

Abstract

Phenotypic screening of a Medicines for Malaria Venture compound library against Mycobacterium tuberculosis (Mtb) identified a cluster of pan-active 2-pyrazolylpyrimidinones. The biology triage of these actives using various tool strains of Mtb suggested a novel mechanism of action. The compounds were bactericidal against replicating Mtb and retained potency against clinical isolates of Mtb. Although selected MmpL3 mutant strains of Mtb showed resistance to these compounds, there was no shift in the minimum inhibitory concentration (MIC) against a mmpL3 hypomorph, suggesting mutations in MmpL3 as a possible resistance mechanism for the compounds but not necessarily as the target. RNA transcriptional profiling and the checkerboard board 2D-MIC assay in the presence of varying concentrations of ferrous salt indicated perturbation of the Fe-homeostasis by the compounds. Structure–activity relationship studies identified potent compounds with good physicochemical properties and in vitro microsomal metabolic stability with moderate selectivity over cytotoxicity against mammalian cell lines.
Introduction
Tuberculosis (TB) caused by Mycobacterium tuberculosis (Mtb) continues to be one of the leading causes of death and morbidity globally, claiming 1.2 million lives in 2018.1 The emergence and spread of multi drug-resistant (MDR) and extensively drug-resistant (XDR) strains of Mtb that are resistant to the first- and second-line drugs have further exacerbated the situation.2 The rise in anti-microbial resistance warrants the search for new drugs with unique modes of action that can bypass existing modes of resistance or can be used as adjunctive therapy to compensate for those that are vulnerable to promoting resistance. While considerable progress has been made toward establishing a TB drug pipeline, the high attrition rate in clinical development reinforces the need to continually replenish the pipeline with high-quality leads that act through the inhibition of novel targets.3
In light of the above, we have been engaged in early TB drug discovery to identify and progress new chemical series with potentially novel modes of action lacking cross-resistance with existing drugs.4,5 One approach to identify compounds with a novel mode of action is to phenotypically screen compound libraries against Mtb growing under different media conditions with different carbon sources followed by a biology triage process to weed out actives hitting frequently encountered targets such as the respiratory system (e.g., QcrB) and cell–wall synthesis (e.g., MmpL3 and DprE1), as well as DNA damaging agents. Herein, we describe structure–activity relationship (SAR) and target identification studies of one novel chemical series, 2-pyrazolylpyrimidinones—represented in Figure 1. This series was identified through high-throughput screening of a Medicines for Malaria Venture (MMV) compound library.
Figure 1.

Generic structure of pyrazolylpyrimidones.
Results and Discussion
Phenotypic Hits with a Novel Mode of Action
A high-throughput screen of a ∼530,000 diverse set of compounds from MMV compound library against Mtb was conducted at the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIAID/NIH, U.S.). Reconfirmed hits were followed up for determination of minimum inhibitory concentration (MIC) on multiple media that identified a cluster of pan-active pyrazolylpyrimidinones represented by compounds 1 and 2 (Table 1). The biology triage of these actives (described in the target identification section), suggestive of a unique mechanism of action (MoA), generated further interest in the exploration of these hits. We recognized that pyrazolylpyrimidinones potentially possess iron-chelating properties by virtue of their two sp2 heteroatoms in a 1,4-relationship on adjacent rings, which might play a crucial role in the SAR and mode of action.6 During these studies, we came across the related pyrazolopyrimidinone (PZP, Figure 2a) compound reported as an intracellular iron chelator in Mtb.7 PZP is reported to chelate iron through the pyrimidinone carbonyl and 2-position pyrazole nitrogen. Similarly, bidentate iron chelation by the pyrazolylpyrimidinones from the pyrimidinone nitrogen and pyrazole nitrogen as depicted in Figure 2b may be hypothesized. A single-crystal X-ray structure of the pyrazolylpyridines–Fe complex is reported in the literature which supports this hypothesis.8,9 This aspect of metal chelation by the compounds was considered while planning further SAR exploration and target identification studies with the series.
Table 1. Properties of the Hits from the MMV Library Screena.

| Properties | 1 | 2 |
|---|---|---|
| MIC 7H9/ADC/Tw (μM) | 0.6 | 2 |
| MIC GAST-Fe (μM) | 0.02 | 0.02 |
| MIC 7H9/Glu/BSA/Tx (μM) | 1.5 | 2 |
| MIC 7H9/Glu/Cas/Tx (μM) | 0.3 | 0.4 |
| MIC 7H9/DPCC/Cas/Tx (μM) | 0.4 | 0.2 |
| MIC 7H9/DPCC/Chol/BSA/Tx (μM) | 1.5 | 6 |
| Vero IC50 (μM) | 8 | 1.2 |
| HepG2 IC50 (Glu/Gal μM) | 4/4 | ND |
| SolubilitypH7.4 (μM) | 10 | <5 |
| LogDpH7.4 | 2.5 | 2.2 |
| Molecular Weight | 266.304 | 295.346 |
| Microsomal stability H/R/Mo %remaining | 98/69/22 | 73/76/78 |
MIC—minimum inhibitory concentration against Mtb H37Rv; 7H9—Middlebrook 7H9; ADC—albumin–dextrose–catalase supplement; Tw—Tween 80; GAST-Fe—glycerol-alanine-salts with Tween and iron salts; Glu—glucose; BSA—bovine serum albumin; Tx—Tyloxapol; Cas—Casitone; DPCC—dipalmitoylphosphatidylcholine and cholesterol; Chol—cholesterol; H/R/Mo—human/rat/mouse; IC50—50% inhibitory concentrations; solubilitypH7.4—aqueous solubility at pH 7.4.
Figure 2.

(a) Structure of PZP; and (b) depiction of metal chelation by pyrazolylpyrimidinone 1.
Synthesis
In order to explore the SAR profiles of the series, various routes were explored toward the synthesis of diverse range of analogues as represented by compounds 3–54 in Tables 2–5. To facilitate exploration of a broad array of substituents off the pyrazole ring and pyrimidinone core, a concise and scalable synthesis leading to an advanced intermediate was desired. As summarized in Scheme 1, this was achieved by the ring formation reaction of 6-substituted-2-thiomethyl-pyrimidin-4-one 55 via condensation of the appropriate β-keto-ester and thiourea, and subsequent methylation of the thiol.10 Intermediates 55a–g were reacted with hydrazine hydrate in refluxing ethanol to give the versatile 2-hydrazinylpyrimidin-4(1H)-ones 56a–g, which gave access to analogues for SAR exploration on the pyrazole ring and the R1 substituent on the pyrimidinone core. Similarly, substitution of a phenyl group at the R2 position was achieved through bromination at C5 of 6-methyl-2-thiomethyl-pyrimidin-4-one (55, R1 = CH3) followed by a Suzuki cross-coupling reaction with phenyl boronic acid to provide 55h, which was then treated with hydrazine to form 56h.
Table 2. Pharmacophore Evaluation of Pyrazolylpyrimidinonesa.

MIC were measured in Middlebrook 7H9/Glu/BSA/Tyloxapol media at day 7; IC50—50% inhibitory concentrations; solubility—aqueous solubility measured at pH 7.4; CHO—an epithelial cell-line derived from the ovary of the Chinese hamster; and V—Vero cell-line; SI—selectivity index between CHO IC50 and Mtb MIC.
Table 5. SAR at the Pyrazole R3 Positiona.

MIC—were measured in Middlebrook 7H9/Glu/BSA/Tyloxapol media at day 7; IC50—50% inhibitory concentrations; solubility—aqueous solubility at pH 7.4; CHO—an epithelial cell-line derived from the ovary of the Chinese hamster; and SI—selectivity index between CHO IC50 and Mtb MIC.
Scheme 1. Synthetic Route to Explore R1 and R2 on the Pyrimidinone and Polar Modifications at R5 on the Pyrazole.

Reagents and conditions: (i) (a) thiourea, KOH, EtOH, reflux (b) iodomethane, NaOH, H2O/EtOH; (ii) NH2NH2·H2O, EtOH, reflux; (iii) Br2, AcOH, 60 °C; (iv) phenylboronic acid, Pd(PPh3)2Cl2, 1,4-dioxane, K2CO3; (v) acetylacetone, EtOH, AcOH, reflux; (vi) 3-aminocrotonitrile, EtOH, reflux; (vii) acetyl chloride, pyridine, CH2Cl2; and (viii) ethyl acetoacetate, EtOH, AcOH, reflux.
The Knorr reaction was used to incorporate the 3,5-dimethylpyrazole via an acid-catalyzed condensation of hydrazine precursors 56a–h and acetylacetone. This led to hit compound 1 and related analogues exploring the R1 (11, 12, 19, 20, and 21) and R2 (8–9) positions of the pyrimidinone core as well as quinazolin-4(3H)-one analogue (10). In addition, 2-hydrazinylpyrimidin-4(1H)-one 56a and 56d were reacted with (Z)-3-aminocrotonitrile to afford 5-aminopyrazolylpyrimidinone analogues 14 and 33,11 which allowed for several transformations to be carried out to expand SAR exploration on the pyrazole ring later described in Scheme 7. To explore polar modifications off the pyrazole ring, a mixture of 2-(5-amino-3-methyl-1H-pyrazol-1-yl)-6-phenylpyrimidin-4(3H)-one 14 and pyridine in dichloromethane was treated with acetyl chloride to provide N-acetyl amide 15.2 In addition, hydrazine 56a was heated with ethyl acetoacetate in acidic ethanol to yield pyrazolone 13.
Scheme 7. Synthetic Routes Used to Explore the Pyrazole R5 Position.

Reagents and conditions: (i) diketone, EtOH, AcOH, reflux; (ii) 3-aminocrotonitrile, EtOH, reflux; (iii) acetaldehyde, DMF, AcOH, NaCNBH4, MeOH, 25 °C; (iv) N-(2-methoxyethyl)-3-oxobutanamide, Lawesson’s reagent, THF, 25 °C; and (v) Cs2CO3, Pd2(dba)3, XantPhos, 1,4-dioxane, aryl halide, 120 °C
Syntheses of compounds 3–7 (Table 2) with changes in the central core ring are summarized in Scheme 2. Compounds masking the pyrimidinone amide were synthesized from compound 1 via a Mitsunobu reaction to form methyl ether 3 and N-methylation of 1 with iodomethane using potassium carbonate in N,N-dimethylformamide (DMF) to afford N-methyl pyrimidinone 4. The nucleophilic aromatic substitution reaction of 2,6-dichloro-4-iodopyridine with sodium methoxide gave intermediates 57 and 58 in a 2:1 ratio, which upon separation allowed for the synthesis of compounds 5 and 6, respectively. Briefly, the three-step synthesis from 57 and 58 involved a Suzuki cross-coupling reaction with phenylboronic acid to afford intermediates 59 and 60, respectively, followed by a Buchwald Hartwig amination to introduce the pyrazole (intermediates 61 and 62, respectively), and finally O-demethylation with boron tribromide to afford compounds 5 and 6, respectively.
Scheme 2. Synthetic Routes to Access Core Modifications.

Reagents and conditions: (i) MeOH, PPh3, DIAD, THF, 25 °C; (ii) MeI, K2CO3, DMF, 100 °C; (iii) NaOCH3, MeOH, 80 °C; (iv) phenylboronic acid, Pd(PPh3)2Cl2, K2CO3, 1,4-dioxane, reflux; (v) 3,5-dimethylpyrazole, xanphos, Pd(OAc)2, Cs2CO3, 1,4-dioxane, 130 °C; (vi) BBr3, MeOH; (vii) BBr3, CH2Cl2; (viii) urea, 1,4-dioxane, 140 °C, microwave; (ix) POCl3, DMF; (x) 3,5-dimethylpyrazole, Cs2CO3, 1,4-dioxane, 80 °C, microwave; and (xi) 20% aq NaOH, 80 °C.
Pyrimidin-2(1H)-one 7 was synthesized by the cyclization of ethyl benzoylacetate with urea to provide uracil 63, which was then reacted with phosphorus oxychloride in the presence of a catalytic amount of DMF to give 2,4-dichloropyrimidine 64 (Scheme 2).12 The activated 2,4-dichloropyrimidine readily undergoes nucleophilic substitution with the pyrazole at the 4-chloro position by heating in the microwave at 80 °C (intermediate 65) and subsequent hydrolysis of 2-Cl afforded compound 7, with the carbonyl functionality between the ring nitrogens.
Compounds 16 and 17 (Table 2), in which the pyrazole ring was replaced with other 5-membered heterocycles, were synthesized in a similar manner from 2,4-dichloro-6-phenylpyrimidine 64 (Scheme 3). The alkaline hydrolysis of intermediate 64 with 20% aqueous sodium hydroxide occurred primarily at the 4-Cl position to provide advanced intermediate 66 (isomer ratio 70:30),13 which was reacted with the corresponding 5-membered heterocycle to displace 2-Cl using cesium carbonate under microwave heating at 80 °C (Scheme 3). Compound 18 with an N-methylimidazole was synthesized by condensing 1-methyl-1H-imidazole-2-carboximidamide with ethyl benzoylacetate in low yields.
Scheme 3. Synthetic Route for Pyrazole Replacements.

Reagents and conditions: (i) urea, 1,4-dioxane, 140 °C, microwave; (ii) POCl3, DMF; (iii) 20% aq NaOH, 80 °C (iv) 5-membered heterocycle, Cs2CO3, 1,4-dioxane, 80 °C, microwave; and (v) EtOH, reflux, 1 h.
In order to introduce groups with polar functionalities at the R1 and R2 positions on the pyrimidinone core, 2,4,6-trichloropyrimidine was a useful and accessible starting material when the appropriate β-keto-ester was not readily available to pursue synthetic routes, as shown in Schemes 1 and 3. Treatment of 2,4,6-trichloropyrimidine in aqueous 1,4-dioxane, brought to the alkaline reaction with NaOH, led to hydrolysis at room temperature with the formation of 4,6-dichloro-2-hydroxypyrimidine 67 and 2,6-dichloropyrimidin-4(3H)-one 68, with the former precipitating out as the sodium salt (Scheme 4).14 The filtrate was concentrated to approximately half the volume and cooled to afford a precipitate which was filtered off and acidified to pH 2 using 5 N HCl to give the required regioisomer 68, in approximately 40% yield. The reactivity of each position of the pyrimidine halides follows the general order C6(4) > C2 ≫ C5.15 A strong preference for the C6 position has been observed in Suzuki cross-coupling reactions.16−20 Coupled with the use of a lower temperature for the Suzuki coupling, this allowed for the sequential introduction of different substituents.
Scheme 4. Synthetic Route to Polar Substitutions on R1 of the Pyrimidinone.

Reagents and conditions: (i) aqueous NaOH, dioxane; (ii) boronic acid, Cs2CO3, Pd(OAc)2, dppf, 1,4-dioxane, 70 °C or K2CO3, PdCl2(dppf)2, 1,4-dioxane, 70 °C; and (iii) 3,5-dimethylpyrazole, Cs2CO3, 1,4-dioxane, 130 °C, microwave.
Thus, 2,6-dichloropyrimidin-4(3H)-one 68 underwent a Suzuki cross-coupling with the appropriate boronic acid, using Pd(OAc)2/dppf or PdCl2(dppf)2 as the catalyst to afford compounds 69a–d. Coupling of intermediates 69a–d with 3,5-dimethylpyrazole was achieved by heating to 130 °C under microwave irradiation using 15 mol % Pd2(dba)3, xanphos, and Cs2CO3 in 1,4-dioxane to afford the desired compounds 22–25 (Table 3). A slightly modified procedure, as shown in Scheme 5, was used to prepare amide 26. Methyl 2,6-dichloropyrimidine-4-carboxylate was treated with sodium methoxide at 0 °C to obtain intermediate 70. Intermediate 70 was then reacted with 3,5-dimethylpyrazole under microwave heating using Cs2CO3 as a base to give acid 71, which was followed by subsequent in situ hydrolysis of the methyl ester. Acid 71 was coupled with 4-fluoroaniline using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) to give amide intermediate 72, which was demethylated using boron tribromide in dichloromethane to give compound 26.
Table 3. Polar Substitutions on Pyrimidinonea.

MIC—were measured in Middlebrook 7H9/Glu/BSA/Tyloxapol media at day 7; IC50—50% inhibitory concentrations; solubility—aqueous solubility measured at pH 7.4; CHO—an epithelial cell-line derived from the ovary of the Chinese hamster; and SI—selectivity index between CHO IC50 and Mtb MIC.
Scheme 5. Synthetic Route to Amide 26.
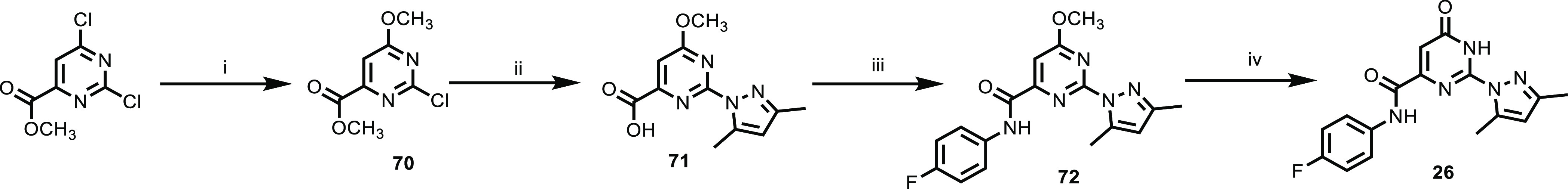
Reagents and conditions: (i) CH3ONa, THF, 0 °C; (ii) 3,5-dimethylpyrazole, Cs2CO3, 1,4-dioxane, 110 °C, microwave; (iii) 4-fluoroaniline, HATU, Et3N, DMF; and (iv) BBr3, CH2Cl2.
To determine if polarity at the R2 position on the pyrimidinone core was tolerated, compounds 27–29 (Table 3) were synthesized using the route, as shown in Scheme 6. 4-Fluorobenzaldehyde was condensed with thiourea and ethyl cyanoacetate using potassium hydroxide in refluxing ethanol to give 2-mecapto-5-cyanopyrimidinone intermediate 73.21 The thiol of intermediate 73 was methylated using methyl iodide in the presence of sodium hydroxide followed by displacement with hydrazine in refluxing ethanol to give 2-hydrazinyl-5-cyanopyrimidinone intermediate 74. This intermediate was then condensed with acetylacetone by heating in acetic acid-ethanol at 85 °C to obtain compound 27, which was hydrolyzed to amide 28 by heating in concentrated sulfuric acid at 85 °C.22 The conversion of amide 28 to acid 29 was achieved via nitrosation by heating with tert-butyl nitrite in acetic acid at 85 °C followed by hydrolysis.23
Scheme 6. Synthetic Route to Polar Substitutions on R2 of the Pyrimidinone.

Reagents and conditions: (i) ethyl 2-cyanoacetate, KOH, ethanol, reflux; (ii) (a) MeI, NaOH, H2O (b) NH2NH2·H2O, EtOH, reflux; (iii) acetylacetone, AcOH, EtOH, 85 °C; (iv) H2SO4, 80 °C; and (v) tert-butyl nitrite, AcOH, 75 °C.
Compounds 30–47 (Table 4) were synthesized to explore the SAR around the R5 position of the pyrazole ring, keeping the C6-position on the pyrimidinone core fixed as trifluoromethyl. Using the same methodology, as shown Scheme 1, the condensation of 56d with the corresponding 1,3-dicarbonyl compound afforded compounds 30–32 exclusively as one regioisomer as confirmed by NOESY NMR (Scheme 7). In addition, 56d provided access to compound 33, which served as a versatile intermediate and allowed for several transformations to form N-substituted pyrazole derivatives. The reductive amination of acetaldehyde with 33 in the presence of sodium cyanoborohydride in methanol gave ethylaminopyrazole 34. Attempts to synthesize the N,N-dialkyl derivative from compound 34 failed as the corresponding derivatives decomposed on standing. Compound 47 was synthesized in the same manner by reductive amination of acetaldehyde with 2-hydrazineyl-6-(3-pyridinyl)pyrimidinone 56g. The synthesis of 5-(aryl-amino)pyrazoles 40–46 was achieved via a Buchwald Hartwig amination of 33 and the corresponding aryl halide.
Table 4. SAR at the Pyrazole R5 Positiona.

MIC—were measured in Middlebrook 7H9/Glu/BSA/Tyloxapol media at day 7; IC50—50% inhibitory concentrations; solubility—aqueous solubility measured at pH 7.4; CHO—an epithelial cell-line derived from the ovary of the Chinese hamster; and SI—selectivity index between CHO IC50 and Mtb MIC.
Compound 38 was synthesized from hydrazine 56d and N-(2-methoxyethyl)-3-oxobutanamide with Lawesson’s reagent, which allows for the efficient installation of substituted-5-amino groups on the pyrazole core in a single step.24 However, this methodology failed in the case of the synthesis of compounds 36, 37, and 39, with 2-(5-hydroxy-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one being obtained as the major product. An alternate highly regioselective route to 5-(substituted-amino)pyrazoles 36, 37, and 39 was achieved following a modified literature procedure.25
As shown in Scheme 8, the displacement of one methylthio group of the α-oxoketene dithioacetal 75(26,27) by the appropriate amine in the presence of acetic acid in ethanol afforded the N,S-acetals 76a-c, which were useful three-carbon 1,3-electrophiles.28 Cyclocondensation with hydrazine 56d in refluxing ethanol, gave access to 5-(substituted-amino)pyrazoles 36, 37, and 39. The regioisomeric products with 3-(substituted-amino)pyrazoles could be isolated in minor quantities and showed much less potent activity against Mtb (data not shown).
Scheme 8. Synthetic Route to Compounds 36–39.

Reagents and conditions: (i) NH2-R, EtOH, reflux; (ii) 33, AcOH, EtOH, 100 °C.
Compound 35, with an O-ethyl group at R5, was synthesized, as shown in Scheme 9. The condensation of ethyl acetoacetate and hydrazine delivered pyrazolone 77, which on subsequent O-ethylation with ethyl iodide gave 78. Intermediate 78 was coupled with 79, synthesized by the oxidation of thiomethyl-pyrimidin-4-one 55d with mCPBA to yield 35.
Scheme 9. Synthesis of Compound 35.

Reagents and conditions: (i) (a) thiourea, KOH, EtOH and (b) MeI, NaOH, H2O; (ii) mCPBA, CH2Cl2, 25 °C; (iii) N2H2. H2O, EtOH; (iv) EtI, K2CO3, DMF, 70 °C; and (v) Cs2CO3, 1,4-Dioxane, 130 °C.
Compounds 48–50 with substitutions on the pyrazole C4 position were synthesized, as shown in Scheme 10. The bromination of compound 12 gave compound 48, which on Suzuki cross-coupling with pyridine-3-boronic acid gave compound 49. Attempts to displace C4-Br of 48 with morpholine to synthesize compound 50 were unsuccessful. In an alternative route, the morpholine ring was preinstalled on acetylacetone via bromination with N-bromosuccinimide (intermediate 80) followed by the reaction with morpholine in the presence of triethylamine to give intermediate 81. This intermediate was then condensed with hydrazine 56d by refluxing in ethanol-acetic acid to give compound 50.
Scheme 10. Synthesis of Compounds with Pyrazole C4 Substitutions.

Reagents and conditions: (i) Br2, AcOH, 100 °C, 2h; (ii) pyridine-3-boronic acid, K2CO3, Pd(dppf)Cl2, THF/water (10:1), 70 °C, 16h; (iii) N-bromosuccinamide, p-toluene sulphonic acid, CH2Cl2; 0 °C; (iv) morpholine, Et3N, CH2Cl2; and (v) EtOH/AcOH reflux, 16 h.
Compound 51 was synthesized in a similar fashion to compound 33, by condensing hydrazine 56d with 3-cyclopropyl-3-oxopropanenitrile (82) (Scheme 11). The synthesis of compounds 52–54 was accomplished from 83, as shown in Scheme 11. The reaction of 83 with 85 under microwave heating gave acid 53. This compound was then coupled with dimethylamine using HATU to give compound 54. Pyrazole ester 83 was converted to the corresponding dimethylaminomethyl derivative 84 via a standard two-step protocol involving the reduction of the ester to the aldehyde followed by reductive amination with dimethylamine. Intermediate 84 was reacted with 2-methylsulphonylpyrimidinone 79 under microwave heating to obtain compound 52.
Scheme 11. Synthesis of Compounds with Pyrazole C3 Modifications.

Reagents and conditions. (i) DMF, 130 °C, 2 h; (ii) Cs2CO3, 1,4-dioxane, 130 °C, microwave; (iii) N,N-dimethylamine, HATU, Et3N, DMF; (iv) (a) DIBALH, CH2Cl2, −78 °C; (b) N,N-dimethylamine, NaCNBH3, THF, RT, 24 h; and (v) Cs2CO3, 1,4-dioxane, 130 °C, microwave.
Formal Hit Assessment to Determine the Pharmacophore Critical for Antitubercular Activity
Although the initial hits, 1 and 2 (Table 1), showed reasonably potent antitubercular activity (MIC < 2 μM), they suffered from unfavorable physicochemical properties including low solubility, low to moderate microsomal metabolic stability (MS), and a low selectivity index (SI) between Mtb MIC and mammalian cell toxicity as determined by measuring 50% inhibitory concentrations (IC50) determined against a CHO cell-line (an epithelial cell-line derived from the Chinese-hamster ovary). A formal hit assessment study was undertaken to determine the minimum pharmacophoric features critical for antitubercular potency and to understand the SAR features needed to improve the SI. Initially, compounds with single point modifications on the pyrimidinone and pyrazole portions of the molecule were synthesized and profiled in a first wave of assays comprising Mtb MIC, CHO IC50, and aqueous solubility. The substituents on the pyrimidinone and pyrazole rings were labeled R1-R5, as shown Figure 1 (see Table 2). Masking of the pyrimidinone amide as −OCH3 (3) or N-CH3 (4) abolished the Mtb activity, indicating the essentiality of the NH, which might be involved in the critical hydrogen bond (HB) donor–acceptor interactions with the target. Each of the nitrogens of the pyrimidinone ring were found to be essential for maintaining Mtb activity (5 and 6). Interestingly, compound 7 with the carbonyl functionality between the ring nitrogens pyrimidin-2(1H)-one vs pyrimidin-4(3H)-one as in 1 maintained similar Mtb activity. The additional SAR explorations in this work were limited to the pyrimidin-4(3H)-one core as in compound 1, considering the ease of synthesis.
Compound 8 with a methyl group added at the pyrimidinone C5 (R2) position, while keeping a phenyl substituent at C6 (R1), retained a good MIC value (2 μM) while improving aqueous solubility (120 μM) and the SI (12.5) for cytotoxicity. However, isomeric compound 9 with C5-phenyl (R2) and C6-methyl (R1) showed a poor MIC value (9 μM) with a lower SI of 1, whereas the fusion of the phenyl ring with the pyrimidinone ring to form the corresponding quinazolinone (compound 10) retained potency but with a low SI of only 2. A phenyl substituent at the R1 (pyrimidinone C6) position was necessary to maintain Mtb activity. Compounds 11 and 12 with CH3 and CF3 groups, respectively, at R1 instead of the phenyl substituent showed a 10–15-fold loss in activity. As compound 12 with the CF3 group at R1 showed higher solubility and SI relative to 1, this modification was included in further explorations for potency and selectivity. Overall, these SAR observations suggested the requirement for hydrophobic substitutions on the pyrimidinone ring for the retention of potent in vitro activity against Mtb.
A few polar modifications on the pyrazole ring were attempted in order to evaluate the scope of reducing lipophilicity and improving drug-like properties. Changing the C5′-methyl (R5) on the pyrazole to oxygen abolished the activity as observed for compound 13, whereas an amino group at the C5′-position as in compound 14 resulted in the retention of activity comparable to compound 1 (3 μM) and significantly improved solubility (180 μM). Unfortunately, this polar modification did not improve the SI. N-acetyl amide derivative 15 retained the activity and SI profile similar to parent amine 14. Compound 16, with an unsubstituted pyrazole, lost potency considerably (MIC 50 μM). A similar loss in potency was observed when the pyrazole ring was replaced with other 5-membered rings like triazole or imidazole, as in compounds 17 and 18, respectively. This suggests the critical nature of an appropriately substituted pyrazole moiety for maintaining antitubercular activity.
The SAR observations regarding the critical pharmacophoric features required for maintaining and/or improving antitubercular activity along with the scope for further SAR studies are summarized in Figure 3. In summary, the pyrimidin-4(3H)-one ring was retained for further SAR studies considering the critical nature of the cyclic amide as well as the ring nitrogens for maintaining the antitubercular activity. The SAR observations suggest the important role of hydrophobic substituents at the R1 and R2 positions for potency. We explored the scope of polar interactions further away from the main scaffold by extending polar groups from the aryl ring at the R1 position. In addition, we also explored a few polar groups at the R2 position instead of a methyl group, which could influence HB-donor acceptor interactions of the pyrimidinone ring amide. The 3,5-disubstituted pyrazole ring was critical for maintaining activity, but there was a clear scope to extend substituents from the R5 position to improve potency and physicochemical properties. The scope of substitution at the R4-postion remained to be explored.
Figure 3.
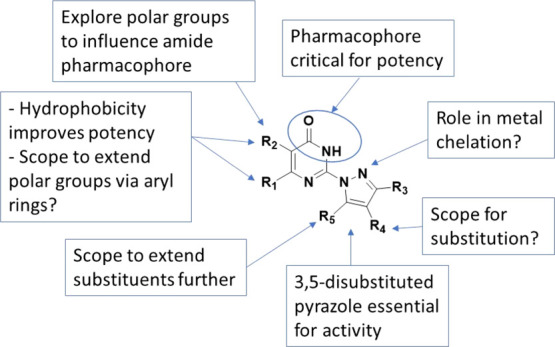
Essential pharmacophoric features and scope for further SAR studies.
SAR of Polar Groups at R1 and R2 Positions of the Pyrimidinone Core
Several polar R1 and R2 substituents on the pyrimidinone ring were incorporated in order to expand the scope relative to activity (Table 3) and increase solubility and SI. Compound 19 with a 4-fluorophenyl group at the R1 position retained activity and cytotoxicity similar to compound 1. The replacement of the phenyl ring with pyridine rings considerably improved solubility, as observed for compounds 20 and 21, although with some loss in Mtb activity. Compound 20, with a 4-pyridyl ring, was more potent (MIC 9 μM) than compound 21 with a 3-pyridyl ring (MIC 50 μM) leading to the speculation of the role of HB-interactions through the 4-pyridine nitrogen. Hence, compounds 22 to 25 with various polar substitutions at the para-position of the phenyl ring at the R1 position were prepared. Unfortunately, all these compounds lost activity. Next, we considered the option of inserting a polar linker between the pyrimidinone and phenyl rings. Among the various groups attempted, only compound 26 with a 4-flurophenyl group extended by a carboxamide linker at the R1 position, retained weak activity (25 μM).
Based on the encouraging data for compound 8 with a C5-methyl group on the pyrimidinone, we explored adding a few polar groups such as CN, CONH2, and CO2H at this position, but unfortunately this led to the loss of activity (compounds 27, 28, and 29). Overall, these observations indicated very limited scope for adding polar substitutions on the pyrimidinone ring. Hence, we turned our attention to exploring possible interactions from the pyrazole ring.
SAR of the Pyrazole Ring
Further exploration on the pyrazole ring was planned, maintaining the CF3 as the R1 substituent on the pyrimidinone, as it increased the solubility and SI exemplified for compound 12. In addition, the smaller CF3 group kept the molecular weight and lipophilicity on the low side compared to the phenyl. The addition of bulkier alkyls like ethyl and cyclopropyl (compounds 30 and 31) at the R5 position on the pyrazole were tolerated for Mtb activity relative to 12 but with a deterioration of the SI, whereas a slight loss in activity was observed for compound 32 with the phenyl group at the R5 position. Isomers of these compounds with bulkier groups at the R3 position could not be synthesized because of the selectivity observed in the condensation reaction of hydrazine with requisite diketones favoring the placement of bulkier groups at the R5 position. Compound 33 with a NH2 at the R5 position maintained a profile similar to compound 12, whereas compound 34 with an aminoethyl at the R5 position showed increased toxicity against CHO cell lines without a significant improvement in activity. Compound 35 with an O-ethyl group at the R5 position had a potency and selectivity profile similar to compound 12. The extension of polar groups like −NH2 or −OH via a short ethylamino linker from the R5 position as in compounds 36 and 37, respectively, was detrimental to activity against Mtb. Compounds 38 and 39, with the neutral ether substituents like methoxyethylamino and pyranamino, respectively, at the R5 position, showed more potent activity (MIC 1.5–2 μM) as compared to 12 while the SI was about the same. Based on these observations, it is speculated that the R5 position lies in a more hydrophobic region of the compound binding site within the target in Mtb. Hence, we decided to examine aryl groups, some with polarity, at R5 toward maintaining activity while improving solubility with polar substituents placed distant from the main scaffold.
Compound 40 with a 4-F-aniline at the R5 position showed Mtb potency and a SI profile similar to 39, with an aminopyran at position R5, but had lower aqueous solubility (15 μM) as expected because of the overall increase in lipophilicity with an additional aryl ring. Compound 41 with a 2,4-difluoroanilino group at the R5 position was the most potent compound (MIC 0.2 μM) obtained so far in these studies but was poorly soluble (<5 μM) in aqueous media. The addition of polar groups like CN and dimethylcarboxamide (42 and 43) at the para-position of the R5-anilino groups decreased activity (MIC 9 and >50 μM, respectively). Similarly, compound 44 with a 4-pyridylamino at the R5 position was inactive against Mtb up to the highest concentration tested (MIC > 50 μM). Interestingly, compound 45 with a 2-pyridylamino group at the same R5 position was quite potent with a MIC of ≤1 μM. The compound had moderate aqueous solubility (25 μM) with a modest SI of 25 for CHO cell-line toxicity, thus making it a valuable compound for MoA studies (see below). A few compounds with polar substituents para to the amino group on the 2-pyridyl ring were synthesized and screened with a view to improving physicochemical properties. For example, compound 46 with a 4-morpholino-2-pyridylamino group at the R5 position retained good activity (MIC = 2 μM) with improved solubility (180 μM) but was significantly more cytotoxic (SI 0.6).
Compound 47, with a 3-pyridyl at the R1 position and an ethylamino group at the R5 position, showed modest activity with an MIC of 6.25 μM and CHO IC50 of >50 μM.
A limited SAR exploration at the R4 position on the pyrazole was conducted to evaluate the scope for further improvement in potency and selectivity. Compound 48 with 4-bromopyrazole showed only moderate activity (MIC 25 μM, CHO IC50 > 50 μM) and was used as an intermediate for further synthesis. Replacing the bromine with a 3-pyridyl ring (49) abolished Mtb activity (MIC >50 μM), whereas the morpholine ring at this position (50) was well tolerated with an MIC = 6.25 μM and a cleaner cytotoxicity profile (CHO IC50 > 50 μM). In comparison, the R3 position on the pyrazole showed a very limited scope for further modifications. In general, a cyclopropyl group as in compound 51 was well tolerated for Mtb potency but larger groups in general gave weakly active compounds. Polar groups like dimethylaminomethyl, carboxylic acid, and amide as in compounds 52, 53, and 54, respectively, led to considerable loss of activity against Mtb (MICs > 50 μM).
In summary, various modification on the pyrimidinone and pyrazole portions of the scaffold showed potential for improvement in potency against Mtb in vitro and MICs as low as 200 nM could be achieved. However, in general there was a narrow scope to improve the SI against mammalian cell-line toxicity. The potential impact of both these activities in vivo remains to be determined.
Physicochemical, DMPK, and Safety Profiles
Presented in Table 6 are physicochemical and in vitro DMPK properties of representative compounds, including solubility, microsomal MS, and human plasma protein binding (PPB). Compounds showed moderate to high aqueous solubility with excellent in vitro microsomal stability, with the exception of compound 8 that was only moderately stable in microsomes. A small number of compounds that were tested for the inhibition of liver cytochrome P450 enzymes did not show significant inhibition up to 20 μM (data not shown), indicating minimal possibility of metabolism-based drug–drug interactions of these compounds, which is a desirable attribute of a TB drug.29,30 The compounds tested in the series were highly bound to the human plasma most likely due to the albumin binding of acidic pyrimidinone.31 The safety profiles of compounds 1 and 12 were evaluated across a panel of 21 liability targets (39 functional assays), which included cell-based GPCRs and ion channels in both agonist and antagonist readout, and biochemical functional assays for nuclear hormone receptors and phosphodiesterases.32 Both compounds showed no significant inhibition or activation of the enzyme/receptors at 10 μM test concentration. Both compounds showed hERG IC50 > 30 μM. In summary, the in vitro safety profile of the compounds indicated no obvious safety liability even though the reason for mammalian cytotoxicity observed was not clear.
Table 6. In Vitro ADME Properties of Selected Compoundsd.
| compound | 8 | 12 | 39 | 40 | 45 | 47 | 50 |
|---|---|---|---|---|---|---|---|
| clog Pa | 1.82 | 1.36 | 1.30 | 2.52 | 1.46 | 0.66 | 1.54 |
| clog Da | 1.82 | 1.36 | 1.09 | 2.52 | 1.46 | 1.09 | 1.40 |
| solubility (μM)b | 120 | 200 | 50 | 15 | 25 | 90 | 160 |
| MSc H/R/Mo % remaining after 30 min | 65/54/45 | >99/92/>99 | >99/>99/93 | 98/98/96 | >99/96/>99 | 80/95/94 | ND |
| PPBd human fu | ND | 0.006 | 0.014 | 0.013 | ND | 0.009 | 0.03 |
Calculated log P/log D (StarDrop).
Kinetic solubility measured at pH 7.4.
Microsomal stability H: human, R: rat Mo: mouse.
PPB; fu—fraction unbound.
The pharmacokinetic parameters of representative compounds, 12, 47, and 40 were measured from mouse blood at intravenous doses of 2 mg/kg and oral doses of 20 mg/kg in the mouse (Table 7). The compounds were well tolerated following either route of administration, with no obvious effects noted in the animals over the course of the exposure. Compounds 12 and 40 showed a low volume of distribution and low rate of clearance with moderate oral bioavailability of approximately 40%. Compound 47 showed rapid blood clearance and high volume of distribution with an oral bioavailability of 33%. Further analysis of mouse blood samples from the PK studies of 47 showed the glucuronide as the major metabolite. However, further optimization of physicochemical properties to improve pharmacokinetic properties, along with improved SI, is warranted in order to identify a compound suitable for in vivo efficacy studies in TB animal models.
Table 7. Pharmacokinetic Parameters in Male C57/BL6 Mouse Blooda.
| route of administration | i.v. | oral | i.v. | oral | i.v. | oral |
|---|---|---|---|---|---|---|
| 12 | 47 | 40 | ||||
| dose (mg/kg) | 2 | 20 | 2 | 20 | 2 | 20 |
| apparent t1/2 (h) | 3.1 | 4 | 5.6 | 4.4 | 4.0 | 6.1 |
| CLtotal (mL/min/kg) | 2.7 | 80.9 | 0.4 | |||
| CLint (mL/min/kg) | 476 | >4000 | 34 | |||
| Vd (L/kg) | 0.7 | 37.4 | 0.1 | |||
| Cmax (μM) | 29.1 | 65.6 | 2.4 | 2.9 | 33.1 | 104.5 |
| Tmax (h) | 0.5 | 0.5 | 1.0 | |||
| AUC0–∞ (min·μM) | 2928 | 12004 | 91 | 313 | 15373 | 65417 |
| oral bioavailability F (%) | 41 | 33 | 44 | |||
i.v.: intravenous; t1/2: elimination half-life; CLtotal: total blood clearance; CLint: intrinsic clearance; Vd: volume of distribution during elimination phase; Cmax, maximum (peak) plasma concentration following oral administration; and AUC: area under the curve.
Bactericidality and Activity against Clinical Isolates
The compounds were found to be bactericidal against replicating Mtb showing 2 log CFU reduction at 1–2× MIC over the time period of 7 days (Fig. S1). Importantly, we also evaluated the activity of 1, 12, 15, and 20 against a panel of clinical isolates (Table S1). All the tested compounds retained MICs against clinical isolates within the 4-fold range of MICs against the drug-sensitive Mtb H37Rv strain.
Target Deconvolution Studies
Biology Triaging
In order to explore the MoA, potent compounds were initially screened against various tool strains to rule out the involvement of known mechanisms and/or targets. The tested compounds retained activity against a QcrB mutant (A396T) and did not show hypersensitivity against a cytochrome-bd oxidase knockout mutant strain (cydKO),33 thereby eliminating them as potential targets. The compounds did not show a positive signal in two standard bioluminescence reporter assays: PiniB-LUX34—detects modulation in the iniB expression, if a compound targets Mtb cell–wall biosynthesis, and PrecA-LUX34—detects modulation in the recA expression, an indicator of genotoxic compounds. A strain carrying a mutation in DprE1 (C387S), which confers resistance to other DprE1 inhibitors, was not resistant, suggesting DprE1 is not the target. However, strains carrying mutations in MmpL3 were cross-resistant to compounds (Table 8).35 The compounds showed an increase of 8–100-fold in MIC values against the strains with either MmpL3F255L or MmpL3V681I or MmpL3G596R, whereas, there was no change in activity against the MmpL3F644L mutant. Most of these mutations lie within the region required for proton translocation.36 Variability in the MICs against these mutant strains is suggestive of the fact that the compounds may bind differently to the protein depending on the mutation. To investigate whether or not compounds 1, 2, 12, 15, 47, 40, and 38 retain target selectivity for MmpL3 in Mtb cells, we asked whether or not conditional depletion of MmpL3 in a mmpL3 hypomorph (Grover et al., manuscript under review) would sensitize Mtb to the growth inhibitory effects of the compounds. However, transcriptional silencing of mmpL3 resulted in no significant change in the MICs of the compounds, confirming that MmpL3 is not the direct target of this series (Table S2). We hypothesize that MmpL3 acts as a transporter of these compounds across the cell membrane as the compounds can form heme-like iron-complexes, and MmpL3 is known to act as a heme transporter.37,38 Hence, MmpL3 is likely to be responsible for building resistance toward compounds through the impairment of transport of compounds across the cell membrane rather than being a direct target. It is to be noted that the compounds were PiniB-LUX negative—indicating a non-cell wall MoA.
Table 8. Cross-Screening against mmpL3 Mutantsa.
| strain | 1 | 2 | 12 | 15 | 47 | 40 | 38 |
|---|---|---|---|---|---|---|---|
| H37Rv WT MIC (μM) | 1.7 | 4.8 | 15 | 27 | 5.8 | 1.2 | 1.2 |
| MmpL3 F255L MIC (μM) | 18 | 55 | 170 | >200 | 41 | >200 | 39 |
| MmpL3 F644L MIC (μM) | 1.9 | 5.4 | 17 | 24 | 5.5 | 1 | 1.9 |
| MmpL3 V681 MIC (μM) | 16 | 45 | >200 | ||||
| MmpL3 G596R MIC (μM) | 40 | 110 | >200 |
WT: wild-type.
Based on the structural similarity of the compounds with the published thymidylate kinase inhibitors,39,40 we asked whether this compound series can inhibit Mtb thymidylate kinase, which is an essential target encoded by Rv3247c. To investigate this, we tested the activity of the compounds 1, 2, and 12 against the Rv3247c hypomorph. However, we did not observe any MIC modulation of the compounds upon silencing of Rv3247c, suggesting that the tested compounds are not the thymidylate kinase inhibitors.
Treatment with 45 and 12 Affects Genes Involved in Fe Homeostasis
In continuation of our efforts to explore MoA, the effects of 45 (at 1 μM and 10 μM) and 12 (at 20 μM) treatment on the gene expression of Mtb was investigated. Mtb cultures were treated with the compound for 6 h, harvested, and the RNA extracted for transcriptional profiling by microarray analysis. The data obtained from the transcriptional profiling of Mtb exposed to 45 and 12 were similar (Table S3). We performed in-depth analysis of the data obtained from samples treated with 45, which was tested at two concentrations. The 50 most upregulated (>4-fold) genes were selected in order to understand their potential contribution in the cellular response based on the MoA of 45 (Table S4). This indicated a transcriptional signature related to iron-sequestration. The upregulated genes included rv2377c-78c-79c-80c-81c-82c-83c-84-85-2386c (mbtA-H)—all of which are associated with mycobactin biosynthesis or regulation,41 which are known to be upregulated in response to iron deprivation.
Another upregulated gene-set rv1342-43-44-45-46-47-48-49 is also associated with the mycobactin biosynthesis or regulation or with iron transport where rv1348 and rv1349 were annotated as iron-regulated transporters, both being essential for growth in vitro.42−44 The esx-3 gene cluster is composed of 11 genes stretching from rv0282 to rv0292. All of these were upregulated 2–5-fold in response to the stress caused by the compound. rv0287 and rv0288 are predicted to be essential for growth in vitro. Interestingly, ESX-3 is known to be required for the mycobactin-mediated siderophore iron uptake pathway,45−48 and it is essential in Mtb but not in Mycobacterium smegmatis, a species which, in addition to mycobactins, produces exochelin that functions independently of ESX-3. However, ESX-3 is still required for mycobactin-mediated iron uptake in the latter species.45,46,48 The nonessential nature of ESX-3 in M. smegmatis has previously proven to be of a great advantage when resolving mechanisms related to ESX-3 and mycobactin iron metabolism.48 Some of the top upregulated genes are involved in lipid metabolism, for example, rv3249-51-52 potentially a transcriptional response to correct damage to fatty acids. Fe has been reported to affect lipid metabolism, specifically because of Fe-catalyzed lipid oxidation. In line with this, we observed that genes, including rv3741-42, (oxidoreductase) were upregulated. The observed upregulation of the rv3269-70 operon encoding a stress response protein and a metal cation transporter, respectively, further supports the notion that the cellular response to 45 is one of the disruption of iron homeostasis. Rv0285—PE5 which was upregulated in our study, is downregulated in the presence of iron.49 Importantly, most of the upregulated genes of our data set were found to be downregulated when Mtb was exposed to an excess of iron.49 Our data set also complements the recently published work by Kurthkoti et al.,50 where authors performed transcriptomics in iron-starved cultures and identified similar upregulated clusters of genes. This strongly suggests that the disruption of Fe homeostasis is an important component of the MoA of 45.
In summary, our data suggest that upon exposure to 2-pyrazolylpyrimidinones, Mtb goes into an iron-starvation state. As expected, it upregulates genes involved in the mycobactin (siderophore) synthesis pathway and its regulation. Siderophores are small, high-affinity iron-chelating compounds secreted by microorganisms such as bacteria and fungi. In the process of cellular uptake, the Fe3+-siderophore complex is subsequently reduced to Fe2+ to release the iron.
Iron Plays a Significant Role in the MoA
To confirm the role of iron in the MoA of this series, an Fe rescue experiment was performed by supplementing standard Middlebrook 7H9/Glu/CAS/Tx media with 200 μM ferric ammonium citrate. The supplementation of Fe3+ resulted in a 4-fold increase in the MIC (Table S5). Under physiological conditions, iron can exist in either the reduced ferrous (Fe2+) form or the oxidized ferric (Fe3+) form. The redox potential of Fe2+/Fe3+ makes iron extremely versatile when it is incorporated into proteins as a catalytic center or as an electron carrier. Thus, iron is important for numerous biological processes, which include the tricarboxylic acid cycle, gene regulation, DNA biosynthesis, and so forth. Although iron is abundant in nature, it does not normally occur in its biologically relevant but aerobically unstable ferrous form. To evaluate this, we supplemented the Mtb culture with 200 μM ferrous sulfate, resulting in an 8-fold increase in the MIC for 45 (Table S5). Next, we confirmed the ability of Fe2+ to rescue the Mtb from 45 toxicity in a checkerboard (2D) assay using GAST (glycerol-alanine-salts-Tween 80) media which is devoid of Fe unlike Middlebrook 7H9/Glu/CAS/Tx (Figure 4A). The inhibition of Mtb growth by 45 in GAST medium supplemented with 12.5 μM Fe+2 (MIC 0.12 μM) decreased in a dose-dependent manner with increasing concentration of Fe2+ and resulted in a ≥32-fold increase in MIC at 100/200 μM Fe2+. To determine that 45 forms a complex with Fe2+, we performed UV–vis scanning of 45 with increasing amounts of Fe2+ (Figure 4B). The band intensity at 270, which was observed for 45 alone, decreased after the addition of Fe2+, whereas the absorbance around 285 is shifted to 300 with the increase in intensity, indicating the formation of the 45-Fe2+ complex.
Figure 4.

Fe2+ plays a critical role in the MoA of 45. (A) Fe2+ rescue of Mtb from growth inhibition mediated by 45. (B) Spectroscopic titration of 45 with Fe2+. The data are representative of two biological replicates performed in duplicate.
In line with the observation of better rescue with ferrous than ferric iron and a preference of heme for the former, there is a strong possibility that the compound binds iron to form a heme-like complex that enters Mtb via MmpL3. This also supports the reason for resistance with some of the MmpL3 mutants as described previously—heme prefers ferrous over ferric. Involvement of a heme-like structure could also explain the associated cytotoxicity of the series, as described earlier. This is because of the toxic nature of heme to eukaryotic cells, which require specific heme-binding proteins to maintain a nontoxic homeostasis.
Conclusions
Whole-cell phenotypic screening of MMV compound library led to the identification of 2-pyrazolylpyrimidinones with potent antitubercular activity and a novel mode of action. Detailed SAR studies identified several compounds with potent activity against Mtb with moderate to high aqueous solubility and excellent in vitro microsomal stability. The compounds were bactericidal against replicating Mtb and retained potency against clinical isolates. Transcriptional profiling suggested that upon exposure to 2-pyrazolylpyrimidinones, Mtb goes into an iron-starvation state that may account for lethality. Iron supplementation using a high concentration of ferrous salts showed shifts in MICs of the compounds, further confirming that 2-pyrazolylpyrimidinones perturbs the Fe-homeostasis in Mtb. Further optimization of pharmacokinetic properties, along with improved SI between MIC and mammalian cytotoxicity, is needed to identify a compound suitable for in vivo efficacy studies in mouse TB models and eventual development as a viable drug useful for the treatment of TB in humans.
Experimental Section
MIC Testing and Triage Assays
Unless otherwise indicated, an Alamar Blue fluorescence-based broth microdilution assay was used to assess MIC of compounds against Mtb strains, as described previously.51,52 Rifampicin was included as a control. Biology triage assays were carried out as described by Naran et al.34
MICs against Mutant Strains
MICs were determined after 5 days of growth in Middlebrook 7H9 medium plus OADC supplement and 0.05% w/v Tween 80 as described previously.53 Growth was monitored by OD. Data were fit using the Levenberg–Marquardt least-squares plot. MIC was defined as the concentration required to inhibit growth by 90%.
RNA Extraction and Transcriptional Profiling
Total RNA was extracted from Mtb H37Rv that had been treated for 6 h with 1× and 10× MIC of the compound or vehicle control as previously described.54 RNA quality was confirmed by UV spectral analysis and Agilent 2100 Bioanalyzer returning a RNA integrity number of 8 or higher. Fluorescent-tagged cDNA was prepared via a direct random-primed labeling method as follows. To 4.0 μg of RNA, 4.5 μg of a random hexamer (Invitrogen# 48190-011) was added, a final volume of 14.5 μL, heat denatured at 70 °C for 5 min then immediately cooled to 0 °C on ice. cDNA synthesis and fluorophore (Cy3 or Cy5) incorporation were carried out using the following reverse transcription reaction components 5 μL of 5× First-Strand buffer, 1.25 μL of 0.1 M DTT, 2.5 μL of dNTP mix (made by 5 mM each of dATP, dGTP), dTTP, 0.5 mM dCTP, plus 1 μL of 200 U/μL SuperScriptIII (Invitrogen# 18080-044), 1 μL of 40 U/μL RNAseOut, and 1 μL of Cy3 or Cy5-dCTP (GE# PA55321) were added for incubation at 25 °C for 5 min and at 48 °C for 90 min. Template RNA was chemically hydrolyzed by 5 μL of 1 M NaOH and heated at 70 °C for 15 min. The hydrolysis reaction was neutralized with 5 μL of 1 M HCl. The labeled cDNA was purified on an Amicon Ultra-0.5 column (Millipor# C82301) following the manufacturer’s recommendations for PCR purification. cDNA yields and dye incorporation were measured with a Nanodrop ND-1000. A mixture of equal amounts (0.7 μg) of Cy3- and Cy5-labeled cDNA was loaded into hybridization chambers for incubation with an Agilent SurePrint G3 4x44K custom oligonuclotide microarray (design number 021966, 021362).
Microarrays were hybridized robotically using a TECAN HS Pro 4800 hybridization station, Agilent 2× gene expression hybridization HI-RPM buffer, and 10× blocking reagent at 65 °C for 17 h and washed with Agilent Gene Expression Wash Buffer 1 at room temperature and Gene Expression Wash Buffer 2 at 37 °C. Then, slides were dried under a nitrogen gas for 3 min at 30 °C. The slides were imaged using an Agilent high-resolution DNA microarray scanner (model G2505C) at 5 μm resolution and 100/10% PMT dual scanning for XDR extended dynamic range. Agilent Feature Extraction software was used for image analysis.
DMPK
All protocols for in vitro DMPK studies and mouse PK studies are available in the supplementary document. Animal studies were conducted following guidelines and policies as stipulated in the UCT Research Ethics Code for Use of Animals in Research and Teaching, after review and approval of the experimental protocol by the UCT Senate Animal Ethics Committee (protocol FHS-AEC 013/032).
Chemistry
All commercial reagents were purchased from Sigma-Aldrich, Combi-Blocks, Enamine, or Fluorochem and were used without further purification. Solvents were used as received unless otherwise stated. Analytical thin-layer chromatography was performed on SiO2 plates on aluminum backing. Visualization was accomplished by UV irradiation at 254 and 220 nm. Flash column chromatography was performed using a Teledyne ISCO flash purification system with SiO2 60 (particle size 0.040–0.055 mm, 230–400 mesh). Purity of all final derivatives for biological testing was confirmed to be >95% as determined using an Agilent 1260 Infinity binary pump, Agilent 1260 Infinity diode array detector (DAD), Agilent 1290 Infinity column compartment, Agilent 1260 Infinity standard autosampler, and Agilent 6120 quadrupole (single) mass spectrometer, equipped with APCI and ESI multimode ionization sources. Using a Kinetex Core C18 2.6 μm column (50 mm × 3 mm); mobile phase B of 0.4% acetic acid, 10 mM ammonium acetate in a 9:1 ratio of HPLC grade methanol and type 1 water, mobile phase A of 0.4% acetic acid in 10 mM ammonium acetate in HPLC grade (type 1) water, with a flow rate of 0.9 mL/min, DAD; or an Agilent UPLC–MS was used: Agilent Technologies 6150 quadrupole, ES ionization, coupled with an Agilent Technologies 1290 Infinity II series UPLC system Agilent 1290 series HPLC at two wavelengths 254 and 290 nm using the following conditions: Kinetex 1.7 μm Evo C18 100A, LC column 50 mm × 2.1 mm, solvent A of 0.1% (formic acid) water, and solvent B of 0.1% (formic acid) acetonitrile. The structures of the intermediates and end products were confirmed by 1H NMR and mass spectrometry. Proton magnetic resonance spectra were determined in an appropriate deuterated solvent on a Varian Mercury spectrometer at 300 MHz or a Varian Unity spectrometer at 400 MHz.
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-phenylpyrimidin-4(3H)-one (1)
A mixture of 2-hydrazinyl-6-phenylpyrimidin-4(3H)-one (56a 0.2 g, 0.989 mmol) and pentane-2,4-dione (0.121 mL, 1.187 mmol) was heated in a mixture of ethanol (0.5 mL) and acetic acid (1.5 ml) at 100 °C for 16 h. After cooling to room temperature, the reaction mixture was poured into ice cooled water and stirred for 30 min. Solids formed were filtered, washed with water, and dried to yield 1 as a white solid (200 mg, 0.744 mmol, 75% yield). HPLC purity: >99%. LC–MS APCI: calcd for C15H14N4O, 266.304; observed m/z [M + H]+, 267.1. 1H NMR (300 MHz, CDCl3): δ 8.01–7.97 (m, 2H), 7.53–7.50 (m, 3H), 6.73 (s, 1H), 6.12 (s, 1H), 2.55 (s, 3H), 2.32 (s, 3H).
6-Methyl-2-(3-methyl-5-(p-tolylamino)-1H-pyrazol-1-yl)pyrimidin-4(3H)-one (2)
Compound was received from the MMV screening library. For synthesis and analytical data, please refer WO2012154880.2
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-methoxy-6-phenylpyrimidine (3)
DIAD (0.110 mL, 0.563 mmol) was added dropwise to a solution of 2-(3,5-dimethyl-1H-pyrazol-1-yl)-6-phenylpyrimidin-4(3H)-one (1) (100 mg, 0.376 mmol), triphenylphosphine (148 mg, 0.563 mmol) in methanol (12.03 mg, 0.376 mmol), and tetrahydrofuran (100 mL), and the reaction mixture was stirred at room temperature for 4 h. The mixture was concentrated under reduced pressure, the residue partitioned between dichloromethane and water, and the layers separated. The aqueous phase was extracted with dichloromethane, the combined organic solutions washed consecutively with water, 2 N aqueous sodium hydroxide, water, and finally brine. The solution was then dried over sodium sulfate and evaporated under reduced pressure. The crude product was purified on an ISCO system using a 4 g silicycle column and eluting with a gradient 0 to 70% ethyl acetate in hexane over 20 min to yield 3 as a white solid (27 mg, 0.09 mmol, 24% yield). HPLC purity: 95%. LC–MS APCI: calcd for C16H16N4O, 280.33; observed m/z [M + H]+, 281.1. 1H NMR (300 MHz, CDCl3): δ 7.99–7.96 (m, 2H), 7.49–7.47 (m, 3H), 6.92 (s, 1H), 6.31 (br s, 1H), 6.08 (s, 1H), 3.56 (s, 3H), 2.50 (s, 3H), 2.33 (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-3-methyl-6-phenylpyrimidin-4(3H)-one (4)
To a stirred solution of 2-(3,5-dimethyl-1H-pyrazol-1-yl)-6-phenylpyrimidin-4(3H)-one (1) (50 mg, 0.188 mmol) in DMF (2 mL) was added potassium carbonate (52 mg, 0.563 mmol). The reaction mixture was stirred for 5 min and iodomethane (35 μL, 0.563 mmol) was added. The reaction mixture was stirred at 100 °C overnight. The reaction mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic layer was washed with LiCl solution (1 × 10 mL) and dried over sodium sulfate, and the solvent was evaporated. The obtained residue was purified by flash chromatography by using 15% ethyl acetate in hexane obtained 4 as a white solid (39 mg, 0.141 mmol, 75% yield). HPLC Purity: 96%. LC–MS APCI: calcd for C16H16N4O, 280.33; observed m/z [M + H]+, 281.1. 1H NMR (300 MHz, CDCl3): δ 8.01–7.97 (m, 2H), 7.53–7.50 (m, 3H), 6.73 (s, 1H), 6.18 (s, 1H), 3.52 (s, 3H), 2.55 (s, 3H), 2.32 (s, 3H).
6-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-phenylpyridin-2(1H)-one (5)
To a cooled solution of crude 61 (0.40 g, 1.43 mmol) in methanol (7 mL) was added boron tribromide (1 M solution in dichloromethane) (4.2 mL, 4.29 mmol) and stirred for 16 h at room temperature. Water was added to the reaction mixture and extracted with dichloromethane. The combined organic phase was dried over sodium sulfate, concentrated under vacuum, and purified by preparative HPLC to give 5 as a white solid (0.08 g, 21%). HPLC purity: 98.6%. LC–MS APCI: calcd for C16H15N3O, 265.122; observed m/z [M + H]+, 266.2. 1H NMR (400 MHz, CDCl3): δ 9.38 (br s, 1H), 7.64–7.62 (m, 2H), 7.51–7.47 (m, 3H), 6.76 (s, 1H), 6.70 (s, 1H), 6.07 (s, 1H), 2.58 (s, 3H), 2.3 (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-phenylpyridin-4(1H)-one (6)
Synthesized from intermediate 62 with a protocol similar to the one described for compound 5. Yield 25% (white solid); HPLC purity: 98.9%. LC–MS APCI: calcd for C16H15N3O, 265.122; observed m/z [M + H]+, 266.2. 1H NMR (400 MHz, CDCl3): δ 10.49 (br s, 1H), 7.96 (d, J = 6.4 Hz, 2H), 7.46–7.40 (m, 4H), 7.11 (s, 1H), 6.07 (s, 1H), 2.70 (s, 3H), 2.35 (s, 3H).
6-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-phenylpyrimidin-2(1H)-one (7)
To the solution of 2-chloro-4-(3,5-dimethyl-1H-pyrazol-1-yl)-6-phenylpyrimidine (intermediate 65, 0.16 g, 0.56 mmol) in THF (2 mL) was added 2 M aqueous NaOH solution (2 mL) at room temperature, and the mixture was heated at 80 °C for 4 h. The reaction mixture was then cooled in ice-bath and acidified with glacial acetic acid. The precipitated solid was filtered under vacuum, washed with chilled water, and dried to get 6-(3,5-dimethyl-1H-pyrazol-1-yl)-4-phenylpyrimidin-2(1H)-one (7, 0.12 g, 80% yield) as a white crystalline powder. HPLC purity: 99.2%. LC–MS APCI: calcd for C15H14N4O, 266.117; observed m/z [M + H]+, 267.2. 1H NMR (400 MHz, DMSO-d6): δ 8.11 (m, 2H), 7.53 (m, 3H), 6.92 (s, 1H), 6.26 (s, 1H), 2.71 (s, 3H), 2.23 (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-5-methyl-6-phenylpyrimidin-4(3H)-one (8)
Synthesized from intermediate 56b following a protocol similar to compound 1. Yield 10% (white solid). HPLC purity: 97%. LC–MS APCI: calcd for C16H16N4O, 280.331; observed m/z [M + H]+, 281.1. 1H NMR (300 MHz, CDCl3): δ 7.68–7.58 (m, 2H), 7.55–7.39 (m, 3H), 6.05 (s, 1H), 2.69 (s, 3H), 2.30 (s, 3H), 2.19 (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-methyl-5-phenylpyrimidin-4(3H)-one (9)
Synthesized from intermediate 56h following a protocol similar to compound 1. Yield 10% (white solid). HPLC purity: 98.9%. LC–MS APCI: calcd for C16H16N4O, 280.132; observed m/z [M + H]+, 281.0. 1H NMR (400 MHz, CDCl3): δ 10.59 (s, 1H), 7.27–7.44 (m, 5H), 6.05 (s, 1H), 2.71 (s, 3H), 2.26 (s, 3H), 2.22 (s, 3H). 13C NMR (100 MHz, CDCl3): 13.63, 15.08, 22.99, 111.38, 122.18, 127.70, 128.36, 130.17, 133.72, 143.40, 145.66, 151.82, 161.01, 161.19.
2-(3,5-Dimethyl-1H-pyrazol-1-yl)quinazolin-4(3H)-one (10)
Synthesized from 2-hydrazineylquinazolin-4(3H)-one following a protocol similar to compound 1. Yield 47% (white solid). HPLC purity: 96.3%. LC–MS APCI: calcd for C13H12N4O, 240.10; observed m/z [M + H]+, 241.1. 1H NMR (400 MHz, DMSO-d6): δ 11.92 (s, 1H), 8.11 (dd, J = 7.9, 1.5 Hz, 1H), 7.81 (ddd, J = 8.5, 7.2, 1.6 Hz, 1H), 7.65 (d, J = 8.1 Hz, 1H), 7.53–7.43 (m, 1H), 6.27 (s, 1H), 2.69 (s, 3H), 2.26 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 160.68, 151.04, 147.19, 143.23, 139.59, 135.19, 126.65, 126.46, 118.79, 115.59, 111.34, 14.74, 13.85.
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-methylpyrimidin-4(3H)-one (11)
Synthesized from intermediate 56c following a protocol similar to compound 1. Yield 78% (white solid). HPLC purity: 96%. LC–MS APCI: calcd for C10H12N4O, 204.23; observed m/z [M + H]+, 205.1 1H NMR (300 MHz, CDCl3): δ 6.73 (s, 1H), 6.12 (d, 1H), 2.55 (s, 3H), 2.32 (s, 3H), 2.23 (d, 1H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (12)
Synthesized from intermediate 56d following a protocol similar to compound 1. Yield 70% (white solid). HPLC purity: 98.9%. LC–MS APCI: calcd for C10H9F3N4O, 258.07; observed m/z [M + H]+, 259.0. 1H NMR (400 MHz, DMSO-d6): δ 6.79 (s, 1H), 6.27 (s, 1H), 2.56 (s, 3H), 2.23 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 13.83, 14.77, 111.89, 119.62, 122.34, 143.71, 152.04.
2-(3-Methyl-5-oxo-2,5-dihydro-1H-pyrazol-1-yl)-6-phenylpyrimidin-4(1H)-one (13)
A mixture of 2-hydrazineyl-6-phenylpyrimidin-4(1H)-one (56a, 150 mg, 0.742 mmol) and ethyl acetoacetate (0.114 mL, 0.890 mmol) was heated in a mixture of ethanol: acetic acid, 1:3, under reflux for 5 h. The solid product obtained after cooling and pouring onto ice-cold water was filtered off, washed with water, and crystallized from ethanol to give 13 as a white solid (39 mg, 0.148 mmol, 20% yield). HPLC purity 93%. LC–MS APCI: calcd for C14H12N4O2, 268.27; observed m/z [M + H]+, 269.1. 1H NMR (400 MHz, DMSO-d6): δ 8.22–8.20 (m, 2H), 7.54–7.51 (m, 2H), 6.74 (s, 1H), 5.31 (s, 1H), 2.29 (s, 3H).
2-(5-Amino-3-methyl-1H-pyrazol-1-yl)-6-phenylpyrimidin-4(1H)-one (14)
2-Hydrazineyl-6-phenylpyrimidin-4(1H)-one 56a (0.1 g, 0.495 mmol) and 3-aminocrotonitrile (0.042 g, 0.519 mmol) were stirred in ethanol (5 mL) and refluxed for 5 h. The solvent was removed, and the crude product was recrystallized from methanol to afford 14 as a white solid (55 mg, 0.742 mmol, 42% yield). HPLC purity: 97%. LC–MS APCI: calcd for C14H13N5O, 267.29; observed m/z [M + H]+, 268.1. 1H NMR (300 MHz, DMSO-d6): δ 8.04–7.92 (m, 2H), 7.60–7.49 (m, 3H), 6.91 (s, 2H), 6.73 (s, 1H), 5.35 (s, 1H), 2.13 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 192.03, 152.67, 151.14, 136.44, 131.20, 129.40, 127.22, 89.28, 14.37.
N-(3-Methyl-1-(4-oxo-6-phenyl-1,4-dihydropyrimidin-2-yl)-1H-pyrazol-5-yl)acetamide (15)
A mixture of 2-(5-amino-3-methyl-1H-pyrazol-1-yl)-6-phenylpyrimidin-4(1H)-one (14, 100 mg, 0.374 mmol) and pyridine (0.04 mL, 0.442 mmol) in dichloromethane (2 mL) was treated with a solution of acetyl chloride (0.04 mL, 0.0448 mmol) in dichloromethane (1 mL). The reaction mixture was stirred at room temperature for 24 h before it was diluted with dichloromethane (5 mL). The mixture was washed with 2 N HCl (10 mL), 5% aqueous solution of NaHCO3, and brine. The organic phase was dried over sodium sulfate, filtered, and concentrated in vacuo to give 15 as a white solid (45 mg, 0.145 mmol, 39% yield). HPLC purity: 98%. LC–MS APCI: calcd for C16H15N5O2, 309.33; observed m/z [M + H]+, 310.1. 1H NMR (300 MHz, DMSO-d6): δ 11.55 (s, 1H), 8.13–8.03 (m, 2H), 7.63–7.52 (m, 3H), 6.95 (s, 1H), 6.64 (s, 1H), 2.22 (d, J = 15.1 Hz, 6H).
6-Phenyl-2-(1H-pyrazol-1-yl)pyrimidin-4(3H)-one (16)
In a 10 mL microwave vial, 2-chloro-6-phenylpyrimidin-4(3H)-one (66, 0.1 g, 0.484 mmol), 1H-pyrazole (0.066 g, 0.968 mmol), and cesium carbonate (0.473 g, 1.452 mmol) were mixed in 1,4-dioxane (1 mL). The vial was capped and microwaved at 100 °C for 1 h. LCMS indicated two products of the same mass (isomers from 70:30 ratio of the starting material). The reaction mixture was concentrated and purified by preparative HPLC to afford 16 as a white solid (29 mg, 0.121 mmol, 25% yield). HPLC purity: 96%. LC–MS APCI: calcd for C13H10N4O, 238.25; observed m/z [M + H]+, 239.1. 1H NMR (300 MHz, DMSO-d6): δ 8.78 (d, 1H, J = 3 Hz), 8.24–8.20 (m, 2H), 7.89 (d, 1H, J = 1.2 Hz), 7.56–7.53 (m, 3H), 7.07 (s, 1H), 6.65 (dd, 1H, J = 3 and 1.8 Hz).
2-(3,5-Dimethyl-1H-1,2,4-triazol-1-yl)-6-phenylpyrimidin-4(3H)-one (17)
Synthesized from intermediate 66 and 3,5-dimethyl-1H-1,2,4-triazole following a protocol similar to compound 16. Yield 28% (white solid). HPLC purity: 99%. LC–MS APCI: calcd for C14H13N5O, 267.29; observed m/z [M + H]+, 268.1. 1H NMR (300 MHz, DMSO-d6): δ 7.95–7.93 (m, 2H), 7.63–7.57 (m, 3H), 7.22 (s, 1H), 2.80 (s, 3H), 2.32 (s, 3H).
2-(1-Methyl-1H-imidazole-2-yl)-6-phenylpyrimidin-4(1H)-one (18)
1-methyl-1H-imidazole-2-carboximidamide (0.075 g, 0.61 mmol) and ethyl benzoylacetate (0.12 g, 0.61 mmol) were refluxed in EtOH for 2 h. The reaction was cooled, solvent was evaporated under vacuo, and the residue was purified on preparative HPLC to give 2-(1-methyl-1H-imidazol-2-yl)-6-phenylpyrimidin-4(1H)-one (18, 15 mg, Yield 10%) as a brown solid. HPLC purity: 94.2%. LC–MS APCI: calcd for C14H12N4O, 252.101; observed m/z [M + H]+, 253.4, 1H NMR (400 MHz, DMSO-d6): δ 7.99 (t, J = 3.6 Hz, 2H), 7.52–7.49 (m, 3H), 7.21 (s, 1H), 7.14 (s, 1H), 6.83 (s, 1H), 4.29 (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-(4-fluorophenyl)pyrimidin-4(3H)-one (19)
Synthesized from intermediate 56e following a protocol similar to compound 1. Yield 33% (white solid). HPLC purity: 93%. LC–MS APCI: calcd for C15H13FN4O, 284.29; observed m/z [M + H]+, 285.1. 1H NMR (300 MHz, DMSO-d6): δ 8.18–8.14 (m, 2H), 7.58–7.32 (m, 2H), 6.89 (s, 1H), 6.26 (s, 1H), 2.71 (s, 3H), 2.24 (s, 3H), 2.09 (br s, 1H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-(pyridin-4-yl)pyrimidin-4(3H)-one (20)
Synthesized from intermediate 56f following a protocol similar to compound 1. Yield 45% (white solid). HPLC purity: 96%. LC–MS APCI: calcd for C14H13N5O, 267.29; observed m/z [M + H]+, 268.1. 1H NMR (300 MHz, DMSO-d6): δ 8.76 (d, 1H, J = 1.5 Hz), 8.74 (d, 1H, J = 1.5 Hz), 8.04 (d, 1H, J = 1.8 Hz), 8.02 (d, 1H, J = 1.8 Hz), 7.07 (s, 1H), 6.28 (s, 1H), 2.72 (s, 3H), 2.25 (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-(pyridin-3-yl)pyrimidin-4(3H)-one (21)
Synthesized from intermediate 56g following a protocol similar to compound 1. Yield 56% (white solid). HPLC purity: 99%. LC–MS APCI: calcd for C14H13N5O, 267.29; observed m/z [M + H]+, 268.1. 1H NMR (300 MHz, DMSO-d6): δ 9.27 (dd, 1H, J = 3 and 1.5 Hz), 8.72 (dd, 1H, J = 6 and 3 Hz), 8.45 (dt, 1H, J = 9 and 3 Hz), 7.56 (ddd, 1H, J = 9, 6 and 1.5 Hz), 7.02 (s, 1H), 6.27 (s, 1H), 2.71 (s, 3H), 2.24 (s, 3H).
N-(4-(2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-oxo-1,6-dihydropyrimidin-4-yl)phenyl) Methanesulfonamide (22)
In 1,4-dioxane (2 mL) solution of N-(4-(2-chloro-6-oxo-1,6-dihydropyrimidin-4-yl)phenyl)methanesulfonamide (69a 0.09 g, 0.300 mmol), under a nitrogen atmosphere, cesium carbonate (0.147 g, 0.450 mmol), 3,5-Dimethylpyrazole (0.035 g, 0.360 mmol), XantPhos (0.052 g, 0.090 mmol), and Tris(dibenzylideneacetone)dipalladium(0) (0.041 g, 0.045 mmol) were added, and the mixture was heated at 130 °C for 18 h in a sealed tube. The reaction mixture was cooled to room temperature, concentrated in vacuo, filtered, and washed with DCM/MeOH, (80:20) and the filtrate concentrated to yield the crude product. The crude product was dissolved in 1 mL of water and acidified with 1 M HCl, then concentrated and purified by preparative HPLC to afford 22 as an off-white solid (3 mg, 8.01 μmol, 3% yield). HPLC purity: 96%. LC–MS APCI: calcd for C16H17N5O3S, 359.40; observed m/z [M + H]+, 360.1. 1H NMR (300 MHz, DMSO-d6): δ 8.00 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 8.3 Hz, 2H), 6.57 (s, 1H), 6.14 (s, 1H), 3.03 (s, 3H), 2.65 (s, 3H), 2.21 (s, 3H).
4-(2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-oxo-3,6-dihydropyrimidin-4-yl)-N-methylbenzamide (23)
Synthesis protocol was the same as for compound 22, starting from intermediate 4-(2-chloro-6-oxo-1,6-dihydropyrimidin-4-yl)-N-methylbenzamide (69b). Yield: 38% (white solid). HPLC purity: 97%; LC–MS APCI: calcd for C17H17N5O2, 323.128; observed m/z [M + H]+, 324.1. 1H NMR (400 MHz, DMSO-d6): δ 8.27 (d, J = 8.5 Hz, 2H), 7.96 (d, J = 8.5 Hz, 2H), 6.67 (s, 1H), 6.18 (s, 1H), 2.80 (s, 3H), 2.73 (s, 3H), 2.20 (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-(4-(methylsulfonyl)phenyl)pyrimidin-4(1H)-one (24)
Synthesis protocol was the same as for compound 22, starting from intermediate 2-chloro-6-(4-(methylsulfonyl)phenyl)pyrimidin-4(3H)-one (69c). Yield: 32% (white solid). HPLC purity: 98%; LC–MS APCI: calcd for C16H16N4O3S, 344.39; observed m/z [M + H]+, 345.1. 1H NMR (300 MHz, DMSO-d6): δ 8.52–8.42 (m, 2H), 8.09 (d, J = 8.3 Hz, 2H), 6.77 (s, 1H), 6.20 (s, 1H), 3.29 (s, 3H), 2.75 (s, 3H), 2.22 (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-(4-morpholinophenyl)pyrimidin-4(1H)-one (25)
Synthesis protocol was the same as for compound 22, starting from intermediate 2-chloro-6-(4-morpholinophenyl)pyrimidin-4(3H)-one (69d). Yield: 3% (white solid). HPLC purity: 95%; LC–MS APCI: calcd for C19H21N5O2, 351.41; observed m/z [M + H]+, 352.1.1H NMR (300 MHz, Methanol-d4): δ 8.07 (d, J = 8.8 Hz, 2H), 7.09 (d, J = 8.8 Hz, 2H), 6.63 (s, 1H), 6.14 (s, 1H), 3.86 (s, 6H), 2.76 (s, 3H), 2.27 (s, 3H), 1.91 (s, 2H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-N-(4-fluorophenyl)-6-oxo-1,6-dihydropyrimidine-4-carboxamide (26)
2-(3,5-dimethyl-1H-pyrazol-1-yl)-N-(4-fluorophenyl)-6-methoxypyrimidine-4-carboxamide 72 (40 mg, 0.117 mmol) was dissolved in dichloromethane (2 mL) under N2 and BBr3, 1 M in dichloromethane (0.352 mL, 0.352 mmol) was added and stirred under N2 at room temperature for 48 h. The reaction was quenched with dilute HCl and diluted with dichloromethane. The dichloromethane layer was separated, and the aqueous layer was again extracted with dichloromethane. Organic layers were combined, washed with brine, and concentrated under vacuum. The residue was purified by column chromatography using 0–20% methanol in dichloromethane to give 26 as an off-white solid (12 mg, 0.035 mmol, 30% yield). HPLC purity: 96.5% LC–MS ESI: calcd for C16H14FN5O2, 327.113; observed m/z [M + H]+, 328.10. 1H NMR (300 MHz, DMSO-d6): δ 12.80 (br s, 1H), 10.15 (br s, 1H), 7.81–7.86 (m, 2H), 7.24 (t, J = 9 Hz, 2H), 6.94 (s, 1H), 6.28 (s, 1H), 2.72 (s, 3H), (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-(4-fluorophenyl)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (27)
A mixture of 4-(4-fluorophenyl)-2-hydrazinyl-6-oxo-1,6-dihydropyrimidine-5-carbonitrile 74 (50 mg, 0.204 mmol) and pentane-2,4-dione (0.025 mL, 0.245 mmol) was heated in a mixture of ethanol (0.5 mL) and acetic acid (1.5 mL) at 85 °C for 3 h. The reaction was cooled, and a yellow crystalline precipitate formed was filtered under vacuum, triturated with a small amount of ethanol, and dried to afford 27 as a light-yellow solid (37 mg, 0.120 mmol, 59% yield). HPLC purity: 95.8%. LC–MS ESI: calcd for C16H12FN5O, 309.103; observed m/z [M – H]+, 308.10. 1H NMR (300 MHz, DMSO-d6): δ 8.05–8.10 (m, 2H), 7.46 (t, J = 9 Hz, 2H), 6.35 (s, 1H), 2.65 (s, 3H), 2.27 (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-(4-fluorophenyl)-6-oxo-1,6-dihydropyrimidine-5-carboxamide (28)
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-(4-fluorophenyl)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile 27 (50 mg, 0.162 mmol) was heated with H2SO4 (0.5 mL, 9.38 mmol) for 2 h at 80 °C. The reaction mixture was then poured slowly on ice with vigorous stirring. A white precipitate was formed which was filtered, washed with cold water and cold ethanol, and dried to give 28 as a light-yellow solid (32 mg, 0.098 mmol, 60% yield). HPLC purity: >98% LC–MS ESI: calcd for C16H14FN5O2, 327.113; observed m/z [M – H]+, 326.10. 1H NMR (300 MHz, DMSO-d6): δ 7.87–7.92 (m, 2H), 7.83 (br s, 1H), 7.50 (br s, 1H), 7.32 (t, J = 9 Hz, 2H), 6.25 (s, 1H), 2.62 (s, 3H), 2.24 (s, 3H).
2-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-(4-fluorophenyl)-6-oxo-1,6-dihydropyrimidine-5-carboxylic Acid (29)
In a 7 mL reaction vial, 2-(3,5-dimethyl-1H-pyrazol-1-yl)-4-(4-fluorophenyl)-6-oxo-1,6-dihydropyrimidine-5-carboxamide 28 (20 mg, 0.061 mmol) and tert-butyl nitrite (0.022 mL, 0.183 mmol) were stirred in acetic acid (0.5 mL) a 75 °C for 3 h. A white precipitate was formed. It was centrifuged out, washed with cold ethanol, and dried in an oven to give 29 as a white solid (12 mg, 0.037 mmol, 60% yield). HPLC purity: >99% LC–MS ESI: calcd for C16H13FN4O3, 328.097; observed m/z [M – H]+, 327.10. 1H NMR (300 MHz, DMSO-d6): δ 7.77–7.81 (m, 2H), 7.35 (t, J = 9 Hz, 2H), 6.27 (s, 1H), 2.62 (s, 3H), 2.24 (s, 3H).
2-(5-Ethyl-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (30)
Synthesized from 56d and hexane-2,4-dione following a protocol similar to compound 1. Yield: 35% (white solid). HPLC purity: 99%. LC–MS APCI: calcd for C11H11F3N4O, 272.088; observed m/z [M + H]+, 272.8. 1H NMR (400 MHz, DMSO-d6): δ 6.71 (s, 1H), 6.27 (s, 1H), 3.04 (q, J = 1.60 Hz, 2H), 2.58 (s, 1H), 2.24 (s, 2H), 1.22 (t, J = 7.20 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 13.22, 13.38, 13.89, 21.53, 108.02, 110.15, 110.32, 149.74, 151.87.
2-(5-Cyclopropyl-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (31)
Synthesized from 56d and 1-cyclopropylbutane-1,3-dione following a protocol similar to compound 1. Yield: 30% (white solid). HPLC purity: 98.4%. LC–MS APCI: calcd for C12H11F3N4O, 284.088; observed m/z [M + H]+, 285.4. 1H NMR (400 MHz, DMSO-d6): δ 6.75 (s, 1H), 6.09 (s, 1H), 2.20 (s, 3H), 0.94–0.99 (m, 2H), 0.70–0.72 (m, 2H).
2-(3-Methyl-5-phenyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (32)
Synthesized from 56d and 1-phenylbutane-1,3-dione following a protocol similar to compound 1. Yield: 39% (white solid). HPLC purity: 98.5%. LC–MS APCI: calcd for C15H11F3N4O, 320.088; observed m/z [M + H]+, 320.8. 1H NMR (400 MHz, CDCl3): δ 13.59 (s, 1H), 7.34–7.42 (m, 5H), 6.84 (s, 1H), 6.59 (s, 1H), 2.51 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 13.82, 108.53, 111.52, 119.39, 121.94, 128.40, 128.81, 129.18, 130.59, 130.59, 146.20, 151.83, 153.0.
2-(5-Amino-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (33)
To a solution of 56d (0.70 g, 1.28 mmol) in DMF was added 3-aminocrotonitrile (0.70 g, 8.43 mmol) and heated to 100 °C for 16 h. Water was added, extracted with ethyl acetate, and the combined organic layer was washed with saturated sodium bicarbonate solution, water, and brine solution, dried over sodium sulfate, and concentrated under vacuum. The crude product was purified by preparative HPLC to yield 33 as a white solid (0.076 g, 23%). HPLC purity: 95.5%. LC–MS APCI: calcd for C9H8F3N5O, 259.068; observed m/z [M + H]+, 259.8. 1H NMR (400 MHz, DMSO-d6): δ 7.15 (s, 2H), 6.30 (s, 1H), 5.23 (s, 1H), 2.08 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 14.34, 88.68, 105.72, 120.24, 122.97, 151.14, 151.32, 151.55, 151.88, 156.03, 168.05.
2-(5-(Ethylamino)-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (34)
To a solution of 33 (0.20 g, 0.77 mmol), acetic acid (0.14 g, 2.30 mmol) in DMF (5 mL) and acetaldehyde (0.10 g, 2.31 mmol) were added and the reaction was allowed to stir at room temperature for 16 h. Sodium cyanoborohydride (0.24 g, 3.86 mmol) in methanol (2 mL) was added and stirred at room temperature overnight. The reaction mixture was concentrated, water was added, and extracted with ethyl acetate. The organic layer was concentrated and purified by preparative HPLC to give 34 as a white solid (0.07 g, 32%). HPLC purity: 95.2%; LC–MS APCI: calcd for C11H12F3N5O, 287.099; observed m/z [M + H]+, 288.0. 1H NMR (400 MHz, CDCl3): δ 7.14 (s, 1H), 6.48 (s, 1H), 5.19 (s, 1H), 3.23 (q, J = 6.40 Hz, 2H), 2.19 (s, 3H), 1.32 (t, J = 7.20 Hz, 3H); 13C NMR (100 MHz, CD3OD): δ 12.56, 13.40, 39.09, 104.82, 119.62, 122.34, 152.30, 152.51, 152.85, 153.14, 155.01.
2-(5-Ethoxy-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (35)
To solution of sulphonyl intermediate 79 (0.50 g, 2.06 mmol) and cesium carbonate (1.6 g, 5.16 mmol) in 1,4-dioxane was added 78 (0.52 g, 4.12 mmol) in sealed tube and heated to 100 °C for 16 h. Solids were filtered through Celite and the filtrate concentrated under vacuum. The crude product was purified by preparative HPLC to afford 35 as a white solid (0.07 g, 12%). HPLC purity: 95%; LC–MS APCI: calcd for C11H11F3N4O2, 288.083; observed m/z [M + H]+, 288.8. 1H NMR (400 MHz, CDCl3): δ 10.43 (s, 1H), 6.53 (s, 1H), 5.80 (s, 1H), 4.28 (q, J = 7.20 Hz, 2H), 2.67 (s, 3H), 1.43 (t, J = 7.20 Hz, 3H).
2-(5-((2-(Dimethylamino)ethyl)amino)-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (36)
To a solution of 76a (0.10 g, 0.49 mmol) in ethanol (2.5 mL) and acetic acid (7.5 mL) was added 2-hydrazinyl-6-(trifluoromethyl)pyrimidin-4(3H)-one (56d, 0.09 g, 0.49 mmol) and heated to reflux for 5 h. The solvent was evaporated under vacuum. The crude product was purified with preparative HPLC to yield 36 as a white solid (0.06 g, 0.181 mmol, 37% yield). HPLC purity: 99.2%. LC–MS APCI: calcd for C13H17F3N6O, 330.142; observed m/z [M + H]+, 331.0. 1H NMR (300 MHz, DMSO-d6): δ 7.65 (s, 1H), 6.48 (s, 1H), 5.17 (s, 1H), 3.21 (q, J = 6.00 Hz, 2H), 2.60 (t, J = 6.00 Hz, 2H), 2.31 (s, 6H), 2.19 (s, 3H).
2-(5-((2-Hydroxyethyl)amino)-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (37)
A protocol similar to compound 36. Yield: 16% (white solid). HPLC purity: 97.28%; LC–MS APCI: calcd for C11H12F3N5O2, 303,094; observed m/z [M + H]+, 304.0. 1H NMR (400 MHz, CDCl3): δ 7.45 (s, 1H), 6.68 (s, 1H), 5.44 (s, 1H), 3.58 (t, J = 5.20 Hz, 2H), 3.21 (t, J = 5.20 Hz, 2H), 2.16 (s, 3H).
2-(5-((2-Methoxyethyl)amino)-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (38)
Step 1: To a −5 °C cooled solution of 4-methyleneoxetan-2-one (1.0 g, 11.90 mmol) in tetrahydrofuran (10 mL) was added 2-methoxyethylamine (0.90 g, 11.30 mmol) and stirred for 30 min. The solvent was evaporated under vacuum. The crude product was purified by column chromatography using 3–5% methanol in dichloromethane to afford N-(2-methoxyethyl)-3-oxobutanamide (1.2 g, 7.50 mmol, 63%). LC–MS APCI: calcd for C7H13NO3, 159.19; observed m/z [M + H]+, 160.2. 1H NMR (300 MHz, DMSO-d6): δ 8.13 (s, 1H), 3.29–3.36 (m, 4H), 3.20–3.27 (m, 5H), 2.13 (s, 3H). Step 2: To a solution of N-(2-methoxyethyl)-3-oxobutanamide (0.50 g, 3.14 mmol) in tetrahydrofuran (10 mL) was added Lawesson’s reagent (1.40 g, 3.45 mmol) and stirred for 30 min at room temperature under a nitrogen atmosphere. Hydrazine 33 (0.61 g, 3.45 mmol) was added to the reaction mixture and stirred at room temperature for 24 h. The reaction was quenched with saturated sodium bicarbonate solution and extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over sodium sulfate, and concentrated under vacuum. The crude product was purified by preparative HPLC to yield 38 as a white solid (0.065 g, 0.22 mmol. 7% yield). HPLC purity: 98.9%. LC–MS APCI: calcd for C12H14F3N5O2, 317.110; observed m/z [M + H]+, 318.0. 1H NMR (400 MHz, DMSO-d6): δ 7.71 (s, 1H), 6.45 (s, 1H), 5.37 (s, 1H), 3.52 (t, J = 5.20 Hz, 2H), 3.27–3.33 (m, 5H), 2.13 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 14.44, 44.66, 58.52, 70.51, 87.07, 106.26, 120.03, 122.75, 151.28, 152.24, 154.78, 166.47.
2-(3-Methyl-5-((tetrahydro-2H-pyran-4-yl)amino)-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (39)
A protocol similar to compound 36. Yield: 20% (white solid). HPLC purity: 97%. LC–MS APCI: calcd for C14H16F3N5O2, 343.126; observed m/z [M + H]+, 344.1. 1H NMR (400 MHz, CDCl3): δ 10.35 (s, 1H), 7.54 (s, 1H), 6.53 (s, 1H), 5.23 (s, 1H), 3.95–4.00 (m, 2H), 3.55–3.61 (m, 2H), 0.42–3.44 (m, 1H), 2.22 (s, 3H), 2.06–2.09 (m, 2H), 1.60–1.68 (m, 2H).
2-(5-((4-Fluorophenyl)amino)-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (40)
In a sealed tube 33 (0.25 g, 0.96 mmol), 1-bromo-4-fluoro benzene (0.33 g, 1.92 mmol) and cesium carbonate (0.93 g, 2.88 mmol) were taken in dioxane (10 mL). The mixture was purged with nitrogen gas for 20 min. To this tris(dibenzylideneacetone)dipalladium(0) (0.079 g, 0.08 mmol) and XantPhos (0.07 g, 0.13 mmol) was added and sealed. The reaction mixture was stirred at 120 °C for 4 h. The reaction mixture filtered through Celite and filtrate was concentrated under vacuum. The crude product was purified by preparative HPLC to obtain 40 as a white solid (0.09 g, 0.51 mmol, 26% yield). HPLC purity: 95.4%; LC–MS APCI: calcd for C15H11F4N5O, 353.090; observed m/z [M + H]+, 354.0. 1H NMR (400 MHz, CD3OD): δ 7.22–7.34 (m, 2H), 6.97–7.09 (m, 2H), 6.40 (s, 1H), 5.87 (s, 1H), 2.24 (s, 3H). 13C NMR (400 MHz, CD3OD): δ 14.00, 90.34, 105.40, 118.74, 121.42, 122.71, 124.14, 130.36, 138.72, 148.73, 152.72, 158.26, 160.06, 160.64, 176.68.
2-(5-((2,4-Difluorophenyl)amino)-3-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (41)
A protocol similar to compound 36. Yield: 8% (white solid). HPLC purity: 97.5%; LC–MS APCI: calcd for C15H10F5N5O, 371.081; observed m/z [M + H]+, 372.4. 1H NMR (400 MHz, CDCl3): δ 10.46 (s, 1H), 9.81 (s, 1H), 7.31–7.34 (m, 1H), 6.92–6.99 (m, 2H), 6.58 (s, 1H), 5.81 (s, 1H), 2.27 (s, 3H).
4-((3-Methyl-1-(6-oxo-4-(trifluoromethyl)-1,6-dihydropyrimidin-2-yl)-1H-pyrazol-5-yl)amino)benzonitrile (42)
Protocol similar to compound 36. Yield 29% (white solid). HPLC purity: 99.9%; LC–MS APCI: calcd for C16H11F3N6O, 360.30; observed m/z [M + H]+, 361.0. 1H NMR (400 MHz, DMSO-d6): δ 12.43 (s, 1H), 7.73 (d, J = 8.80 Hz, 2H), 7.17 (d, J = 8.80 Hz, 2H), 6.17 (s, 1H), 6.06 (s, 1H), 2.21 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 14.46, 92.36, 101.50, 105.83, 116.12, 120.01, 123.59, 134.42, 143.47, 145.22, 149.10, 159.53, 171.58.
N,N-Dimethyl-4-((3-Methyl-1-(6-oxo-4-(trifluoromethyl)-1,6-dihydropyrimidin-2-yl)-1H-pyrazol-5-yl)amino)benzamide (43)
Protocol similar to compound 36. Yield 30% (white solid). HPLC purity: 97.9%. LC–MS APCI: calcd for C18H17F3N6O2, 406.137; observed m/z [M + H]+, 407.0. 1H NMR (400 MHz, DMSO-d6): δ 11.89 (s, 1H), 7.40 (d, J = 8.00 Hz, 2H), 7.09 (d, J = 8.00 Hz, 2H), 6.07 (s, 1H), 6.06 (s, 1H), 2.97 (s, 6H), 2.19 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 14.47, 91.18, 115.60, 128.27, 129.59, 142.49, 144.92, 149.29, 170.44, 171.90.
2-(3-Methyl-5-(pyridin-4-ylamino)-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (44)
Protocol similar to compound 36. Yield 12% (white solid). HPLC purity 99.8%; LC–MS APCI: calcd for C14H11F3N6O, 336.28; observed m/z [M + H]+, 337.0. 1H NMR (400 MHz, DMSO-d6): δ 12.29 (s, 1H), 8.33 (d, J = 6.40 Hz, 2H), 6.98 (d, J = 6.40 Hz, 2H), 6.18 (s, 1H), 6.05 (s, 1H), 5.32 (s, 1H), 2.21 (s, 3H).
2-(3-Methyl-5-(pyridin-2-ylamino)-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (45)
Protocol similar to compound 36. Yield 38% (white solid). HPLC purity: 98.2%. LC–MS APCI: calcd for C14H11F3N6O, 336.095; observed m/z [M + H]+, 336.8. 1H NMR (400 MHz, CDCl3): δ 10.55 (s, 1H), 8.36 (d, J = 3.20 Hz, 1H), 7.64 (t, J = 7.20 Hz, 1H), 6.91–6.94 (m, 2H), 6.75 (d, J = 7.48 Hz, 1H), 6.59 (s, 1H), 2.32 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 14.3, 96.13, 108.63, 111.78, 116.90, 137.89, 148.0, 151.95.
2-(3-Methyl-5-((5-morpholinopyridin-2-yl)amino)-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (46)
A protocol similar to compound 36. Yield 15% (white solid). HPLC purity: 99.6%. LC–MS APCI: calcd for C18H18F3N7O2, 421.147; observed m/z [M + H]+, 422.4. 1H NMR (400 MHz, CDCl3): δ 10.37 (s, 1H), 8.03 (s, 1H), 7.32 (s, 1H), 6.79 (s, 1H), 6.73 (d, J = 8.00 Hz, 1H), 6.57 (s, 1H), 3.91 (t, J = 4.80 Hz, 4H), 3.13 (t, J = 4.40 Hz, 4H), 2.30 (s, 3H).
2-(5-(Ethylamino)-3-methyl-1H-pyrazol-1-yl)-6-(pyridin-3-yl)pyrimidin-4(3H)-one (47)
To a solution of 2-(5-amino-3-methyl-1H-pyrazol-1-yl)-6-(pyridin-3-yl)pyrimidin-4(3H)-one (0.10 g, 0.37 mmol), acetic acid (0.07 g, 1.11 mmol) in DMF (5 mL) and acetaldehyde (0.05 g, 1.11 mmol) were added, and reaction mixture stirred at room temperature for 16 h. Sodium cyanoborohydride (0.11 g, 1.86 mmol) in methanol (2 mL) was added, and the reaction mixture stirred at room temperature overnight. The reaction mixture was concentrated, water was added, and extracted with ethyl acetate. The organic layer was concentrated and purified by preparative HPLC to give 47 as a white solid (0.03 g, 0.299 mmol, 27%). HPLC purity 97.53%; LC–MS APCI: calcd for C15H16N6O, 296.139; observed m/z [M + H]+, 297.2. 1H NMR (400 MHz, CD3OD): δ 9.42 (s, 1H), 8.77–8.82 (m, 2H), 7.82 (q, J = 5.20 Hz, 1H), 7.15 (s, 1H), 3.42 (q, J = 7.20 Hz, 2H), 2.40 (s, 3H), 1.39 (t, J = 7.20 Hz, 3H).
2-(4-Bromo-3,5-dimethyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (48)
To a solution of 12 (1.0 g, 3.87 mmol) in acetic acid (20 mL) was added bromine (0.24 mL, 4.65 mmol) at 20 °C and stirred at room temperature for 3 days. Water was added to the reaction mixture. Solids formed were filtered, washed with water, and dried. The crude product was purified by preparative HPLC to yield 48 as a white solid (0.3 g, 1.07 mmol, 23.0%). HPLC purity: 99.3%; LC–MS APCI: calcd for C10H8BrF3N4O, 335.983; observed m/z [M – H]+, 337.0; 1H NMR (400 MHz, DMSO-d6): δ 13.58 (s, 1H), 6.93 (s, 1H), 2.58 (s, 3H), 2.24 (s, 3H).
2-(3,5-Dimethyl-4-(pyridin-3-yl)-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (49)
A solution of 48 (0.50 g, 1.48 mmol), pyridine-3-boronic acid (0.22 g, 1.78 mmol) and potassium carbonate (0.41 g, 2.96 mmol) in tetrahydrofuran/water (20/2 mL) was repeatedly purged with nitrogen. Pd(dppf)Cl2 (0.18 g, 0.22 mmol) was added and heated at 70 °C for 16 h. The reaction mixture was cooled, water was added, and extracted with ethyl acetate. Combined organic layers were washed with water and brine solution, dried over sodium sulfate, and concentrated under vacuum. The crude product was purified by preparative HPLC to yield 49 as a white solid (0.10 g, 0.36 mmol, 20% yield). HPLC purity: 99.8%; LC–MS APCI: calcd for C15H12F3N5O, 335.099; observed m/z [M – H]+, 336.1. 1H NMR (400 MHz, CDCl3): δ 400 MHz, CDCl3: δ 8.68 (d, J = 4.88 Hz, 1H), 8.58 (s, 1H), 7.66 (d, J = 7.60 Hz, 1H), 7.48 (t, J = 7.28 Hz, 1H), 6.64 (s, 1H), 2.68 (s, 3H), 2.29 (s, 3H).
2-(3,5-Dimethyl-4-morpholino-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (50)
Step 1: 3-Bromopentane-2,4-dione (80)—To a solution of acetylacetone (1.0 g, 10.0 mmol) in acetic acid (15 mL) was added bromine (1.9 g, 12.0 mmol) at 0 °C. The reaction was heated at 60 °C for 3 h. Saturated sodium bicarbonate solution was added, and the layer was separated. The combined organic layer was washed with water, dried over sodium sulfate, and concentrated under vacuum to yield 80 as a light-yellow solid (1.2 g, 67.0%). This was taken as such to the next step without further analysis. Step 2: 3-Morpholinopentane-2,4-dione (81)—To a solution of morpholine (0.58 g, 6.70 mmol) and triethylamine (1.1 g, 11.17 mmol) in dichloromethane (10 mL) was added 80 (1.0 g, 5.58 mmol) dissolved in dichloromethane (10 mL) at 0 °C. The reaction was stirred at room temperature for 2 h. Water was added, and the layers separated. The combined organic layers were washed with water, dried over sodium sulfate, and concentrated under vacuum to yield 81 (0.80 g, 78%) as viscous brown oil. LC–MS APCI: calcd for C9H15NO3, 185.22; observed m/z [M + H]+, 186.2. 1H NMR (400 MHz, DMSO-d6): δ 4.31 (s, 1H), 3.57–3.62 (m, 4H), 2.84 (d, J = 4.40 Hz, 2H), 2.60 (d, J = 4.80 Hz, 2H), 2.18 (s, 6H). Step 3: 2-(3,5-Dimethyl-4-morpholino-1H-pyrazol-1-yl)-6-trifluoromethylpyrimidin-4(3H)-one (50)—A solution of 81 (0.30 g, 1.62 mmol) and 56d (0.31 g, 1.62 mmol) in ethanol/acetic acid mixture (15/5 mL) was refluxed for 16 h. The solvent was evaporated under vacuum. Crude product was purified by preparative HPLC to yield 50 (0.07 g, 13%). HPLC purity: 99.9%; LC–MS APCI: calcd for C14H16F3N5O2, 343.126; observed m/z [M + H]+, 344.1.1H NMR (400 MHz, DMSO-d6): δ 13.10 (s, 1H), 6.79 (s, 1H), 3.69 (d, J = 2.80 Hz, 4H), 2.96 (d, J = 3.60 Hz, 4H), 2.51 (s, 3H), 2.30 (s, 3H).
2-(5-Amino-3-cyclopropyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (51)
To a solution of 2-hydrazinyl-6-(trifluoromethyl)pyrimidin-4(3H)-one (56d, 0.25, 1.28 mmol) in DMF (5 mL) was added 3-cyclopropyl-3-oxopropanenitrile (0.15 g, 1.41 mmol) and heated at 130 °C in a microwave for 1 h. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer washed with plenty of water and brine solution, dried over sodium sulfate, and concentrated in vacuo. The crude mass was purified by prep HPLC to give 2-(5-amino-3-cyclopropyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one 51 as a white solid (51 mg, 0.179 mmol, 14% yield). HPLC purity: 96%; LC–MS APCI: calcd for C11H10F3N5O, 285.084; observed m/z [M + H]+, 286.0. 1H NMR (400 MHz, CDCl3): δ 6.54 (s, 1H), 5.79 (br s, 2H), 5.25 (s, 1H), 1.82 (t, J = 4.40 Hz, 1H), 1.00 (d, J = 8.00 Hz, 2H), 0.82 (d, J = 2.00 Hz, 2H).
2-(3-((Dimethylamino)methyl)-5-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl)pyrimidin-4(3H)-one (52)
Step 1: ethyl 2,4-dioxopentanoate—To a solution of sodium metal (0.86 g, 37.67 mmol) dissolved in ethanol (15 mL) was added a mixture of diethyl oxalate (5.0 g, 34.24 mmol) and acetone (2.0 g, 34.24 mmol) and heated at 60 °C for 16 h. The reaction mass was concentrated under vacuum, diluted with water, and the product was extracted with ethyl acetate. The organic phase was washed with water and brine, dried over sodium sulfate, and concentrated. The crude product was then purified by column chromatography using silica gel (230–400 mesh) with 10–20% ethyl acetate in petroleum ether as an eluent to yield ethyl 2,4-dioxopentanoate as yellow oil (2.1 g, 13.93 mmol, 37%). 1H NMR (300 MHz, CDCl3): δ 14.40 (s, 1H), 6.39 (s, 1H), 4.37 (q, J = 7.20 Hz, 2H), 2.28 (s, 3H), 1.40 (t, J = 7.20 Hz, 3H). Step 2: Ethyl 5-methyl-1H-pyrazole-3-carboxylate (83)—To a solution of ethyl 2,4-dioxopentanoate (2.0 g, 12.65 mmol) in ethanol (10 mL) and acetic acid (20 mL) was added hydrazine hydrate (0.75 g, 15.18 mmol). The reaction mixture was heated at the reflux for 16 h. Solvents were evaporated under vacuum. Water was added, and the product was extracted with ethyl acetate. The organic layer washed with saturated sodium bicarbonate solution, water, and brine, dried over sodium sulfate, and concentrated under vacuum to afford 83 an off-white solid (1.3 g, 8.6 mmol, 68%). LC–MS APCI: calcd for C7H10N2O2, 154.17; observed m/z [M + H]+, 154.8. 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H), 6.46 (s, 1H), 4.25 (q, J = 7.20 Hz, 2H), 2.26 (s, 3H), 1.27 (t, J = 6.80 Hz, 3H). Step 3: 5-methyl-1H-pyrazole-3-carbaldehyde—To a solution of 83 (4.0 g, 25.97 mmol) in dichloromethane (50 mL) at −78 °C was added di-isobutylaluminiumhydride (1 M in dichloromethane, 52.0 mL, 51.94 mmol) dropwise under a nitrogen atmosphere. The reaction mixture was stirred at −78 °C for 2 h. It was then quenched with methanol at −78 °C, warmed to room temperature, and washed with water. The organic phase was dried over sodium sulfate and concentrated under vacuum to yield 5-methyl-1H-pyrazole-3-carbaldehyde. This material was taken as such for the next step without purification. Step 4: N,N-Dimethyl-1-(5-methyl-1H-pyrazol-3-yl)methanamine (84)—To a solution of 5-methyl-1H-pyrazole-3-carbaldehyde (1.0 g, 9.09 mmol) in methanol was added N,N-dimethyl amine (2a M in tetrahydrofuran, 23 mL, 45.45 mmol) and stirred for 24 h at room temperature. Sodium cyanoborohydride (1.7 g, 27.27 mmol) was added to the reaction and stirred at room temperature for another 4 h. The reaction mixture was concentrated, the crude material diluted with water, and the product was extracted with ethyl acetate. The organic layer washed with water and brine solution, dried over sodium sulfate, and concentrated under vacuum to afford 84 as a white solid (0.60 g, 4.31 mmol, 48%). LC–MS APCI: calcd for C7H13N3, 139.20; observed m/z [M + H]+, 139.8. Step 5: 2-(3-((Dimethylamino)methyl)-5-methyl-1H-pyrazol-1-yl)-6-(trifluoromethyl) pyrimidin-4(3H)-one (52)—A solution of 84 (0.60 g, 4.31 mmol), sulfonyl intermediate 79 (0.48 g, 2.15 mmol) and cesium carbonate (2.0 g, 6.37 mmol) in 1,4-dioxane was heated in sealed tube at 130 °C for 16 h. The reaction mixture was filtered through Celite and the filtrate was concentrated under vacuum. The crude product was then purified by preparative HPLC to yield the free base as a gum. It was dissolved in dichloromethane and stirred with hydrogen chloride in 1,4-dioxane (in excess) for 2 h. Solvents were evaporated under vacuum, formed solids were washed with diethyl ether, and dried under vacuum to afford 52 (0.06 g, 9.38%) as a white hydrochloride salt. HPLC purity: 99.8%. LC–MS APCI: calcd for C12H14F3N5O, 301.115; observed m/z [M + H]+, 302.2. 1H NMR (400 MHz, CD3OD): δ 6.83 (s, 1H), 6.59 (s, 1H), 4.43 (s, 2H), 2.98 (s, 6H), 2.75 (s, 3H).
1-(4-(4-Fluorophenyl)-6-oxo-1,6-dihydropyrimidin-2-yl)-5-methyl-1H-pyrazole-3-carboxylic Acid (53)
To solution of 6-(4-fluorophenyl)-2-(methylsulfonyl)pyrimidin-4(3H)-one (0.3 g, 1.118 mmol) 85 (Synthesized according to protocol for compound 79) and cesium carbonate (0.911 g, 2.80 mmol) in DMF was added 83 (0.172 g, 1.118 mmol) in a sealed tube and heated to 130 °C in a microwave for 2 h. The reaction mixture was concentrated under vacuum and then was purified by preparative HPLC to yield 53 as an off-white solid (60 mg, 0.179 mmol, 16% yield). HPLC purity: 94%. LC–MS APCI: calcd for C15H11FN5O3, 314.27; observed m/z [M + H]+, 315.1. 1H NMR (300 MHz, Methanol-d4): δ 8.19–8.08 (m, 2H), 7.26 (t, J = 8.8 Hz, 2H), 6.77 (s, 1H), 6.66 (d, J = 1.1 Hz, 1H), 2.93–2.82 (m, 3H).
1-(4-(4-Fluorophenyl)-6-oxo-1,6-dihydropyrimidin-2-yl)-N,N,5-trimethyl-1H-pyrazole-3-carboxamide (54)
To a solution of HATU (0.036 g, 0.095 mmol), 53 (0.02 g, 0.064 mmol) and dimethylamine (0.017 mL, 0.191 mmol) in DMF was added triethylamine (0.018 mL, 0.127 mmol) and reaction mixture was stirred at 35 °C overnight. Upon completion, the reaction mixture was concentrated in vacuo, washed with 10% LiCl, and the resulting precipitate was filtered to yield 54 as a white solid (5 mg, 0.014 mmol, 22% yield). HPLC purity: 96%. LC–MS APCI: calcd for C17H16FN5O2, 341.34; observed m/z [M + H]+, 342.1.1H NMR (300 MHz, DMSO-d6): δ 8.21 (dd, J = 8.9, 5.6 Hz, 2H), 7.37 (t, J = 8.9 Hz, 2H), 7.08 (s, 1H), 6.64 (d, J = 1.0 Hz, 1H), 3.27 (s, 3H), 3.01 (s, 3H), 2.74 (d, J = 0.9 Hz, 3H).
Acknowledgments
MMV, Meyrin, Switzerland for donating the compound library. Dr. Sridevi Bashayam, Dr. Jayan Joseph, Dr. Jayanth Thiruvellore, and Rajkumar Dhinakaran, Syngene, India for profound chemistry support. Ronnett Seldon from TB biology, H3D for performing MIC and biology triage assays. Dr. Nicole Cardoso from TB biology, H3D for helping in Fe-chelation experiment. Prof. Digby Warner, University of Cape Town for useful discussions on biology triage assays, Technical staff at in vitro DMPK and parasitology divisions of H3D for excellent technical assistance in generating all in vitro DMPK and cytotoxicity data, Marianna de Kock and Claudia Spies, SAMRC and Stellenbosch University, South Africa for excellent technical assistance in generating MICs against clinical TB isolates. AbbVie Incorporation, North Chicago, IL, USA for in vitro safety screening. Lina Castro, Shilah Bonnett and Megha Gupta from Infectious Disease Research Institute for technical assistance and support.
Glossary
Abbreviations
- AUC
area under the curve
- BuOH
butanol
- CS2
carbon disulfide
- DIAD
diisopropyl azodicarboxylate
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- EtOAc
ethyl acetate
- EtOH
ethanol
- fu
fraction unbound
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- MIC
minimum inhibitory concentration
- mCPBA
meta-chloroperbenzoic acid
- NaBH4
NaCNBH3, sodium cyanoborohydride
- POCl3
phosphorus(V) oxychloride
- RT
room temperature
- SAR
structure–activity relationship
- SOCl2
thionyl chloride
- TB
tuberculosis
- THF
tetrahydrofuran
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01727.
Synthesis and analytical data of intermediates, microbiology assays, MIC data against clinical isolates, screening data against MmpL3 under-expressing strain of Mtb, in vitro DMPK assays, and mouse pharmacokinetic studies (PDF)
LCMS HPLC and NMRs of lead compounds (PDF)
Data from RNA microarray experiment (XLSX)
Data from RNA microarray experiment (XLSX)
Molecular formula strings and some data (CSV)
Author Present Address
‡‡ Center for Global Infectious Disease Research, Seattle Children’s Research Institute, Seattle, WA 98101, United States.
Author Contributions
C.S.d.M. and V.S. contributed equally to the work. Chemical synthesis, compound design, and SAR expansion were performed by C.S.d.M., S.B.S., and S.R.G. Computational and compound design support was provided by C.J.E. Iron complementation experiment and the analysis was performed by V.S. Analysis and interpretation of the biology data from target deconvolution studies was performed by V.S. and A.M. In vivo PK experiments including MetID studies were performed by D.T. and C.B. In vitro DMPK data were analyzed by N.L. The cytochrome P450 inhibition experiment was performed by N.L. Screening against MmpL3 conditional knockdown mutant was conducted by C.E. and the data were analyzed by C.E. and D.S. Screening against MmpL3 mutant strains was carried out by A.K. with data analysis by A.K. and T.P. The screening of MMV compound library and RNA microarray experiment were performed by H.I.M.B. Screening conceptualization and funding support by C.E.B. Agilent microarray protocols were developed by Q.S. and T.G.M. The synthesis of initial hits was performed by K.I.B. Data analysis and compound acquisition performed by T.B. Compound design and data analysis performed by P.C.R. data analysis and manuscript review by S.R.G.. Scientific concept and funding acquisition by P.G.W. G.S.B. provided scientific and strategic guidance for the work and edited the manuscript. Introduction and target deconvolution sections of the manuscript were written by V.S. SAR, DMPK and part of synthesis and experimental sections were written by S.R.G. Synthesis and chemistry experimental sections were written by C.S.d.M, and K.C. provided inputs on compound design, SAR analysis, and edited the manuscript. All the authors have given approval to the final version of the manuscript.
The project was funded through Global Health Grants (numbers OPP1066878 and OPP1024038) received from the Bill and Melinda Gates Foundation, the Division of Intramural Research of the NIAID/NIH and the Strategic Health Innovation Partnerships (SHIP) unit of the South African Medical Research Council (SAMRC). The University of Cape Town, SAMRC, and South African Research Chairs Initiative of the Department of Science and Technology, administered through the South African National Research Foundation are gratefully acknowledged for support (K.C.).
The authors declare no competing financial interest.
Author Status
†† AM deceased in November 2019.
Supplementary Material
References
- World Health Organization . Global Tuberculosis Report; World Health Organization: Geneva, 2019.
- Mabhula A.; Singh V. Drug-resistance in Mycobacterium tuberculosis: where we stand. Medchemcomm 2019, 10, 1342–1360. 10.1039/c9md00057g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.newtbdrugs.org/pipeline (accessed March 18, 2019).
- Soares de Melo C.; Feng T.-S.; van der Westhuyzen R.; Gessner R. K.; Street L. J.; Morgans G. L.; Warner D. F.; Moosa A.; Naran K.; Lawrence N.; Boshoff H. I. M.; Barry C. E. 3rd; Harris C. J.; Gordon R.; Chibale K. Aminopyrazolo[1,5-a]pyrimidines as potential inhibitors of Mycobacterium tuberculosis: structure activity relationships and ADME characterization. Bioorg. Med. Chem. 2015, 23, 7240–7250. 10.1016/j.bmc.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Wilson C. R.; Gessner R. K.; Moosa A.; Seldon R.; Warner D. F.; Mizrahi V.; Soares de Melo C.; Simelane S. B.; Nchinda A.; Abay E.; Taylor D.; Njoroge M.; Brunschwig C.; Lawrence N.; Boshoff H. I. M.; Barry C. E. 3rd; Sirgel F. A.; van Helden P.; Harris C. J.; Gordon R.; Ghidelli-Disse S.; Pflaumer H.; Boesche M.; Drewes G.; Sanz O.; Santos G.; Rebollo-Lopez M. J.; Urones B.; Selenski C.; Lafuente-Monasterio M. J.; Axtman M.; Lelièvre J.; Ballell L.; Mueller R.; Street L. J.; Ghorpade S. R.; Chibale K. Novel antitubercular 6-dialkylaminopyrimidine carboxamides from phenotypic whole-cell high throughput screening of a softfocus library: structure-activity relationship and target identification studies. J. Med. Chem. 2017, 60, 10118–10134. 10.1021/acs.jmedchem.7b01347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Labadie S.; Zhang B.; Ortwine D. F.; Patel S.; Vinogradova M.; Kiefer J. R.; Mauer T.; Gehling V. S.; Harmange J.-C.; Cummings R.; Lai T.; Liao J.; Zheng X.; Liu Y.; Gustafson A.; Van der Porten E.; Mao W.; Liederer B. M.; Deshmukh G.; An L.; Ran Y.; Classon M.; Trojer P.; Dragovich P. S.; Murray L. From a novel HTS hit to potent, selective, and orally bioavailable KDM5 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2974–2981. 10.1016/j.bmcl.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Dragset M. S.; Poce G.; Alfonso S.; Padilla-Benavides T.; Ioerger T. R.; Kaneko T.; Sacchettini J. C.; Biava M.; Parish T.; Argüello J. M.; Steigedal M.; Rubin E. J. A novel antimycobacterial compound acts as an intracellular iron chelator. Antimicrob. Agents Chemother. 2015, 59, 2256–2264. 10.1128/aac.05114-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L.; Liu T.; Chen J.; Zhang X. Bis{tris-[3-(2-pyrid-yl)-1H-pyrazole]nickel(II)} dodeca-molybdo(V,VI)phosphate hexa-hydrate. Acta Crystallogr., Sect. E: Struct. Rep. Online 2010, 66, m319–m320. 10.1107/s1600536810005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajnák C.; Schäfer B.; Šalitroš I.; Fuhr O.; Ruben M.; Boča R. Influence of the charge of the complex unit on the SCO properties in pyrazolyl-pyridinyl-benzimidazole based Fe(II) complexes. Polyhedron 2017, 135, 189–194. 10.1016/j.poly.2017.06.035. [DOI] [Google Scholar]
- Attaby F. A.; Eldin S. M. Synthesis of pyrimidine, thiazolopyrimidine, pyrimidotriazine and triazolopyrimidine derivatives and their biological evaluation. Z. Naturforsch., B: J. Chem. Sci. 1999, 54, 788–798. 10.1515/znb-1999-0614. [DOI] [Google Scholar]
- Tait B. P.; Noel A.; Cullen M.. Proteostasis Regulators for Treating Cystic Fibrosis and other Protein Misfolding Diseases. WO2012154880 A1, 2012.
- Burgula L. N.; Radhakrishnan K.; Kundu L. M. Synthesis of modified uracil and cytosine nucleobases using a microwave-assisted method. Tetrahedron Lett. 2012, 53, 2639–2642. 10.1016/j.tetlet.2012.03.056. [DOI] [Google Scholar]
- Zhang Z.; Wallace M. B.; Feng J.; Stafford J. A.; Skene R. J.; Shi L.; Lee B.; Aertgeerts K.; Jennings A.; Xu R.; Kassel D. B.; Kaldor S. W.; Navre M.; Webb D. R.; Gwaltney S. L. Design and synthesis of pyrimidinone and pyrimidinedione inhibitors of dipeptidyl peptidase IV. J. Med. Chem. 2011, 54, 510–524. 10.1021/jm101016w. [DOI] [PubMed] [Google Scholar]
- Kazimierczuk Z.; Lipski M.; Shugar D. Intermediates in the synthesis of purines and pteridines: selective hydrolysis of chloropyrimidines. Acta Biochim. Pol. 1972, 19, 359–366. [PubMed] [Google Scholar]
- Peng Z.-H.; Journet M.; Humphrey G. A highly regioselective amination of 6-aryl-2,4-dichloropyrimidine. Org. Lett. 2006, 8, 395–398. 10.1021/ol052578p. [DOI] [PubMed] [Google Scholar]
- Cocuzza A. J.; Hobbs F. W.; Arnold C. R.; Chidester D. R.; Yarem J. A.; Culp S.; Fitzgerald L.; Gilligan P. J. 4-aryl-2-anilinopyrimidines as corticotropin-releasing hormone (CRH) antagonists. Bioorg. Med. Chem. Lett. 1999, 9, 1057–1062. 10.1016/s0960-894x(99)00132-8. [DOI] [PubMed] [Google Scholar]
- Gong Y.; Pauls H. W. A convenient synthesis of heteroaryl benzoic acids via Suzuki reaction. Synlett 2000, 829–831. [Google Scholar]
- Gronowitz S.; Hornfeldt A. B.; Kristjansson V.; Musil T. On the synthesis of various thienylpyrimidines and selenienylpyrimidines. Chem. Scr. 1986, 26, 305–309. [Google Scholar]
- Jiang B.; Yang C.-g. Synthesis of indolylpyrimidines via cross-coupling of indolylboronic acid with chloropyrimidines: facile synthesis of meridianin D. Heterocycles 2000, 53, 1489–1498. 10.3987/com-00-8890. [DOI] [Google Scholar]
- Schomaker J. M.; Delia T. J. Arylation of halogenated pyrimidines via a Suzuki coupling reaction. J. Org. Chem. 2001, 66, 7125–7128. 10.1021/jo010573+. [DOI] [PubMed] [Google Scholar]
- Basavaraja H. S.; Nagamani J. E.; Kumar M. M.; Padmashali B. Antimicrobial evaluation of novel substituted pyrimidinopyrazoles and pyrimidinotriazoles. J. Adv. Pharm. Technol. Res. 2010, 1, 236–244. [PMC free article] [PubMed] [Google Scholar]
- Patel A.; Pasha T. Y.; Kothari R. Synthesis and biological evaluation of some novel heterocyclic compounds as protein tyrosine phosphatase (PTP-1B) Inhibitor. J. Chem. Pharmaceut. Res. 2015, 7, 509–517. [Google Scholar]
- Yedage S. L.; Bhanage B. M. tert-Butyl nitrite-mediated synthesis of N-nitrosoamides, carboxylic acids, benzocoumarins, and isocoumarins from amides. J. Org. Chem. 2017, 82, 5769–5781. 10.1021/acs.joc.7b00570. [DOI] [PubMed] [Google Scholar]
- Dodd D. S.; Martinez R. L. One-pot synthesis of 5-(substituted-amino)pyrazoles. Tetrahedron Lett. 2004, 45, 4265–4267. 10.1016/j.tetlet.2004.04.017. [DOI] [Google Scholar]
- Peruncheralathan S.; Yadav A. K.; Ila H.; Junjappa H. Highly regioselective synthesis of 1-aryl-3 (or 5)-alkyl/aryl-5 (or 3)-(N-cycloamino)pyrazoles. J. Org. Chem. 2005, 70, 9644–9647. 10.1021/jo051478u. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Aggarwal V.; Ila H.; Junjappa H. A novel and convenient synthesis of 2-amino-4-(N-alkyl-N-arylamino)-pyrimidines using polarized ketene S,S-acetals and S,N-acetals. Synthesis 1980, 748–751. 10.1055/s-1980-29200. [DOI] [Google Scholar]
- Rahman A.; Vishwakarma J. N.; Yadav R. D.; Ila H.; Junjappa H. Reaction of alpha-ketoketene S,N-acetals with hydroxylamine - a facile general-route to 5-aryl-3-(N-arylamino, N-alkylamino, or N-azacycloalkyl)-isoxazoles. Synthesis 1984, 247–249. 10.1055/s-1984-30793. [DOI] [Google Scholar]
- Singh O. M.; Junjappa H.; Ila H. Reaction of lithioamino anions with alpha-oxoketene dithioketals: an improved and a new general method for the synthesis of alpha-oxoketene S,N- and N,N-ketals. J. Chem. Soc., Perkin Trans. 1 1997, 23, 3561–3566. 10.1039/a701435j. [DOI] [Google Scholar]
- Grange J. M.; Winstanley P. A.; Davies P. D. O. Clinically Significant Drug Interactions with Antituberculosis Agents. Drug Saf. 1994, 11, 242–251. 10.2165/00002018-199411040-00003. [DOI] [PubMed] [Google Scholar]
- Sriwijitalai W.; Wiwanitkit V. Drug–drug interaction analysis: Antituberculosis drugs versus antiretroviral drugs. Biomed. Biotechnol. Res. J. 2019, 3, 101. 10.4103/bbrj.bbrj_52_19. [DOI] [Google Scholar]
- Gleeson M. P. Plasma protein binding affinity and its relationship to molecular structure: an in-silico analysis. J. Med. Chem. 2007, 50, 101–112. 10.1021/jm060981b. [DOI] [PubMed] [Google Scholar]
- Lynch J. J. III; Van Vleet T. R.; Mittelstadt S. W.; Blomme E. A. G. Potential functional and pathological side effects related to off-target pharmacological activity. J. Pharmacol. Toxicol. Methods 2017, 87, 108–126. 10.1016/j.vascn.2017.02.020. [DOI] [PubMed] [Google Scholar]
- Moosa A.; Lamprecht D. A.; Arora K.; Barry C. E. 3rd; Boshoff H. I. M.; Ioerger T. R.; Steyn A. J. C.; Mizrahi V.; Warner D. F. Susceptibility of Mycobacterium tuberculosis cytochrome bd oxidase mutants to compounds targeting the terminal respiratory oxidase, cytochrome c. Antimicrob. Agents Chemother. 2017, 61, e01338 10.1128/aac.01338-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naran K.; Moosa A.; Barry C. E. 3rd; Boshoff H. I. M.; Mizrahi V.; Warner D. F. Bioluminescent Reporters for rapid mechanism of action assessment in tuberculosis drug discovery. Antimicrob. Agents Chemother. 2016, 60, 6748–6757. 10.1128/aac.01178-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioerger T. R.; O’Malley T.; Liao R.; Guinn K. M.; Hickey M. J.; Mohaideen N.; Murphy K. C.; Boshoff H. I. M.; Mizrahi V.; Rubin E. J.; Sassetti C. M.; Barry C. E. 3rd; Sherman D. R.; Parish T.; Sacchettini J. C. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS One 2013, 8, e75245 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Li J.; Yang X.; Wu L.; Zhang J.; Yang Y.; Zhao Y.; Zhang L.; Yang X.; Yang X.; Cheng X.; Liu Z.; Jiang B.; Jiang H.; Guddat L. W.; Yang H.; Rao Z. Crystal Structures of membrane transporter MmpL3, an anti-TB drug target. Cell 2019, 176, 636–648. 10.1016/j.cell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- Owens C. P.; Chim N.; Goulding C. W. Insights on how the Mycobacterium tuberculosis heme uptake pathway can be used as a drug target. Future Med. Chem. 2013, 5, 1391–1403. 10.4155/fmc.13.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullius M. V.; Harmston C. A.; Owens C. P.; Chim N.; Morse R. P.; McMath L. M.; Iniguez A.; Kimmey J. M.; Sawaya M. R.; Whitelegge J. P.; Horwitz M. A.; Goulding C. W. Discovery and characterization of a unique mycobacterial heme acquisition system. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 5051–5056. 10.1073/pnas.1009516108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H.; Carrero-Lérida J.; Silva A. P. G.; Whittingham J. L.; Brannigan J. A.; Ruiz-Pérez L. M.; Read K. D.; Wilson K. S.; González-Pacanowska D.; Gilbert I. H. Synthesis and evaluation of alpha-thymidine analogues as novel antimalarials. J. Med. Chem. 2012, 55, 10948–10957. 10.1021/jm301328h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik M.; Raichurkar A.; Bandodkar B. S.; Varun B. V.; Bhat S.; Kalkhambkar R.; Murugan K.; Menon R.; Bhat J.; Paul B.; Iyer H.; Hussein S.; Tucker J. A.; Vogtherr M.; Embrey K. J.; McMiken H.; Prasad S.; Gill A.; Ugarkar B. G.; Venkatraman J.; Read J.; Panda M. Structure guided lead generation for M. tuberculosis thymidylate kinase (Mtb TMK): discovery of 3-cyanopyridone and 1,6-naphthyridin-2-one as potent inhibitors. J. Med. Chem. 2015, 58, 753–766. 10.1021/jm5012947. [DOI] [PubMed] [Google Scholar]
- Cole S. T.; Brosch R.; Parkhill J.; Garnier T.; Churcher C.; Harris D.; Gordon S. V.; Eiglmeier K.; Gas S.; Barry C. E. 3rd; Tekaia F.; Badcock K.; Basham D.; Brown D.; Chillingworth T.; Connor R.; Davies R.; Devlin K.; Feltwell T.; Gentles S.; Hamlin N.; Holroyd S.; Hornsby T.; Jagels K.; Krogh A.; McLean J.; Moule S.; Murphy L.; Oliver K.; Osborne J.; Quail M. A.; Rajandream M.-A.; Rogers J.; Rutter S.; Seeger K.; Skelton J.; Squares R.; Squares S.; Sulston J. E.; Taylor K.; Whitehead S.; Barrell B. G. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- DeJesus M. A.; Gerrick E. R.; Xu W.; Park S. W.; Long J. E.; Boutte C. C.; Rubin E. J.; Schnappinger D.; Ehrt S.; Fortune S. M.; Sassetti C. M.; Ioerger T. R. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio 2017, 8, e02133 10.1128/mbio.02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J. E.; Gawronski J. D.; Dejesus M. A.; Ioerger T. R.; Akerley B. J.; Sassetti C. M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Serafini A.; Boldrin F.; Palú G.; Manganelli R. Characterization of a Mycobacterium tuberculosis ESX-3 conditional mutant: essentiality and rescue by iron and zinc. J. Bacteriol. 2009, 191, 6340–6344. 10.1128/jb.00756-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini A.; Pisu D.; Palù G.; Rodriguez G. M.; Manganelli R. The ESX-3 secretion system is necessary for iron and zinc homeostasis in Mycobacterium tuberculosis. PLoS One 2013, 8, e78351 10.1371/journal.pone.0078351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist M. S.; Steigedal M.; Ahmad R.; Mehra A.; Dragset M. S.; Schuster B. M.; Philips J. A.; Carr S. A.; Rubin E. J. Mycobacterial Esx-3 requires multiple components for iron acquisition. mBio 2014, 5, e01073 10.1128/mbio.01073-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist M. S.; Unnikrishnan M.; McConnell M. J.; Borowsky M.; Cheng T.-Y.; Siddiqi N.; Fortune S. M.; Moody D. B.; Rubin E. J. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 18792–18797. 10.1073/pnas.0900589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez G. M.; Voskuil M. I.; Gold B.; Schoolnik G. K.; Smith I. ideR, An essential gene in mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect. Immun. 2002, 70, 3371–3381. 10.1128/iai.70.7.3371-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurthkoti K.; Amin H.; Marakalala M. J.; Ghanny S.; Subbian S.; Sakatos A.; Livny J.; Fortune S. M.; Berney M.; Rodriguez G. M. The capacity of Mycobacterium tuberculosis To survive iron starvation might enable it to persist in iron-deprived microenvironments of human granulomas. mBio 2017, 8, e01092 10.1128/mbio.01092-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Brecik M.; Mukherjee R.; Evans J. C.; Svetlíková Z.; Blaško J.; Surade S.; Blackburn J.; Warner D. F.; Mikušová K.; Mizrahi V. The complex mechanism of antimycobacterial action of 5-fluorouracil. Chem. Biol. 2015, 22, 63–75. 10.1016/j.chembiol.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Singh V.; Donini S.; Pacitto A.; Sala C.; Hartkoorn R. C.; Dhar N.; Keri G.; Ascher D. B.; Mondésert G.; Vocat A.; Lupien A.; Sommer R.; Vermet H.; Lagrange S.; Buechler J.; Warner D. F.; McKinney J. D.; Pato J.; Cole S. T.; Blundell T. L.; Rizzi M.; Mizrahi V. The inosine monophosphate dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infect. Dis. 2017, 3, 5–17. 10.1021/acsinfecdis.6b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollinger J.; Bailey M. A.; Moraski G. C.; Casey A.; Florio S.; Alling T.; Miller M. J.; Parish T. A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PLoS One 2013, 8, e60531 10.1371/journal.pone.0060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boshoff H. I. M.; Myers T. G.; Copp B. R.; McNeil M. R.; Wilson M. A.; Barry C. E. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism novel insights into drug mechanisms of action. J. Biol. Chem. 2004, 279, 40174–40184. 10.1074/jbc.m406796200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.