

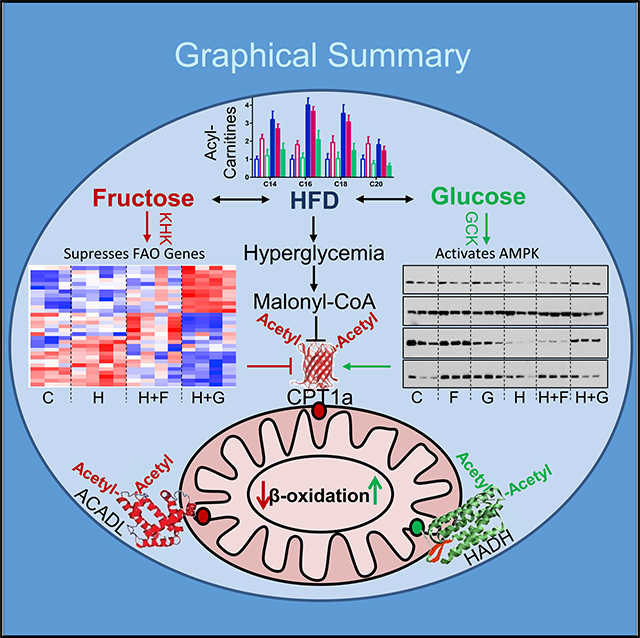
SUMMARY
Dietary sugars, fructose and glucose, promote hepatic de novo lipogenesis and modify the effects of a high-fat diet (HFD) on the development of insulin resistance. Here, we show that fructose and glucose supplementation of an HFD exert divergent effects on hepatic mitochondrial function and fatty acid oxidation. This is mediated via three different nodes of regulation, including differential effects on malonyl-CoA levels, effects on mitochondrial size/protein abundance, and acetylation of mitochondrial proteins. HFD- and HFD plus fructose-fed mice have decreased CTP1a activity, the rate-limiting enzyme of fatty acid oxidation, whereas knockdown of fructose metabolism increases CPT1a and its acylcarnitine products. Furthermore, fructose-supplemented HFD leads to increased acetylation of ACADL and CPT1a, which is associated with decreased fat metabolism. In summary, dietary fructose, but not glucose, supplementation of HFD impairs mitochondrial size, function, and protein acetylation, resulting in decreased fatty acid oxidation and development of metabolic dysregulation.
In Brief
Softic et al. show that in addition to its well-described effects to increase hepatic fatty acid synthesis and malonyl-CoA levels, consumption of fructose-sweetened water decreases dietary fat oxidation. This is mediated by impaired expression of fatty acid oxidation genes and by modifying the mitochondrial proteome including post-translational modification of mitochondrial proteins, such as acetylation.
Graphical Abstract
INTRODUCTION
Increased caloric intake and decreased energy expenditure are risk factors for development of obesity and its metabolic complications. Along with high consumption of fat in typical western diets, increased consumption of sugar-sweetened beverages has emerged as an important risk factor for development of obesity, type 2 diabetes, and non-alcoholic fatty liver disease (NAFLD) (de Ruyter et al., 2012; Ebbeling et al., 2012; Ma et al., 2015). Dietary sugar is predominantly consumed as sucrose or high-fructose corn syrup, both of which contain fructose and glucose di- or monosaccharides. The effects of sugar intake, especially fructose, on metabolic complications have largely been attributed to increased hepatic de novo lipogenesis (Samuel, 2011; Softic et al., 2016b). Fructose robustly promotes lipogenesis, not only by serving as a substrate for fatty acid synthesis but also by stimulating the expression of enzymes involved in de novo lipogenesis via activation of the lipogenic transcription factors SREBP1c and ChREBP (Janevski et al., 2012; Kim et al., 2016; Softic et al., 2017).
Increased de novo lipogenesis is usually accompanied by decreased fatty acid oxidation (FAO); however, how this occurs is less clear and may involve multiple mechanisms. Dietary sugars increase levels of hepatic malonyl-CoA, which is an intermediate in de novo lipogenesis. Malonyl-CoA also inhibits carnitine palmitoyl transferase 1 (CPT1), which catalyzes the conversion of fatty acyl-CoAs to their cognate acylcarnitines, e.g., palmitoyl-CoA to palmitoylcarnitine. This allows movement of acylcarnitines from the cytosol into the mitochondrial intermembrane space, thus serving as the rate-limiting step of FAO (McGarry et al., 1977). Since fructose induces higher rates of de novo lipogenesis than glucose (Zakim, 1972), it is reasonable to expect that fructose leads to a more profound decrease in FAO. Indeed, subjects consuming fructose-sweetened beverages have decreased post-prandial fat oxidation and resting energy expenditure as compared to subjects consuming calorically equivalent glucose-sweetened drinks (Cox et al., 2012). Interestingly, fructose treatment of isolated mitochondria results in decreased FAO, suggesting that fructose may also directly inhibit mitochondrial beta-oxidation (Prager and Ontko, 1976), independent of its effects on malonyl-CoA and thus CPT1 activity. Consistent with this observation, fructose-induced inhibition of palmitate oxidation is similar to its inhibition of palmitoylcarnitine oxidation (Prager and Ontko, 1976). Malonyl-CoA-independent regulation of FAO may be particularly important in the liver, as the liver CPT1a isoform is 30- to 100-fold less sensitive to malonyl-CoA inhibition than its heart and muscle counterpart CPT1b (Shi et al., 2000). Thus, in the liver, fructose may also induce its effects by decreasing expression of enzymes regulating FAO (Roglans et al., 2007; Vilà et al., 2008) and by promoting mitochondrial dysfunction.
In addition to substrate and transcriptional regulation, there is growing evidence that nutrients can regulate mitochondrial function via acylation of mitochondrial proteins, including acetylation, succinylation, malonylation, and propionylation (Choudhary et al., 2014; Kim et al., 2006). Acetylation, the best studied of these modifications, involves covalent addition of acetyl groups to a lysine residue. This neutralizes the positive charge of the epsilon-amino group, which can affect protein structure and activity (Glozak et al., 2005). Acetylation is dependent on nutrient availability, as fasting or caloric restriction increases oxidation of fatty acids, thereby producing acetyl-CoA that may serve as a substrate for mitochondrial protein acetylation (Kim et al., 2006; Schwer et al., 2009; Weinert et al., 2015). The level of acetylation is also dependent on the length of nutrient exposure, as long-term, but not short-term, high-fat diet (HFD) feeding increases mitochondrial protein acetylation in livers of fasted mice (Hirschey et al., 2011; Kendrick et al., 2011). Another major regulator of protein acetylation in the mitochondrion is Sirt3, which can remove acetyl groups (Bharathi et al., 2013; Lombard et al., 2007). Although high-fat intake and Sirt3 activity are known to modify protein acetylation, the mechanisms by which dietary sugars might affect this post-translational modification are largely unknown.
In this study, we have examined the effects of fructose or glucose supplementation on mitochondrial function and FAO in mice fed chow or HFD. We find that these two hexoses differentially affect mitochondrial size, mitochondrial protein acetylation, and mitochondrial functions, particularly FAO. These differences in the effects of fructose and glucose on mitochondrial protein acetylation and function may contribute to differences in metabolic homeostasis and the propensity for development of fatty liver disease.
RESULTS
Fructose and Glucose Supplementation Affect FAO
Cohorts of 6-week-old male C57Bl6/J mice were fed chow (21.6% calories from fat) or HFD (60.0% calories from fat) and given ad libitum access to water or water containing 30% (w/v) fructose or glucose for 10 weeks. As previously reported (Softic et al., 2017) and summarized in Figure S1A, mice on the chow diet supplemented with fructose (chow + F) or glucose (chow + G) gained about 30% more weight than mice drinking regular water (chow), but remained normoglycemic. On the HFD, all groups of mice developed obesity, with an average 40%–60% increase in body weight and increased liver weight with histologic evidence of hepatic steatosis. Mice on the HFD plus regular water (HFD) or water supplemented with fructose (HFD + F) also developed insulin resistance and hyperglycemia, and exhibited a 2-fold increase in insulin levels. In contrast, mice supplemented with glucose (HFD + G) gained a similar amount of weight but remained insulin sensitive, as reflected by decreased insulin levels and an improved response to exogenous insulin administration, both in terms of increased Akt phosphorylation and a greater decrease in blood glucose. This occurred despite no difference in total caloric intake, i.e., calories from solid food and the sugar water, between the fructose- and glucose-supplemented mice in both the chow and HFD groups. Mechanistically, fructose supplementation increased endogenous fatty acid synthesis and induced expression of the lipogenic transcription factors Srebp1c and Chrebp-β, whereas glucose supplementation increased triglyceride (TG) synthesis and the expression of Chrebp-β and total Chrebp protein (Softic et al., 2017).
To determine how fructose and glucose supplementation might interact with HFD to affect fatty acid metabolism, we performed targeted mass spectrometry (MS)-based hepatic acylcarnitine profiling. Livers of mice on chow supplemented with fructose or the HFD showed increased levels of saturated long-chain acylcarnitines (C14:0, C16:0, C18:0, and C20:0) by 2- to 4-fold (Figure 1A). Interestingly, while similarly elevated acylcarnitines were observed in the HFD + F group, mice fed the HFD + G exhibited lower levels of long-chain acylcarnitines than the HFD or HFD + F mice, similar to those in chow-fed mice. In addition, several short-chains, odd-chain acylcarnitines, derived primarily from amino acid catabolism, were significantly lower in mice fed the HFD + G (Figure 1B).
Figure 1. Fructose and Glucose Supplementation Modifies Fatty Acid Oxidation.

(A and B) Long- (A) and short-chain (B) acylcarnitine profiles measured from the livers of mice collected in the fed state after 10 weeks on respective diets. The data are represented as mean ± SEM, n = 6 mice per group. Complete names of acylcarnitines are provided in Table S2.
(C) Cpt1a mRNA expression and protein levels.
(D) CPT1a activity in isolated mitochondria and hepatic malonyl-CoA levels from the same mice. n = 8 mice per group. # denotes the significant difference as compared to the chow control, whereas * signifies the significant difference between the groups under the crossbar.
(E and F) Hepatic pAMPK and pACC1 levels.
(G) Serum FFA and β-hydroxybutyrate levels in overnight fasted mice.
(H) Oxygen consumption rate in AML-12 hepatocytes pretreated with glucose or fructose for 48 h.
The rate-limiting step in hepatic FAO is CPT1a-mediated acylcarnitine production, which enables transport of fatty acids to mitochondria. Cpt1a mRNA was increased by glucose supplementation and the HFD, and these effects were additive (Figure 1C). CPT1a protein expression was also increased by the HFD, but not by glucose supplementation. CPT1a enzymatic activity, on the other hand, was decreased in isolated mitochondria from livers of the HFD- and HFD + F-fed mice but restored toward normal levels in the HFD + G group. Thus, decreased CPT1a activity cannot explain the elevated acylcarnitine profile in the HFD and HFD + F groups, which likely is secondary to their decreased consumption. CPT1a activity inversely correlated with the levels of malonyl-CoA, as hepatic malonyl-CoA levels were increased by 50% in HFD and HFD + F mice. Supplementation of HFD with glucose normalized both malonyl-CoA and CPT1a activity (Figure 1D).
Malonyl-CoA is an intermediate in fatty acid synthesis, produced by the action of acetyl-CoA carboxylase alpha (ACACA/ACC1). ACC1 activity is inhibited by phosphorylation catalyzed by 5′-AMP kinase (AMPK) (Hardie and Pan, 2002). Phosphorylation of AMPK on threonine-172 and ACC1 on serine-79 were decreased in both HFD and HFD + F groups, but their phosphorylation was restored to the level of chow-fed controls in the HFD + G group (Figures 1E and 1F). These findings correlate with the degree of insulin resistance in these mice, as HFD- and HFD + F-fed, but not HFD + G-fed, mice developed hyperinsulinemia with increased basal glycemia and fasting HOMA-IR score and impaired glucose tolerance (Figure S1A) (Softic et al., 2017). Increased insulin levels lead to lower AMPK phosphorylation as insulin strongly inhibits AMPK activity by reducing phosphorylation on threonine-172 (Jeon, 2016). Increased insulin sensitivity and lower insulin levels in the HFD + G mice, as compared to HFD and HFD + F groups (Figure S1A) are, at least in part, mediated by increased catabolism of branched-chain amino acids, leading to reduced hepatic (Softic et al., 2017) and serum levels of valine and leucine/isoleucine (Figure S1B). Taken together, these data indicate that mice fed the glucose, but not fructose, supplemented HFD have increased AMPK activity, leading to increased ACC1 phosphorylation and less active ACC1, thus producing less malonyl-CoA and increased mitochondrial CPT1a activity. Although malonyl-CoA is known to negatively regulate CPT1a activity (McGarry et al., 1977), malonyl-CoA is likely diluted away in the process of mitochondrial isolation, suggesting that additional mitochondria-intrinsic regulatory mechanisms are involved in mediating the effects of glucose or fructose supplementation on FAO.
The differential effects of fructose versus glucose on hepatic fatty acid metabolism were reflected in serum free fatty acid (FFA) and ketone levels. After an overnight fast, chow-fed mice supplemented with glucose had higher serum FFA and β-hydroxybutyrate levels than mice on fructose or regular water. Similarly, HFD + G feeding increased FFA and β-hydroxybutyrate levels relative to HFD or HFD + F feeding (Figure 1G). These findings suggest that glucose supplementation, in either the chow- or HFD-fed context, leads to higher levels of FFA in the plasma and a higher rate of FAO than observed with fructose supplementation. Increased FFA and β-hydroxybutyrate levels with glucose supplementation of chow and HFD-fed mice can also be explained by decreased insulin levels in these mice.
In agreement with increased CPT1a activity and greater ketogenesis with glucose supplementation of HFD, hepatic expression of cytoplasmic fatty acid transport protein 2 (Fatp2) was increased with glucose supplementation in chow and further increased in glucose-supplemented HFD mice (Figure S1C). The expression of fatty acid-binding protein 1 (Fabp1) and fatty acid transporter CD36, on the other hand, was increased by both glucose and fructose supplementation of chow- or HFD-fed mice and by the HFD. Expression of fatty acid transport protein 5 (Fatp-5) was not altered by sugar supplementation but was increased in all groups on HFD.
To investigate the effects of glucose versus fructose on FFA oxidation in vitro, AML-12 cells were pretreated with 25 mM glucose or fructose for 48 h, and FFA oxidation was assessed using a Seahorse flux analyzer. Interestingly, fructose-treated hepatocytes had a 50% lower oxygen consumption rate (OCR) when exposed to a fatty acid mixture of oleate and palmitate than glucose-pretreated hepatocytes (Figure 1H). This difference persisted following inhibition of glucose utilization by 2-deoxyglucose treatment but was abolished after CPT1a inhibition by etomoxir, indicating that fructose and glucose treatment induced different rates of mitochondrial FFA oxidation, even under insulin-free conditions in cell culture.
Dietary Sugars Regulate Expression of FAO Genes
FAO, ketogenesis, and Cpt1a expression are known to be activated by FGF21. Hepatic Fgf21 mRNA was elevated in mice receiving hypercaloric diets with the highest increase observed with glucose supplementation or HFD plus glucose or fructose (Figure S2A). Serum FGF21 levels almost exactly paralleled the hepatic mRNA expression (Figure S2B). FGF21 acts via FGF receptor 2 (FGFR2) and its co-receptor, β-klotho. Both of these transcripts were not affected by sugar supplementation but were increased in all HFD-fed mice compared to chow-fed groups (Figure S2C). The expression of FGF receptor 4 (Fgfr4), the most abundant FGF receptor in the liver, was increased in mice fed the HFD, with and without fructose, but was decreased to chow diet levels in the HFD + G group. A decrease in hepatic FGFR4 has been previously associated with increased Fgf21 expression and enhanced FAO (Ge et al., 2014; Yu et al., 2013).
To assess the impact of sugar supplementation on hepatic mRNA expression of FGFs and FAO genes, we performed RNA sequencing (RNA-seq) analysis after 10 weeks on chow, HFD, HFD + F, and HFD + G diets (Figures 2A and 2B) (BioProject: PRJNA391187). As compared to chow, HFD-fed mice showed upregulation of calmodulin-2 and −3 (Calm2 and Calm3), which regulate calcium levels required to support FGF21 signaling (Moyers et al., 2007). HFD + F-fed mice had increased expression of protein kinase cAMP-dependent type II regulatory subunit beta (Prkar2b) and adenylate cyclase 7 (Adcy7), involved in conversion of ATP to cAMP, also required for FGF21 signaling (Hondares et al., 2011), while the HFD + G group showed increased expression of Fgf1 and Akt1, both of which have been associated with improved metabolism. Furthermore, in agreement with our qPCR data, mice in the HFD + G group showed decreased Fgfr4 expression. These alterations in FGF signaling were associated with changes in genes regulating fatty acid metabolism. Thus, the HFD group had increased mRNA levels of elongation of very long-chain fatty acids, protein 3 (Elovl3) and 5 (Elovl5), which regulate elongation of fatty acids. The HFD + F group had increased expression of Acaca/Acc1 and fatty acid synthase (Fasn), which produce endogenous fatty acid, including malonyl-CoA, whereas the HFD + G group had increased Cpt1a, Acadl, and Hadha expression, which mediate mitochondrial FAO.
Figure 2. Dietary Sugars Regulate Expression of FAO Genes.

(A and B) RNA-seq analysis of genes involved in (A) Fgf receptor signaling and (B) fatty acid metabolism pathways after 10 weeks on respective diets.
(C and D) Fgf21 expression (C) and serum levels (D) as well as Cpt1α expression (E) in mice treated with control or siRNA against Khk for the last 4 weeks of a 10-week study.
(F) Western blot analysis of CPT1a and ACC1 following KHK siRNA treatment.
(G and H) Hepatic metabolomic analysis of (G) unsaturated long-chain and (H) short- and medium-chain acylcarnitines in control or KHK-siRNA-treated mice.
The data are represented as mean ± SEM, n = 8 mice per group. # denotes the significant difference as compared to the chow control, whereas * signifies the significant difference between the groups under the crossbar. Complete names of acylcarnitines are provided in Table S2.
qPCR analysis confirmed the RNA-seq data and demonstrated increased expression of Acadl and Acadm, as well as trifunctional enzyme Hadha, in the HFD + G-, but not HFD + F-fed, mice. (Figure S2D). Malonyl-CoA decarboxylase (Mcd) mRNA expression (Figure S2D), which catalyzes breakdown of malonyl-CoA, thereby increasing CPT1a activity, was also increased with glucose supplementation on both chow and the HFD. Combined RNA-seq and metabolomic analysis revealed that one of the top three most significantly altered pathways between fructose and glucose supplementation of HFD was mitochondrial beta-oxidation of long-chain saturated fatty acids. Fructose supplementation of HFD was not different from HFD, both in terms of gene expression and acylcarnitine levels (Figure S2E). Conversely, glucose supplementation of HFD showed increased expression of genes involved in mitochondrial FAO with concomitant decrease in acylcarnitine intermediates of beta-oxidation. Direct comparison of HFD + F to HFD + G groups showed decreased gene expression and an increased acylcarnitine profile, suggestive of decreased FAO with fructose, as compared to glucose supplementation of HFD.
The effect of fructose on the expression of FAO genes and acylcarnitine intermediates was directly tested by feeding mice HFD, HFD + F, and HFD + G diets for 6 weeks, followed by treatment with small interfering RNA (siRNA) directed at ketohexokinase (Khk) or control siRNA for an additional 4 weeks. Khk catalyzes the first step of fructose metabolism, and we have previously shown siRNA can achieve over 90% knockdown of hepatic Khk mRNA and protein levels (Softic et al., 2017). Interestingly, knockdown of KHK resulted in further increase in Fgf21 expression in HFD- and HFD + F-fed mice, but not in HFD + G mice, which had the highest levels of Fgf21 (Figure 2C). Again, serum FGF21 levels correlated closely with liver Fgf21 mRNA expression (Figure 2D). Cpt1a mRNA was also increased in HFD and HFD + F groups following KHK knockdown, while it did not increase in the HFD + G group, which already had the highest Cpt1a expression (Figure 2E). CPT1a protein was robustly increased in all three groups of mice on HFD treated with KHK siRNA, whereas phosphorylation of ACC1 was not affected by KHK knockdown (Figure 2F). The long-chain acylcarnitine profile increased with KHK knockdown only in the HFD + F group (Figure 2G), while short- and medium-chain acylcarnitine profile was increased in livers of mice treated with KHK siRNA on all three HFDs (Figure 3H). The increased acylcarnitine profile positively correlates with increased CPT1a protein levels and likely represents increased CPT1a activity following KHK knockdown.
Figure 3. Fructose Supplementation Alters Mitochondrial Morphology.

(A and B) Mitochondrial number/area (A) and morphology (B) as assessed by electron microscopy from the livers of mice after 10 weeks on the diets.
(C and E) Western blot (C) and quantification (E) of mitochondrial fission and fusion proteins in the liver.
(E and F) Number and the percent of small mitochondria (E) and mitochondrial morphology (F) in HFD + F-fed mice treated with control or siRNA against Khk.
(G and H) Western blot (G) and quantification (H) of proteins involved in mitophagy from the same mice.
The data are represented as mean ± SEM, n = 3–6 mice per group. # denotes the significant difference as compared to the chow control, whereas * signifies the significant difference between the groups under the crossbar.
Thus, while HFD supplemented with either fructose or glucose resulted in increased Fgf21 expression and serum levels, only glucose supplementation was associated with increased expression of genes involved in FAO. These effects were dependent on fructose metabolism, as knockdown of KHK increased Fgf21 and Cpt1a expression in HFD- and HFD + F-fed mice. Furthermore, CPT1a protein was increased on all three HFDs following KHK knockdown, which was associated with increased acylcarnitine levels. Thus, knockdown of KHK led to an increase in mRNA expression and protein levels of enzymes regulating beta-oxidation, which suggests transcriptional control of FAO by fructose.
Fructose Supplementation Alters Mitochondrial Morphology
As sugar supplementation modified malonyl-CoA levels and expression of FAO genes, we investigated whether fructose or glucose supplementation also had an effect on mitochondrial morphology. Quantitative analysis of electron microscopy images of livers from mice after 10 weeks feeding of the chow diet supplemented with fructose or glucose revealed increased mitochondrial number with either sugar supplementation (Figure 3A). HFD did not significantly affect mitochondrial number, whereas HFD + F mice showed a further increase in mitochondrial number, while HFD + G mice did not, as compared to HFD group. Mitochondrial number was the highest in the HFD + F group, which was increased 3-fold over both chow- and HFD-fed groups (Figure 3A). Mitochondrial area, on the other hand, was decreased in mice on all hypercaloric diets, with the smallest mitochondria observed in the HFD + F group (Figure 3A). Thus, while mitochondrial number was highest in the HFD + F group, average mitochondrial area was the lowest, suggestive of increased mitochondrial fission. Indeed, when plotted according to the mitochondrial size, the largest percent of small mitochondria, i.e., mitochondria less than 500 nm in length, was found in fructose-supplemented mice (Figures S3A and S3B).
Mitochondrial size is regulated by a dynamic interplay between mitochondrial fission and fusion. Consistent with the changes in mitochondrial size, chow + F and chow + G mice had 60%–80% increased levels of mitochondrial fission protein, FIS1 (Figures 3C and 3D). HFD or HFD + G did not result in increased FIS1 levels, but HFD + F mice exhibited a 60% increase in FIS1 protein. OPA1, a mitochondrial protein that regulates fusion, was increased by 30% (n = 6; p < 0.05) in chow + F and chow + G mice. However, opposite to FIS1 findings, HFD + G-fed, but not HFD + F-fed, mice had a significant 20% increase in OPA1 levels (n = 6; p < 0.05). Inadequate mitochondrial remodeling leads to mitophagy and a compensatory increase in mitochondrial biogenesis. Consistent with this, PGC1α, a protein involved in mitochondrial biogenesis, increased by 60% in the chow + F group and doubled in the HFD + F group but was not changed in the chow + G or HFD + G groups (Figure 3C). Likewise, levels of the mitochondrial pore protein VDAC were significantly increased in the HFD + F group and tended to be increased in chow + F group, indicative of higher mitochondrial mass in these groups.
The changes in mitochondrial number and size observed in mice on the HFD + F diet were dependent on fructose metabolism, as knockdown of KHK with siRNA resulted in normalization of mitochondrial number and a decreased percent of small mitochondria, as compared to control siRNA-treated mice (Figures 3E and 3F). Mitochondrial integrity was also dependent on fructose metabolism as PINK1, a marker of damaged mitochondria, was increased following KHK knockdown in HFD- and HFD + F-fed mice, but not in HFD + G-fed mice, which had the lowest levels of PINK1 (Figures 3G and 3H). In healthy mitochondria, PINK1 is cleaved and we observed increased cleaved PINK1 in HFD- and HFD + F-fed mice following KHK knockdown. Accumulation of full-length PINK1 leads to recruitment and activation of PARKIN, which triggers mitophagy (Ding and Yin, 2012). PARKIN was increased in HFD following KHK knockdown, and there was a trend toward increased levels in HFD + F-fed mice treated with KHK siRNA, but not in HFD + G-fed mice, which had correspondingly low levels of PINK1 (Figure S3C). ULK1 is a nutrient-dependent kinase that is involved in early steps of autophagosome biogenesis (Ding and Yin, 2012). Its protein levels were lowest in HFD and HFD + F groups, and they increased to the levels observed in HFD + G group upon KHK knockdown (Figures 3G and 3H). Similarly, the autophagy adaptor protein p62 was the lowest in HFD and HFD + F groups, as compared to the HFD + G group, and it also increased following KHK knockdown in these groups, but not in HFD + G group, which already had elevated levels. p62 directly interacts with LC3B to recruit autophagosomal membranes to the mitochondria, and we find reduced LC3B in HFD and HFD + F mice, but not in HFD + G-fed mice (Figure S3C). Taken together, these data indicate that damaged mitochondria accumulate in HFD- and HFD + F-fed mice, as exemplified by increased PINK1 in these groups, but not in HFD + G-fed mice. In spite of increased PINK1, mitophagy was not activated as ULK1, p62, and LC3B were lower in these groups, as compared to the HFD + G group. Knockdown of KHK resulted in increased levels of total PINK1 and PARKIN, along with concomitant increases in ULK1, p62, and LC3B to levels similar to that in the HFD + G group. Thus, increased removal of damaged mitochondria in the HFD + F-fed group could explain the decreased mitochondrial number in this group following KHK knockdown.
Dietary Manipulations Affect Abundance of Mitochondrial Proteins
To more thoroughly assess the effect of diets on liver mitochondria, we performed unbiased, quantitative proteomic analysis (Gillet et al., 2012) on isolated mitochondria from livers of mice after 10 weeks on a diet. Of the 838 proteins quantified in the 30 samples (5 biological replicates from 6 diets), 275 proteins showed statistically significant changes in relative abundance in at least one condition. Heatmaps of Z scores for the quantified proteins are shown in Figure 4A and Meyer et al. (2018). When normalized to the chow group, 13 mitochondrial proteins were increased and 23 were decreased in the fructose-supplemented mice, while 20 proteins were increased and 14 decreased in the glucose-supplemented mice (Figure 4B). Relative to chow, HFD diet alone decreased 94 proteins, while 7 were increased. This effect was further potentiated in HFD + F mice, such that 205 proteins were decreased and 24 were increased. By contrast, the mitochondrial protein profile was partially rescued in HFD + G mice, with only 57 proteins decreased and 22 proteins increased.
Figure 4. Dietary Manipulations Affect Abundance of Mitochondrial Proteins.

(A) Heatmap of 279 significantly changed mitochondrial proteins.
(B) Up- and downregulated proteins as compared to the chow group.
(C and D) A volcano plot analysis and western blot conformation of proteins altered in (C) chow versus HFD and (D) HFD + F versus HFD + G groups.
(E) Significantly changed proteins clustered into functional categories in mice on any HFD as compared to chow.
A volcano plot comparing chow and HFD shows that proteins involved in FAO, such as type II hydroxyacyl-CoA dehydrogenase (HADH-II), CPT1a, and 3-hydroxybutyrate dehydrogenase 1 (BDH1), were among the most upregulated by HFD, while several cytochrome P450 proteins, such as CYP2c29, CYP1a2, and CYP3a11, were among the most downregulated in response to HFD (Figure 4C). Western blot analysis confirmed that CPT1a and BDH1 were increased on the HFD, whereas CYP3a was increased on the chow diet. Comparing effects of fructose and glucose in HFD-fed mice, three proteins were specifically increased in the HFD + F group: succinate-CoA ligase α-subunit (SUCLG1), protein tyrosine phosphatase mitochondrial 1 (PTPMT1), and cytochrome P450 2c polypeptide 29 (CYP2c29) (Figure 4D). SUCLG1 catalyzes the conversion of succinyl-CoA and ADP to succinate and ATP, thus producing ATP without utilizing canonical mitochondrial oxidative phosphorylation. PTPMT1 dephosphorylates mitochondrial proteins and is required for cardiolipin biosynthesis, thereby playing an essential role in ATP production and mitochondrial morphology, while CYP2c29 produces superoxide. Together these could contribute to decreased ATP production in fructose-supplemented mice. Conversely, in HFD + G mice, many of the increased proteins were involved in reactive oxygen species (ROS) detoxification, including carbonic anhydrase 3 (Ca3), glutathione s-transferase alpha 3 and Pi 1 (GSTA3 and GSTP1), and heat shock 70-kDa protein 5 (HSP5a), suggesting that glucose supplementation decreases mitochondrial ROS levels. Proteomic analysis was confirmed by western blot, which showed increased HSPA5 and GSTA3 in the HFD + G group, whereas SUCLG2, β-subunit, was increased in the HFD + F group.
Functional-term enrichment analysis of significantly altered mitochondrial proteins identified in any of the three HFD groups, compared to the control group, revealed several functional clusters (Figure 4E). These include two clusters involved in metabolism of lipids and ketone body metabolism, a cluster involved in steroid hormone biosynthesis, a cluster involved in gluconeogenesis, and several clusters involved in amino acid metabolism. Interestingly, although proteomics was performed on mitochondrial fractions, we also identified a number of significantly altered ribosomal proteins. These could represent ribosomes associated with mitochondria for co-translational import of mitochondrial proteins (Lesnik et al., 2014). The altered ribosomal proteins included structural ribosome subunits and proteins involved in RNA binding, mRNA processing, and translational elongation (Figure S4A). Out of 80 structurally distinct ribosomal proteins, 23 proteins compromising the 40S subunit, 28 proteins in the 60S subunit, and 3 elongation factors were decreased in the HFD + F group (Figure S4B). About half of these proteins were also decreased in the HFD group, while glucose addition to the HFD protected against many of these changes. A decrease in ribosomal proteins may lead to decreased protein translation and partially explain the decreased number of total mitochondrial proteins observed in HFD and HFD + F, but not HFD + G, groups.
In Vitro Sugar Supplementation Replicates Mitochondrial Phenotype in Mice
To determine if the in vivo effects on liver mitochondrial function were due to cell-autonomous effects of hepatic sugar metabolism, AML-12 hepatocytes were cultured in low-glucose DMEM medium and supplemented with either 25 mM fructose or 25 mM glucose for 48 h in the presence of BSA loaded with FFAs or BSA alone. Analogous to their in vivo effects, fructose and glucose supplementation increased mitochondrial volume, as measured by MitoTracker green fluorescence (Figure 5A). Addition of 0.5 mM oleate/palmitate mixture also increased mitochondrial number, an effect that was enhanced by co-addition of glucose or fructose. Furthermore, intracellular TGs increased by 50%–60% with fructose and glucose supplementation of culture medium, compared to cells cultured in the same medium with no added sugars. Addition of FFA for 48 h doubled TG accumulation compared to the control, and this effect was additive to the effect of sugar, such that fructose and glucose supplementation in the presence of FFA increased TG accumulation to 2.5- to 3-fold over the control (Figure S5A). These effects were confirmed by BODIPY staining (Figure 5B) and are in agreement with increased lipid accumulation in mice supplemented with fructose and glucose on chow or HFD, as previously reported (Softic et al., 2017). Interestingly, ROS levels, as measured by the superoxide dye dihydroethidium (DHE), were increased by FFA and fructose, but not glucose supplementation (Figure 5C). Analogous to in vitro findings, increased DHE staining was also observed in the livers of chow + F and HFD + F mice, whereas chow + G and HFD + G mice had lower in vivo ROS staining (Figure S5B). This is in agreement with proteomic analysis, showing an increased amount of proteins involved in ROS detoxification in HFD + G as compared to HFD + F mice.
Figure 5. In Vitro Sugar Supplementation Replicates Mitochondrial Phenotype in Mice.

(A–C) Fluorescence in AML-12 hepatocytes following 24 h treatment with fructose, glucose, or FFA mixture, visualized by (A) MitoTracker green, (B) BODIPY, or (C) dihydroethidium. Scale bar, 100 μm.
(D) Oxygen consumption rate in primary hepatocytes acutely treated with 25 mM glucose or fructose followed by treatment with oligomycin, FCCP, and rotanone plus antimycin. The data are represented as mean ± SEM, n = 10 wells per group, * denotes significant difference between the groups under crossbar.
(E) NAD(P)H levels in primary hepatocytes treated with fructose or glucose for 10 min. Scale bar, 10 μm. Results represent two independent experiments.
(F) KHK and CPT1a protein in liver homogenates, primary mouse hepatocytes, and AML-12 immortalized hepatocytes. n = 3 lanes per group.
(G) KHK and CPT1a protein in AML-12 immortalized hepatocytes grown in media containing FBS or in serum-free media for 7 days. n = 4 lanes per group.
Seahorse analysis of primary hepatocytes exposed to 25 mM fructose also showed 20% lower OCR compared to primary hepatocytes treated with 25 mM glucose (Figure 5D). Following treatment with oligomycin, which blocks ATP synthesis, fructose-treated hepatocytes had 25% less ATP production, as compared to glucose-treated cells. NAD(P)H provides electrons for ATP synthesis, and NAD(P)H levels, as measured by multiphoton imaging in isolated primary hepatocytes, were also decreased within 10 min following addition of 25 mM fructose but were unchanged after addition of equimolar glucose (Figures 5E and S5C). Thus, in both in vivo and in vitro studies, fructose induces increases in mitochondrial number, lowers ATP and NADH levels, and increases ROS level, suggesting that fructose promotes mitochondrial dysfunction via mechanisms that are not activated by glucose.
Utilizing AML-12 cells, we wanted to confirm that KHK knockdown can increase CPT1a protein in vitro. Unfortunately, KHK protein levels were already low in AML-12 cells, as compared to liver homogenates and primary hepatocytes. However, an inverse relationship between KHK and CPT1a was preserved, so that KHK was high and CPT1a low in the liver, whereas AML-12 cells had low KHK and high CPT1a protein (Figure 5F). Indeed, cultured AML-12 cells loose many proteins that are found in mature hepatocytes, as is the case for KHK, while culturing them in serum-free media can lead to an increase in some of these mature hepatocyte markers toward normal (Wu et al., 1994). Indeed, when AML-12 cells were cultured in serum-free media for seven days, they showed more epithelial appearance with cord-like structures and had increased ceruloplasmin, decreased alpha-fetoprotein, and unchanged haptoglobulin levels, indicative of more mature hepatocytes (Figure S5D). However, AML-12 cells grown in serum-free media showed decreased KHK levels in spite of showing this more mature phenotype (Figure 5G). Nonetheless, an inverse relationship between KHK and CPT1a levels was preserved in AML-12 cells grown in fetal bovine serum (FBS) and serum-free media. Decreased KHK expression in AML-12 cells grown in serum-free media is likely mediated by lower insulin signaling, as we find decreased insulin receptor (IR) and Akt phosphorylation in these cells.
Macronutrient Composition Drives Mitochondrial Protein Acetylation
Reversible acetylation of lysine residues on mitochondrial proteins is being increasingly recognized as a post-translational modification that can affect protein function (Glozak et al., 2005; Hirschey et al., 2010, 2011). Western blotting of isolated mitochondrial proteins from liver using anti-acetyl lysine antibody showed increased protein acetylation in chow + F and chow + G mice, as compared to the chow group (Figure 6A). HFD alone had little effect on protein acetylation, whereas HFD + F caused a decrease and HFD + G feeding an increase in acetylation of several mitochondrial proteins. This could not be explained by changes in Sirt3, the major mitochondrial deacetylase, which was increased in mice fed chow + F, HFD + F, or HFD alone. The acetylation pattern of mitochondrial proteins was similar to that observed in whole cell lysates, although more acetylated proteins were apparent in whole cell lysate (Figure 6A).
Figure 6. Macronutrient Composition Dictates Mitochondrial Protein Acetylation.

(A) Western blot analysis of mitochondrial lysates using anti-acetyl lysine antibody from the mice after 10 weeks on diets.
(B) Acetylation profile measured by nano-liquid chromatography-tandem mass spectrometry in mice on diets as compared to mice with global Sirt3 loss. n = 5 mice per group.
(C) Heatmap analysis of mitochondrial acetylation profile.
(D and E) A volcano plot analysis of acetylated lysine sites in (D) chow versus HFD and (E) HFD + F versus HFD + G groups.
(F) Acetylated sites on Opa1 protein.
(G) Acetylated lysine sites on proteins involved in the fatty acid beta-oxidation pathway.
To more precisely define the nature of mitochondrial protein acetylation induced by diets, we performed quantitative MS following anti-acetyl lysine immunoprecipitation of trypsin-digested mitochondrial proteins from liver (Meyer et al., 2017). A total of 1,155 acetylated sites on 343 proteins were identified. Of these, 857 acetylated sites (74%) on 308 proteins showed a statistically significant (FDR < 0.01) fold change > 1.5 induced by at least one diet, when corrected to total protein levels. These data show that sugar and HFD are strong drivers of mitochondrial protein acetylation (Figure 6B) (Meyer et al., 2018). Consistent with the fact that changes in acetylation did not correlate with Sirt3 levels, only a fraction of these had been previously identified as Sirt3-regulated sites in mice (Dittenhafer-Reed et al., 2015; Rardin et al., 2013). Also consistent with the immunoblots, both fructose and glucose supplementation in chow-fed mice produced increased acetylation of multiple mitochondrial proteins, while high-fat feeding produced a modest decrease in acetylation of mitochondrial proteins (Figure 6C). Interestingly, fructose supplementation of the HFD produced a further decrease in acetylation, whereas glucose supplementation increased acetylation of many mitochondrial proteins toward normal chow levels. The changes in protein acetylation were not driven by the changes in the amount of protein, as site-level acetylation changes were normalized for each mitochondrial protein (also see Meyer et al., 2018)
A volcano plot of acetylation sites comparing the HFD to chow identified proteins involved in FAO as the most hyperacetylated by the HFD. These included K234 on acetyl-CoA acyltransferase 2 (ACCA2), K459 of enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase (EHHADH), and K73 on HADH (Figure 6D). On the other hand, acetylation of proteins regulating ATP and NADH levels, such as K181 on adenylate kinase 2 (AK2), K250 on ubiquinol-cytochrome C reductase core protein II (UQCRC2), and K106 on NADH:ubiquinone oxidoreductase subunit A8 (NDUFA8), were among the most significantly decreased in mice on the HFD (Figure 6D). Comparison of acetylated sites between HFD + F and HFD + G groups identified five hyperacetylated sites specific to the fructose-supplemented group (Figure 6E). The most significantly changed of these was K446 of prolyl 4-hydroxylase subunit beta (P4HB), the enzyme that hydroxylates prolyl residues of preprocollagen. Interestingly, increased collagen formation has been reported in livers of mice supplemented with fructose on the HFD (Kohli et al., 2010).
A large number of significantly increased acetylated sites were found in HFD + G compared to the HFD + F group (Figure 6E). Some of the most significantly increased sites in the HFD + G group were found on the proteins essential for mitochondrial network organization and ATP synthesis. These include acetylation on K423 of the ATPase family, AAA domain containing 3a (ATAD3), K46 of mitochondrial-encoded ATP synthase 8 (MTATP8), K617 of heat shock 70-kDa protein 9 (HSPa9), and K194 of pyruvate dehydrogenase complex component X (PDHX), which links glycolysis to the Krebs cycle. While both sugar and the HFD provide calories, they exert unique and often opposite effects on mitochondrial protein acetylation. For example, K834 on Opa1 was exclusively hyperacetylated with fructose or glucose supplementation on the chow diet, while the HFD induced hypoacetylation on K596, regardless of sugar intake (Figure 6F).
Mitochondrial proteins regulating essential metabolic functions, such as FAO, ATP synthesis, and ROS production showed robust acetylation changes in response to diet. Proteins involved in FAO, such as the alpha and beta subunits of HADH (HADHA and HADHB); short, medium, long, and very long-chain ACAD; and methylmalonyl-CoA mutase (MUT1), were almost uniformly hyperacetylated in chow-fed mice on fructose or glucose supplementation and almost uniformly hypoacetylated in mice fed on the HFD or HFD + F (Figure 6G). As with many other metabolic features, mice fed the HFD + G showed a unique response, with increased acetylation at specific sites in these proteins. Similarly, acetylation of proteins involved in regulating mitochondrial ROS levels, such as members of the heat shock families HSP10, HSPE1, HSP60, HSPD1, HSP70, HSPA9, and HSPA5, as well as catalase (CAT) and mitochondrial superoxide dismutase (SOD2) were hyperacetylated in chow + F and chow + G mice and almost uniformly hypoacetylated in HFD and HFD + F groups, but not in the HFD + G group (Figure S6B). Finally, proteins involved in ATP synthesis also had increased acetylation in chow + F and chow + G mice, while acetylation was greatly decreased in HFD and HFD + F mice, but not in the HFD + G-fed group (Figure S6C).
Protein Acetylation Correlates with Enzymatic Activity and In Vivo Metabolic Phenotype
To further define these diet-induced acetylation changes on enzymes regulating FAO, we performed immunoprecipitation followed by immunoblot analysis with an anti-acetyl lysine antibody. In agreement with MS data, immunoprecipitation and western blotting demonstrated increased acetylation of the alpha subunit of HADHA by fructose and glucose supplementation in chow-fed mice and by glucose supplementation of the HFD (Figure S7A). Similar changes in acetylation pattern were observed for the beta subunit of HADHB (Figure S7B). Immunoprecipitation of acetyl lysine residues followed by immunoblot for ACADL demonstrated that ACADL was uniquely hyperacetylated with fructose- but not glucose-supplemented HFD (Figures 7A and S7C). Hyperacetylation of ACADL is known to decrease its enzymatic activity (Hirschey et al., 2010), which is in agreement with our data showing that HFD + F mice have lower beta-hydroxybutyrate levels, as compared to HFD + G mice. In contrast to the immunoblot data, however, MS analysis did not show increased acetylation of ACADL in the HFD + F group. This is likely due to different antigens recognized by the antibody. Bharathi et al. reported that acetylation of K318/322 in ACADL correlated with decreased activity (Bharathi et al., 2013), and we used this antibody, but K318/322 acetylation sites were not identified in our spectrometry analysis. To confirm that acetylation of ACADL in HFD + F mice is dependent on fructose metabolism, we preformed immunoprecipitation and immunoblot experiments following KHK knockdown in mice on all three HFDs. As shown above, acetylation of ACADL was highest in HFD + F-fed mice as compared to mice on HFD and HFD + G diets. Acetylation of ACADL was decreased in HFD + F mice treated with KHK siRNA, indicating that acetylation of this protein was indeed dependent on fructose metabolism.
Figure 7. Protein Acetylation Correlates with Enzymatic Activity and In Vivo Metabolic Phenotype.

Acetylation intensity of (A) ACADL and (B) CPT1a proteins determined by anti-acetyl lysine immunoprecipitation followed by western blot with anti-ACADL and anti-CPT1a antibody from livers of mice after 10 weeks on the diets. Numbers represent 3 pooled samples from each group. # denotes the significant difference as compared to the chow control, whereas * signifies the significant difference between the groups under the corssbar. The bottom panels show immunoprecipitation with anti-acetyl lysine antibody, followed by western blot for ACADL and CPT1a in mice on all three HFDs for 10 weeks, treated with control or KHK siRNA for the last 4 weeks. Each lane represents 3 pooled samples from each group.
Immunoprecipitation and western blotting also demonstrated that CPT1a showed a trend toward increased acetylation by fructose or glucose supplementation on the chow diet and it was markedly hyperacetylated in HFD- and HFD + F-fed, but not HFD + G-fed, mice (Figures 7B and S7C). In HFD mice, this is in part due to increased total CPT1a protein, as many proteins involved in FAO were increased on the HFD. Hyperacetylation of CPT1a in HFD and HFD + F mice may have contributed to reduced CPT1a enzymatic activity in isolated mitochondria from these mice. We assessed CPT1a acetylation following KHK knockdown and found that acetylation was increased in mice on all three HFDs treated with KHK siRNA. This is likely due to increased CPT1a protein in these groups following KHK knockdown, in agreement with findings above (Figure 2F).
Some differences in estimated levels of acetylation occurred depending on the method of analysis. There are several reasons for this. First, there is a fundamental difference between the two methods. Immunoblotting depends on the relative affinity of the anti-acetyl lysine antibody for the different acetylated groups in the protein, but otherwise cannot distinguish site-specific acetylation or polyacetylation. MS, on the other hand, identifies peptides from each acetylated version of the protein as separate chemical entities but for methodological reasons may fail to identify all of the acetylated sites. In addition, in the MS analysis, we normalized the changes in acetylation of each peptide based on measurement of changes in protein, as estimated by MS. In addition, some of the differences may also arise from changes in total protein levels of individual mitochondrial proteins in the liver of mice on the chow versus HFD. These three factors likely contribute to differences in estimated acetylation of CTP1a. For example, MS analysis shows acetylation at K195 and K634 to be low on HFD, low on HFD + F, and lowest on HFD + G, whereas western blot analysis shows total levels of acetylated CPT1a to be high on HFD, high on HFD + F, and mildly elevated in the HFD + G group. Since CPT1a total protein increases on HFD, western blot image of CPT1a acetylation is expected to be higher on the HFD. Conversely, acetylation of CPT1a is expected to be lower on MS analysis since acetylated protein is divided by the total amount of protein. This was also observed following KHK knockdown where increased total CPT1a protein likely drives an increase in acetylated CPT1a. These differences between the methods are to some extent unavoidable and have been discussed in several reviews (Aebersold et al., 2013; Bluemlein and Ralser, 2011).
Since acetyl-CoA is the primary donor for acetylation, it is worth noting that hepatic acetyl-CoA levels were elevated with fructose supplementation on the chow diet but were reduced by the HFD alone (Figure S7D) (Meyer et al., 2018). However, acetylation of many mitochondrial proteins was largely independent of hepatic acetyl-CoA levels, as well as Sirt3 levels, indicating that fat and sugar intake may regulate mitochondrial protein acetylation by mechanisms independent of these variables. While acetylation changes induced by different diets are robust, further studies will be needed to determine how the acetylation on a specific lysine residue correlates with a protein’s enzymatic activity. Taken together, our data show that fructose and glucose supplementation have profound effects on mitochondrial protein acetylation, and these sugars can also modify the acetylation response to an HFD. Acetylation effects induced by sugar supplementation, thus, represent a new regulatory pathway by which sugar metabolism affects mitochondrial FAO.
DISCUSSION
In the present study, we have further explored the mechanisms by which the two most abundant dietary monosaccharides, fructose and glucose, can alter the risk of developing obesity and its metabolic complications. We previously reported that fructose and glucose can modify the effect of a HFD on de novo lipogenesis where fructose supplementation leads to enhanced fatty acid synthesis and insulin resistance, while glucose supplementation is associated with increased TG synthesis and protection from some obesity-associated metabolic derangements (Softic et al., 2017). Here, we have explored the effects of fructose and glucose supplementation on mitochondrial function and hepatic FAO in the context of both chow and HFD. We find that chronic fructose supplementation superimposed on the HFD, as compared to calorically equivalent glucose supplementation, leads to decreased FAO, through at least three different nodes of regulation including effects on malonyl-CoA levels, mitochondrial number and function, and post-translational modifications of mitochondrial proteins.
First, fructose supplementation of HFD and HFD alone result in increased hepatic malonyl-CoA, a well-known endogenous inhibitor of FAO (McGarry et al., 1977). This is not observed in HFD + G group, whose malonyl-CoA levels are similar to chow. Increased malonyl-CoA results in decreased CPT1a activity, which is associated with increased accumulation of hepatic long-chain acylcarnitines, commonly observed with decreased FAO. These changes explain decreased serum β-hydroxybutyrate levels, a ketone body predominantly produced in the liver by oxidation of fatty acids, in HFD + F and HFD groups. These effects on malonyl-CoA levels appear to be mediated by decreased phosphorylation and inactivation of AMPK, leading to decreased phosphorylation and activation of ACC1, which catalyzes the carboxylation of acetyl-CoA to produce malonyl-CoA (Kim et al., 2017). Decreased AMPK phosphorylation in HFD and HFD + F mice likely reflects hyperinsulinemia, which develops in these mice, as insulin strongly decreases AMPK activation (Jeon, 2016). In this way, this may also represent another case of selective insulin resistance where insulin in HFD and HFD + F mice is unable to control blood glucose but is still able to stimulate de novo lipogenesis and increase malonyl-CoA levels. In vitro, fructose as compared to glucose pretreatment of cultured hepatocytes is also associated with lower FAO as measured by the O2 consumption rate of cells acutely exposed to fatty acids. In summary, fructose and glucose supplementation results in different hepatic malonyl-CoA levels via their effects on systemic insulin resistance/hyperinsulinemia, de novo lipogenesis, and AMPK activation.
A second mode of regulation by which fructose and glucose exert their discrete effects on FAO is through their effects on mitochondrial size and function. Fructose supplementation of the HFD increased mitochondrial number but decreased mitochondrial size, likely mediated by increased fission and decreased fusion. Thus, FIS1 is increased with fructose and OPA1 is increased with glucose supplementation of HFD. Mitochondrial size is also regulated by impaired mitophagy in HFD + F mice, resulting in accumulation of small and damaged mitochondria. These effects are dependent on fructose metabolism as knockdown of KHK restored mitophagy, normalized mitochondrial number, and decreased the percentage of small mitochondria in HFD + F mice. The effects on mitochondrial size directly correlate with mitochondrial protein mass, as quantified by mitochondrial proteome analysis, which is reduced in the HFD + F compared to the HFD + G group. This is accompanied by a robust decrease in many proteins comprising large and small ribosomal subunits associated with the mitochondria, likely reflecting a decrease in synthesis of mitochondrial proteins. Decreases in mitochondrial proteins correlated with a decreased mRNA level of enzymes involved in FAO such as Cpt1a, Acadl, and Hadha with fructose as compared to glucose supplementation. These changes are also dependent on fructose metabolism as KHK knockdown increased expression and release of FGF21 as well as mRNA expression and protein levels of CPT1a. CPT1a enzymatic activity is also likely increased, as we find increased acylcarnitine profile following KHK knockdown. Increased mRNA and protein levels of enzymes regulating FAO following KHK knockdown suggests transcriptional control of FAO by fructose. Reduced mitochondrial size and protein mass correlated negatively with mitochondrial function, such that fructose supplementation led to a decrease in ATP and NADH levels but also an increase in ROS production. Taken together, fructose, as compared to glucose supplementation, results in mitochondrial dysfunction characterized by a decrease in mitochondrial size and protein mass, including a decrease in proteins involved in the FAO pathway, which is restored following KHK knockdown.
The third, and perhaps most interesting, node of regulation is through differential effects of fructose and glucose supplementation on mitochondrial protein acetylation and function. Acetylation of ChREBP on K672 has been reported to increase its transcriptional activity (Bricambert et al., 2010), and we previously reported that ChREBP is hyperacetylated in chow + F, chow + G, and HFD + G groups, while HFD and HFD + F mice show decreased ChREBP acetylation (Softic et al., 2017). Here, we show that HADHA/B is hyperacetylated in response to glucose on both chow and HFD, which may increase its enzymatic activity. HADHA is known to be hyperacetylated by caloric restriction (Schwer et al., 2009), a condition that increases FAO. We also find increased levels of ketogenesis, FGF21 signaling, and increased expression of genes regulating FAO in the glucose-supplemented groups, confirming that the hyperacetylation of HADHA/B is occurring in the setting of increased FAO. Whereas hyperacetylation of HADHA/B may increase its enzymatic activity, hyperacetylation of ACADL has been shown to decrease its activity (Hirschey et al., 2010). We find increased ACADL acetylation in HFD + F mice, which show decreased ketogenesis, suggesting that hyperacetylation of ACADL may contribute to a decrease in FAO. Hyperacetylation of ACADL in the HFD + F group is dependent on fructose metabolism as knockdown of KHK decreased its acetylation. Additionally, we find increased acetylation of CPT1a in HFD and HFD + F groups, in concert with decreases in its enzymatic activity. However, acetylation of CPT1a is further increased following KHK siRNA treatment, in concert with increased CPT1a protein, suggesting that transcriptional regulation of CPT1a, along with changes in acetylation, play a role in determination of CPT1a activity. Thus, increased acetylation of HADHA and HADHB with glucose and increased acetylation of CPT1a and ACADL with fructose supplementation on the HFD represents a third node of regulation by which sugar intake modifies FAO. Our findings may help explain studies that have suggested that in addition to its effects on malonyl-CoA levels, fructose-mediated inhibition of FAO must also occur within the β-oxidation cycle (Prager and Ontko, 1976).
Whereas previous studies have shown that acetylation of mitochondrial proteins in the liver is increased in conditions with limited nutrient availability such as fasting (Kim et al., 2006) and caloric restriction (Schwer et al., 2009), we find major increases in mitochondrial protein acetylation with caloric excess created by fructose and glucose supplementation for 10 weeks. By contrast, caloric excess from HFD over the same time span tends to decrease acetylation of some, but not all, proteins. The acetylation changes induced by sugar intake appear to be independent of the major mitochondrial deacetylase Sirt3,- since Sirt3 levels do not change in response to the different dietary regimens. This is analogous to changes in acetylation induced by ethanol intake, which are also independent of Sirt3 levels (Fritz et al., 2013). Our findings in HFD are in contrast to previous studies showing increased acetylation after 13–16 weeks of the HFD (Hirschey et al., 2011; Kendrick et al., 2011). Several methodologic differences might account for the differences between these studies, including the length of dietary exposure, the time of day when samples were collected, whether samples were collected in the fed versus fasted state, and the different sucrose content of HFDs used in the studies, as each of these can affect protein acetylation. In addition to modifying overall acetylation levels, sugar supplementation affects acetylation on different lysine residues than those modified by HFD. This was observed with OPA1 acetylation, where sugar supplementation increased acetylation on lysine 834, while HFD decreased acetylation on lysine 596. Sugar intake can also further modify the acetylation pattern induced by the HFD. For example, OPA1 acetylation was decreased by the HFD and further decreased by the addition of fructose but not by the addition of glucose to the HFD, indicating the regulatory role of specific sugars. In addition to acetylation, dietary composition and sugar supplementation also have broad effects on protein succinylation (Meyer et al., 2018), indicating that fructose and glucose exert effects on multiple post-translational modifications.
In both humans (Boesiger et al., 1994) and mice (van den Berghe et al., 1977), hepatic ATP levels decrease upon acute fructose injection, and this effect is thought to be secondary to rapid fructose phosphorylation, leading to ATP depletion. Here, we find that fructose-pretreated hepatocytes produce less ATP but also have decreased hepatic NAD(P)H levels and increased ROS production, as compared to glucose-treated cells. This decrease in hepatic NAD(P)H levels cannot be explained by rapid fructose phosphorylation and may be explained by decreased mitochondrial function, including decreased FAO, as etomoxir, a CPT1a inhibitor, is known to decrease both NADH and ATP levels (Pike et al., 2011). In addition to well-documented effects of fructose to decrease mitochondrial ATP production (Boesiger et al., 1994; van den Berghe et al., 1977) and increase ROS levels (Zhang et al., 2015), our findings showing that fructose intake also affects hepatic mitochondrial morphology and function can potentially account for mitochondrial dysfunction observed in patients with NAFLD who have high fructose intake (Abdelmalek et al., 2010; Ouyang et al., 2008; Thuy et al., 2008). Development of fatty liver disease in humans is associated with mitochondrial dysfunction characterized by increased mitochondrial number (Li et al., 2014), loss of mitochondrial cristae with paracrystalline inclusions (Sanyal et al., 2001), and an increased hepatic long-chain acylcarnitine profile (Lake et al., 2015). Furthermore, patients with NAFLD also have decreased hepatic ATP levels (Cortez-Pinto et al., 1999) and increased ROS levels (Seki et al., 2002). This is analogous to the mitochondrial phenotype observed in our HFD + F mice. Our study shows that fructose supplementation on HFD leads to mitochondrial dysfunction and decreased FAO, which in addition to the effect of fructose to promote de novo lipogenesis is likely an important risk factor for development and progression of NAFLD in patients with increased fructose consumption (Abdelmalek et al., 2010; Ouyang et al., 2008; Thuy et al., 2008).
In conclusion, sugar supplementation can modify the effects of the HFD on liver lipid composition and insulin resistance. Fructose supplementation increased fatty acid synthesis mediated via upregulation of SREBP1c and ChREBP-β where glucose supplementation increased TG synthesis associated with upregulation of ChREBP (Kim et al., 2016; Softic et al., 2017). In this manuscript, we also show that fructose and glucose supplementation of the HFD has dichotomous effects on mitochondrial function and FAO, which are mediated via at least three unique modes of regulation. Fructose, as compared to glucose supplementation, increases hepatic malonyl-CoA levels, exerts different effects on mitochondrial size and function, and alters acetylation of proteins involved in the FAO pathway. Indeed, fructose supplementation on HFD increases acetylation of ACADL and CPT1a, while glucose supplementation results in increased acetylation of HADA/B, uncovering a novel pathway by which sugar intake affects protein post-translational modifications that are associated with changes in hepatic FAO. These data indicate that along with its well-known effects to promote fatty acid synthesis, fructose supplementation in the presence of an HFD decreases mitochondrial metabolism, which may contribute to the association of high fructose intake with the development of obesity and its metabolic complications.
Limitations of Study
In rodent studies, all obesogenic HFDs contain some amount of sugar as a major ingredient. The HFD used in this study (D12492) contains the lowest amount of sucrose (a dimer of glucose and fructose) that we could identify, but it still adds 8.9% of sucrose per gram of diet consumed. Thus, fructose or glucose supplementation of the HFD provided additional sugar to a diet that already contains some amount of sugar. In light of this understanding, we can reason that drinking additional fructose has similar metabolic effects as eating the HFD since there were no major differences between HFD and HFD + F groups. On the other hand, drinking glucose-sweetened water exhibits similar effects as decreasing the percent of calories from fat in the diet, which in essence occurred when we provided sugar calories in water to mice consuming the HFD. Since all HFDs contained some fructose as part of the sucrose content, it is not unexpected that knockdown of KHK can lead to metabolic improvements and an increase in CPT1a on all three diets. The smallest incremental improvement was observed in the HFD + G group, which had the lowest percent of total calories from fructose. In order to make unequivocal observations about metabolic effects of fructose or glucose-sweetened drinks, it would be ideal to perform the experiments using a custom HFD that does not contain any sugar. Future studies will also be needed to determine the minimal amount of fructose/sucrose in the diet that can induce metabolic derangements, and our data suggest that this is lower than 9%.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, C. Ronald Kahn (c.ronald.kahn@joslin.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals and Diets
All protocols were approved by the IACUC of the Joslin Diabetes Center and were in accordance with NIH guidelines. Mice were housed at 20–22°C on a 12 h light/dark cycle with ad libitum access to food and water. C57Bl6, male mice at 6 weeks of age were purchased from Jackson Laboratory and fed either chow diet (Mouse Diet 9F, PharmaServ) or high fat diet (Research diets, D12492) for 10 weeks. Caloric composition of chow diet consisted of 23% protein, 21.6% fat and 55.4% carbohydrates, while HFD had 20% protein, 60% fat and 20% carbohydrates. Mice were provided either tap water or 30% (weight/volume) fructose or 30% glucose solution in water. Mice were weighed, and their food intake was recorded once per week. Mice were sacrificed from 8 to 11am, and one mouse from each cage, i.e. dietary group, was utilized before sacrificing the next mouse in the same cage. This was repeated until all four mice per cage were sacrificed. Serum parameters were determined by the Joslin Diabetes and Endocrinology Research Center assay core using commercial colorimetric assays for beta-hydroxybutyrate and free fatty acids (Softic et al., 2016a).
METHOD DETAILS
mRNA, qPCR and RNA Sequence Analysis
mRNA was extracted by homogenizing liver tissue in trizol, treating with chloroform and participating in 70% ethanol. mRNA was purified using RNeasy Mini Kit columns (Quiagen, #74106). cDNA was made using High Capacity cDNA Revers Transcription Kit (Applied Biosystems, #4368813). qPCR was performed utilizing C1000 Thermal Cycler (Bio Rad, # CFX384). Primer sequences used are listed in supplemental figures. HTG EdgeSeq mRNA sequence analysis was performed by BioPolymers Facility at Harvard Medical School. Data was analyzed using Bioinformatics Core at Joslin Diabetes Center.
Protein Extraction, Immunoblot and Immunoprecipitation
Tissues were homogenized in RIPA buffer (EMD Millipore) with protease and phosphatase inhibitor cocktail (Biotools). For immunoprecipitation, tissue was homogenized in non-denaturing buffer, 2 mg of protein lysate and 2 μg of primary antibody was agitated overnight in cold room and protein was pulled with protein A/G magnetic beads (Biotool, # B23201). Proteins were separated using SDS-PAGE and transferred to PVDF membrane (Millipore). Immunoblotting was achieved using the indicated antibodies listed in resources section. In Figure 1 Cpt1a antibody was used generated by Dr. Herrero (Herrero et al., 2005). Quantification of immunoblots was performed using ImageJ.
Metabolomic Analysis
50 mg of previously frozen liver tissue was homogenized in 1 mL of 50% aqueous acetonitrile, containing 0.3% formic acid. Liver amino acids, acyl CoAs and organic acids, and acylcarnitines were analyzed using stable isotope dilution techniques by GC/MS and MS/MS as described previously (White et al., 2016).
Mitochondrial Histology and Immunofluorescence
A piece of liver tissue measuring 1 × 2 mm was fixed in 2.5% paraformaldehyde, 5.0% glutaraldehyde, 0.06% picric acid in 0.2M Cacodylate buffer. The samples were processed, and electron microscopy pictures were taken by Harvard Medical School Electron Microscopy Facility. Mitochondrial number and size were assessed from 5000x images using Image J software. Mitochondrial mass was assessed in AML-12 hepatocytes cultured on glass chamber slides (ThermoFisher Scientific, 154534) and exposed to the experimental conditions for 48 hours. Cells were washed with PBS and fixed in 4% paraformaldehyde and incubated with either, MitoTracker green (Life Technologies, M7514), BODIPY (Invitrogen, D3922) or DHE (Sigma, D7008) and protected from light. The slides were countered stained with DAPI mounting media (H-1500, Vector Laboratories) and immunofluorescence was recorded using Olympus BX60 fluorescence microscope
Seahorse Cellular Flux Measurements
Mitochondrial oxidative phosphorylation and FFA oxidation were measured using SeaHorse Bioscience XF24 Cellular Flux Analyzer (Agilent Technologies). Oxidative phosphorylation was measured in primary mouse hepatocytes cultured in low glucose, DMEM media and acute treated with 25 mM glucose or fructose followed by exposure to standard ox/phos perturbations with oligomycin, FCCP, and rotenone plus antimycin. Primary hepatocytes could not survive prolonged fast needed to induce FFA oxidation, thus this was performed in cultured hepatocytes. AML-12 hepatocytes were grown in DMEM media treated with 25mM fructose or glucose for 48 hours. Cells were then cultured in DMEM media without glucose, but supplemented with 1mM sodium pyruvate and 2mM Glutamax for 18 to 24 hours. The media was changed to Krebs-Henseleit Buffer and oxygen consumption rate was measured following exposure to 0.4mM FFA mixture (2:1 oleate to palmitate), 2-deoxyglucose and etomoxir.
Cpt1a Enzymatic Activity
CPT1 activity in 20 μg of protein from mitochondrial-enriched cell fractions was determined by the radiometric method as previously described (Herrero et al., 2005) with minor modifications. The substrates were L-[methyl-3H] carnitine and palmitoyl-CoA. Enzyme activity was assayed for 4 min at 30°C in a total volume of 200 mL. For malonyl-CoA inhibition assay, 20 μg of mitochondrion-enriched cell fractions was pre-incubated for 1.5 min at 30°C with 100 μM malonyl-CoA prior to CPT1 activity assay.
Multiphoton Imaging of Mitochondrial NAD(P)H
For in vitro NAD(P)H quantification, cultured hepatocytes were grown on glass bottom plates to confluency and were mounted on the Zeiss 710 microscope stage. Images were collected with a 63x, 1.2 NA Zeiss C-Apochromat objective on a Zeiss-LSM-710 microscope with the Cameleon infrared laser using 710 nm for excitation of NADH. Emission fluorescence was collected de-scanned 410–490 nm.
Liver Specific Khk Knockdown
Liver specific knockdown was achieved utilizing a small interfering RNA (siRNA) conjugated to N-acetylgalactoseamine (GalNAc) (Softic et al., 2017). Alnylam Pharmaceuticals synthesized siRNA to specifically target mouse Khk mRNA. A GalNAc conjugate siRNA targeting transthyretin (TTR) was used as a negative control. Maximal knockdown is achieved within first week and it remains down for two weeks. Mice on HFD, HFD+F and HFD+G diets were injected subcutaneously with 20mg/kg of Khk or control siRNA at 6 and 8 weeks on the diet and were sacrificed after 10 weeks on the diets.
Mitochondria Protein Digestion and Acetylated Peptide Enrichment
Mitochondria protein was quantified using the BCA assay, and mitochondria containing 1 mg of protein were lysed with addition of 100 μL of 8M Urea, 10 μL of 10% maltoside, 10 μL of 1M TEAB buffer (pH 8.5), and water to a final volume of 200 μL. Proteins were reduced for 30 minutes at 37° Celsius by addition of DTT to a final concentration of 4.5 mM. Reduced protein solutions were then cooled to room temperature and alkylated by the addition of iodoacetamide to a final concentration of 10 mM for 30 minutes at room temperature. The alkylated protein solution was then diluted to 1 mL final volume using 50 mM TEAB, pH 8.5, and trypsin was added at a ratio of 1 part trypsin per 50 parts mitochondrial protein (wt:wt, 20 μg per sample). Trypsin digestion proceeded overnight for 18 hours at 37° Celsius. Digestion was quenched by addition of formic acid to a final concentration of 1%, and insoluble material was precipitated by centrifugation at 1,800 relative centrifugal force (RCF) for 15 minutes at room temperature. Soluble peptides were then desalted using Waters’ Oasis HLB Vacuum cartridges (30 mg sorbent, 1 cc volume). Peptides were eluted with 1.2 mL of 80% acetonitrile (ACN)/0.2% FA/19.8% water, and dried to completion using a vacuum centrifugal concentrator. Desalted peptides were then resuspended in 1.4 mL of IAP buffer (50mM MOPS, 10 mM Na2PO4, 50 mM NaCl) for immunoprecipitation. Acetylated peptides were enriched using anti-acetyl lysine antibody-bead conjugated PTMScan (Cell Signaling Technologies) using 10 μL of beads per sample (1/4 of the manufacturer-supplied tube per sample, approximately 62.5 μg of conjugated antibody). Immunoprecipitation proceeded overnight at 4° Celsius with rocking. The next day beads were washed twice with 1 mL of ice-cold IAP buffer, three times with ice-cold water (Burdick and Jackson HPLC-grade), and then eluted using 100 μL of 0.15% TFA (55 μL followed by a second elution using 45 μL). Peptides eluted from the enrichment were desalted directly using C18 reversed phase StageTips (two eighteen-gauge disks per StageTip), and resuspended in 7 μL of 0.2% formic acid for mass spectrometry analysis.
Nanoflow Liquid Chromatography – Tandem Mass Spectrometry
Peptide separations were carried out using mobile phase A consisting of 97.95% water/0.05% FA/2% acetonitrile, and mobile phase B consisting of 98% acetonitrile/1.95% water/0.05% FA. Samples were loaded onto the first of two sequential C18 column chips (75 μm x 15 cm ChromXP C18-CL chip, 3 μm particles, 300 Å) using an Eksigent cHiPLC system for 30 minutes at a flow of 0.6 μL per min of mobile phase A. Separation was performed over two 75 μm x 15 cm ChromXP C18-CL analytical chip (30 cm total length) using a gradient from 5% to 40% mobile phase B over 80 minutes. The column chip was washed by increasing to 80% B over 5 minutes that was maintained for 8 minutes, followed by a return to 5% mobile phase B over two minutes that was maintained for 25 minutes to re-equilibrate the column. Eluting peptides were directly electrosprayed into a TripleTOF 5600 mass spectrometer (SCIEX) and analyzed by SWATH data-independent acquisition (DIA). Every SWATH cycle consisted of a 250 ms precursor ion scan from 400–1,250 m/z followed by fragmentation of all ions between 400–1,200 m/z using 64 variable width precursor isolation windows. The SWATH window definitions used for enrichments of acetylated peptides were determined based on the frequency of acetylated peptide identifications in each m/z region using previous data-dependent acquisitions for 42 ms each, resulting in a total cycle time of approximately 3 sec. Non-enriched peptides were also analyzed by SWATH to determine protein-level changes. The SWATH window definitions used to collect data for protein-level changes were based on the cross-lab SWATH study (Valderhaug et al., 2016). Fragment ion spectra were collected from 100–2,000 m/z.
QUANTIFICATION AND STATISTICAL ANALYSIS
Proteome and Acetylome Data Analysis
Data for protein-level analysis was analyzed using Spectronaut (Bruderer et al., 2015) with our in-house spectral library from DDA runs of pooled protein samples. DIA data from acetyl-peptide enrichment was processed using PIQED (Meyer et al., 2017), which can automatically identify and quantify site-level PTMs using only DIA-MS data, using settings described in the original manuscript. All the acetylation data is available on Panorama: https://panoramaweb.org/labkey/project/Schilling/Hyperacetylation%20of%20Mitochondrial%20Proteins/begin.view. PIQED analysis used protein-level quantities determined using Spectronaut for correction of site-level changes by protein-level changes. Additional analysis was carried out using custom scripts written in R (R Development Core Team, 2008). Functional analysis of protein changes was performed using ClueGO within Cytoscape (Christiaens et al., 2016; Ossowski et al., 2016).
Statistical Analyses
All data are presented as mean ± SEM. The data analysis, comparing the effects of pelleted diet and sugar-supplemented water, was first analyzed using two-way analysis of variance (ANOVA) and for statistically significant overall results, post hoc t-tests were carried out to determine significant differences between the individual groups. Significant difference between diet types as compared to the Chow group is noted with a number sign (#) and significant difference within the diet groups is designated by an asterisk (*). For both signs, the single symbol represent a p value of <0.05, two symbols represent a p value of <0.01, and three symbols denote a p value of <0.001, and four symbols stand for a p value of <0.0001 throughout the study.
DATA AND CODE AVAILABILITY
The accession number for the RNAseq reported in this paper is BioProject: PRJNA391187 or SRA: SRP109839. Proteomics data has been newly deposited and is publically available. The Massive repository MSV000081902 (ftp://massive.ucsd.edu or ftp://MSV000081902@massive.ucsd.edu username: MSV000081902, password: dietstudy) contains all the raw mass spectrometry data files used for quantification of proteins, and protein acetylation, as well as the raw files used to generate a sample-specific protein-level spectral library for quantification using Spectronaut. Acetylation data is available on the Panorama: https://panoramaweb.org/targetedms/Schilling/Meyer_acylomes_diets/showPrecursorList.view?id=26538.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal Anti-KHK | Sigma | HPA007040 |
| Mouse monoclonal Anti-CPT1a | Abcam | 128568 |
| Mouse monoclonal Anti-HADHA/HADHB | Abcam | 110302; RRID: AB_11141632 |
| Mouse monoclonal Anti -VDAC | Abcam | 14734 |
| Rabbit polyclonal Anti-PINK1 | Abcam | 23707 |
| Mouse monoclonal Anti-PARKIN | Santa Cruz | 32282 |
| Rabbit polyclonal Anti-GAPDH | Santa Cruz | 25778 |
| Rabbit polyclonal Anti-FIS1 | Santa Cruz | 98900 |
| Mouse monoclonal Anti-OPA1 | Santa Cruz | 393296 |
| Rabbit polyclonal Anti-PGC1a | Santa Cruz | 13067 |
| Goat polyclonal Anti-ACADL | Santa Cruz | 82466 |
| Rabbit polyclonal Anti-pACC/TotACC | Cell Signaling Technology | 3661/3662 |
| Rabbit Policlonal Anti-172pAMPK/TotAMPK | Cell Signaling Technology | 2535/2532 |
| Rabbit monoclonal Anti-ULK1 | Cell Signaling Technology | 8054 |
| Rabbit polyclonal Anti-p62 | Cell Signaling Technology | 5114 |
| Rabbit polyclonal Anti-LC3B | Cell Signaling Technology | 2775 |
| Rabbit polyclonal Acetyl-Lysine | Cell Signaling Technology | 9441 |
| PTMScan Acety-Lysine Motif [Ac-K] Kit, Immunoaffinity Beads | Cell Signaling Technology | 13416 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Halt protease inhibitor | Thermo Fisher | 78430 |
| Trichostatin A | Sigma Aldrich | T1952 |
| Nicotinamide | Sigma Aldrich | N0636-100G |
| Dithiothreitol | Sigma Aldrich | D9779 |
| Iodoacetamide | Sigma Aldrich | I1149 |
| Sequencing Grade modified Trypsin (frozen) | Promega | V5113 |
| LC-MS grade Formic Acid | Promega | 94318 |
| Oasis HLB 1cc vac cartridges 30mg | Waters | 186003908 |
| Empore sorbent disks | 3M | 98060402173 |
| Critical Commercial Assays | ||
| Rat Insulin ELISA | EMD Millipore | EZRMI-13K |
| Free Fatty Acid Quantification Kit | Sigma | MAK044-1KT |
| b-Hydroxybutyrate Assay Kit | Sigma | MAK041-1KT |
| Deposited Data | ||
| RNA seq | NCBI BioProject database | BioProject: PRJNA391187 or SRA: SRP109839 |
| All raw mass spectrometry data | https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=cff90185a8234e1b89c010e5a84ae109 | MSV000081902 |
| Acetylated peptide spectral library and processed quantitative data | https://panoramaweb.org/targetedms/Schilling/Meyer_acylomes_diets/showPrecursorList.view?id=26538 | N/A |
| Experimental Models: Cell Lines | ||
| AML-12; mouse hepatocyte | ATCC | Cat# CRL-2254; Lot# 61449103 |
| Primary mouse hepatocytes | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57Bl/6j | The Jackson Laboratory | Stock# 000664 |
| Oligonucleotides | ||
| See Table S1 | N/A | N/A |
| Software and Algorithms | ||
| Spliced Transcripts Alignment to a Reference | http://code.google.com/p/rna-star/ | N/A |
| Spectronaut | https://www.biognosys.com/ | N/A |
| PIQED | https://github.com/jgmeyerucsd/PIQEDia | N/A |
| R | https://www.r-project.org/; https://github.com/jgmeyerucsd/pRoteomics | N/A |
Context and Significance.
Intake of sugar-sweetened beverages accelerates the development of obesity and its complications. Researchers at Harvard Medical School compared the effects of the two most commonly consumed sugars, fructose and glucose, in mice on regular and high-fat diets. Building on their previous findings that fructose promotes accumulation and formation of body fat, here they show that fructose-sweetened drinks also impair fat burning. These effects of fructose are accomplished by suppressing the expression of genes involved in fat oxidation and modifying mitochondrial proteins related to fat metabolism. Thus, in addition to being used in fat synthesis, dietary fructose also propagates signals to decrease fat metabolism. These findings imply that consuming fructose-sweetened drinks, while also eating a high-fat diet, has additive negative effects.
Highlights.
Addition of fructose to a high-fat diet increases hepatic malonyl-CoA more than glucose
Knockdown of the fructose metabolizing gene ketohexokinase increases CTP1a levels
Fructose supplementation alters mitochondrial size and function
Dietary fructose induces acetylation of ACADL and CPT1a to modify fat oxidation
ACKNOWLEDGMENTS
The authors would like to thank Jonathan Dreyfuss and Hui Pan at Joslin Diabetes Center Bioinformatics Core for analyzing RNA-seq data. This work was supported in part by NIH grants R01 DK031036 and R01 DK033201 to C.R.K.; R01 DK056626, R01 DK103046, and R37 DK048873 to D.E.C.; R24 DK085610 to C.R.K., C.B.N., and B.W.G.; and K12 HD000850, P30 DK40561, and NASPGHAN Foundation Young Investigator Award to S.S. M.K.G. was supported by JDRF advanced post-doctoral award 3-APF-2017-393-A-N, the Joslin DRC grant P30DK034834, the Ministry of Spain (SAF2013-45887-R to L.H. and SAF2014-52223-C2-1-R to D.S. cofunded by the European Regional Development Fund [ERDF]), the CIBERobn grant CB06/03/0001 to D.S., the Government of Catalonia (2014SGR465 to D.S.), and the Fundació La Marató de TV3 to D.S. J.G.M. was supported by T32 AG000266. We acknowledge support from the NIH shared instrumentation grant for the TripleTOF system at the Buck Institute (1S10 OD016281 to B.W.G.). This work was also supported, in part, by a grant from Alnylam Pharmaceuticals Inc., awarded to S.S. and C.R.K. Other authors have no relevant financial interests to disclose.
Footnotes
DECLARATION OF INTERESTS
J.W. and K.F. are employees and shareholders of Alnylam Pharmaceuticals. Alnylam Pharmaceuticals, Inc., holds a patent on KHK siRNA.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2019.09.003.
REFERENCES
- Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, Johnson RJ, and Diehl AM; Nonalcoholic Steatohepatitis Clinical Research Network (2010). Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 51, 1961–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R, Burlingame AL, and Bradshaw RA (2013). Western blots versus selected reaction monitoring assays: time to turn the tables? Mol. Cell. Proteomics 12, 2381–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharathi SS, Zhang Y, Mohsen AW, Uppala R, Balasubramani M, Schreiber E, Uechi G, Beck ME, Rardin MJ, Vockley J, et al. (2013). Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site. J. Biol. Chem. 288, 33837–33847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluemlein K, and Ralser M (2011). Monitoring protein expression in whole-cell extracts by targeted label- and standard-free LC-MS/MS. Nat. Protoc. 6, 859–869. [DOI] [PubMed] [Google Scholar]
- Boesiger P, Buchli R, Meier D, Steinmann B, and Gitzelmann R (1994). Changes of liver metabolite concentrations in adults with disorders of fructose metabolism after intravenous fructose by 31P magnetic resonance spectroscopy. Pediatr. Res. 36, 436–440. [DOI] [PubMed] [Google Scholar]
- Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, and Dentin R (2010). Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Invest. 120, 4316–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruderer R, Bernhardt OM, Gandhi T, Miladinović SM, Cheng LY, Messner S, Ehrenberger T, Zanotelli V, Butscheid Y, Escher C, et al. (2015). Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 14, 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Weinert BT, Nishida Y, Verdin E, and Mann M (2014). The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550. [DOI] [PubMed] [Google Scholar]
- Christiaens O, Prentice K, Pertry I, Ghislain M, Bailey A, Niblett C, Gheysen G, and Smagghe G (2016). RNA interference: a promising biopesticide strategy against the African Sweetpotato Weevil Cylas brunneus. Sci. Rep. 6, 38836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, and Diehl AM (1999). Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA 282, 1659–1664. [DOI] [PubMed] [Google Scholar]
- Cox CL, Stanhope KL, Schwarz JM, Graham JL, Hatcher B, Griffen SC, Bremer AA, Berglund L, McGahan JP, Havel PJ, et al. (2012). Consumption of fructose-sweetened beverages for 10 weeks reduces net fat oxidation and energy expenditure in overweight/obese men and women. Eur. J. Clin. Nutr. 66, 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ruyter JC, Olthof MR, Seidell JC, and Katan MB (2012). A trial of sugar-free or sugar-sweetened beverages and body weight in children. N. Engl. J. Med. 367, 1397–1406. [DOI] [PubMed] [Google Scholar]
- Ding WX, and Yin XM (2012). Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, Fotuhi Siahpirani A, Kemmerer ZA, Prolla TA, Roy S, Coon JJ, and Denu JM (2015). SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. 21, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbeling CB, Feldman HA, Chomitz VR, Antonelli TA, Gortmaker SL, Osganian SK, and Ludwig DS (2012). A randomized trial of sugar-sweetened beverages and adolescent body weight. N. Engl. J. Med. 367, 1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz KS, Green MF, Petersen DR, and Hirschey MD (2013). Ethanol metabolism modifies hepatic protein acylation in mice. PLoS One 8, e75868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, Zhang J, Gong Y, Gupte J, Ye J, Weiszmann J, Samayoa K, Coberly S, Gardner J, Wang H, et al. (2014). Fibroblast growth factor receptor 4 (FGFR4) deficiency improves insulin resistance and glucose metabolism under diet-induced obesity conditions. J. Biol. Chem. 289, 30470–30480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, and Aebersold R (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, and Seto E (2005). Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23. [DOI] [PubMed] [Google Scholar]
- Hardie DG, and Pan DA (2002). Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 30, 1064–1070. [DOI] [PubMed] [Google Scholar]
- Herrero L, Rubí B, Sebastián D, Serra D, Asins G, Maechler P, Prentki M, and Hegardt FG (2005). Alteration of the malonyl-CoA/carnitine palmitoyltransferase I interaction in the beta-cell impairs glucose-induced insulin secretion. Diabetes 54, 462–471. [DOI] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stančáková A, Goetzman E, Lam MM, Schwer B, et al. (2011). SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondares E, Iglesias R, Giralt A, Gonzalez FJ, Giralt M, Mampel T, and Villarroya F (2011). Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J. Biol. Chem. 286, 12983–12990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janevski M, Ratnayake S, Siljanovski S, McGlynn MA, Cameron-Smith D, and Lewandowski P (2012). Fructose containing sugars modulate mRNA of lipogenic genes ACC and FAS and protein levels of transcription factors ChREBP and SREBP1c with no effect on body weight or liver fat. Food Funct. 3, 141–149. [DOI] [PubMed] [Google Scholar]
- Jeon SM (2016). Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 48, e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, Watson PA, Birdsey N, Bao J, Gius D, et al. (2011). Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 433, 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. (2006). Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618. [DOI] [PubMed] [Google Scholar]
- Kim MS, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, Noh HL, Kang HJ, Meissen JK, Blatnik M, et al. (2016). ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Invest. 126, 4372–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, Burgess SC, Li C, Ruddy M, Chakravarthy M, et al. (2017). Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 26, 394–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, Tang PH, Miles L, Miles MV, Balistreri WF, et al. (2010). High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 52, 934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake AD, Novak P, Shipkova P, Aranibar N, Robertson DG, Reily MD, Lehman-McKeeman LD, Vaillancourt RR, and Cherrington NJ (2015). Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids 47, 603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnik C, Cohen Y, Atir-Lande A, Schuldiner M, and Arava Y (2014). OM14 is a mitochondrial receptor for cytosolic ribosomes that supports co-translational import into mitochondria. Nat. Commun. 5, 5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, et al. (2014). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 32, 834–841. [DOI] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. (2007). Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Fox CS, Jacques PF, Speliotes EK, Hoffmann U, Smith CE, Saltzman E, and McKeown NM (2015). Sugar-sweetened beverage, diet soda, and fatty liver disease in the Framingham Heart Study cohorts. J. Hepatol. 63, 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry JD, Mannaerts GP, and Foster DW (1977). A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Invest. 60, 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JG, Mukkamalla S, Steen H, Nesvizhskii AI, Gibson BW, and Schilling B (2017). PIQED: automated identification and quantification of protein modifications from DIA-MS data. Nat. Methods 14, 646–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JG, Softic S, Basisty N, Rardin MJ, Verdin E, Gibson BW, Ilkayeva O, Newgard CB, Kahn CR, and Schilling B (2018). Temporal dynamics of liver mitochondrial protein acetylation and succinylation and metabolites due to high fat diet and/or excess glucose or fructose. PLoS One 13, e0208973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyers JS, Shiyanova TL, Mehrbod F, Dunbar JD, Noblitt TW, Otto KA, Reifel-Miller A, and Kharitonenkov A (2007). Molecular determinants of FGF-21 activity-synergy and cross-talk with PPARgamma signaling. J. Cell. Physiol. 210, 1–6. [DOI] [PubMed] [Google Scholar]
- Ossowski ZM, Skrobot W, Aschenbrenner P, Cesnaitiene VJ, and Smaruj M (2016). Effects of short-term Nordic walking training on sarcopenia-related parameters in women with low bone mass: a preliminary study. Clin. Interv. Aging 11, 1763–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, Johnson RJ, and Abdelmalek MF (2008). Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 48, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LS, Smift AL, Croteau NJ, Ferrick DA, and Wu M (2011). Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta 1807, 726–734. [DOI] [PubMed] [Google Scholar]
- Prager GN, and Ontko JA (1976). Direct effects of fructose metabolism on fatty acid oxidation in a recombined rat liver mitochondria-hish speed supernatant system. Biochim. Biophys. Acta 424, 386–395. [DOI] [PubMed] [Google Scholar]
- R Development Core Team (2008). R: A language and environment for statistical computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, et al. (2013). Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. USA 110, 6601–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roglans N, Vilà L, Farré M, Alegret M, Sánchez RM, Vázquez-Carrera M, and Laguna JC (2007). Impairment of hepatic Stat-3 activation and reduction of PPARalpha activity in fructose-fed rats. Hepatology 45, 778–788. [DOI] [PubMed] [Google Scholar]
- Samuel VT (2011). Fructose induced lipogenesis: from sugar to fat to insulin resistance. Trends Endocrinol. Metab. 22, 60–65. [DOI] [PubMed] [Google Scholar]
- Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, and Clore JN (2001). Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120, 1183–1192. [DOI] [PubMed] [Google Scholar]
- Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, Giallourakis C, Comb MJ, Alt FW, and Lombard DB (2009). Calorie restriction alters mitochondrial protein acetylation. Aging Cell 8, 604–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, and Wakasa K (2002). In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 37, 56–62. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhu H, Arvidson DN, and Woldegiorgis G (2000). The first 28 N-terminal amino acid residues of human heart muscle carnitine palmitoyltransferase I are essential for malonyl CoA sensitivity and high-affinity binding. Biochemistry 39, 712–717. [DOI] [PubMed] [Google Scholar]
- Softic S, Boucher J, Solheim MH, Fujisaka S, Haering MF, Homan EP, Winnay J, Perez-Atayde AR, and Kahn CR (2016a). Lipodystrophy due to adipose tissue-specific insulin receptor knockout results in progressive NAFLD. Diabetes 65, 2187–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic S, Cohen DE, and Kahn CR (2016b). Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 61, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, Willoughby J, Harbison C, Fitzgerald K, Ilkayeva O, et al. (2017). Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Invest. 127, 4059–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuy S, Ladurner R, Volynets V, Wagner S, Strahl S, Königsrainer A, Maier KP, Bischoff SC, and Bergheim I (2008). Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J. Nutr. 138, 1452–1455. [DOI] [PubMed] [Google Scholar]
- Valderhaug TG, Aasheim ET, Sandbu R, Jakobsen GS, Småstuen MC, Hertel JK, and Hjelmesæth J (2016). The association between severity of King’s Obesity Staging Criteria scores and treatment choice in patients with morbid obesity: a retrospective cohort study. BMC Obes. 3, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berghe G, Bronfman M, Vanneste R, and Hers HG (1977). The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem. J. 162, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilà L, Roglans N, Alegret M, Sánchez RM, Vázquez-Carrera M, and Laguna JC (2008). Suppressor of cytokine signaling-3 (SOCS-3) and a deficit of serine/threonine (Ser/Thr) phosphoproteins involved in leptin transduction mediate the effect of fructose on rat liver lipid metabolism. Hepatology 48, 1506–1516. [DOI] [PubMed] [Google Scholar]
- Weinert BT, Moustafa T, Iesmantavicius V, Zechner R, and Choudhary C (2015). Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J. 34, 2620–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White PJ, Lapworth AL, An J, Wang L, McGarrah RW, Stevens RD, Ilkayeva O, George T, Muehlbauer MJ, Bain JR, et al. (2016). Branchedchain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol. Metab. 5, 538–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JC, Merlino G, and Fausto N (1994). Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc. Natl. Acad. Sci. USA 91, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XX, Watts LM, Manchem VP, Chakravarty K, Monia BP, McCaleb ML, and Bhanot S (2013). Peripheral reduction of FGFR4 with antisense oligonucleotides increases metabolic rate and lowers adiposity in diet-induced obese mice. PLoS One 8, e66923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakim D (1972). The effect of fructose on hepatic synthesis of fatty acids. Acta Med. Scand. Suppl. 542, 205–214. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang JH, Chen XY, Hu QH, Wang MX, Jin R, Zhang QY, Wang W, Wang R, Kang LL, et al. (2015). Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid. Redox Signal. 22, 848–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNAseq reported in this paper is BioProject: PRJNA391187 or SRA: SRP109839. Proteomics data has been newly deposited and is publically available. The Massive repository MSV000081902 (ftp://massive.ucsd.edu or ftp://MSV000081902@massive.ucsd.edu username: MSV000081902, password: dietstudy) contains all the raw mass spectrometry data files used for quantification of proteins, and protein acetylation, as well as the raw files used to generate a sample-specific protein-level spectral library for quantification using Spectronaut. Acetylation data is available on the Panorama: https://panoramaweb.org/targetedms/Schilling/Meyer_acylomes_diets/showPrecursorList.view?id=26538.