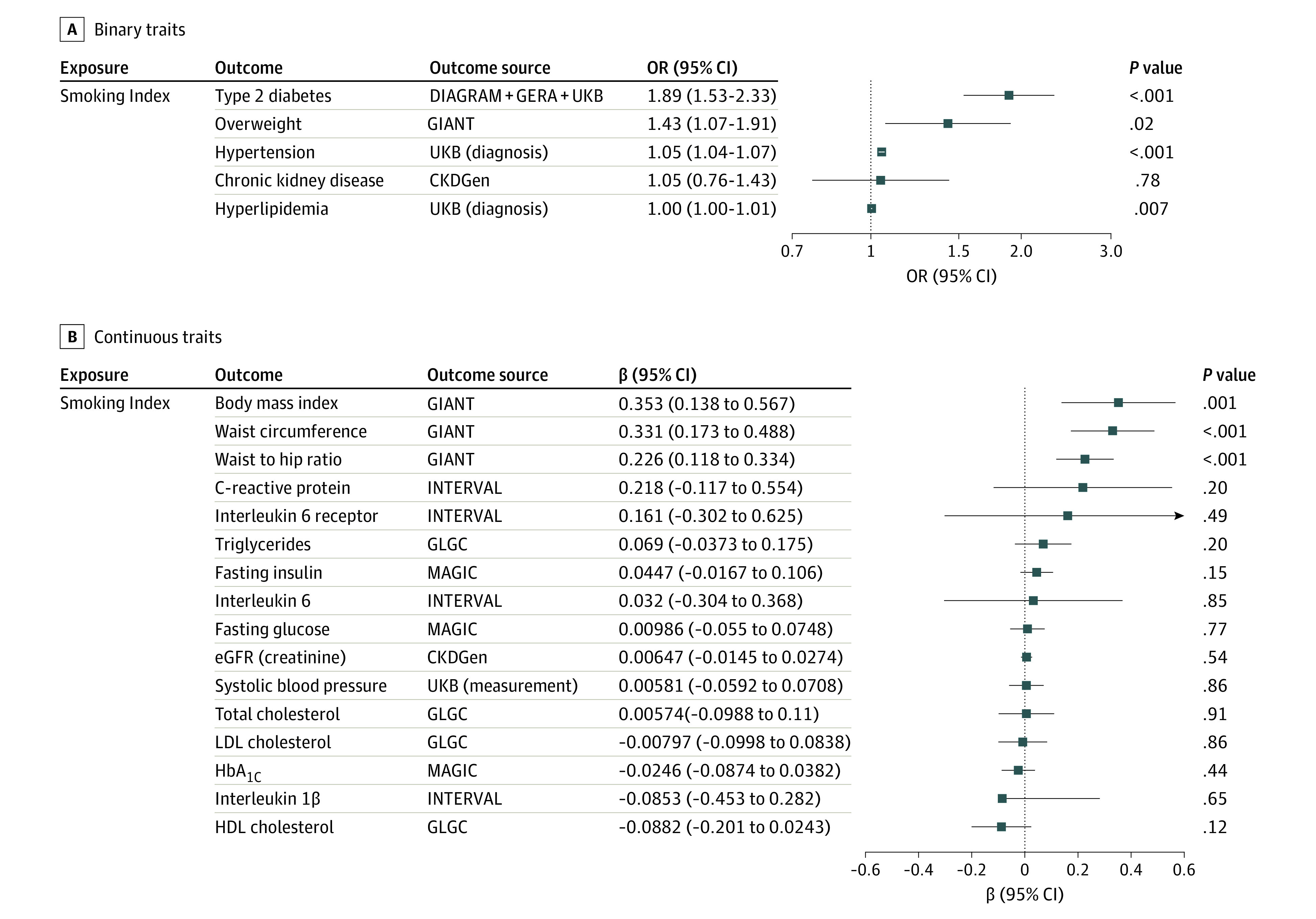
Inverse-variance–weighted mendelian randomization was performed to determine whether genetic liability to smoking altered risk of cardiometabolic risk factors for atherosclerotic cardiovascular disease. Genetic liability to smoking increased risk of both (A) binary traits and (B) continuous traits that are common risk factors for cardiometabolic disease. Effect estimates are expressed per 1 SD increase in lifetime smoking index. CKDGen indicates Chronic Kidney Disease Genetics Consortium; DIAGRAM, Diabetes Genetics Replication and Meta-analysis consortium; eGFR, estimated glomerular filtration rate; GERA, Genetic Epidemiology Research on Aging; GIANT, Genetic Investigation of Anthropometric Traits Consortium; GLGC, Global Lipid Genetics Consortium; HbA1C, hemoglobin A1C; HDL, high-density lipoprotein; LDL, low-density lipoprotein; MAGIC, Meta-Analyses of Glucose and Insulin-Related Traits Consortium; OR, odds ratio; and UKB, UK Biobank.
Influence of Traditional Cardiometabolic Risk Factors on Smoking-ASCVD Relationship
To evaluate whether increasing genetic liability to smoking was directly associated with increased risk of ASCVD, or whether the association was attenuated after accounting for traditional cardiovascular risk factors (type 2 diabetes, lipids, body mass index, and systolic blood pressure), we performed multivariable MR. Increasing genetic liability to smoking was associated with increased risk of PAD, CAD, and stroke, after accounting for effects of smoking on each risk factor independently, and in a combined model considering all risk factors (Figure 4). The point estimates for the association of smoking with ASCVD were not substantially attenuated after accounting for traditional risk factors (PAD: OR, 2.84 [95% CI, 1.28-6.32]; P = .01; CAD: OR, 1.47 [95% CI, 0.52-4.18]; P = .47; stroke: OR, 3.44 [95% CI, 0.65-18.20]; P = .15), or in a model further including interleukin 6 as a marker of inflammatory risk of ASCVD (PAD: OR, 2.83 [95% CI, 1.26-6.35]; P = .01; CAD: OR, 1.48 [95% CI, 0.51-4.24]; P = .47; stroke: OR, 3.58 [95% CI, 0.69-18.70]; P = .13) compared with the univariate estimates.
Figure 4. Direct Effect Sizes for Associations Between Smoking and Risk of Peripheral Artery Disease (PAD), Coronary Artery Disease (CAD), and Stroke.

Multivariable mendelian randomization was performed to estimate the direct effect of the association of smoking with atherosclerotic cardiovascular disease after accounting for the effects of smoking on other cardiovascular risk factors. The estimated direct effects of smoking on PAD, CAD, and stroke are not substantially attenuated in models adjusting for traditional cardiovascular risk factors (type 2 diabetes, body mass index, lipids [high-density lipoprotein cholesterol, low-density lipoprotein cholesterol, and triglycerides], and systolic blood pressure), or inflammation (interleukin 6 [IL-6] levels). Odds ratios (ORs) are expressed per 1 SD increase in lifetime smoking index.
Discussion
Using MR, we leveraged population-scale human genetics to estimate a potentially causal association between smoking, cardiometabolic risk factors, and ASCVD outcomes across diverse vascular beds (CAD, PAD, and stroke). By drawing from several large GWASs, we were able to consider more cases by an order of magnitude for smoking, cardiometabolic risk factors, and ASCVD outcomes compared with previous observational studies. Although observational studies remain at risk of bias due to residual confounding, the genetic instrumental variables used here in the MR framework provided effect estimates that were less susceptible to reverse causality and confounding from environmental factors. Because genetic variants are randomly assorted during meiosis, mimicking randomization in a clinical trial, we were able to estimate potentially causal relationships between smoking and cardiometabolic traits.
Our results suggested that smoking had a direct atherogenic effect that varied across vascular beds. This finding is largely consistent with prior investigations of smoking on ASCVD. Although the association between smoking and ASCVD has been shown previously in observational studies, our MR analysis provides strong evidence that may be consistent with a causal relationship. Our finding that smoking appears to more strongly influence the risk of PAD compared with CAD or stroke is consistent with recent results from the ARIC (Atherosclerosis Risk in Communities) study cohort, in which the effect of smoking was greatest for PAD, and a recent MR analysis of UK Biobank participants demonstrating a strong effect of smoking on PAD.4,40 Although the mechanism behind the stronger relationship between smoking and PAD is not clear, structural and functional differences within the vascular beds and the complex interplay between smoking and other ASCVD risk factors may contribute.41,42 For example, although both acute PAD and CAD events typically result from luminal thrombosis, and both diseases have atherosclerotic manifestations, typical acute CAD lesions occur in the setting of atherothrombosis, while acute PAD-associated lesions more typically result from in situ thrombosis or embolism.43 Indeed, a strong association between the factor V Leiden variant (F5 p.R506Q) and PAD has been identified; however, this association is not present for CAD, raising the possibility that smoking-related changes in coagulation may explain some of the differential associations between smoking and ASCVD outcomes.9,44 Further understanding of the mechanistic differences in ASCVD pathophysiology across vascular beds may ultimately lead to more targeted prevention and treatment strategies.
Genetic liability to smoking is also associated with cardiometabolic traits that are themselves risk factors ASCVD. The MR finding that increasing genetic liability to smoking is associated with type 2 diabetes is consistent with recent observational and MR studies.6,45,46,47,48 We also identified increasing genetic liability to smoking as a risk factor for hypertension and increased waist circumference, body mass index, and waist to hip ratio, although prior studies have identified conflicting effects of smoking on these traits.49,50,51,52,53,54 A prior single-sample MR analysis from the Nord-Trøndelag Health Study (HUNT Study) found a protective effect of smoking on body mass index, waist circumference, and hip circumference but found no associations with blood pressure, levels of lipids, or glucose levels.51 Their study may have been limited by the single-sample design, modest study size, and weak single single-nucleotide variant (rs1051730) instrument for smoking, which all may have contributed to bias toward observational estimates.12 More recent MR studies have corroborated our finding that smoking traits are associated with increased body mass index.48 Conflict among observational studies may be related to residual confounding or reverse causality. Mendelian randomization assumes that genetic variants proxying an exposure produce similar effects to the exposure itself, although this assumption may not always be valid. For example, lifetime exposure to adverse genetics may have different health consequences when compared with more concentrated environmental exposures, highlighted by the much larger protective effects of genetically lower low-density lipoprotein cholesterol level and systolic blood pressure on risk of coronary heart disease in comparison with effect estimates from randomized trials of treatments for these risk factors.55,56
The effect of increasing genetic liability to smoking on ASCVD outcomes appears to be independent from the effects of smoking on traditional cardiovascular risk factors. The point estimate of the direct effect of smoking (when jointly considering smoking and cardiometabolic risk factors) was similar (or greater) than the total effect, suggesting the possibility of causal interaction between smoking and traditional risk factors, which could be investigated using factorial MR in a single-sample setting.57 Proposed mechanisms by which smoking may independently contribute to cardiovascular events include among others hypercoagulability, increased myocardial work, decreased oxygen delivery (due to elevated carboxyhemoglobin levels), coronary vasoconstriction, and increased catecholamine levels.57
The finding that smoking confers strong independent risk for ASCVD even when considering other traditional cardiovascular risk factors has important public health implications. More precise estimation of the effect of smoking on ASCVD outcomes may help calibrate the expected benefit of smoking cessation initiatives, and efforts to reduce the burden of cardiovascular disease should continue to focus on smoking cessation. Further, public awareness of ASCVD varies across outcomes and is particularly low for PAD.58 The current analysis provides strong genetic evidence for the effects of smoking and other ASCVD risk factors on CAD, PAD, and stroke and may be useful in raising public understanding of these risk factor–outcome relationships.
Limitations
The current study must be interpreted within the context of its limitations. The study focused primarily on individuals of European ancestry, which may limit generalization to other populations, highlighting the need for genomic studies in diverse ancestral groups. The MR framework relies on a key assumption that the risk conferred by an exposure is equivalent whether mediated by genetics or environment, and that genetic risk is conferred through the exposure of interest rather than via pleiotropic effects.12 Although findings were consistent in sensitivity analyses using MR methods robust to the presence of pleiotropy, there may be gene-environment interactions, such as those previously shown at the ADAMTS7 locus for CAD and at the CHRNA3 locus for PAD, that modify and alter the relationship between smoking and ASCVD outcomes.9,59 Although differences in the underlying structure of the ASCVD studies could affect the estimate of differential risk between the ASCVD outcomes, the 2-sample MR framework tends to bias causal estimates toward the null, lending further confidence in our overall finding that smoking was strongly associated with increased risk of all ASCVD outcomes. Similarly, differences in the ascertainment of ASCVD and cardiometabolic traits (eg, inclusion of incident vs prevalent disease) may lead to biased estimates owing to prevalence-incidence or to selective-survival bias although the analyzed cohorts in the present study included predominately prevalent disease, which would be expected to bias estimates toward the null. Finally, future study of additional smoking-related traits, such as duration or quantity of smoking and smoking cessation, and other MR methods may provide additional insight into potential differential effects of these traits in different vascular beds, clarifying recent observational findings that these traits may affect ASCVD risk.4,60
Conclusions
In this MR study using genetic data from large studies of smoking, atherosclerosis, and cardiometabolic disease, genetic liability to smoking was associated with increased risk of ASCVD, with the strongest association between smoking and PAD, independent from traditional cardiovascular risk factors.
eFigure 1. MR of Lifetime Smoking Index to GSCAN Smoking Traits
eFigure 2. Primary Smoking-ASCVD MR Funnel Plots
eFigure 3. Primary Smoking-ASCVD MR Leave-One-Out Analysis
eFigure 4. Primary Smoking-ASCVD MR Single-SNP Analysis
eFigure 5. Primary Smoking-ASCVD MR Single-SNP Analysis
eFigure 6. MR of Smoking Initiation to ASCVD Outcomes
eFigure 7. MR of Smoking Initiation to Cardiometabolic Risk Factors
eTable 1. Smoking Index exposure SNPs and Corresponding SNP Effects for CAD, PAD, and Stroke Outcomes
eTable 2. Egger Bias Intercept Test for Smoking Index-ASCVD Analysis
eTable 3. MR Steiger Directionality Test for Smoking-Cardiometabolic Risk Factor Analysis
References
- 1.Lubin JH, Couper D, Lutsey PL, Woodward M, Yatsuya H, Huxley RR. Risk of cardiovascular disease from cumulative cigarette use and the impact of smoking intensity. Epidemiology. 2016;27(3):395-404. doi: 10.1097/EDE.0000000000000437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Price JF, Mowbray PI, Lee AJ, Rumley A, Lowe GDO, Fowkes FGR. Relationship between smoking and cardiovascular risk factors in the development of peripheral arterial disease and coronary artery disease: Edinburgh Artery Study. Eur Heart J. 1999;20(5):344-353. doi: 10.1053/euhj.1998.1194 [DOI] [PubMed] [Google Scholar]
- 3.Gordon T, Kannel WB. Predisposition to atherosclerosis in the head, heart, and legs. The Framingham study. JAMA. 1972;221(7):661-666. doi: 10.1001/jama.1972.03200200011003 [DOI] [PubMed] [Google Scholar]
- 4.Ding N, Sang Y, Chen J, et al. Cigarette smoking, smoking cessation, and long-term risk of 3 major atherosclerotic diseases. J Am Coll Cardiol. 2019;74(4):498-507. doi: 10.1016/j.jacc.2019.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658-665. doi: 10.1002/gepi.21758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention (US); National Center for Chronic Disease Prevention and Health Promotion (US); Office on Smoking and Health (US) . How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General. Centers for Disease Control and Prevention (US); 2010. [PubMed] [Google Scholar]
- 7.Minami J, Ishimitsu T, Matsuoka H. Effects of smoking cessation on blood pressure and heart rate variability in habitual smokers. Hypertension. 1999;33(1 Pt 2):586-590. doi: 10.1161/01.HYP.33.1.586 [DOI] [PubMed] [Google Scholar]
- 8.Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121-1130. doi: 10.1038/ng.3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klarin D, Lynch J, Aragam K, et al. ; VA Million Veteran Program . Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat Med. 2019;25(8):1274-1279. doi: 10.1038/s41591-019-0492-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malik R, Chauhan G, Traylor M, et al. ; AFGen Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium; International Genomics of Blood Pressure (iGEN-BP) Consortium; INVENT Consortium; STARNET; BioBank Japan Cooperative Hospital Group; COMPASS Consortium; EPIC-CVD Consortium; EPIC-InterAct Consortium; International Stroke Genetics Consortium (ISGC); METASTROKE Consortium; Neurology Working Group of the CHARGE Consortium; NINDS Stroke Genetics Network (SiGN); UK Young Lacunar DNA Study; MEGASTROKE Consortium . Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(4):524-537. doi: 10.1038/s41588-018-0058-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wootton RE, Richmond RC, Stuijfzand BG, et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study. Psychol Med. 2020;50(14):2435-2443. doi: 10.1017/S0033291719002678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies NM, Holmes MV, Davey Smith G. Reading mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi: 10.1136/bmj.k601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burgess S, Thompson DJ, Rees JMB, Day FR, Perry JR, Ong KK. Dissecting causal pathways using mendelian randomization with summarized genetic data:Application to age at menarche and risk of breast cancer. Genetics. 2017;207(2):481-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu M, Jiang Y, Wedow R, et al. ; 23andMe Research Team; HUNT All-In Psychiatry . Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51(2):237-244. doi: 10.1038/s41588-018-0307-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shim H, Chasman DI, Smith JD, et al. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS One. 2015;10(4):e0120758. doi: 10.1371/journal.pone.0120758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP; STROBE Initiative . The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. PLoS Med. 2007;4(10):e296. doi: 10.1371/journal.pmed.0040296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pattaro C, Teumer A, Gorski M, et al. ; ICBP Consortium; AGEN Consortium; CARDIOGRAM; CHARGe-Heart Failure Group; ECHOGen Consortium . Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat Commun. 2016;7:10023. doi: 10.1038/ncomms10023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willer CJ, Schmidt EM, Sengupta S, et al. ; Global Lipids Genetics Consortium . Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274-1283. doi: 10.1038/ng.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue A, Wu Y, Zhu Z, et al. ; eQTLGen Consortium . Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun. 2018;9(1):2941. doi: 10.1038/s41467-018-04951-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558(7708):73-79. doi: 10.1038/s41586-018-0175-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Locke AE, Kahali B, Berndt SI, et al. ; LifeLines Cohort Study; ADIPOGen Consortium; AGEN-BMI Working Group; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium . Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197-206. doi: 10.1038/nature14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berndt SI, Gustafsson S, Mägi R, et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet. 2013;45(5):501-512. doi: 10.1038/ng.2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manning AK, Hivert M-F, Scott RA, et al. ; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium; Multiple Tissue Human Expression Resource (MUTHER) Consortium . A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44(6):659-669. doi: 10.1038/ng.2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shungin D, Winkler TW, Croteau-Chonka DC, et al. ; ADIPOGen Consortium; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GEFOS Consortium; GENIE Consortium; GLGC; ICBP; International Endogene Consortium; LifeLines Cohort Study; MAGIC Investigators; MuTHER Consortium; PAGE Consortium; ReproGen Consortium . New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518(7538):187-196. doi: 10.1038/nature14132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi: 10.1161/CIR.0000000000000678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridker PM, Everett BM, Thuren T, et al. ; CANTOS Trial Group . Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119-1131. doi: 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 27.Stone NJ, Robinson JG, Lichtenstein AH, et al. ; American College of Cardiology/American Heart Association Task Force on Practice Guidelines . 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25, pt B):2889-2934. doi: 10.1016/j.jacc.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 28.Swerdlow DI, Holmes MV, Kuchenbaecker KB, et al. ; Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium . The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379(9822):1214-1224. doi: 10.1016/S0140-6736(12)60110-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Georgakis MK, Malik R, Gill D, Franceschini N, Sudlow CLM, Dichgans M; INVENT Consortium,CHARGE Inflammation Working Group . Interleukin-6 signaling effects on ischemic stroke and other cardiovascular outcomes: a Mendelian Randomization study. Circ Genom Precis Med. 2020;13(3):e002872. doi: 10.1161/CIRCGEN.119.002872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife. 2018;7:e34408. doi: 10.7554/eLife.34408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verbanck M, Chen CY, Neale B, Do R. Widespread pleiotropy confounds causal relationships between complex traits and diseases inferred from mendelian randomization. bioRxiv 157552. Preprint posted online June 30, 2017. doi: 10.1101/157552 [DOI]
- 32.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304-314. doi: 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693-698. doi: 10.1038/s41588-018-0099-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burgess S, Labrecque JA. Mendelian randomization with a binary exposure variable: interpretation and presentation of causal estimates. Eur J Epidemiol. 2018;33(10):947-952. doi: 10.1007/s10654-018-0424-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altman DG, Bland JM. Interaction revisited: the difference between two estimates. BMJ. 2003;326(7382):219. doi: 10.1136/bmj.326.7382.219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. 2016;40(7):597-608. doi: 10.1002/gepi.21998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13(11):e1007081. doi: 10.1371/journal.pgen.1007081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanderson E, Davey Smith G, Windmeijer F, Bowden J. An examination of multivariable mendelian randomization in the single-sample and two-sample summary data settings. Int J Epidemiol. 2019;48(3):713-727. doi: 10.1093/ije/dyy262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burgess S, Thompson SG. Multivariable mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181(4):251-260. doi: 10.1093/aje/kwu283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larsson SC, Mason AM, Bäck M, et al. ; Million Veteran Program . Genetic predisposition to smoking in relation to 14 cardiovascular diseases. Eur Heart J. 2020;41(35):3304-3310. doi: 10.1093/eurheartj/ehaa193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steenman M, Espitia O, Maurel B, et al. Identification of genomic differences among peripheral arterial beds in atherosclerotic and healthy arteries. Sci Rep. 2018;8(1):3940. doi: 10.1038/s41598-018-22292-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGill HC Jr, McMahan CA, Herderick EE, et al. ; PDAY Research Group . Effects of coronary heart disease risk factors on atherosclerosis of selected regions of the aorta and right coronary artery. Arterioscler Thromb Vasc Biol. 2000;20(3):836-845. doi: 10.1161/01.ATV.20.3.836 [DOI] [PubMed] [Google Scholar]
- 43.Narula N, Olin JW, Narula N. Pathologic disparities between peripheral artery disease and coronary artery disease. Arterioscler Thromb Vasc Biol. 2020;40(9):1982-1989. doi: 10.1161/ATVBAHA.119.312864 [DOI] [PubMed] [Google Scholar]
- 44.Mahmoodi BK, Tragante V, Kleber ME, et al. Association of factor V Leiden with subsequent atherothrombotic events: a GENIUS-CHD study of individual participant data. Circulation. 2020;142(6):546-555. doi: 10.1161/CIRCULATIONAHA.119.045526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thom CS, Ding Z, Voight BF. Initiation of smoking and susceptibility to type 2 diabetes: a mendelian randomization study. medRxiv 2020.01.30.20019737. Preprint posted online February 3, 2020. doi: 10.1101/2020.01.30.20019737 [DOI]
- 46.Liu X, Bragg F, Yang L, et al. ; China Kadoorie Biobank Collaborative Group . Smoking and smoking cessation in relation to risk of diabetes in Chinese men and women: a 9-year prospective study of 0·5 million people. Lancet Public Health. 2018;3(4):e167-e176. doi: 10.1016/S2468-2667(18)30026-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan S, Larsson SC. A causal relationship between cigarette smoking and type 2 diabetes mellitus: a mendelian randomization study. Sci Rep. 2019;9(1):19342. doi: 10.1038/s41598-019-56014-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thom CS, Ding Z, Levin MG, et al. ; VA Million Veteran Program . Genetic determinants of increased body mass index mediate the effect of smoking on increased risk for type 2 diabetes but not coronary artery disease. Hum Mol Genet. 2020;29(19):3327-3337. doi: 10.1093/hmg/ddaa193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freitas SRS, Alvim RO. Smoking and blood pressure phenotypes: new perspective for an old problem. Am J Hypertens. 2017;30(6):554-555. doi: 10.1093/ajh/hpx039 [DOI] [PubMed] [Google Scholar]
- 50.Dare S, Mackay DF, Pell JP. Relationship between smoking and obesity: a cross-sectional study of 499,504 middle-aged adults in the UK general population. PLoS One. 2015;10(4):e0123579. doi: 10.1371/journal.pone.0123579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Åsvold BO, Bjørngaard JH, Carslake D, et al. Causal associations of tobacco smoking with cardiovascular risk factors: a mendelian randomization analysis of the HUNT Study in Norway. Int J Epidemiol. 2014;43(5):1458-1470. doi: 10.1093/ije/dyu113 [DOI] [PubMed] [Google Scholar]
- 52.Chiolero A, Jacot-Sadowski I, Faeh D, Paccaud F, Cornuz J. Association of cigarettes smoked daily with obesity in a general adult population. Obesity (Silver Spring). 2007;15(5):1311-1318. doi: 10.1038/oby.2007.153 [DOI] [PubMed] [Google Scholar]
- 53.Slagter SN, van Vliet-Ostaptchouk JV, Vonk JM, et al. Associations between smoking, components of metabolic syndrome and lipoprotein particle size. BMC Med. 2013;11(1):195. doi: 10.1186/1741-7015-11-195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winsløw UC, Rode L, Nordestgaard BG. High tobacco consumption lowers body weight: a mendelian randomization study of the Copenhagen General Population Study. Int J Epidemiol. 2015;44(2):540-550. doi: 10.1093/ije/dyu276 [DOI] [PubMed] [Google Scholar]
- 55.Burgess S, Butterworth A, Malarstig A, Thompson SG. Use of mendelian randomisation to assess potential benefit of clinical intervention. BMJ. 2012;345:e7325. doi: 10.1136/bmj.e7325 [DOI] [PubMed] [Google Scholar]
- 56.Burgess S, Harshfield E. Mendelian randomization to assess causal effects of blood lipids on coronary heart disease: lessons from the past and applications to the future. Curr Opin Endocrinol Diabetes Obes. 2016;23(2):124-130. doi: 10.1097/MED.0000000000000230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benowitz NL, Gourlay SG. Cardiovascular toxicity of nicotine: implications for nicotine replacement therapy. J Am Coll Cardiol. 1997;29(7):1422-1431. doi: 10.1016/S0735-1097(97)00079-X [DOI] [PubMed] [Google Scholar]
- 58.Hirsch AT, Murphy TP, Lovell MB, et al. ; Peripheral Arterial Disease Coalition . Gaps in public knowledge of peripheral arterial disease: the first national PAD public awareness survey. Circulation. 2007;116(18):2086-2094. doi: 10.1161/CIRCULATIONAHA.107.725101 [DOI] [PubMed] [Google Scholar]
- 59.Saleheen D, Zhao W, Young R, et al. Loss of cardioprotective effects at the ADAMTS7 locus as a result of gene-smoking interactions. Circulation. 2017;135(24):2336-2353. doi: 10.1161/CIRCULATIONAHA.116.022069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duncan MS, Freiberg MS, Greevy RA Jr, Kundu S, Vasan RS, Tindle HA. Association of smoking cessation with subsequent risk of cardiovascular disease. JAMA. 2019;322(7):642-650. doi: 10.1001/jama.2019.10298 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eFigure 1. MR of Lifetime Smoking Index to GSCAN Smoking Traits
eFigure 2. Primary Smoking-ASCVD MR Funnel Plots
eFigure 3. Primary Smoking-ASCVD MR Leave-One-Out Analysis
eFigure 4. Primary Smoking-ASCVD MR Single-SNP Analysis
eFigure 5. Primary Smoking-ASCVD MR Single-SNP Analysis
eFigure 6. MR of Smoking Initiation to ASCVD Outcomes
eFigure 7. MR of Smoking Initiation to Cardiometabolic Risk Factors
eTable 1. Smoking Index exposure SNPs and Corresponding SNP Effects for CAD, PAD, and Stroke Outcomes
eTable 2. Egger Bias Intercept Test for Smoking Index-ASCVD Analysis
eTable 3. MR Steiger Directionality Test for Smoking-Cardiometabolic Risk Factor Analysis