

Abstract
Introduction
Presenilin enhancer2 (Pen‐2) is an essential subunit of γ‐secretase, which is a key protease responsible for the cleavage of amyloid precursor protein (APP) and Notch. Mutations on Pen‐2 cause familial Alzheimer disease (AD). However, it remains unknown whether Pen‐2 regulates neuronal survival and neuroinflammation in the adult brain.
Methods
Forebrain neuron‐specific Pen‐2 conditional knockout (Pen‐2 cKO) mice were generated for this study. Pen‐2 cKO mice expressing Notch1 intracellular domain (NICD) conditionally in cortical neurons were also generated.
Results
Loss of Pen‐2 causes astrogliosis followed by age‐dependent cortical atrophy and neuronal loss. Loss of Pen‐2 results in microgliosis and enhanced inflammatory responses in the cortex. Expression of NICD in Pen‐2 cKO cortices ameliorates neither neurodegeneration nor neuroinflammation.
Conclusions
Pen‐2 is required for neuronal survival in the adult cerebral cortex. The Notch signaling may not be involved in neurodegeneration caused by loss of Pen‐2.
Keywords: Alzheimer disease, astrogliosis, microgliosis, neurodegeneration, presenilin enhancer2
This study reports a novel mouse model, which exhibits age‐related neurodegeneration. Forebrain neuron‐specific deletion of Pen‐2 leads to early astrogliosis followed by age‐dependent neuronal loss and severe inflammatory responses in the adult cerebral cortex. The Notch signaling does not play a critical role in Pen‐2‐dependent neuronal survival.
Highlights.
Conditional deletion of Pen‐2 in the forebrain causes age‐related cortical atrophy in mice.
Deletion of Pen‐2 leads to early astrogliosis in the adult cortex.
Deletion of Pen‐2 results in age‐related neurodegeneration and neuroinflammation.
The expression of NICD rescues neither neuronal loss nor inflammatory responses in the cortex.
1. INTRODUCTION
Alzheimer disease (AD) is the most common form of dementia and has become a global problem for the elderly. AD is clinically characterized by progressive cognitive deficits and morphologically by neuronal loss and the presence of amyloid plaques and neurofibrillary tangles. 1 , 2 Based on the start time of symptoms, AD can be categorized into two subtypes, early‐onset, and late‐onset. Major genetic causes for early‐onset of AD include mutations on APP (amyloid precursor protein), Psen1 (presenilin1), and Psen2 (presenilin2). 3 , 4 , 5 These mutations cause familial AD that displays clinical symptoms in midlife. 1 It is believed that Aβ accumulation, 6 tau hyperphosphorylation, 7 loss of presenilin function, 8 , 9 metabolic dysfunction, 10 , 11 and epigenetic abnormality 12 , 13 play critical roles in the pathogenesis of AD. So far, there are still no validated prognostic or diagnostic tests. 14
γ‐Secretase is a well‐known protease responsible for the intramembranous cleavage of APP and Notch to produce Aβ and Notch intracellular domain (NICD). 15 , 16 It contains four essential subunits including presenilin, presenilin enhancer2 (Pen‐2), nicastrin, and anterior pharynx defective1 (Aph‐1). 17 The Pen‐2 gene, located on chromosome 19q13, is composed of 4 exons. 18 It has been reported that downregulation of Pen‐2 is associated with reduced levels of presenilin and impaired maturation of nicastrin. 19 Interestingly, production of N‐ and C‐terminal fragments of presenilin1 is abolished by downregulation of Pen‐2 and is enhanced by overexpression of Pen‐2. 20 Therefore, Pen‐2 is required for the endoproteolysis of presenilin. Although previous evidence has demonstrated that Pen‐2 may protect zebrafish embryos from apoptosis, 21 , 22 physiological functions of Pen‐2 in the adult brain remain largely unknown due to early embryonic lethality in germ‐line Pen‐2 knockout mice. 23 Recently, we employed conditional knockout (cKO) techniques to overcome the lethality problem caused by germ‐line deletion of Pen‐2. Functional analyses on neural progenitor cells (NPCs)‐specific Pen‐2 KO mice reveal that Pen‐2 controls the fate switch of intermediate progenitors from radial glial progenitors in the developing cortex through a Notch‐dependent mechanism, 24 suggesting that Pen‐2 is critical for the maintenance of neural stem cells.
Recent evidence has shown that mutations on Pen‐2 are associated with familial AD. 25 , 26 , 27 Thus, Pen‐2 is involved in the pathogenesis of AD. However, it remains unknown whether Pen‐2 regulates neuronal survival in the adult cerebral cortex. To address this question, we generated forebrain neuron‐specific Pen‐2 cKO mice by crossing floxed Pen‐2 24 with a transgenic mouse expressing Cre under the promoter of α‐calcium‐calmodulin‐dependent kinase II (CaMKIIα). 28 , 29 , 30 We show that loss of Pen‐2 leads to early astrogliosis, age‐related reduction on the cortical size, and age‐dependent neurodegeneration. We observe enhanced inflammatory responses in Pen‐2 cKO mice. Moreover, we find that expression of NICD does not improve neurodegenerative phenotypes in Pen‐2 cKO mice. Overall, this study highlights an essential role of Pen‐2 in neuronal survival in the adult cortex.
2. MATERIALS AND METHODS
2.1. Animals
We employed a strategy similar to those previously reported 31 , 32 to generate forebrain neuron‐specific Pen‐2 cKO mice for this study. We bred Pen‐2f/+ 24 with the CaMKIIα‐Cre mouse 29 , 30 to obtain Pen‐2f/+;CaMKIIα‐Cre, which was then bred to Pen‐2f/f to produce Pen‐2f/f (control), Pen‐2f/+;CaMKIIα‐Cre (control), and Pen‐2f/f;CaMKIIα‐Cre (Pen‐2 cKO) mice. To generate Pen‐2 cKO animals expressing NICD in the forebrain, Pen‐2f/+;CaMKIIα‐Cre mice were crossed to Pen‐2f/f; LSL‐N1ICD to produce Pen‐2f/+; CaMKIIα‐Cre;LSL‐N1ICD (control), Pen‐2f/f; CaMKIIα‐Cre (Pen‐2 cKO) and Pen‐2f/f;CaMKIIα‐Cre;LSL‐N1ICD (Pen‐2 cKO;N1ICD). Tail DNAs were used for genotyping by PCR. To detect the floxed Pen‐2 allele, the following primers were used: GACCCGTAGAAGAGCAGTCAGT (forward) and ATAAAGAATAGGCTGGGTGGTG (reverse). To detect N1ICD gene, the following primers were used: AAGTGCAGGTGCCAGAACAT (forward) and GCAGCATCTGAACGAGAGTA (reverse).
The genetic background of the mice was C57BL/6, and both genders were used in this study. The mice were group‐housed (4‐5 per cage) and had free access to food and water. They were maintained in an SPF room in the core facility of the Model Animal Research Center (MARC) at Nanjing University. The animal room was maintained under constant humidity and temperature (25 ± 1°C). The light‐cycle of the animal room was automatically controlled. Mouse breeding was conducted under an animal protocol approved by the IACUC in the MARC of Nanjing University. All the experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of Nanjing University.
2.2. Nissl staining and measurement on the thickness of the cortex
Mice at 2, 3, 6, or 9 months were euthanized by CO2 and were then perfused with PBS. The brain was fixed in 4% paraformaldehyde (PFA) overnight at 4°C, followed by dehydration using ethanol. After being embedded in paraffin, four brains were placed into one block, which was sectioned sagittally at the thickness of 10 μm using a microtome (Leica Microsystems, Bannockburn, IL, United States). A total of 5 sagittal brain sections, spaced at 300 μm apart, were used for Nissl staining. Sections were deparaffinized in xylene and rehydrated in ethanol. Sections were treated with 0.1% cresyl‐violet for 1 minute and were then washed with distilled water. Dried sections were sealed using neutral resin (Sinopharm Chemical Reagent Co. Ltd., Shanghai). We used a method described by Acx and colleagues (2017) to measure the thickness of the cortex. First, Nissl‐stained images were captured using an Olympus BX53 microscope. Second, the thickness of the parietal‐occipital cortex overlying the hippocampus was measured and averaged for mice at different ages (3 mice per group per age).
2.3. Immunohistochemistry (IHC)
We used a method described recently. 33 , 34 Sections were first deparaffinized and were then rehydrated using ethanol, boiled in 0.01 mol/L sodium citrate buffer (pH = 6.0) for 25 minutes. Sections were blocked by PBS containing 5% bovine serum albumin (BSA) for 30 minutes and incubated overnight at 4°C with the following antibodies: anti‐NeuN (ABN781:1000; Millipore), anti‐MAP2 (MAB3418, 1:500; Millipore), anti‐GFAP (A14673; 1:500; Abclonal), and anti‐Iba1 (019‐19741; 1:500; Wako). On the next day, the sections were incubated for 1 hour at room temperature in PBS with secondary antibodies (1:500) including Alexa Fluor 488 goat anti‐mouse/anti‐rabbit and Alexa Fluor 594 goat anti‐mouse/anti‐rabbit (Invitrogen). Images were captured and analyzed using a ZEISS LSM‐880 confocal laser‐scanning microscope.
2.4. Cell counting
We used a method described recently by Acx and colleagues (2017). Briefly, three sagittal sections spaced 300 μm apart were used for IHC and at least three mice were included for each genotype at each age. For counting of NeuN‐positive (NeuN+), GFAP+ or Iba1+ cells in the cortex, IHC images for the parietal‐occipital cortex overlying the hippocampus morphologically were captured under the 10 × objective lens of an Olympus BX53 microscope. ImageJ was used to count NeuN+, GFAP+, or Iba1+ cells in each image. The number of cells in each image (876 × 660 μm2) was then averaged across 3 sections.
2.5. Tissue preparations for Western analysis
Mice were euthanized by CO2. 35 , 36 Tissues for different brain regions were freshly collected from mice at 2, 3, 6, or 9 months and were quickly placed into liquid nitrogen. Samples were stored at −80°C until use. Tissues from the cortex were homogenized in cold radio immunoprecipitation assay (RIPA) lysis buffer (1% NP40, 0.1% SDS, 0.5% sodium deoxycholate and 1 mmol/L EDTA in TBS) containing protease and phosphatase inhibitors (Thermo) and were then centrifugated at 12 000 g for 15 minutes. The RIPA‐soluble supernatants were used as total protein lysates. Protein concentrations were analyzed using a BSA method described previously. 24 , 32 Normalized cortical protein lysates with a total of 40 μg protein were separated on a 10% SDS–PAGE (Invitrogen) and then transferred to nitrocellulose membrane. The latter was blocked with 5% dry milk for 1 hour and then incubated with primary antibodies overnight. The membrane was washed with TBS for three times and then reacted with a Li‐Cor IRDye infrared dye‐coupled secondary antibody. Membranes were scanned, and data were analyzed using the Odyssey Infrared Imaging System (Li‐Cor). The following primary antibodies were used: anti‐Pen‐2 (A15172; 1:1000; Abclonal), anti‐nicastrin (34‐9200; 1:500; Invitrogen), anti‐APP (A8717; 1:1000; Sigma‐Aldrich), anti‐Iba1 (382207; 1:1000; ZENBIO), anti‐GFAP (A14673; 1:1000; Abclonal), anti‐GPADH (CW0100; 1:10,000; CWBIO), and anti‐β‐actin (CW0096; 1:10,000, CWBIO).
2.6. RNA Extraction and quantitative real‐time PCR
Total RNAs from mouse cortices were purified using the TRIzol reagent (Invitrogen) according to the manufacturer's instructions. RNA integrity was confirmed by the detection of 28S and 18S rRNA bands in agarose gel electrophoresis. RNA concentration was measured using a Nanodrop spectrophotometer. Equal amounts (1 μg) of RNAs were reverse transcribed into cDNA using PrimeScript RT reagent Kit (Takara). Quantitative real‐time PCR (qPCR) was performed by the ABI StepOne Plus system. Gapdh was used as the internal control in each PCR reaction. The PCR reactions were performed three times independently, and each sample was loaded in duplicates. The average CT values were used to calculate relative levels. The primer sequences used were as follows. For Pen‐2: TGGATTTGCGTTCCTGCCTTTTCT (forward) and ATGAAGTTGTTAGGGAGTGCC (reverse). For GFAP: AGGTGG AGAGGGACAACTTT (forward) and TCTGCCTCCTGTCTATACGC (reverse). For Iba1: GCTTTTGGACTGCTGAAGGC (forward) and GTTTGGACGGCAGATCC TCA (reverse). For IL1β: GCAACTGTTCCTGAACTCAACT (forward) and ATCT TTTGGGGTCCGTCAACT (reverse). For TNFα: CCTCCCTCTCATCAGTTCTAT GG (forward) and GGCTACAGGCTTGTCACTCG (reverse). For N1ICD: TGTCA ATGTTCGAGGACCAG (forward) and GCAGCATCTGAACGAGAGTA (reverse). For Gapdh: AATGTGTCCGTCGTGGATCT (forward) and CCCTGTTGCTGTAG CCGTAT (reverse).
2.7. Statistical Analysis
Data were presented as the mean ± the standard error of the mean (SEM). All the data were subject to Normality test using the Shapiro‐Wilk method in SPSS. While three sets of data including Western blotting on nicastrin (for Pen‐2 cKO) and qPCR analyses on IL1β (for Pen‐2 cKO at 6 months) and NICD (for control) did not exhibit normal distribution (P < 0.05), the remaining showed normal distribution (P > 0.05). For data without normal distribution, P values were obtained from Univariate Analysis of Variance (ANOVA), by which we analyzed genotype effects between different groups. For data with normal distribution, Student t test (two‐tailed, unpaired) was conducted to analyze genotype effects. P < 0.05 was considered statistically significant and P < 0.01 highly significant.
3. RESULTS
3.1. Generation of forebrain neuron‐specific Pen‐2 cKO mice
Pen‐2f/f 24 mice were used to generate Pen‐2 cKO mice in which Cre is expressed in excitatory neurons in the postnatal forebrain. To visualize the expression pattern of Cre, we crossed a tdTomato reporter line to the CaMKIIα‐Cre mouse. Abundant NeuN+/tdTomato+ cells were observed specifically in the cortex and the hippocampus of CaMKIIα‐Cre;LSL‐tdTomato mice (Figure 1A). To quantify the inactivation efficiency of Pen‐2, we analyzed cortical samples from Pen‐2 cKO mice at 3 months. First, Western analysis showed a significant reduction on protein levels of Pen‐2 in mutants compared with controls (Figure 1B and Figure (S1)). Second, qPCR analysis revealed significantly decreased levels of Pen‐2 mRNAs in mutants (Figure 1C). The residual amount of Pen‐2 in Pen‐2 cKO cortices likely came from glia, interneurons, and some neurons in which Cre was not expressed (Figure 1A,B). As expected, conditional deletion of Pen‐2 did not affect the expression of APP in postnatal neurons in the cortex (Figure 1D and Figure S1). We observed massive accumulation of the C‐terminal fragment of APP (APP‐CTF) (control = 100 ± 6%; cKO = 3812 ± 183%; n = 3 mice per group; P < 0.001) and significant reduction on nicastrin protein in Pen‐2 cKO cortical samples compared with controls (Figure 1D and Figure S1). Overall, loss of Pen‐2 significantly impaired γ‐secretase activity in the brain.
Figure 1.
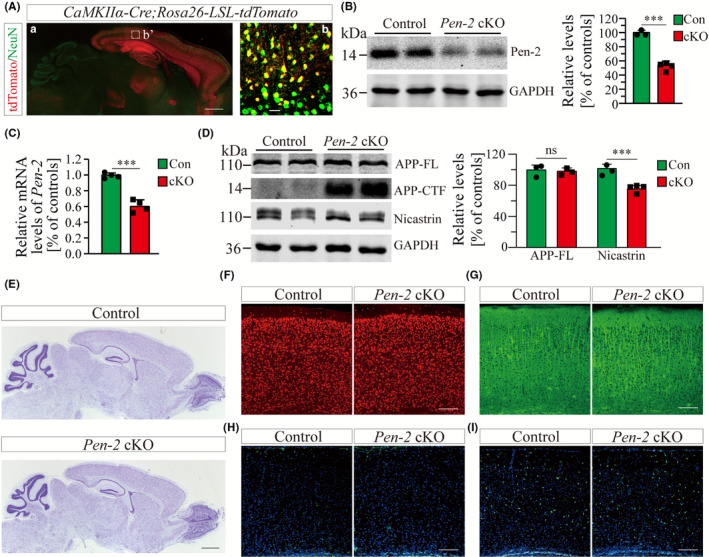
Molecular characterization of forebrain neuron‐specific Pen‐2 cKO mice. A, Co‐staining for NeuN and tdTomato. CaMKIIα‐Cre;Rosa26‐LSL‐tdTomato mice were used to collect brain sections. There was wide expression of tdTomato in the cortex, the hippocampus, the olfactory bulb, and the striatum but not the cerebellum. Most NeuN+ cells were positive for tdTomato. The boxed area in (a) was enlarged as (b). Scale bar is 500 μm in (a) but 25 μm in (b). B, Western analysis on Pen‐2. Cortical protein samples for control (Con) and Pen‐2 cKO (cKO) mice at 3 mo were used. There was significant difference on Pen‐2 levels between control and Pen‐2 cKO mice (***P < 0.001; n = 3‐4 mice per group). C, Relative Pen‐2 mRNA levels. There was significant difference between control (Con) and Pen‐2 cKO (cKO) mice (***P < 0.001; n = 4 per group). D, Western analyses for APP‐FL, APP‐CTF and nicastrin. Cortical samples at 3 mo were used. There was no significant difference on protein levels of APP‐FL between control (Con) and Pen‐2 cKO (cKO) mice (P > 0.6; n = 3 per group). There was significant difference on protein levels of nicastrin between control and Pen‐2 cKO mice (***P < 0.001; n = 3‐4 per group). E, Nissl staining. There was comparable brain size in control and Pen‐2 cKO mice at 2 mo. Scale bar is 1 mm. F‐I, Representative fluorescence IHC images for NeuN (F), MAP2 (G), GFAP (H) and Iba1 (I). Images were taken from cortices of control and Pen‐2 cKO mice at 2 mo. Scale bar is 200 μm
To find out whether conditional inactivation of Pen‐2 affected the general morphology of the cortex, we conducted Nissl staining using brain sections from mice at 2 months. We found that the architecture of the cortex and the hippocampus was comparable between control and Pen‐2 cKO mice (Figure 1E). Moreover, IHC experiments revealed no detectable change on the immuno‐reactivity of NeuN, MAP2, GFAP, or Iba1 in Pen‐2 cKO mice compared with controls (Figure 1F‐I). Together, cortical development in general was unaffected by deletion of Pen‐2.
3.2. Age‐dependent cortical atrophy and neuronal loss in Pen‐2 cKO mice
To determine whether neuronal survival was affected in aged Pen‐2 cKO mice, Nissl staining (Figure 2A) and NeuN IHC (Figure 2B) were performed using brain sections from mice at ages ranging from 3 to 9 months. First of all, Nissl staining revealed a thin cortex in Pen‐2 cKO mice at 6 or 9 months compared with littermate controls (Figure 2A), and quantification results confirmed significantly reduced thickness of the cortex (Figure 2C). In contrast, the thickness of the cortex was not different at 3 months (Figure 2C). Secondly, NeuN IHC also showed thinner cortex in Pen‐2 cKO mice at 6 and 9 months than in controls (Figure 2B). The number of NeuN+ cells was not changed in Pen‐2 cKO mice at 3 months compared with controls, but it was significantly decreased at 6 and 9 months (Figure 2D), indicating age‐related neurodegeneration in the cortex of Pen‐2 cKO mice.
Figure 2.
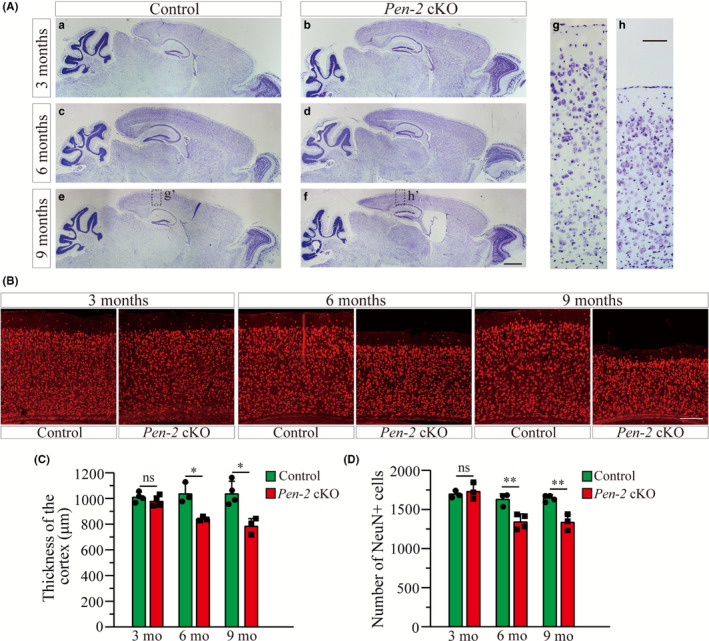
Age‐dependent neurodegeneration in Pen‐2 cKO mice. A, Nissl staining. Brain sections of mice at 3, 6, and 9 mo were used. There was cortical atrophy and enlarged lateral ventricle in Pen‐2 cKO mice at 6 and 9 mo compared with controls. Boxed areas in (e) or (f) were enlarged as (g) and (h), respectively. Scale bar is 1 mm for (a‐f) or 100 μm for (g‐h). B. Representative fluorescence images for NeuN IHC taken from cortices. Brain sections from mice at 3, 6, and 9 mo were used. Scale bar is 200 μm. C, Cortical thickness. There was no significant difference on the size of the cortex between control and Pen‐2 cKO mice at 3 mo. There was significant decrease in Pen‐2 cKO mice at 6 (*P < 0.05; n = 3 mice per group) or 9 mo (*P < 0.05; n = 3‐4 per group). D, Averaged number of NeuN+ cells. There were significant decreases on NeuN+ cells in Pen‐2 cKO mice at 6 (**P < 0.01; n = 3‐4 per group) and 9 (**P < 0.01; n = 3‐4 per group)
3.3. Progressive astrogliosis in Pen‐2 cKO mice
To examine whether deletion of Pen‐2 affected astrocytes, we performed IHC on GFAP using mice at different ages (Figure 3A). While the immuno‐reactivity of GFAP was increased in the cortex of Pen‐2 cKO mice at 3 months compared with controls, it was rigorously detected at 6 and 9 months (Figure 3A). Cell counting results showed significant increase on the number of GFAP+ cells in Pen‐2 cKO cortices at each age (Figure 3B). Moreover, we conducted Western analysis on GFAP using total cortical homogenates prepared from mice at the above different ages (Figure 3C and Figure S2). While GFAP protein exhibited a small but significant increase in Pen‐2 cKO mice at 3 months of age, it was robustly increased at 6 and 9 months (Figure 3C). Finally, we performed qPCR to examine GFAP mRNAs, which were significantly increased in Pen‐2 cKO mice at 6 and 9 months compared with controls (data not shown). Thus, inactivation of Pen‐2 caused progressive astrogliosis in the cortex.
Figure 3.
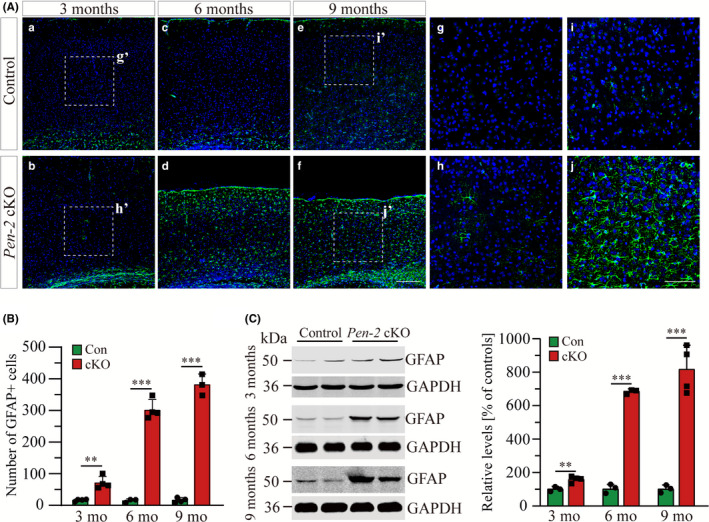
Progressive astrogliosis in Pen‐2 cKO mice. A, Representative images for fluorescence IHC on GFAP in the cortex. Brain sections of mice at 3, 6, and 9 mo were used. The immuno‐reactivity of GFAP was increased in Pen‐2 cKO mice since 3 mo of age compared with controls. Boxed areas in (a), (b), (e), and (f) were enlarged as (g), (h), (i), and (j), respectively. Scale bar is 200 μm for (a‐f) or 100 μm for (g‐j). B. Averaged number of GFAP+ cells in the cortex. There was significant difference between control (Con) and Pen‐2 cKO (cKO) mice at each age (3 mo: **P < 0.01; 6 mo: ***P < 0.001; 9 mo: ***P < 0.001; n = 3‐4 mice per group). Analysis of variance (ANOVA) revealed significant Age (F = 115.3, df2/16, P < 0.001) and Age × Genotype effects (F = 116.5, df2/16, P < 0.001), suggesting progressive astroglial activation. C, Western analysis on GFAP. Cortical samples at 3, 6, and 9 mo were used. GFAP levels in Pen‐2 cKO (cKO) mice differed from those in controls (Con) at each age (P < 0.05; n = 3‐4 per group). The increase in GFAP levels was above 6‐fold in Pen‐2 cKO cortices compared with controls at 6 or 9 mo
3.4. Age‐related microgliosis and neuroinflammatory responses in Pen‐2 cKO mice
We next analyzed microglia by performing IHC on Iba1. First, the immuno‐reactivity of Iba1 was comparable in control and Pen‐2 cKO cortices at 3 months, but it was increased in Pen‐2 cKO mice at 6 and 9 months (Figure 4A). Quantification results showed that the number of Iba1+ cells was not changed in the cortex of Pen‐2 cKO mice at 3 months (Figure 4B) and that it was highly increased at 6 and 9 months (Figure 4B). Thus, cell counting results were consistent with those from IHC. Next, we performed Western analysis on Iba1 (Figure 4C and Figure S3). Indeed, relative Iba1 levels were unaltered in Pen‐2 cKO mice at 3 months (P > 0.4) but were increased at 6 and 9 months compared with controls (Figure 4C). Finally, we found that mRNA levels for Iba1 were significantly increased in Pen‐2 cKO mice at 6 or 9 months (Figure 4D). Overall, there was striking age‐related microgliosis in Pen‐2 cKO mice.
Figure 4.
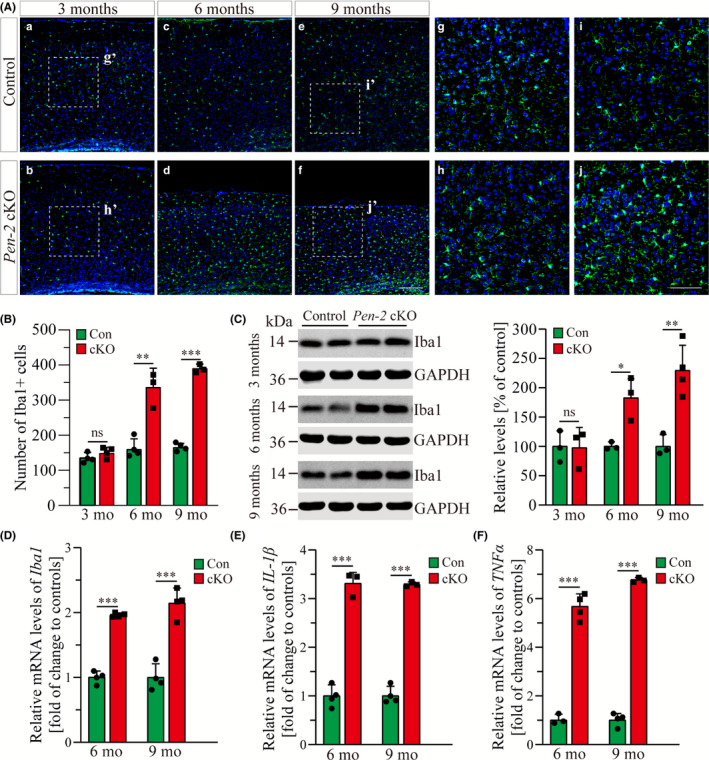
Age‐related microgliosis in Pen‐2 cKO mice. A, Representative images for fluorescence IHC on Iba1 in the cortex. Mice at 3, 6, and 9 mo were examined. Note that the immuno‐reactivity of Iba1 was not increased in Pen‐2 cKO mice at 3 mo compared with controls. Boxed areas in (a), (b), (e), and (f) were enlarged as (g), (h), (i), and (j), respectively. Scale bar is 200 μm for (a‐f) or 100 μm for (g‐j). B, Averaged number of Iba1+ cells in the cortex. There was significant difference between control (Con) and Pen‐2 cKO (cKO) mice at each age (3 mo: P > 0.2; 6 mo: **P < 0.01; 9 mo: ***P < 0.001; n = 3‐4 mice per group). C, Western analysis on Iba1. Note that Iba1 levels in Pen‐2 cKO (cKO) mice did not differ from those in controls (Con) at 3 mo of age but were increased at 6 and 9 mo (3 mo: not significant; 6 mo: *, P < 0.05; 9 mo: **P < 0.01; n = 3‐4 per group). D‐F, qPCR analyses for Iba1 (D), IL‐1β (E), and TNFα (F). Data for 6 and 9 mo were presented. There was significant increase on levels for Iba1 (D), IL‐1β (E), and TNFα (F) in Pen‐2 cKO (cKO) mice compared with controls (Con) at either age (***P < 0.001; n = 3‐4 per group)
Next, we examined expression for neuroinflammation markers such as IL‐1β and TNFα using cortical RNA samples. There was no significant change on IL‐1β or TNFα mRNA levels in Pen‐2 cKO cortices at 3 months compared with littermate controls (data not shown). In contrast, there was remarkable increase on mRNA levels for IL‐1β (Figure 4E) and TNFα (Figure 4F) in Pen‐2 cKO cortices. Overall, the above results suggest striking inflammatory responses in Pen‐2 cKO cortices.
3.5. Nonessential role of the Notch signaling in Pen‐2‐dependent neuronal survival
Notch is a major target of γ‐secretase. 17 We previously demonstrated that expression of NICD rescues decreased population of neural stem cells caused by deletion of Pen‐2. 24 To investigate whether the Notch signaling also plays a critical role in Pen‐2‐dependent neuronal survival, we crossed Pen‐2 cKO mice to N1ICD transgenics to obtain Pen‐2 cKO mice expressing NICD in Cre+ neurons in the adult cortex (Figure 5A).
Figure 5.
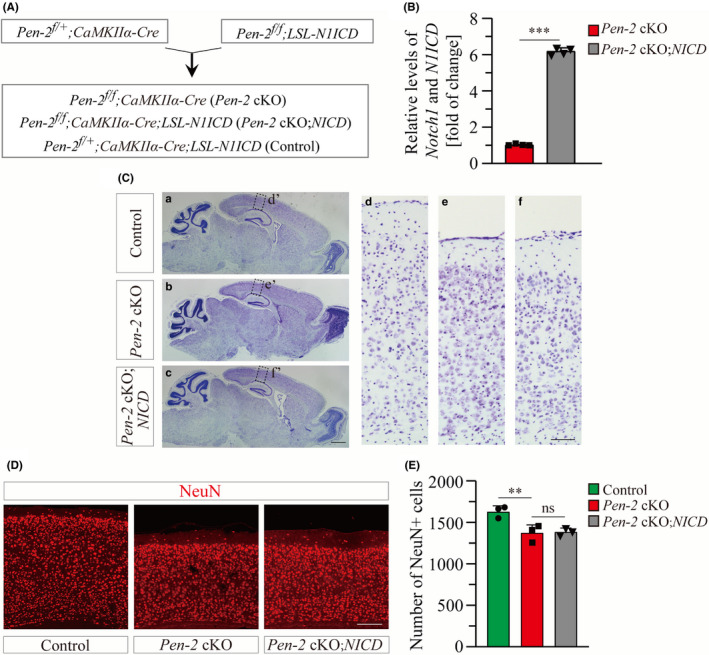
NICD does not ameliorate neuronal loss in Pen‐2 cKO mice. A, Experimental plan for the generation of Pen‐2 cKO mice expressing N1ICD conditionally in a Cre‐dependent manner. B, qPCR analysis on Notch1 and N1ICD. Relative mRNA levels of Notch1 and N1ICD were highly significantly increased in the cortex of Pen‐2 cKO;NICD mice as compared to Pen‐2 cKOs (***P < 0.001; n = 4 mice per group). C, Nissl staining. Brain sections of control, Pen‐2 cKO, and Pen‐2 cKO;NICD mice at 5‐7 mo were used. Note that there was decreased cortical thickness in Pen‐2 cKO mice with and without NICD expression compared with controls. Boxed areas in (a‐c) were enlarged as (d‐f). Scale bar is 1 mm in (a‐c) or 100 μm in (d‐f). D, Representative images for fluorescence IHC on NeuN. Images were taken from the cortex. Note that there was decreased cortical thickness in Pen‐2 cKO and Pen‐2 cKO;NICD mice compared with controls. Scale bar is 200 μm. E, Averaged number of NeuN+ cells in the cortex. There was no significant difference between Pen‐2 cKO and Pen‐2 cKO;NICD mice. There was significant difference between Pen‐2 cKO and control mice (ns, not significant; **P < 0.01; n = 3 per group)
First of all, qPCR analysis on total cortical RNA samples showed highly significant increase on N1ICD mRNA levels in Pen‐2 cKO mice expressing NICD compared with Pen‐2 cKOs without NICD expression (Figure 5B). Secondly, we examined brain morphology of mice at 5‐7 months by Nissl staining (Figure 5C), which revealed comparable thickness of the cortex between Pen‐2 cKO mice with and without NICD expression. Thirdly, the number of NeuN+ cells did not differ between Pen‐2 cKO mice expressing NICD and those without NICD expression (Figure 5D,E). It was much less in Pen‐2 cKO mice with and without NICD expression than in controls (Figure 5D,E).
Moreover, we performed IHC on GFAP and Iba1 using brain sections from Pen‐2 cKO mice expressing NICD (Figure 6A,B). While the number of GFAP+ cells in the cortex did not differ between Pen‐2 cKO and Pen‐2 cKO;NICD mice (Figure 6C), it was significantly increased in Pen‐2 cKO with and without NICD expression compared with controls (Figure 6C). Similar results were obtained for Iba1 IHC. We did not find any difference on the number of Iba1+ cells in the cortex in Pen‐2 cKO mice with and without NICD expression (Figure 6D). Finally, Western analyses confirmed no significant difference on protein levels of GFAP (cKO = 399 ± 17%; cKO; NICD = 413 ± 35%; n = 3‐4 mice per group; P > 0.6) or Iba1 (cKO = 183 ± 21%; cKO; NICD = 175 ± 18%; n = 3 per group; P > 0.6) between Pen‐2 cKO mice with and without NICD expression (Figure 6E and Figure S4). Together, NICD rescued neither neurodegeneration nor neuroinflammation in Pen‐2 cKO cortices.
Figure 6.
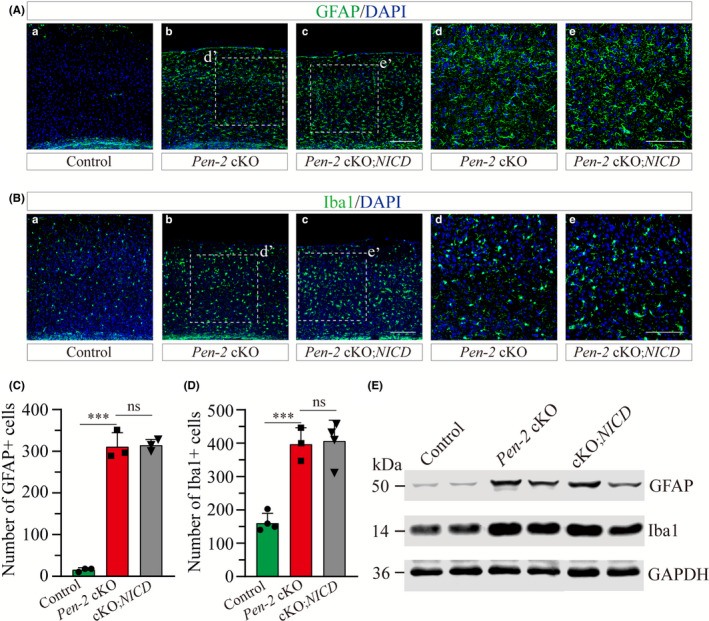
NICD does not reduce gliosis in Pen‐2 cKO mice. A‐B, Representative images for fluorescence IHC on GFAP (A) and Iba1 (B). Images were taken from the cortex of control, Pen‐2 cKO or Pen‐2 cKO;NICD mice. Boxed areas in (b,c) were enlarged as (d,e). Scale bar is 200 μm in (a‐c) or 100 μm in (d‐e). C, Averaged number of GFAP+ cells in the cortex. There was no significant difference between Pen‐2 cKO and Pen‐2 cKO;NICD mice. There was significant difference between Pen‐2 cKO and control mice (ns, not significant; ***P < 0.001; n = 3 mice per group). D, Averaged number of Iba1+ cells in the cortex. There was no significant difference between Pen‐2 cKO and Pen‐2 cKO;NICD mice. There was significant difference between Pen‐2 cKO and control mice (ns, not significant; ***P < 0.001; n = 3‐4 per group). E, Western blotting on GFAP and Iba1. Cortical samples of control, Pen‐2 cKO and Pen‐2 cKO;NICD mice were used. Blots for GFAP or Iba1 were comparable between Pen‐2 cKO and Pen‐2 cKO;NICD mice
4. DISCUSSION
Recent evidence has shown that Pen‐2 is implicated in familial AD. 25 , 26 Since global deletion of Pen‐2 causes early embryonic lethality in mice, 23 this precludes the possibility to use Pen‐2−/− mice to study whether Pen‐2 plays a role in neuronal survival in the adult cortex. To overcome this problem, we employed the Cre‐LoxP techniques and generated viable forebrain neuron‐specific Pen‐2 cKO mice (Figure 1). As expected, there is significant reduction on Pen‐2 expression in Pen‐2 cKO cortices (Figure 1). Nicastrin is another essential subunit of γ‐secretase, 17 and it exhibits significant decrease in Pen‐2 cKO mice as well (Figure 1). We reason that deletion of Pen‐2 prevents the formation of the γ‐secretase complex so that the stability of other subunits is impaired in cortical neurons. In line with this view, forebrain neuron‐specific inactivation of presenilins or nicastrin causes decreased levels of Pen‐2. 37 Moreover, previous in vitro studies have also shown that knockdown of one γ‐secretase subunit could significantly reduce protein levels of other subunits. 19 , 38 Overall, γ‐secretase subunits may depend on each other for stability in mature neurons.
While we have focused on Pen‐2 in this study, a number of recent studies have investigated the role of other γ‐secretase subunits in the adult cortex. First, it has been shown that conditional inactivation of presenilins causes memory loss, synaptic impairments, and age‐dependent cortical neurodegeneration. 39 , 40 Second, nicastrin was the second subunit identified to be critical for neuronal survival in the cortex. 37 , 41 , 42 , 43 Third, there is age‐related neuronal loss in forebrain neuron‐specific Aph‐1 cKO mice. 44 Together, the above evidence suggests that each γ‐secretase subunit may be equally important for the maintenance of mature neurons in the cortex.
Numerous mutations on Psen1, Psen2, nicastrin, and Pen‐2 have been reported in familial AD, 8 , 25 , 26 , 27 , 45 , 46 but the relevant mechanisms are not clear. It has been proposed that loss of presenilin mechanism may play a key role in AD. 8 , 9 , 47 However, since presenilins possess γ‐secretase‐dependent 17 and γ‐secretase‐independent functions, 8 , 48 it remains unsolved which type of function may protect mature cortical neurons. Tremendous efforts have been made by independent groups to search for mechanisms underlying neuronal survival regulated by γ‐secretase subunits. First, Saura et al (2004) showed that presenilins may promote neuronal survival via Notch‐dependent activation of CREB‐binding protein (CBP). Since neuron‐specific inactivation of CBP does not cause age‐related neuronal loss, 49 it is unlikely that CBP is involved in presenilins‐dependent neuronal survival. Second, apoptotic cell death has been reported in nicastrin cKO mice. 37 In contrast, TUNEL+ cells are not detected in Pen‐2 cKO cortices aged at 6 or 9 months (data not shown), suggesting that neuronal loss may not be caused by apoptosis. The discrepancy on TUNEL data for nicastrin cKO 37 and Pen‐2 cKO mice may be due to different CaMKIIα‐Cre lines used to generate these two different cKO mice. For example, the expression of Cre begins around P18 in nicastrin cKO mice 50 but 6 weeks in Pen‐2 cKOs. 30
Molecular mechanisms underlying neurodegeneration caused by loss of γ‐secretase subunit are largely unclear. Since APP is one of the most important targets of γ‐secretase, 17 recent studies have examined whether APP‐CTF, a cleavage product of APP by γ‐secretase, plays an important role in neuronal survival in the adult brain. Since it has been shown that accumulation of APP‐CTF is detrimental to neurons, 51 Acx and colleagues (2017) generated Aph‐1 cKO mice on the APP null background. However, there is no significant difference on the number of cortical neurons between Aph‐1 cKO and Aph‐1 cKO;APP−/− mice. 44 Thus, deletion of APP and prevention of APP‐CTF do not ameliorate neurodegeneration in Aph‐1 cKO mice. This finding excludes the possibility that neurodegeneration is due to accumulation of APP‐CTF. Although the Notch signaling is critical for the fate determination of NPCs, 52 , 53 the present study shows that conditional expression of NICD in forebrain neurons is insufficient to improve neurodegenerative phenotypes in Pen‐2 cKO mice. Overall, neither APP‐CTF nor the Notch signaling serves as a key mediator for γ‐secretase subunit‐dependent neuronal survival in the adult cortex.
Reactive astrogliosis is a common response in injured brain associated with diseases such as trauma, infection, neurodegeneration, and ischemia. 54 We observe significant astrogliosis but not microgliosis in Pen‐2 cKO mice at 3 months of age, suggesting that astroglial activation occurs prior to microgliosis and neuronal loss in this neurodegenerative model. In line with this finding, previous studies showed early astrogliosis in nicastrin cKO mice at 3 months. 37 , 42 , 43 Moreover, it has been shown that deletion of presenilins or Aph‐1 causes enhanced astrogliosis in the cortex. 39 , 44 , 55 These results strongly suggest that inflammatory responses may play a pivotal role in neurodegeneration caused by loss of γ‐secretase subunit. Consistent with these findings, it has been proposed that uncontrolled inflammation drives the progression of neurodegeneration. 56 The novel Pen‐2 cKO model generated in this study may serve as an excellent tool to test the anti‐inflammation strategy for neurodegenerative diseases and to study mechanisms underlying neuronal maintenance dependent on normal γ‐secretase function. Answers to these questions may not only provide insights on the pathogenesis of AD but also promote identification of potential therapeutic targets.
5. CONCLUSIONS
Functional analysis on forebrain neuron‐specific Pen‐2 cKO mice reveals that Pen‐2 plays an essential role in the survival of mature cortical neurons. Early astrogliosis takes place prior to the occurrence of evident neurodegeneration in Pen‐2 cKO cortices, followed by severe astrogliosis and microgliosis. The failure on rescue of neurodegeneration and gliosis in Pen‐2 cKO cortices by NICD suggests that the Notch signaling is not critical for Pen‐2‐dependent maintenance of neurons in the adult cortex.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank Shanshan Cheng and Jinxing Hou for technical help.
Bi H‐R, Zhou C‐H, Zhang Y‐Z, et al. Neuron‐specific deletion of presenilin enhancer2 causes progressive astrogliosis and age‐related neurodegeneration in the cortex independent of the Notch signaling. CNS Neurosci Ther.2021;27:174–185. 10.1111/cns.13454
Bi and Zhou are contributed equally to this work.
Contributor Information
Gui‐Quan Chen, Email: chenguiquan@nju.edu.cn.
Yi‐Min Hu, Email: guyueym@njmu.edu.cn.
DATA AVAILABILITY STATEMENT
The data presented in this article are available from corresponding authors upon request (guyueym@njmu.edu.cn and chenguiquan@nju.edu.cn).
REFERENCES
- 1. Ballard C, Gauthier S, Corbett A, et al. Alzheimer's disease. Lancet. 2011;377(9770):1019‐1031. [DOI] [PubMed] [Google Scholar]
- 2. Palmer AM. Neurochemical studies of Alzheimer's disease. Neurodegeneration. 1996;5(4):381‐391. [DOI] [PubMed] [Google Scholar]
- 3. Levylahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome‐1 familial Alzheimers‐disease locus. Science. 1995;269(5226):973‐977. [DOI] [PubMed] [Google Scholar]
- 4. Schellenberg GD, Bird TD, Wijsman EM, et al. Genetic‐linkage evidence for a familial Alzheimers‐disease locus on chromosome‐14. Science. 1992;258(5082):668‐671. [DOI] [PubMed] [Google Scholar]
- 5. Goate A, Chartierharlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimers‐disease. Nature. 1991;349(6311):704‐706. [DOI] [PubMed] [Google Scholar]
- 6. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353‐356. [DOI] [PubMed] [Google Scholar]
- 7. Small SA, Duff K. Linking Abeta and tau in late‐onset Alzheimer's disease: A dual pathway hypothesis. Neuron. 2008;60(4):534‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shen J, Kelleher RJ 3rd. The presenilin hypothesis of Alzheimer's disease: evidence for a loss‐of‐function pathogenic mechanism. Proc Natl Acad Sci USA. 2007;104(2):403‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Strooper B. Loss‐of‐function presenilin mutations in Alzheimer disease ‐ Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8(2):141‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rahman MH, Peng SL, Hu XY, et al. A network‐based bioinformatics approach to identify molecular biomarkers for type 2 diabetes that are linked to the progression of neurological diseases. Int J Environ Res Public Health. 2020;17(3):1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beglopoulos V, Tulloch J, Roe AD, et al. Early detection of cryptic memory and glucose uptake deficits in pre‐pathological APP mice. Nat Commun. 2016;7:11761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li X, Yao B, Chen L, et al. Ten‐eleven translocation 2 interacts with forkhead box O3 and regulates adult neurogenesis. Nat Commun. 2017;8:15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chouliaras L, Mastroeni D, Delvaux E, et al. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol Aging. 2013;34(9):2091‐2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Atluri G, Padmanabhan K, Fang G, et al. Complex biomarker discovery in neuroimaging data: finding a needle in a haystack. NeuroImage‐Clin. 2013;3:123‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Francis R, McGrath G, Zhang J, et al. Aph‐1 and Pen‐2 are required for Notch pathway signaling, γ‐secretase cleavage of βAPP, and presenilin protein accumulation. Dev Cell. 2002;3(1):85‐97. [DOI] [PubMed] [Google Scholar]
- 16. Fortini ME. γ‐Secretase‐mediated proteolysis in cell‐surface‐receptor signalling. Nat Rev Mol Cell Biol. 2002;3(9):673‐684. [DOI] [PubMed] [Google Scholar]
- 17. De Strooper B. Aph‐1, pen‐2, and nicastrin with presenilin generate an active γ‐secretase complex. Neuron. 2003;38(1):9‐12. [DOI] [PubMed] [Google Scholar]
- 18. Crystal AS, Morais VA, Pierson TC, et al. Membrane topology of γ‐secretase component PEN‐2. J Biol Chem. 2003;278(22):20117‐20123. [DOI] [PubMed] [Google Scholar]
- 19. Steiner H, Winkler E, Edbauer D, et al. PEN‐2 is an integral component of the γ‐secretase complex required for coordinated expression of presenilin and nicastrin. J Biol Chem. 2002;277(42):39062‐39065. [DOI] [PubMed] [Google Scholar]
- 20. Luo WJ, Wang H, Li H, et al. PEN‐2 and APH‐1 coordinately regulate proteolytic processing of presenilin 1. J Biol Chem. 2003;278(10):7850‐7854. [DOI] [PubMed] [Google Scholar]
- 21. Zetterberg H, Campbell W, Yang H, et al. The cytosolic loop of the gamma‐secretase component presenilin enhancer 2 protects Zebrafish embryos from apoptosis. J Biol Chem. 2006;281(17):11933‐11939. [DOI] [PubMed] [Google Scholar]
- 22. Campbell WA, Yang H, Zetterberg H, et al. Zebrafish lacking Alzheimer presenilin enhancer 2 (Pen‐2) demonstrate excessive p53‐dependent apoptosis and neuronal loss. J Neurochem. 2006;96(5):1423‐1440. [DOI] [PubMed] [Google Scholar]
- 23. Bammens L, Chávez‐Gutiérrez L, Tolia A, et al. Functional and topological analysis of Pen‐2, the fourth subunit of the γ‐secretase complex. J Biol Chem. 2011;286(14):12271‐12282. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24. Cheng S, Liu T, Hu Y, et al. Conditional inactivation of Pen‐2 in the developing neocortex leads to rapid switch of apical progenitors to basal progenitors. J Neurosci. 2019;39(12):2195‐2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andreoli V, Trecroci F, La Russa A, et al. Presenilin enhancer‐2 gene: Identification of a novel promoter mutation in a patient with early‐onset familial Alzheimer's disease. Alzheimers Dement. 2011;7(6):574‐578. [DOI] [PubMed] [Google Scholar]
- 26. Albani D, Batelli S, Pesaresi M, et al. A novel PSENEN mutation in a patient with complaints of memory loss and a family history of dementia. Alzheimers Dement. 2007;3(3):235‐238. [DOI] [PubMed] [Google Scholar]
- 27. Sala Frigerio C, Piscopo P, Calabrese E, et al. PEN–2 gene mutation in a familial Alzheimer’s disease case. J Neurol. 2005;252(9):1033‐1036. [DOI] [PubMed] [Google Scholar]
- 28. Cheng S, Hou J, Zhang C, et al. Minocycline reduces neuroinflammation but does not ameliorate neuron loss in a mouse model of neurodegeneration. Sci Rep. 2015;5:10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng S, Zhang C, Xu C, et al. Age‐dependent neuron loss is associated with impaired adult neurogenesis in forebrain neuron‐specific Dicer conditional knockout mice. Int J Biochem Cell Biol. 2014;57:186‐196. [DOI] [PubMed] [Google Scholar]
- 30. Fukaya M, Kato A, Lovett C, et al. Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci USA. 2003;100(8):4855‐4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu M, Han X, Liu R, et al. PDK1 deficit impairs the development of the dentate gyrus in mice. Cereb Cortex. 2019;29(3):1185‐1198. [DOI] [PubMed] [Google Scholar]
- 32. Xu C, Yu L, Hou J, et al. Conditional deletion of PDK1 in the forebrain causes neuron loss and increased apoptosis during cortical development. Front Cell Neurosci. 2017;11:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han X, Wei Y, Wu X, et al. PDK1 regulates transition period of apical progenitors to basal progenitors by controlling asymmetric cell division. Cereb Cortex. 2020;30:406‐420. [DOI] [PubMed] [Google Scholar]
- 34. Peng Y, Sun J, Hon S, et al. L‐3‐n‐butylphthalide improves cognitive impairment and reduces amyloid‐beta in a transgenic model of Alzheimer's disease. J Neurosci. 2010;30(24):8180‐8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang C, Liu T, Wang Q, et al. Loss of PP2A disrupts the retention of radial glial progenitors in the telencephalic niche to impair the generation for late‐born neurons during cortical development. Cereb Cortex. 2020;30(7):4183‐4196. [DOI] [PubMed] [Google Scholar]
- 36. Peng Y, Xu SF, Chen GQ, et al. l‐3‐n‐butylphthalide improves cognitive impairment induced by chronic cerebral hypoperfusion in rats. J Pharmacol Exp Ther. 2007;321(3):902‐910. [DOI] [PubMed] [Google Scholar]
- 37. Tabuchi K, Chen G, Sudhof TC, et al. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age‐related neurodegeneration. J Neurosci. 2009;29(22):7290‐7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Edbauer D, Winkler E, Haass C, et al. Presenilin and nicastrin regulate each other and determine amyloid b‐peptide production via complex formation. Proc Natl Acad Sci USA. 2002;99(13):8666‐8671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saura CA, Choi S‐Y, Beglopoulos V, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age‐dependent neurodegeneration. Neuron. 2004;42(1):23‐36. [DOI] [PubMed] [Google Scholar]
- 40. Feng R, Wang H, Wang J, et al. Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer's presenilin‐1 and presenilin‐2. Proc Natl Acad Sci USA. 2004;101(21):8162‐8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sesele K, Thanopoulou K, Paouri E, et al. Conditional inactivation of nicastrin restricts amyloid deposition in an Alzheimer’s disease mouse model. Aging Cell. 2013;12(6):1032‐1040. [DOI] [PubMed] [Google Scholar]
- 42. Hou JX, Cheng SS, Chen L, et al. Astroglial activation and tau hyperphosphorylation precede to neuron loss in a neurodegenerative mouse model. CNS Neurosci Ther. 2016;22(3):244‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu T‐T, Ye X‐L, Zhang J‐P, et al. Increased adult neurogenesis associated with reactive astrocytosis occurs prior to neuron loss in a mouse model of neurodegenerative disease. CNS Neurosci Ther. 2017;23(11):885‐893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Acx H, Serneels L, Radaelli E, et al. Inactivation of γ‐secretases leads to accumulation of substrates and non‐Alzheimer neurodegeneration. EMBO Mol Med. 2017;9(8):1088‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Helisalmi S, Dermaut B, Hiltunen M, et al. Possible association of nicastrin polymorphisms and Alzheimer disease in the Finnish population. Neurology. 2004;63(1):173‐175. [DOI] [PubMed] [Google Scholar]
- 46. Amtul Z, Lewis PA, Piper S, et al. A presenilin 1 mutation associated with familial frontotemporal dementia inhibits gamma‐secretase cleavage of APP and notch. Neurobiol Dis. 2002;9(2):269‐273. [DOI] [PubMed] [Google Scholar]
- 47. Zhang C, Wu B, Beglopoulos V, et al. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460(7255):632‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tu H, Nelson O, Bezprozvanny A, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease‐linked mutations. Cell. 2006;126(5):981‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen G, Zou X, Watanabe H, et al. CREB binding protein is required for both short‐term and long‐term memory formation. J Neurosci. 2010;30:13066‐13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yu H, Saura CA, Choi SY, et al. APP processing and synaptic plasticity in presenilin‐1 conditional knockout mice. Neuron. 2001;31(5):713‐726. [DOI] [PubMed] [Google Scholar]
- 51. Yankner BA, Dawes LR, Fisher S, et al. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989;245(4916):417‐420. [DOI] [PubMed] [Google Scholar]
- 52. Ables JL, Breunig JJ, Eisch AJ, et al. Not(ch) just development: Notch signalling in the adult brain. Nat Rev Neurosci. 2011;12(5):269‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zamanian JL, Xu L, Foo LC, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32(18):6391‐6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beglopoulos V, Sun X, Saura CA, et al. Reduced β‐amyloid production and increased inflammatory responses in presenilin conditional knock‐out mice. J Biol Chem. 2004;279(45):46907‐46914. [DOI] [PubMed] [Google Scholar]
- 56. Gao H‐M, Hong J‐S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29(8):357‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data presented in this article are available from corresponding authors upon request (guyueym@njmu.edu.cn and chenguiquan@nju.edu.cn).