

Abstract
STK17B is a member of the death-associated protein kinase family and has been genetically linked to the development of diverse diseases. However, the role of STK17B in normal and disease pathology is poorly defined. Here we present the discovery of thieno[3,2-d]pyrimidine SGC-STK17B-1 (11s), a high-quality chemical probe for this understudied “dark” kinase. 11s is an ATP-competitive inhibitor that showed remarkable selectivity over other kinases including the closely related STK17A. X-ray crystallography of 11s and related thieno[3,2-d]pyrimidines bound to STK17B revealed a unique P-loop conformation characterized by a salt bridge between R41 and the carboxylic acid of the inhibitor. Molecular dynamics simulations of STK17B revealed the flexibility of the P-loop and wide range of R41 conformations available to the apo-protein. The isomeric thieno[2,3-d]pyrimidine SGC-STK17B-1N (19g) was identified as a negative control compound. The >100-fold lower activity of 19g on STK17B was attributed to the reduced basicity of its pyrimidine N1.
Graphical Abstract

Introduction
Protein kinases are a large family of enzymes that catalyze the transfer of the γ-phosphate from the cofactor ATP to a serine, threonine, or tyrosine residue in their substrate proteins. Structural studies show that these enzymes contain a β-sheet rich N-lobe and an α-helix rich C-lobe that is connected by a flexible hinge into which ATP is bound.1 Sequence analysis of the human genome has identified over 500 members of the kinase protein family2 that have been organized into nine groups based on sequence similarity. One of these groups is the calcium/calmodulin regulated kinases (CAMK), which contains the death-associated protein kinases (DAPK) family of five serine/threonine kinases: namely, the closely-related DAPK1–3 and the more distant STK17A and STK17B (also known as DRAK1 and DRAK2, respectively).3 The five members of the DAPK family are characterized by a highly conserved N-terminal catalytic domain, but show large differences in their flanking regulatory domains. DAPK1 is a regulator of caspase-dependent and independent cell death. It is necessary for apoptosis and is annotated as a tumor suppressor whose expression is lost in many cancers. DAPK2 and DAPK3 share many of these biological functions as integrators of cell death signaling.
Much less is known about the role of STK17A and STK17B in cell signaling. STK17A expression was associated with an increase in head and neck squamous cell carcinoma through inhibition of TGFβ1-mediated tumor suppression.4 STK17A was also overexpressed in glioblastoma multiforme (GBM) and associated with decreased patient survival. Knockdown of STK17A decreased cellular proliferation, motility, and invasion in multiple GBM cell lines.4 STK17B may also have a role in cancer. It is overexpressed in hepatocellular carcinoma and breast cancer, and silencing of STK17B in cells suggested that it may have utility in treatment of these malignancies.4 Notably, STK17B is abundant in lymphoid organs and predominantly expressed in mature T-cells and B-cells.5 STK17B knock out mice were remarkably resistant to autoimmune challenge when crossed onto a non-obese diabetic mouse strain6 (a model of Type 1 diabetes) or challenged with experimental autoimmune encephalomyelitis7 (a model of Multiple Sclerosis). This autoimmune protection appeared to be driven by reduction in the survival of autoreactive T-cells that were responsible for cell and tissue damage. Thus, STK17B may have a unique role within the DAPK family as an immune regulatory target.
Although STK17B may be an interesting therapeutic target in oncology and autoimmunity, there have been no selective inhibitors available to probe the sequelae of decreasing its catalytic activity in cells or animals. The only reported STK17B inhibitors are either promiscuous, show poor selectivity within the DAPK family, or lack activity in cells.4 Recent efforts in broad screening have surfaced a range of scaffolds that bind to STK17B, but none with all the properties required for a chemical probe. Among the non-selective STK17B inhibitors are quercetin 1, which is a family-wide CAMK inhibitor,7, 8 and the receptor tyrosine kinase inhibitor dovitinib 2, which has micromolar cross-reactivity with both STK17A/B9 (Figure 1). Several other inhibitors have shown up to 20-fold selectivity for STK17B within the DAPK family, but none of them have been reported to be cell active.10-12 Benzofuranone 3 is the only inhibitor with modest STK17B selectivity that was also reported to be active in cells. In a phenotypic assay, 3 protected islet β-cells from apoptosis induced by free fatty acid palmitate.13
Figure 1.

Chemical structures of STK17B inhibitors.
Almost all known kinase inhibitors are heterocycles that are competitive with the cofactor ATP. The targeting of a conserved cofactor binding site can make development of highly selective kinase inhibitors a challenge. Nonetheless, medicinal chemists have been successful in exploiting some distinctive features in the ATP binding site among protein kinases.14, 15 Approaches commonly used to gain selectivity include a) targeting a hydrophobic back pocket that is not penetrated by ATP, but which is accessible in those kinases with a small gate keeper residue (V, T, A, or G); b) exploiting the intrinsic flexibility of the hinge region between the N- and C-lobes that contains several glycine residues; c) development of covalent inhibitors that capture cysteine residues that are found in a subset of kinases; or d) binding into an extended pocket formed when kinases adopt an inactive conformation, called DFG-out, thus blocking ATP from access to the catalytic domain. Using one or more of these approaches, several high-quality chemical probes have been developed for protein kinases.
The kinase N-lobe contains a highly flexible phosphate-binding loop (P-loop; also referred to as the glycine-rich loop) that has received less attention in the development of selective inhibitors. The P-loop makes critical contacts with the γ-phosphate of ATP to support catalysis. It was observed in a kinked conformation in the structure of imatinib with ABL that supported a hydrophobic cage around the drug.16 A rare P-loop conformation was seen in MAP4K4 bound to a selective inhibitor and extended analysis of crystal structures in the protein data bank (PDB) revealed an unusual conformation in 7 additional kinases.17 We now report discovery of a series of potent, selective, and cell active STK17B inhibitors based on the thieno[3,2-d]pyrimidine PFE-PKIS 43 (4), which was uncovered through deep profiling of the kinase inhibitor set known as PKIS2.18 Cocrystal structures of 4 and related thieno[3,2-d]pyrimidines with STK17B uncovered a unique P-loop flip that appears to be the molecular basis of their remarkable potency and selectivity. Optimization of the thieno[3,2-d]pyrimidine chemotype resulted in the first high quality chemical probe for STK17B.
Results
Identification of an STK17B inhibitor:
Several thieno[3,2-d]pyrimidines, previously published as inhibitors of Tpl2 (MAPK3K8, also known as COT),19 were donated by Pfizer to the Structural Genomics Consortium (SGC) kinase chemogenomic set,18 now known as KCGS.20 The compounds were screened at 1 μM through the DiscoverX scanMAX panel of 403 wild type human kinases and three compounds, PFE-PKIS 43 (4), PFE-PKIS 14 (5), and PFE PKIS 9 (6), were identified as narrow spectrum kinase inhibitors with S10(1 μM) ≤ 0.020 (Table 1).18 Full KD determinations performed for all kinases inhibited >75% at 1 μM revealed that 4–6 were potent inhibitors of STK17B with 8–60 fold selectivity over STK17A (Table 1). Notably, 4–6 were each reported to be only weak inhibitors of Tpl2.19 4 was the most potent and selective of the analogs. To probe the molecular basis of its remarkable kinase selectivity a co-crystal structure was obtained with the catalytic domain of STK17B (Figure 2). As expected, 4 occupied the ATP-binding pocket of STK17B (Figure 2A). However, the orientation of the inhibitor was markedly different to the proposed binding mode in Tpl2.19 In the STK17B structure, the carboxylic acid of 4 was directed toward the catalytic lysine (K62) with the hydrophobic 6-aryl substituent extended towards the solvent (Figures 2A and B). Notably, it was the N1 of the thieno[3,2-d]pyrimidine that was observed to bind to the amide backbone of A113 in the hinge, not the N3 as previously proposed for Tpl2.19 In addition to the interaction with K62, the carboxylic acid of 4 formed a second salt bridge with R41 of the P-loop (Figures 2A and B). The salt bridge with R41 was accommodated by a remarkable conformational change in the P-loop, in which R41 had flipped a 180° from its orientation in the structure of STK17B bound to ADP (Figure 2C).21
Table 1.
Thieno[3,2-d]pyrimidines with STK17B activity
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| DiscoverX a |
||||||||
| Entry | Compound | X | R1 | R2 |
scanMAX S10(1 μM) |
STK17A KD (nM) |
STK17B KD (nM) |
Tpl2 b IC50 (nM) |
| 1 | PFE-PKIS 43 (4) | S | SCH3 | H | 0.005 | 230 | 3.8 | 3400 |
| 2 | PFE-PKIS 14 (5) | S | CONH2 | H | 0.020 | 270 | 11 | 1500 |
| 3 | PFE-PKIS 9 (6) | O | ─NHN=C─ | 0.012 | 250 | 30 | 1000 | |
DiscoverX KINOMEscan. scanMAX, S10(1 μM) = number of kinases with 10 % or less of control activity remaining at 1 μM / number of wild type kinases tested (403), n = 1. KD, determined by dose response, n = 2.
Reported by Ni et al (reference 19).
Figure 2.

X-ray structure of the catalytic domain of STK17B. (A) STK17B/4 complex (PDB 6Y6F). STK17B protein backbone is shown in green. The side chains of R41, K62, and the backbone amide of A113 are highlighted. 4 is shown as sticks with atoms colored C pink, N blue, O red, and S yellow. (B) STK17B/4 complex (PDB 6Y6F). Alternate view showing the interaction of the carboxylic acid of 4 with R41 and K62. The unusual conformation of the P-loop is further stabilized by a salt bridge between R41 and E117. (C) STK17B/ADP complex (PDB 6QF4). STK17B protein backbone is shown in light blue. The side chains of R41, K62, E111, E117 and the backbone amide of A113 are highlighted. ADP is shown as sticks with atoms colored C pink, N blue, O red, and P orange.
Although, 4 showed outstanding potency and selectivity as a potential chemical probe for STK17B, additional testing revealed that it had very low stability in liver microsomes. After 30 minutes 100% of the compound was consumed in mice liver microsomes and its t1/2 was determined to be < 5 min (vide infra). To gain insight into the low metabolic stability of 4 its P450 metabolism was modeled by SMARTCyp,22 which identified the sulfur atom of the methyl thioether moiety as the most reactive part of the molecule (SI Figure 1). To improve the metabolic stability of 4 and define the structure activity for STK17B inhibition, we initiated the synthesis of a series of thienopyrimidine analogs targeting three regions of the molecule: the carboxylic acid, the heterocyclic core, and the 6-aryl group.
Synthesis of analogs:
The synthesis 6-arylthieno[3,2-d]pyrimidines 4 and 11a-11ab was adapted from the abbreviated procedures reported by Pfizer (Scheme 1).19 The first step involved the deprotonation and iodination of 4-chlorothieno[3,2-d]pyrimidine (7), resulting in 4-chloro-6-iodothieno[3,2-d]pyrimidine (8) in good yield. Intermediate 8 underwent an SNAr reaction with ethyl 2-mercaptoacetate to obtain 9, which was saponified to 10 in good yield. Compound 10 was a key intermediate that allowed synthesis of 6-arylthieno[3,2-d]pyrimidines 4, 11a–11ac by Suzuki coupling with a range of arylboronic acids. The primary amide 12 was generated by reaction of 4 with ammonia.
Scheme 1 a.
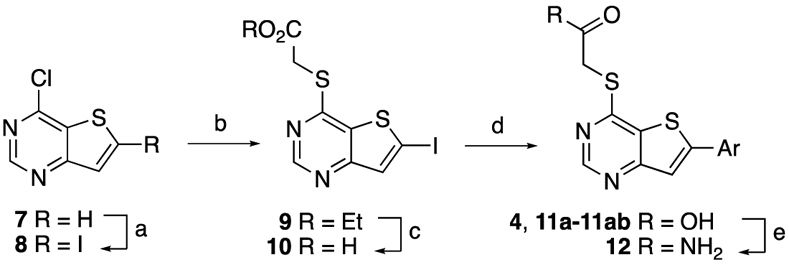
a Reagents and conditions: (a) (i) LDA 1.2 equiv, THF, −78 °C, 20 min, (ii) I2 1.2 equiv, −78 °C, 20 min, rt, 12 h, 68%; (b) (i) ethyl 2-mercaptoacetate 1.5–2.0 equiv, MeOH, 0 °C, (ii) Et3N 3–4 equiv, 0 °C to rt, 12 h, 65%; (c) (i) 1 M LiOH (aq) 2.0 equiv, THF:EtOH 1:1, 70 °C, 1 h, (ii) 1 M HCl (aq) until pH 2 – 3, 0 °C, 80%; (d) (i) ArB(OH)2 1.1 equiv., PdCl2(PPh3)2 5 mol%, K2CO3 2.2 equiv., 1,4-dioxane:water 3:1, 95 °C, 3–12 h, (ii) 1 M HCl (aq) until pH 2 – 3, 0 °C 30-65%; (e) HATU 1.0 equiv, ammonia (aq) excess, N,N-diisopropylethylamine 2.2 equiv, DMF, rt, 12 h, 25%.
Synthesis of side-chain modified analogs (Scheme 2) utilized two additional intermediates that were obtained by the regioselective Suzuki coupling of 8, yielding 4-chloro-6-arylthieno[3,2-d]pyrimidines 13a and 13b. These intermediates were further derivatized by SNAr reaction with ethyl 2-hydroxyacetate and ethyl glycinate, yielding esters 14 and 16a, which were saponified to yield compounds 15 and 16b, respectively.
Scheme 2 a.
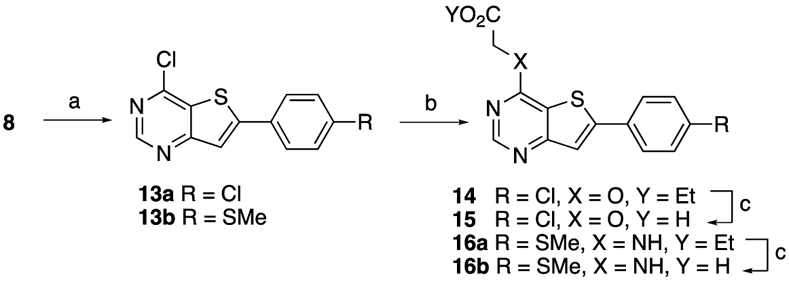
a Reagents and conditions: (a) 4-Chlorophenylboronic acid 1.0 equiv or 4-(methylthio)phenylboronic acid 1.0 equiv, Pd(PPh3)4 5 mol%, NaHCO3 1.0 equiv, CsF 1.0 equiv, 1,4-dioxane:water 3:1, 95 °C, 3 h, 55%; (b) ethyl 2-hydroxyacetate 2.0 equiv, NaH 2.6 equiv. (60% dispersion in mineral oil), anhydrous DMF, 100 °C, 30 min microwave, 45%; or ethyl glycinate hydrochloride 2.5 equiv, TEA 10 equiv, EtOH, 90 °C, 18 h, 78% (c) (i) LiOH 2.2 equiv, THF, 70 °C, 4h, (ii) 1 M HCl (aq) until pH 4-5, 0 °C, 82%.
A series of regioisomeric thieno[2,3-d]pyrimidines were synthesized from the commercially available 6-bromo-4-chlorothieno[2,3-d]pyrimidine (17) (Scheme 3). SNAr reaction with 2-mercaptoacetic acid gave 18 in good yield. Thieno[2,3-d]pyrimidine 17 showed higher reactivity in this substitution reaction than the corresponding thieno[3,2-d]pyrimidine regioisomer 8, which did not react directly with 2-mercaptoacetic acid. Suzuki coupling of 18 with several boronic acids resulted in products 19a–g in moderate yields.
Scheme 3 a.

a Reagents and conditions: (a) (i) 2-mercaptoacetic acid 1.25 equiv, EtOH or CH3CN, 0 °C, (ii) TEA 2.5 equiv, 0 °C to rt, 12 h, (iii) 1 M HCl (aq) until pH 2–3, 0 °C, 75%; (b) (i) ArB(OH)2 1.0 equiv., Pd(PPh3)4 5 mol%, K2CO3 3–4 equiv, 1,4-dioxane:water 3:1, 95 °C, 3 h, (ii) 1 M HCl (aq) until pH 2–3, 0 °C 48-65%.
Structure-activity relationships:
4 and its analogs were initially screened for their STK17A and STK17B activity using a split luciferase KinaseSeeker binding assay23 at a concentration of 1 μM. Any analog showing >70% competition at STK17B was subjected to dose response to determine its IC50 in the binding assay. In addition, all analogs were tested in dose response up to 10 μM by NanoBRET assay24 in HEK293 cells to determine intracellular STK17B target engagement. In the thieno[3,2-d]pyrimidine scaffold A, initial exploration focused on the α-thioacetic acid side chain because of the key interactions observed in the co-crystal structure of 4 with STK17B (Figure 2). In the binding assay 4 was selective for STK17B over STK17A at 1 μM and gave an IC50 of 20 nM on STK17B (Table 2), slightly weaker than the 3.8 nM KD value obtained in the DiscoverX binding assay. 4 also demonstrated robust activity in the live cell NanoBRET assay with an IC50 of 210 nM (Table 2, entry 1). Substitution of −SMe for −Cl in the 6-aryl ring (11a) retained STK17B activity and selectivity in the binding assay (Table 2, entry 2) and showed only a small decrease in activity in cells. The primary amide analog 12 of acid 4 showed a reduced STK17B activity in the binding assay and a lack of activity in cells (Table 2, entry 3). Replacement of sulfur for oxygen in the sidechain in 15 reduced the STK17B activity in the binding assay and the analog was inactive in cells (Table 2, entry 5). Surprisingly, its ester analog 14 had equivalent (albeit low) activity in the STK17B binding assay (Table 2, entry 4), but was also inactive in cells. Double substitution of nitrogen for sulfur and ester for the acid in the side chain resulted in an analog 16a that was devoid of STK17B activity (Table 2, entry 6). The binding affinity for amino acid 16b (Table 2, entry 7) could not be determined due to interference of the compound with the split luciferase assay. However, it was shown to be inactive when screened in cells (IC50 >10 μM). The regioisomeric thieno[2,3-d]pyrimidine analogs 19a and 19b (scaffold B) showed only very low activity in both the STK17A and STK17B binding assays and were inactive in the cell-based assay (Table 2, entries 8 and 9). Overall, these initial results confirmed the importance of the α-thioacetic acid side chain for potent STK17B activity and revealed a rather narrow structure-activity. Only 4 and its 6-chloro analog 11a showed activity in cells, and only one regioisomer of the thienopyrimidine scaffold was active. The sensitivity of the acetic acid side chain to the nature of the α-hetero substituent was surprising, since in COT/Tpl2 substitution of sulfur for oxygen was tolerated,19 although with reduced activity.
Table 2.
Initial SAR
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| KinaseSeeker a | NanoBRET b | ||||||||
| Entry | Compound | Scaffol d |
X | R1 | R2 | STK17 A (% I) |
STK17 B(% I) |
STK17B IC50 (nM) |
STK17B IC50 (nM) |
| 1 | 4 | A | S | OH | SCH3 | 15 | 90 | 20 ± 3.9 | 210 ± 40 |
| 2 | 11a | A | S | OH | Cl | 3 | 90 | 39 ± 8.2 | 680 ± 190 |
| 3 | 12 | A | S | NH2 | SCH3 | 1 | 61 | — | >10,000c |
| 4 | 14 | A | O | OEt | Cl | 4 | 36 | — | >10,000 c |
| 5 | 15 | A | O | OH | Cl | 0 | 36 | — | >10,000 c |
| 6 | 16a | A | NH | OEt | SCH3 | 4 | 0 | — | >10,000 c |
| 7 | 16b | A | NH | OH | SCH3 | d | d | — | >10,000 c |
| 8 | 19a | B | S | OH | SCH3 | 0 | 26 | — | >10,000 c |
| 9 | 19b | B | S | OH | OCH3 | 14 | 18 | — | >10,000 |
KinaseSeeker split luciferase binding assay. % I, inhibition compared to control (DMSO) at 1 μM, n = 2, data ± 10%. IC50, determined by dose response, n = 2 ± SD. — not determined.
Target engagement determined by NanoBRET assay in HEK293 cells, n = 3 ± SE.
n = 1.
not determined due to assay interference.
Since substitution of the 6-aryl ring appeared to be tolerated in STK17B, a second round of analogs sought to explore structure activity in this part of the molecule. Analogs were synthesized in the regioisomeric thienopyrimidines C and D with the α-thioacetic acid functionality in place (Table 3). Analogs in the thieno[3,2-d]pyrimidine scaffold C generally showed good STK17B binding with selectivity over STK17A (Table 3, entries 1–9). Potent activity (>70% inhibition at 1 μM) was seen with electron-donating groups o-OMe, m-SMe, p-SiPr, p-OBn (11b–d, 11j), alkyl groups p-Et, p-vinyl, p-iPr, p-cPr (11e–h) and m,p-dichloro substitution (11i). In the cell based NanoBRET assay the most potent compounds were 11b, 11e-i (Table 3, entries 1, 4–8), which bear substituents in para and meta positions. Surprisingly, analogs 11d and 11j with a p-SiPr and p-OBn substituents were inactive in cells (Table 3, entries 3 and 9) despite having potent binding to STK17B. Compound 11c, substituted in the ortho position, showed moderate activity in cells (Table 3, entry 2). The dichloro analog, 11i showed good activity in cells (Table 3, entry 8). Most notably, 11b, the m-SMe analog of 4, was about 2.5-fold less potent in the STK17B binding and cell assays (Table 3, entry 1). Analogs of the regioisomeric thieno[2,3-d]pyrimidine D (Table 3, entries 10–13) again showed low activity on STK17A and STK17B, and showed no activity against STK17B in cells. This last result confirmed the initial finding in Table 2, which pointed to the lack of STK17B activity in the thieno[2,3-d]pyrimidine scaffold.
Table 3.
SAR of 6-aryl substituents
 | |||||||
|---|---|---|---|---|---|---|---|
| KinaseSeeker a | NanoBRET b | ||||||
| Entry | Compound | Scaffold | Ar | STK17A (% I) |
STK17B (% I) |
STK17B IC50 (nM) |
STK17B IC50 (nM) |
| 1 | 11b | C |  |
12 | 94 | 49 ± 12 | 500 ± 20 |
| 2 | 11c | C |  |
30 | 90 | 110 ± 12 | 1700 ± 400 |
| 3 | 11d | C |  |
0 | 90 | 49 ± 7.9 | >10,000 |
| 4 | 11e | C |  |
0 | 90 | 41 ± 6.9 | 380 ± 62 |
| 5 | 11f | C |  |
8 | 96 | 28 ± 3.4 | 120 ± 6 |
| 6 | 11g | C |  |
0 | 88 | 59 ± 8.0 | 550 ± 100 |
| 7 | 11h | C |  |
0 | 83 | 62 ± 9.0 | 530 ± 46 |
| 8 | 11i | C |  |
0 | 91 | 92 ± 6.7 | 310 ± 30 |
| 9 | 11j | C |  |
0 | 81 | 110 ± 12 | >10,000 |
| 10 | 19c | D |  |
23 | 43 | — | >10,000 c |
| 11 | 19d | D |  |
0 | 18 | — | >10,000 c |
| 12 | 19e | D |  |
6 | 46 | 880 ± 120 | >10,000 c |
| 13 | 19f | D |  |
0 | 3 | — | >10,000 c |
KinaseSeeker split luciferase binding assay. % I, inhibition compared to control (DMSO) at 1 μM, n = 2, data ± 10%. IC50, determined by dose response, n = 2 ± SD. — not determined.
Target engagement determined by NanoBRET assay in HEK293 cells, n = 3 ± SE.
n = 1.
To explore the tolerance for polarity in the 6-aryl ring additional analogs were synthesized in the thieno[3,2-d]pyrimidine scaffold C (Table 4). In the binding assay, these compounds showed robust STK17B activity with good selectivity over STK17A regardless of the type and position of the substituent around the phenyl ring (Table 4, entries 1–7). In the cell-based assay, amides and sulfoxide (11k–m) were inactive (Table 4, entries 1-3). Analogs with a p-Ac (11n) and m-morpholine (11o) showed moderate activity in cells (Table 4, entries 4-5). Interestingly, compound 11p bearing a p-morpholine demonstrated sub-micromolar cell potency (Table 4, entry 6). In contrast, the acetylpyridine 11q was inactive in cells (Table 4, entry 7).
Table 4.
SAR of polar 6-aryl substituents
 | ||||||
|---|---|---|---|---|---|---|
| KinaseSeeker a | NanoBRET b | |||||
| Entry | Compound | Ar | STK17A (% I) |
STK17B (% I) |
STK17B IC50 (nM) |
STK17B IC50 (nM) |
| 1 | 11k |  |
7 | 93 | 23 ± 2.4 | >10,000 c |
| 2 | 11l | 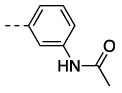 |
24 | 94 | 35 ± 4.5 | >10,000 c |
| 3 | 11m |  |
11 | 95 | 7 ± 0.72 | >10,000 c |
| 4 | 11n |  |
21 | 96 | 13 ± 1.6 | 1500 ± 210 |
| 5 | 11o |  |
20 | 92 | 83 ± 8.4 | 2900 ± 340 |
| 6 | 11p | 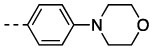 |
37 | 98 | 14 ± 1.1 | 720 ± 60 |
| 7 | 11q |  |
9 | 93 | 37 ± 3.5 | >10,000 c |
KinaseSeeker split luciferase binding assay. % I, inhibition compared to control (DMSO) at 1 μM, n = 2, data ± 10%. IC50, determined by dose response, n = 2 ± SD.
Target engagement determined by NanoBRET assay in HEK293 cells, n = 3 ± SE.
n = 1.
The next series of analogs explored the reduction of electron density in the 6-aryl ring using a thiophene modification or addition of a mildly electron withdrawing group in the para position (Table 5). Two analogs where the 6-phenyl ring was replaced by thiophene (11r) or benzothiophene (11s) (Table 5, entries 1 and 2), yielded compounds with potent and selective STK17B binding and robust activity in cells. In contrast, the more electron negative p-CN (11t) and p-CO2Et (11u) derivatives were less potent in cells (Table 5, entries 3 and 4).
Table 5.
SAR of lipophilic 6-substituents
 | ||||||
|---|---|---|---|---|---|---|
| KinaseSeeker a | NanoBRET b | |||||
| Entry | Compound | Ar | STK17A (% I) |
STK17B (% I) |
STK17B IC50 (nM) |
STK17B IC50 (nM) |
| 1 | 11r |  |
13 | 91 | 63 ± 5.1 | 650 ± 30 |
| 2 | 11s |  |
0 | 93 | 43 ± 3.5 | 190 ± 61 |
| 3 | 11t |  |
13 | 93 | 35 ± 3.4 | 3200 ± 800 |
| 4 | 11u |  |
1 | 95 | 40 ± 5.7 | 870 ± 360 |
KinaseSeeker split luciferase binding assay. % I, inhibition compared to control (DMSO) at 1 μM, n = 2, data ± 10%. IC50, determined by dose response, n = 2 ± SD.
Target engagement determined by NanoBRET assay in HEK293 cells, n = 3 ± SD.
For the final series of analogs, the effect of fluorine substitution on the 6-aryl ring was explored with a goal to identify modifications that would be less prone to P450 oxidation (Table 6). The mono- and difluorophenyl analogs (11v–x) were active in the binding assay but showed only modest activity in cells (Table 6, entries 1–3). Reintroduction of the p-SMe substituent in 11y resulted in a boost in cell activity (Table 6, entry 4). The p-OMe analog 11z (Table 6, entry 5) showed a 1.3-fold drop in cell potency. Compounds 11aa and 11ab bearing a trifluoromethyl and a trifluoromethoxy substituent respectively, were found to have good binding to STK17B and sub-μM cell potency (Table 6, entries 6–7).
Table 6.
SAR of fluorine substituted 6-aryl substituents
 | ||||||
|---|---|---|---|---|---|---|
| KinaseSeeker a | NanoBRET b | |||||
| Entry | Compound | Ar | STK17A (% I) |
STK17B (% I) |
STK17B IC50 (nM) |
STK17B IC50 (nM) |
| 1 | 11v |  |
8 | 94 | 49 ± 7.8 | 980 ± 410 |
| 2 | 11w |  |
22 | 90 | 60 ± 17 | 1200 ± 370 |
| 3 | 11x |  |
1 | 83 | 74 ± 19 | 820 ± 100 |
| 4 | 11y |  |
0 | 93 | 27 ± 5.1 | 530 ± 450 |
| 5 | 11z |  |
0 | 94 | 18 ± 2.6 | 700 ± 260 |
| 6 | 11aa |  |
0 | 92 | 33 ± 5.2 | 340 ± 40 |
| 7 | 11ab |  |
13 | 75 | 280 ± 25 | 500 ± 100 |
KinaseSeeker split luciferase binding assay. % I, inhibition compared to control (DMSO) at 1 μM, n = 2, data ± 10%. IC50, determined by dose response, n = 2 ± SD.
Target engagement determined by NanoBRET assay in HEK293 cells, n = 3 ± SD.
In summary, the structure-activity study identified a series of 6-arylthieno[3,2-d]pyrimidine-4-thioacetic acids with good binding selectivity for STK17B over STK17A. Analogs with the best cell potency as measured by NanoBRET target engagement were often substituted on the 6-aryl group with small para-substituents, which could be either lipophilic (4, 11e, 11f, 11g, 11h, 11i, and 11y) or mildly electron withdrawing (11a, 11u and 11aa). In addition, analogs with thiophene (11r), benzothiophene (11s), p-morpholinophenyl (11p), or m-trifluromethoxyphenyl (11ab) showed good cell potency.
Metabolic stability:
Since initial evaluation of 4 had shown it to possess poor metabolic stability, likely due to P450 metabolism of the p-SMe substituent, a series of the cell active 6-arylthieno[3,2-d]pyrimidines were selected for metabolic stability studies. In parallel, the compounds were tested for aqueous solubility and their LogD at pH 7.0 was calculated. Table 7 shows the results of this study with compounds sorted by their metabolic stability after 30 min of incubation in mouse liver microsomes. 4 (Table 7, entry 1), which showed no compound remaining after a 30 min incubation in mouse liver microsomes, had a calculated LogD in the drug-like range25 and modest aqueous solubility. Replacement of p-SMe by p-CO2Et (11u) did not improve metabolic stability (Table 7, entry 2). Addition of a m-F substituent (11y) to the ring resulted in a small improvement in metabolic stability (Table 7, entry 3). Removal of the p-SMe substituent further improved metabolic stability as seen with 11c, 11g, 11f, 11w, and 11r (Table 7, entries 4–8). 11p, 11s, 11h and 11o bearing p-morpholino, benzothiophene, p-cyclopropyl, and m-morpholino group, respectively, showed even greater metabolic stability, each with >50% of the compound remaining after 30 min (Table 7, entries 9–12). The fluorinated analogs 11z, 11aa, and 11x showed the best metabolic stability in the series (Table 7, entries 13–15). None of the analogs showed improved aqueous solubility over 4 despite changes in their calculated LogD. The benzothiophene 11s had lower solubility, but this did not affect its activity in cells.
Table 7.
Microsomal stability and physiochemical properties
 | |||||
|---|---|---|---|---|---|
| Entry | Compound | Ar | Microsomal stability (%) b |
Aq. solubility (μg/mL) b |
cLogD c |
| 1 | 4 |  |
0 | 42 | 1.27 |
| 2 | 11u |  |
0 | 59 | 0.90 |
| 3 | 11y |  |
18 | 52 | 1.28 |
| 4 | 11c |  |
24 | 69 | 0.33 |
| 5 | 11g |  |
35 | 23 | 1.95 |
| 6 | 11f |  |
38 | 74 | 1.41 |
| 7 | 11w | 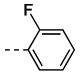 |
45 | 51 | 0.77 |
| 8 | 11r |  |
47 | 56 | 0.33 |
| 9 | 11p |  |
63 | 61 | 0.61 |
| 10 | 11s |  |
63 | 5 | 1.65 |
| 11 | 11h |  |
66 | 55 | 1.49 |
| 12 | 11o |  |
73 | 54 | 0.60 |
| 13 | 11z |  |
88 | 49 | 0.47 |
| 14 | 11aa |  |
88 | 57 | 1.49 |
| 15 | 11x |  |
95 | 65 | 0.72 |
% compound remaining in mouse liver microsomes at 30 min.
Kinetic solubility at pH 7.4.
Calculated LogD at pH 7.0.
Selection of a chemical probe:
After consideration of the binding, cell potency, and metabolic stability data 11s was selected as a potential chemical probe. Despite its lower kinetic aqueous solubility (5 μg/mL), 11s demonstrated the highest cell potency in the NanoBRET target engagement assay (IC50 = 190 nM) among the analogs that showed <50% metabolism in the liver microsome assay. To further characterize its kinome-wide selectivity, 11s was profiled at 1 μM across the DiscoverX scanMAX panel of 403 wild type human kinase assays (SI File 1). Eight kinases, including STK17B, were annotated with >75% displacement of the immobilized ligand at 1 μM (Table 8). KD determinations were performed by dose response for these eight kinases plus STK38L (71% I at 1 μM) and STK17A (27% I at 1 μM). 11s gave a KD = 5.6 nM on STK17B in the DiscoverX binding assay, but showed some cross-activity with CAMKK2, CAMKK1, AURKB, STK17A, and PIM2. Annotations for MET, NEK6, WEE1, and STK38L were shown to be false positives in the initial scanMAX since they did not confirm in the dose response experiment. To further explore the selectivity profile of 11s, enzyme inhibition assays were performed in the presence of ATP for all 10 kinases annotated in the initial scanMAX experiment. In the enzyme assays 11s showed potent inhibition of STK17B with an IC50 = 34 nM and >100-fold selectivity over STK17A. It was also >100-fold selective over all of the other kinases tested, with only STK17A, CAMKK1, and AURKB generating a measurable dose-response (IC50 5–9 μM). Robust inhibition was not seen on CAMKK2 in the enzyme assay despite its activity in the DiscoverX binding assay. As a final confirmation of the kinase selectivity of 11s, live cell NanoBRET assays were run on STK17A, CAMKK2, AURKB, and STK38L (Table 8). 11s showed an IC50 = 2.4 μM on CAMKK2 but >10 μM on the other kinases. Thus, 11s is a potent STK17B inhibitor with remarkable selectivity over STK17A across multiple assay formats. It had outstanding kinome-wide selectivity, with cross-reactivity on CAMKK1 and CAMKK2 as the only potential confounding activities noted in the binding assays. Importantly, 11s is the most potent and selective cell active STK17B inhibitor reported to date and will be highly useful as a chemical probe for this dark kinase.
Table 8.
Kinase selectivity of chemical probe (11s)
| KINOMEscan a | Enzyme b | NanoBRET c | ||
|---|---|---|---|---|
| Kinase | % I | KD | IC50 (nM) | IC50 (nM) |
| MET | 100 | >10,000 | >10,000 | — |
| NEK6 | 100 | >10,000 | >10,000 | — |
| PIM2 | 100 | 1500 | >10,000 | — |
| WEE1 | 100 | >10,000 | >10,000 | — |
| STK17B | 99 | 5.6 | 34 | 190 |
| CAMKK2 | 92 | 21 | >10,000 | 2,400 |
| AURKB | 86 | 110 | 8900 | >10,000 |
| CAMKK1 | 86 | 49 | 5200 | — |
| STK38L | 71 | >10,000 | >10,000 | >10,000 |
| STK17A | 27 | 440 | 4700 | >10,000 |
DiscoverX KINOMEscan. % I, scanMAX % inhibition of control at 1 μM, n = 1. KD, determined by dose response, n = 2.
IC50 for enzyme inhibition at Km ATP determined at Eurofins, n = 2.
Target engagement IC50 determined by NanoBRET assay in HEK293 cells. — not determined.
Identification of a negative control analog:
To augment the utility of 11s as a chemical probe, a close analog was designed as an inactive control26 (Table 9). Building on the structure activity data that showed the regioisomeric thieno[2,3-d]pyrimidines were inactive as STK17B inhibitors, the analog 19g was synthesized (Scheme 3). When profiled across the DiscoverX scanMAX panel at 1 μM no kinase was identified where 19g showed >75% displacement of the immobilized ligand (SI File 1). However, 19g did displace >65% of the immobilized ligand on 5 kinases. KD determinations indicated that 4 of these were false positives, and only HIPK4 gave a measurable KD = 290 nM. In addition, KD on STK17B and STK17A were measured as 0.91 μM and 9.8 μM, respectively. In the enzyme inhibition assay 19g had an IC50 = 4.8 μM on STK17B and IC50 >10 μM on STK17A. 19g was, therefore, >100-fold less active as an inhibitor of STK17B than the chemical probe 11s (Table 9). Furthermore, in cells 19g was inactive at 10 μM in the STK17B NanoBRET cellular target engagement assay. Based on these activity data, and its chemical similarity to the chemical probe, 19g is a suitable negative control for use in biological assays.
Table 9.
STK17B potency and relative basicity of chemical probe (11s) and negative control (19g)
 | ||
|---|---|---|
| STK17B: | ||
| Binding KD (nM) a | 5.6 | 910 |
| Enzyme IC50 (nM) b | 34 | 4,800 |
| Cell IC50 (nM) c | 190 | >10,000 |
| Basicity: | ||
| pKa (N1) d | 2.8 | 0.1 |
| pKa (N3) d | 1.1 | 0.5 |
KD, determined by dose response at DiscoverX, n = 2.
IC50 for enzyme inhibition at Km ATP determined at Eurofins, n = 2.
Target engagement IC50 determined by NanoBRET assay in HEK293 cells, n = 3.
pKa was obtained from density functional theory (DFT) calculations using the molecular electrostatic potential as the descriptor.
The relative inactivity of all thieno[2,3-d]pyrimidine analogs on STK17B was somewhat surprising. Specifically, the >100-fold decreased STK17B activity of thieno[2,3-d]pyrimidine 19g compared to thieno[3,2-d]pyrimidine 11s was not easily explained by a simple analysis of their shape complementarity within the ATP binding pocket of STK17B (data not shown). To further understand the molecular basis of the dramatic difference in activity between the regioisomers, density functional theory (DFT) calculations were performed to evaluate the molecular electrostatic potential as the descriptor of protonation propensity and predict the pKa value for the respective heterocycles.27, 28 A benchmark calculation was first performed on the four regioisomers of thienopyridine. The calculated pKa values were in very close agreement with the potentiometric pKa values reported in the literature (SI Table 1),29 highlighting the dramatically lower basicity of the thieno[2,3-b]pyridine isomer. When applied to the thieno[3,2-d]pyrimidine 11s, DFT calculated that N1 was more basic than N3 (Table 9). However, for thieno[2,3-d]pyrimidine 19g both nitrogens were found to be much less basic and the relative basicity was reversed between N1 and N3. Remarkably, thieno[3,2-d]pyrimidine N1 of the chemical probe 11s was calculated to be 500-fold more basic than thieno[2,3-d]pyrimidine N1 of the negative control 19g. In the active thieno[3,2-d]pyrimidine series, N1 forms the key H-bond with the hinge motif of the ATP-binding pocket. Our computational results suggest that N1 in the negative control thieno[2,3-d]pyrimidine series may lack sufficient basicity to support a similar strength of molecular recognition.30
Structural studies:
The STK17B chemical probe 11s demonstrated remarkable kinase selectivity for an ATP-competitive inhibitor. Most notably, the amino acids lining the ATP-binding pocket of STK17A are 100% identical except for a conservative mutation of K37 (STK17B) to R65 (STK17A) on the outside edge of the solvent accessible region. To explore whether the unusual conformation flip of the P-loop seen in the 4/STK17B cocrystal structure (Figure 2) could contribute to kinase selectivity, the cocrystal structures of STK17B in complexes with the chemical probe 11s and close analog 11h were solved. The structures of 11s/STK17B and 11h/STK17B revealed electron density for the P-loop in two distinct conformations (Figure 3 and SI Figure 2). In the folded conformation A, the P-loop resembled that seen in the 4/STK17B structure, in which R41 formed a salt bridge with the carboxylic acid of the inhibitor (Figure 3B). In the extended conformation B, the P-loop was in an orientation with R41 rotated away from the inhibitor binding pocket similar to that observed in the ADP/STK17B structure (Figure 3C). Detailed inspection of the 11s/STK17B structure revealed only a single moderate (3.0 Å) H-bond interaction between the backbone amide of A113 in the hinge region and N1 of the thieno[3,2-d]pyrimidine inhibitor. In folded conformation A, the carboxylic acid of 11s was held in a sandwich of salt bridges with R41 of the P-loop and the catalytic lysine (K62), and also made tight (2.5 Å) H-bonds to a water molecule and an ethylene glycol solvent molecule (Figure 3B). The 6-benzothiophene substituent sat in a hydrophobic pocket that was open to the solvent and appeared to derive most of its binding affinity through shape complementarity. The phenol of Y112 and terminal amine of K37 were the only polar residues in this region but did not interact with 11s. Y112 also did not appear to form a π-stacking interaction with the inhibitor.
Figure 3.

X-ray structure of the catalytic domain of STK17B. (A) STK17B/11s complex (PDB 6Y6H). STK17B protein backbone is shown with the P-loop conformation A in green and the P-loop conformation B in light blue. The side chains of R41, K62, and the backbone amide of A113 are highlighted. 11s is shown as sticks with atoms colored C pink, N blue, O red, and S yellow. (B) STK17B/11s complex (PDB 6Y6H) conformation A showing the interaction of 11s carboxylic acid with R41 and K62. Conformation A of the P-loop is further stabilized by a salt bridge between R41 and E117. (C) STK17B/11s complex (PDB 6Y6H) conformation B showing the interaction of 11s carboxylic acid with only K62 and A113. (D) STK17B/quercetin complex (PDB 3LM5). STK17B protein backbone is shown in light blue. The side chains of R41, E80, D179 and the backbone amides of E111 and A113 are highlighted. Quercetin is shown as sticks with atoms colored C pink and O red. (E) STK17B/dovitinib complex (PDB 7AKG). STK17B protein backbone is shown in light blue. The side chains of R41, K62 and the backbone amides of E111 and A113 are highlighted. Dovitinib is shown as sticks with atoms colored C pink, N blue, F pale blue and O red.
The STK17B cocrystal structures of two non-selective DAPK family inhibitors, quercetin and dovitinib (Figure 3 and SI Table 2) were also solved. Quercetin is a broad spectrum inhibitor of the CAMK group,8 while dovitinib inhibits the DAPK family with IC50 in the 1–5 μM range.7 Quercetin formed H-bonds to several STK17B residues including the hinge region, D179 of the DFG motif, and E80 of the αC helix. A similar network of H-bonds was observed in the cocrystal structure of quercetin with DAPK1 (PDB 5AUW).4 For dovitinib, the only polar interaction with STK17B were two H-bonds from the pyridinone to E111 and A113 of the hinge region (2.9 and 2.6 Å, respectively). Notably, in both the quercetin/STK17B (Figure 3D) and dovitinib/STK17B (Figure 3E) complexes the P-loop exhibited extended conformation B where neither inhibitor interacted with R41, which was rotated away from the inhibitor pocket. Thus, only in the cocrystal structures with thieno[3,2-d]pyrimidine-4-thioacetic acids 4, 11s, and 11h was the P-loop observed in folded conformation A.
Molecular dynamics:
The folded P-loop conformation of STK17B seen with the thieno[3,2-d]pyrimidines-4-thioacetic acids 4, 11s, and 11h provided an explanation for their remarkable kinome-wide selectivity. However, the near 100-fold selectivity over STK17A was still surprising given the high sequence identity of its kinase domain including the critical P-loop. A docking study with 11s was performed to probe the origin of the STK17B/A selectivity in more detail. Since no X-ray crystal structure of STK17A was available, a homology model was generated with the 3D structure comparative modeling software MODELLER31 using the structure of STK17B/11s (PDB: 6Y6H) as a template (SI File 5). The resulting homology model, which directly compared the binding sites of STK17A and STK17B in complex with the chemical probe 11s, showed that the identity and conformation of the ATP-binding site residues contacting the inhibitor were nearly identical (SI Figure 4). The only difference observed in the model was the conservative change of STK17B K37 to STK17A R16 at the solvent accessible mouth of the pocket, which lies 3.1Å from the terminal benzothiophene ring of 11s. Since multiple analogs of 11s with a range of 6-substituents also showed similar STK17B/A selectivity (e.g. 4–6 in Table 1), we concluded that residues outside of the primary binding pocket must be responsible.
To explore the possibility that residues outside of the ATP-binding pocket could influence the conformational flexibility of the P-loop and explain the remarkable potency and selectivity of 4 and 11s, we performed molecular dynamics (MD) simulations of STK17B in the apo and inhibitor bound forms starting from R41 in both A and B conformations. Microsecond timescale simulation (1.25 μs) of the apo-STK17B with R41 in folded conformation A revealed exceptional flexibility of the P-loop. Starting from folded conformation A, after 1 μs, R41 switched to extended conformation B (Figure 4A, supplementary movie 1). This switch was characterized by an intermediate state (not observed in crystal structures) in which R41 formed a transient network of hydrogen bonds with E117, S120 and E125 in the region encompassing the helix αD as well as packing against the aromatic ring of F119 (Figure 4A). In the process, the distance between E125 and R41 was reduced from 17.1 Å in the crystal structure to within H-bond distance in the intermediate state. On the other hand, 1.25 μs long simulation of the apo-STK17B with R41 starting in extended conformation B did not show a switch back to folded conformation A. Instead R41 maintained the salt bridge with E38, as observed in the crystal structure (Figure 4B, supplementary movie 2).
Figure 4.

MD simulation of apo-STK17B (PDB 6Y6H). R41 and key residues forming interactions are shown as sticks and colored in magenta and salmon, respectively. (A) R41 starting in conformation A. After 1000 ns of the simulation, R41 flips to conformation B. (B) R41 starting in conformation B, where it remains throughout the simulation.
In the inhibitor bound forms, both 4 and 11s stayed bound to the protein during the 250 ns simulation time scale (Figure 5A and SI Movies 3-5). The results showed that 4 adopted two distinct conformations, due to twisting of the thienopyrimidine substituents, as shown by the increase in root mean square deviation (RMSD) at around 85 ns (Figure 5A, upper panel and SI Movie 3) while 11s adopted only a single distinct conformation (Figure 5A, middle and lower panels and SI Movies 4 and 5). In STK17B folded conformation A, the inhibitors formed more hydrogen bonds with the protein (Figure 5B, top and middle panels) than in STK17B extended conformation B (Figure 5B, lower panel). The higher number of hydrogen bonds can be attributed to the interaction of R41 with the inhibitors (Figure 5C, top and middle panels). In STK17B extended conformation B, 11s adopted only a single inhibitor conformation in which it was bound to K62 of the catalytic pocket (SI Movie 5) despite the lack of interaction with R41 (Figure 5C, lower panel). Thus, 11s was bound stably within the ATP-pocket independent of the interaction with R41. However, the additional salt bridge between the inhibitor and R41 in folded conformation A of the P-loop would be expected to increase the enthalpy of binding.
Figure 5.

MD simulation of inhibitor bound STK17B (PDB 6Y6F and 6Y6H). (A) RMSD plot of ligands (4 or 11s) during the course of simulation. GROMACS 2018.1 built-in tool was used to calculate RMSD of the ligand after least square fit using the Cα atoms. (B) Total number of hydrogen bonds between the ligand and STK17B during the simulation. (C) Total number of hydrogen bonds between the ligand and R41 during the simulation.
Discussion
Despite the track record for development of kinase inhibitors as drugs for oncology, inflammation, and fibrosis, the majority of human kinases lack selective inhibitors that can be used as chemical probes to study their pharmacology.18, 32 Many of these dark kinases are known to play an important role in cell signaling as shown by genetic studies.33 STK17B is one such example. The phenotype of knock out mice shows that loss of STK17B protects against autoimmune challenge.5, 34, 35 However, prior to our work no potent, cell active, selective STK17B inhibitor was available to study the role of this dark kinase in immune cell signaling.4
ATP-competitive kinase inhibitors generally show cross-activity with multiple kinases beyond their primary target. As a community project to create a comprehensive collection of inhibitors for every human kinase, we sought donations of molecules from pharmaceutical companies and academic investigators.18, 32 The output of this project, known as the Kinase Chemogenomic Inhibitor Set (KCGS) currently contains selective inhibitors of over 200 kinases.20 The identification 4 as a potent and selective inhibitor of STK17B is a powerful example of this approach. Several thieno[3,2-d]pyrimidines, originally synthesized as inhibitors of the kinase COT/Tpl2, 19 were donated by Pfizer to the project. Deep profiling of these molecules through the DiscoverX scanMAX panel identified activity on STK17B that was generally 100 times greater than on the original Tpl2 target.
The results of a structure-activity study showed the critical role of the α-thioacetic acid for potent activity on STK17B. Replacement of the sulfur with nitrogen was not tolerated and even the switch to oxygen led to a loss in STK17B activity. In contrast, the switch of sulfur for oxygen was tolerated in COT/Tpl2.19 Notably the orientation of 4 was flipped 180° compared to the proposed binding mode in Tpl2. A cocrystal structure of 4 with STK17B revealed the key role of the carboxylic acid in binding to the catalytic pocket of the kinase. The carboxylic acid was sandwiched between two residues; R41 from the P-loop and the catalytic lysine K62. Interaction with the catalytic lysine is not uncommon with polar or acidic kinase inhibitors,36, 37 but the salt bridge with R41 was remarkable, given that it required a large conformational change in the P-loop. This conformational change was also seen in the cocrystal structure of the chemical probe 11s with STK17B. Could the P-loop flip explain the remarkable selectivity of thieno[3,2-d]pyrimidines 4 and 11s for STK17B over its close neighbor STK17A?
Several lines of evidence support the critical role of the P-loop in the kinase selectivity of the thieno[3,2-d]pyrimidines. First, the P-loop is known to be one of the most flexible regions of the kinase catalytic domain and has been implicated previously in selective inhibition of ABL and MAP4K4.16, 17, 38 Likewise, molecular dynamics simulation showed that apo-STK17B can easily access both folded conformation A and extended conformation B of the P-loop. Second, quercetin and dovitinib, which bind with P-loop conformation B in their STK17B structures, show no selectivity over STK17A. The cofactor ADP also crystallized in P-loop conformation B with the position of the ribose such that it would block R41 from adopting folded conformation A (Figure 2C). Finally, all of the residues lining the ATP-binding pocket are identical between STK17A and STK17B making it even more likely that the remarkable selectivity of the thieno[3,2-d]pyrimidine inhibitors is driven by differences in conformational flexibility of the kinases. STK17A and STK17B differ in regions outside of the ATP binding region that might impact inhibitor specificity by altering the conformational flexibility of R41 in the P-loop, as revealed in the molecular dynamics simulation. In particular, E125 and L126 in STK17B are substituted to an aspartate (D153) and arginine (R154), respectively (SI Figure 3). These STK17A specific variations are expected to destabilize the intermediate R41 conformation observed in STK17B (Figure 4A). Thus, while the thieno[3,2-d]pyrimidines can access both conformations, it is only in the unique folded conformation A that the salt bridge with R41 would provide increased binding affinity.
The cocrystal structure of the thieno[3,2-d]pyrimidine-4-α-thioacetic acids 4, and 11s with STK17B showed a single 3.0 Å H-bond interaction between the pyrimidine N1 with the backbone NH of A113 in the hinge loop, which is in the range of distances typically seen for Type I kinase inhibitors.39, 40 The observation of only a single H-bond suggests it is critical for molecular recognition of the thieno[3,2-d]pyrimidines by the STK17B hinge region. Notably, all of the regioisomeric thieno[2,3-d]pyrimidine-4-α-thioacetic acids, including the negative control 19g, were inactive as inhibitors of STK17B and almost all other kinases (SI File 1). The shape of the ATP-binding pocket in STK17B provides no insight as to why the regioisomers showed such dramatic differences in activity. A more plausible explanation for the divergent activity of the thieno[3,2-d]pyrimidine- and thieno[2,3-d]pyrimidine-4-α-thioacetic acids in STK17B was revealed by analysis of their electrostatic potentials. DFT calculations based on the molecular electrostatic potential of the heterocycles unveiled dramatic differences between the regioisomers as reflected by their predicted pKa values. It was already documented that related thienopyridine regioisomers showed differences in their measured pKa values; thieno[2,3-b]pyridine has a much lower pKa compared to the other three regioisomers due to the inductive effect of sulfur adjacent to the pyridine nitrogen.29 Likewise, in the thieno[2,3-d]pyrimidine 19g electron density on N1 was much lower than in thieno[3,2-d]pyrimidine 11s and there was a 500-fold difference in the calculated pKa. The extremely weak basicity of N1 in the thieno[2,3-d]pyrimidine regioisomers may decrease their ability to be recognized by STK17B. Notably, there are some thieno[2,3-d]pyrimidines that have been reported as kinase inhibitors,41 and in one case a thieno[2,3-d]pyrimidine-α-thiopropanoic acid was a potent inhibitor of CSNK2. However, it was also found that changing the salt concentration was sufficient to invert this inhibitor’s binding pose such that N1 no longer interacted with the hinge.42 Thus, while not all thieno[2,3-d]pyrimidines are devoid of kinase activity,41 in the case of STK17B it appears that the more basic thieno[3,2-d]pyrimidine regioisomers are much more active as inhibitors. Importantly, the identification of an inactive analog (19g) that is a simple regioisomer of the chemical probe (11s) will be useful to control for off-target activity in the study of STK17B biology.26
The 6-aryl group on the thieno[3,2-d]pyrimidine sits in the region of the ATP-pocket that reaches towards solvent. Structure-activity studies confirmed that substitution of this ring was tolerated and could be used to optimize the cell activity and increase the metabolic stability of the series. Analogs with small lipophilic substituents on the para-position demonstrated some of the best cell activity, whereas the ortho- and meta-substituents were much less active. The larger thiophene and benzothiophene analogs (11r and 11s) also showed potent cell activity. The most surprising result was seen with the para-morpholine substituent (11p), which had robust cell activity. In contrast, substituents that added an H-bond acceptor or donors were generally less active, as were substitutions that were electron withdrawing (e.g., −CN and −CO2Et). Our ability to measure STK17B target engagement in cells by NanoBRET assay24 was critical in being able to navigate the subtle balance of lipophilicity and polarity required to offset the intrinsic charge of the carboxylic acid. It is notable that none of the acidic thieno[3,2-d]pyrimidines previously optimized as COT/Tpl2 inhibitors were reported to be active in cells.19
Modification of the 6-aryl substituent also addressed the poor metabolic stability of 4. The p-SMe group was a likely site of oxidative metabolism, being predicted to be a P450 hot spot by the SMARTCyp algorithm. Replacement of the −SMe moiety with groups less prone to oxidation yielded analogs with much improved stability in liver microsomes. From the series, we selected 11s as the analog with the best properties to be a high-quality chemical probe,26 given its balance of potency, selectivity, cell activity, and metabolic stability. 11s is a nanomolar STK17B inhibitor that has 100-fold selectivity over other DAPK family members including STK17A. It shows robust target engagement of STK17B in cells at concentrations below 1 μM and has good stability in liver microsomes. The modest aqueous solubility does not impact its use in cell culture experiments.
Like other members of the thieno[3,2-d]pyrimidine series, 11s showed outstanding selectivity across a panel of over 400 human kinases. However, two kinases that emerged as a potential off-targets were CAMKK1 and CAMKK2. The chemical probe showed <10-fold selectivity over these kinases in the DiscoverX binding assay, although the selectivity window was greater in enzyme inhibition and cell-based NanoBRET assays (Table 8). Both the DAPK and the CAMKK families belong to the CAMK branch of the kinome. Although sequence analysis of their ATP-binding pockets predicted that cross-reactivity of Type I inhibitors would not be seen,14 our recent chemocentric review highlights carboxylic acid containing compounds GSK650394 and STO6090 as potent CAMKK2 inhibitors43 suggesting that there are commonalities in the ATP-binding pockets that might support the binding of acidic kinase inhibitors. Using NanoBRET target engagement assays 11s showed >10-fold selectivity for STK17B over CAMKK2 in cells. Thus, the cumulative binding, enzyme inhibition, and target engagement data (Table 8) indicate that at a 1 μM concentration 11s will be a selective STK17B inhibitor in cell-based assays.
Conclusions
SGC-STK17B-1 (11s) has been identified as a high-quality chemical probe for the dark kinase STK17B that demonstrated sub-micromolar cellular activity and remarkable selectivity over other kinases including STK17A. The thieno[3,2-d]pyrimidine-4-α-thioacetic acid chemotype bound to STK17B in a unique conformation that sandwiched the inhibitors between the catalytic lysine and the P-loop arginine. The kinase selectivity demonstrated by these inhibitors was likely due to interaction with unique conformational states sampled by STK17B, which allowed access to the folded P-loop observed in multiple X-ray co-crystal structures. The public availability of SGC-STK17B-1 (11s) as the first truly potent and selective chemical probe of STK17B and its negative control analog SGC-STK17B-1N (19g) will aid in the illumination of the cell biology of this dark kinase.
Experimental Section
General Chemistry.
Solvents were used as ACS or HPLC grade. Anhydrous tetrahydrofuran (THF), lithium diisopropylamide 2.0M solution (THF/heptane/ethylbenzene), commercial reagents, and catalyst were used as received. For air sensitive reactions, the glassware was oven dried and cooled under nitrogen. Low-temperature baths (−78 °C) were made from dry ice and 2-propanol. All the reactions were carried out under positive pressure of N2. NMR spectra were recorded as CDCl3 and DMSO-d6 solutions on a 400, 500 or 700 MHz instrument. The 1H NMR chemical shits are reported as δ values in parts per million (ppm) relative to tetramethylsilane (TMS, δ = 0.00). The residual chloroform signal, CHCl3 (δ = 7.26) was used as reference and for DMSO signal (quintet, centerline δ = 2.50) was used as reference. 13C NMR chemical shifts are reported as δ values in parts per million (ppm) relative to TMS and DMSO signal (septet, centerline δ = 39.52) as reference. Analytical thin layer chromatography (TLC) was performed on silica gel plates, 200 μm with F254 indicator, visualization was accomplished by UV light (254/365 nm) or 3% ethanol solution of phosphomolybdic acid. Flash column chromatography was performed with 50 μm irregular silica or 60 μm spherical silica prepacked cartridges. Yields are reported as pure material after isolation by column chromatography or recrystallization. Analytical LC/MS data was obtained using a Waters Acquity Ultra performance liquid chromatography (UPLC) system equipped with a photodiode array (PDA) detector running acetonitrile/water gradient. Samples for high-resolution mass spectrometry (HRMS) were analyzed with a ThermoFisher Q Exactive HF-X mass spectrometer coupled with a Waters Acquity H-class liquid chromatograph system. Samples were introduced through a heated electrospray source (HESI) at a flow rate of 0.6 mL/min. Electrospray source conditions were set as: spray voltage 3.0 kV, sheath gas (nitrogen) 60 arb, auxiliary gas (nitrogen) 20 arb, sweep gas (nitrogen) 0 arb, nebulizer temperature 375 °C, capillary temperature 380 °C, RF funnel 45 V. The mass range was set to 150–2000 m/z. All measurements were recorded at a resolution setting of 120,000. All compounds were > 95% pure by analytical LC.
4-Chloro-6-iodothieno[3,2-d]pyrimidine (8).
An oven dried 250 mL three neck round-bottom flask provided with a pressure-equalizing addition funnel, was charged with 4-chlorothieno[3,2-d]pyrimidine 7 (1.0 equiv, 20.0 mmol, 3.40 g) was added under positive N2 pressure, followed by the addition of anhydrous THF (65 mL). Solution was cooled down to −78 °C, LDA (2.0M, 1.2 equiv, 24.0 mmol, 12.0 mL) was added portion wise over 2–3 minutes, and the mixture was stirred for 20 min at −78 °C. A solution of I2 (1.2 equiv, 24.0 mmol, 6.08 g) in anhydrous THF (15 mL) was added over 10 min using the equalizing addition funnel. This mixture was stirred for 30 min at −78 °C, allowed to warm up to room temperature over the curse of 1 h then stirred for 2 more hours at this temperature. It was quenched with saturated NH4Cl(aq)/ice water, CHCl3 added (200 mL), stirred, layers separated. The aqueous layer was extracted with CHCl3 two times, organic layers were combined and rinsed with aq. Na2S2O3 two times and brine two times, dried over Na2SO4, concentrated in vacuo, and purified over silica gel with hexanes/EtOAc in a gradient up to 100% of the polar solvent giving a clear brown solid (4.5 g, 65%). 1H NMR (400 MHz, CDCl3) δ 8.90 (s, 1H), 7.84 (s, 1H).
Ethyl 2-((6-iodothieno[3,2-d]pyrimidin-4-yl)thio)acetate (9).
In a 25 mL round bottom flask provided with a rubber septum, a magnetic stir bar, and under positive N2 pressure, 4-chloro-6-iodothieno[3,2-d]pyrimidine 8 (1.0 equiv, 3.5 mmol, 1.05 g) was suspended in EtOH (6.0 mL) at 0°C (ice bath), followed by the dropwise addition of ethyl 2-mercaptoacetate (1.6 equiv, 5.6 mmol, 0.62 mL) and triethylamine (3.2 equiv, 11.2 mmol, 1.6 mL). The reaction mixture was stirred overnight with slow warm up from 0 °C to room temperature. Dichlomethane was added to the reaction crude, solvents evaporated in vacuo, and purified over silica gel with hexanes/EtOAc in a ramp up to 15% EtOAc (the main by-product of this reaction is the des-iodopyrimidine, which elutes right after the main product). After purification, a whitish solid was recovered (800 mg, 62%): 1H NMR (400 MHz, CDCl3) δ 8.83 (s, 1H), 7.78 (s, 1H), 4.24 (q, J = 7.1 Hz, 2H), 4.18 (s, 2H), 1.29 (t, J = 7.1 Hz, 3H).
2-((6-Iodothieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (10).
In a 100 mL round bottom flask provided with a reflux condenser and a magnetic stir bar, ethyl 2-((6-iodothieno[3,2-d]pyrimidin-4-yl)thio)acetate 9 (1.0 equiv, 2.1 mmol, 800 mg) was dissolved in a mixture of EtOH (15 mL) and THF (15 mL), followed by the addition of LiOH (2.0 equiv, 4.2 mmol, 100 mg) pre-dissolved in water (5 mL). This mixture was refluxed at 70 °C for 1h. Solvents were removed under reduced pressure and the white solid obtained was taken into ice/water and the pH adjusted to 2–3 with 2M aq. HCl. The resulting solid was filtrated, thoroughly rinsed with water, and air and vacuum dried yielding a white solid (590 mg, 80%): Melting point 183.5 – 186.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (s, 1H), 7.92 (s, 1H), 3.90 (s, 2H); 13C NMR (176 MHz, DMSO) δ 168.5, 161.4, 159.2, 154.0, 133.8, 132.3, 93.1, 33.5. HRMS (HESI) calcd for C8H6N2O2S2I [M+H]+ 352.8837, found 352.8917.
General procedure A. Synthesis of 6-arylthieno[3,2-d]pyrimidines (4, 11a-11ab).
In a 25 mL round bottom flask provided with a reflux condenser, a rubber septum, a magnetic stir bar, and under positive N2 pressure the following reagent were added: 6-iodothieno[3,2-d]pyrimidin-4-yl)thio)acetic acid 10 (1.0 equiv, 0.21 mmol, 75 mg), K2CO3 (2.2 equiv, 0.47 mmol, 65 mg), Pd(PPh3)Cl2 (5-6 mol%, 8-10 mg) and the corresponding arylboronic acid (1.1 equiv, 0.23 mmol). A previously N2 degassed solution of 1,4-dioxane/water (3/1, 2.5 mL) was added and the reaction mixture was stirred under mild reflux for 8-12 h. After this period, solvent was removed under reduced pressure, solids taken into ice/water, and the pH adjusted to 2-3 with 2M HCl(aq). The resulting solid was filtrated, thoroughly rinsed with water, air and vacuum dried, and purified over silica gel using CH2Cl2/MeOH/HOAc (95/4/1) in a gradient up 5-10% of the polar solvent.
2-((6-(4-(Methylthio)phenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (PFE-PKIS 43) (4).
Prepared using general procedure A, with 4-(methylthio)phenylboronic acid. After purification, a pale yellow solid was obtained (30 mg, 40%): Melting point 194.0 – 197.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.95 (s, 1H), 8.93 (s, 1H), 8.07 (s, 1H), 7.91 (d, J = 8.2 Hz, 2H), 7.40 (d, J = 8.2 Hz, 2H), 4.25 (s, 2H), 2.55 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.4, 160.9, 159.6, 154.1, 151.3, 141.8, 128.1, 127.1, 126.5, 126.0, 119.4, 31.5, 14.2. HRMS (HESI) calcd for C15H12N2O2S3 [M+H]+ 349.0139, found 349.0121.
2-((6-(4-Chlorophenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11a).
Prepared using general procedure A, with (4-chlorophenyl)boronic acid. After purification, a white solid was obtained (40 mg, 55%): Melting point >250.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.95 (s, 1H), 8.96 (s, 1H), 8.16 (s, 1H), 8.01 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 4.26 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.3, 161.3, 159.3, 154.1, 150.1, 135.0, 130.8, 129.4, 128.5, 127.0, 120.8, 31.5.HRMS (HESI) calcd for C14H9ClN2O2S2 [M+H]+ 336.9872, found 336.9849.
2-((6-(3-(Methylthio)phenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11b).
Prepared using general procedure A, with 3-(methylthio)phenylboronic acid. After purification, a pale yellow solid was obtained (48 mg, 65%): Melting point >250.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (s, 1H), 8.11 (s, 1H), 7.78 (t, J = 1.8 Hz, 1H), 7.68 (dt, J = 7.5, 1.3 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.41 – 7.35 (m, 1H), 3.92 (s, 2H), 2.58 (s, 3H); 13C NMR (176 MHz, DMSO) δ 171.0, 158.7, 154.1, 150.3, 144.4, 140.0, 132.8, 129.8, 127.4, 127.0, 123.3, 123.1, 120.7, 28.9, 14.5. HRMS (HESI) calcd for C15H12N2O2S3 [M+H]+ 349.0139, found 349.0124.
2-((6-(2-Methoxyphenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11c).
Prepared using general procedure A, with 2-methoxyphenylboronic acid. After purification, a whitish solid was obtained (35 mg, 50%): Melting point 219.0 – 221.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.93 (s, 1H), 8.93 (s, 1H), 8.11 (s, 1H), 8.07 – 8.04 (m, 1H), 7.51 (ddd, J = 8.6, 7.5, 1.7 Hz, 1H), 7.28 (dd, J = 8.6, 1.1 Hz, 1H), 7.13 (td, J = 7.5, 1.1 Hz, 1H), 4.26 (s, 2H), 4.02 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.4, 160.8, 158.1, 156.1, 153.7, 147.3, 131.6, 129.1, 127.4, 121.2, 121.0, 120.2, 112.6, 56.0, 31.4. HRMS (HESI) calcd for C15H12N2O3S2 [M+H]+ 333.0367, found 333.0345.
2-((6-(4-(Isopropylthio)phenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11d).
Prepared using general procedure A, with 4-(isopropylthio)phenylboronic acid. After purification, a pale yellow solid was obtained (36 mg, 45%): Melting point 234.0 – 237.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (s, 1H), 8.02 (s, 1H), 7.89 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 8.1 Hz, 2H), 3.95 (s, 2H), 3.67 (sept, J = 6.8 Hz, 1H), 1.30 (d, J = 6.8 Hz, 6H); 13C NMR (176 MHz, DMSO) δ 163.5, 158.9, 154.1, 153.6, 150.3, 138.8, 135.8, 129.6, 129.4, 127.1, 126.7, 124.3, 119.8, 40.0, 36.1, 22.7. HRMS (HESI) calcd for C17H16N2O2S3 [M+H]+ 377.0452, found 377.0433.
2-((6-(4-Ethylphenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11e).
Prepared using general procedure A, with 4-ethylphenylboronic acid. After purification, a whitish solid was obtained (28 mg, 40%): Melting point 175.0 – 177.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.94 (s, 1H), 8.05 (s, 1H), 7.89 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 4.26 (s, 2H), 2.68 (q, J = 7.5 Hz, 2H), 1.21 (d, J = 7.5 Hz, 3H); 13C NMR (176 MHz, DMSO) δ 169.3, 160.9, 159.5, 154.0, 151.9, 146.6, 129.4, 128.8, 126.8, 126.6, 119.4, 31.4, 27.9, 15.3. HRMS (HESI) calcd for C16H14N2O2S2 [M+H]+ 331.0574, found 331.0561.
2-((6-(4-Vinylphenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11f).
Prepared using general procedure A, with 4-vinylphenylboronic acid. After purification, a pale yellow solid was obtained (43 mg, 62%): Melting point 198.0 – 201.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.95 (s, 1H), 8.95 (s, 1H), 8.13 (s, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 8.4 Hz, 1H), 6.82 (dd, J = 17.7, 10.9 Hz, 1H), 5.99 (d, J = 17.7 Hz, 1H), 5.39 (d, J = 10.9 Hz, 1H), 4.26 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.3, 161.0, 159.4, 154.1, 151.2, 139.0, 135.7, 131.2, 127.0, 127.0, 126.8, 120.0, 116.1, 31.5. HRMS (HESI) calcd for C16H12N2O2S2 [M+H]+ 329.0418, found 329.0401.
2-((6-(4-Isopropylphenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11g).
Prepared using general procedure A, with 4-isopropylphenylboronic acid. After purification, a whitish solid was obtained (40 mg, 55%): Melting point 154 – 159 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.94 (s, 1H), 8.05 (s, 1H), 7.92 – 7.87 (m, 2H), 7.45 – 7.40 (m, 2H), 4.26 (s, 2H), 3.00 – 2.94 (sept, J = 6.9 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H); 13C NMR (176 MHz, DMSO) δ 169.4, 161.0, 159.5, 154.0, 151.9, 151.1, 129.6, 127.4, 126.8, 126.6, 119.5, 33.3, 31.5, 23.6. HRMS (HESI) calcd for C17H16N2O2S2 [M+H]+ 345.0731, found 345.0707.
2-((6-(4-Cyclopropylphenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11h).
Prepared using general procedure A, with 4-cyclopropylphenylboronic acid. After purification, a whitish solid was obtained (35 mg, 48%): Melting point 184.0 – 187.0 °C. 1H NMR (700 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.93 (s, 1H), 8.03 (s, 1H), 7.84 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 8.3 Hz, 2H), 4.25 (s, 2H), 2.03 – 1.98 (m, 1H), 1.05 – 1.01 (m, 2H), 0.79 – 0.75 (m, 2H); 13C NMR (176 MHz, DMSO) δ 169.3, 160.8, 159.5, 154.0, 151.8, 146.9, 128.9, 126.6, 126.5, 126.2, 119.2, 31.4, 15.1, 10.1. HRMS (HESI) calcd for C17H14N2O2S2 [M+H]+ 343.0574, found 343.0558.
2-((6-(3,4-Dichlorophenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11i).
Prepared using general procedure A, with 3,4-dichlorophenylboronic acid. After purification, a white solid was obtained (31 mg, 40%): Melting point 222.0 – 226.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.96 (s, 1H), 8.98 (s, 1H), 8.32 (d, J = 2.2 Hz, 1H), 8.27 (s, 1H), 7.95 (dd, J = 8.4, 2.2 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 4.27 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.2, 161.5, 159.1, 154.1, 148.5, 132.8, 132.5, 132.3, 131.4, 128.4, 127.2, 126.8, 121.9, 31.5. (HESI) calcd for C14H8Cl2N2O2S2 [M+H]+ 372.9453, found 372.9438.
2-((6-(4-(Benzyloxy)phenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11j).
Prepared using general procedure A, with 4-(Benzyloxy)phenylboronic acid. After purification, a pale yellow solid was obtained (26 mg, 30%): Melting point 192.0 – 195.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.93 (bs, 0.5H), 11.95 (bs, 0.5H), 8.92 (s, 1H), 7.98 (s, 1H), 7.94 – 7.91 (m, 2H), 7.48 (d, J = 7.5 Hz, 2H), 7.42 (dd, J = 8.3, 6.8 Hz, 2H), 7.36 (d, J = 7.5 Hz, 1H), 7.19 – 7.16 (m, 2H), 5.22 (s, 2H), 4.25 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.3, 160.6, 160.1, 159.7, 154.0, 151.7, 136.6, 128.5, 128.3, 127.9, 127.7, 126.4, 124.6, 118.5, 115.6, 69.4, 31.4. HRMS (HESI) calcd for C21H16N2O3S2 [M+H]+ 409.0680, found 409.0671.
2-((6-(4-Acetamidophenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11k).
Prepared using general procedure A, with 4-acetamidophenylboronic acid. After purification, a pale yellow solid was obtained (23 mg, 30%): Melting point 232.0 – 235.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 10.23 (s, 1H), 8.92 (s, 1H), 7.99 (s, 1H), 7.94 – 7.88 (m, 2H), 7.77 – 7.72 (m, 2H), 4.25 (s, 2H), 2.09 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.4, 168.7, 160.8, 159.6, 154.0, 151.7, 141.3, 127.4, 126.4, 126.3, 119.2, 118.7, 31.5, 24.2. HRMS (HESI) calcd for C16H13N3O3S2 [M+H]+ 360.0476, found 360.0463.
2-((6-(3-Acetamidophenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11l).
Prepared using general procedure A, with 3-acetamidophenylboronic acid. After purification, a pale yellow solid was obtained (26 mg, 35%): Melting point 221.0 – 225.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.96 (s, 1H), 10.18 (s, 1H), 8.96 (s, 1H), 8.21 – 8.17 (m, 1H), 8.01 (s, 1H), 7.68 (ddd, J = 11.1, 7.9, 1.9 Hz, 2H), 7.48 (t, J = 7.9 Hz, 1H), 4.27 (s, 2H), 3.32 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.4, 168.7, 161.2, 159.3, 154.1, 151.6, 140.2, 132.2, 130.0, 126.7, 121.4, 120.6, 120.1, 116.7, 31.5, 24.1. HRMS (HESI) calcd for C16H13N3O3S2 [M+H]+ 360.0476, found 360.0454.
2-((6-(4-(Methylsulfinyl)phenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11m).
Prepared using general procedure A, with 4-(methylsulfinyl)phenylboronic acid. After purification, a whitish solid was obtained (34 mg, 45%): Melting point 220.0 – 222.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.98 (s, 1H), 8.24 (s, 1H), 8.20 – 8.16 (m, 2H), 7.87 – 7.83 (m, 2H), 4.27 (s, 2H), 2.82 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.3, 161.4, 159.2, 154.2, 150.2, 148.5, 133.9, 127.5, 127.1, 124.7, 121.4, 43.1, 31.5. HRMS (HESI) calcd for C15H12N2O3S3 [M+H]+ 365.0088, found 365.0071.
2-((6-(4-Acetylphenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11n).
Prepared using general procedure A, with 4-acetylphenylboronic acid. After purification, a whitish solid was obtained (40 mg, 55%): Melting point 193.0 – 197.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.97 (s, 1H), 8.99 (s, 1H), 8.29 (s, 1H), 8.14 (d, J = 8.5 Hz, 2H), 8.10 (d, J = 8.5 Hz, 2H), 4.28 (s, 2H), 2.65 (s, 3H); 13C NMR (176 MHz, DMSO) δ 197.3, 169.3, 161.5, 159.2, 154.2, 150.0, 137.6, 135.9, 129.2, 127.4, 127.0, 121.9, 31.5, 26.9. HRMS (HESI) calcd for C16H12N2O3S2 [M+H]+ 345.0367, found 345.0357.
2-((6-(3-Morpholinophenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11o).
Prepared using general procedure A, with 3-morpholinophenylboronic acid. After purification, a pale orange solid was obtained (29 mg, 35%): Melting point 229.0 – 233.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.95 (s, 1H), 8.13 (s, 1H), 7.46 (t, J = 2.1 Hz, 1H), 7.41 – 7.34 (m, 2H), 7.12 – 7.07 (m, 1H), 4.26 (s, 2H), 3.77 (dd, J = 5.8, 3.8 Hz, 4H), 3.26 – 3.23 (m, 4H); 13C NMR (176 MHz, DMSO) δ 169.3, 161.0, 159.4, 154.0, 152.4, 151.7, 132.6, 130.1, 126.7, 120.0, 117.3, 116.9, 112.7, 66.0, 48.0, 31.4. HRMS (HESI) calcd for C18H17N3O3S2 [M+H]+ 388.0789, found 388.0780.
2-((6-(4-Morpholinophenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11p).
Prepared using general procedure A, with 4-morpholinophenylboronic acid. After purification, an orange solid was obtained (29 mg, 35%): Melting point 205.0 – 208.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.92 (s, 1H), 8.88 (s, 1H), 7.89 (s, 1H), 7.82 (d, J = 8.9 Hz, 2H), 7.06 (d, J = 8.9 Hz, 2H), 4.24 (s, 2H), 3.78 – 3.72 (m, 4H), 3.28 – 3.23 (m, 4H); 13C NMR (176 MHz, DMSO) δ 169.5, 160.2, 159.9, 154.0, 152.5, 152.3, 127.8, 125.9, 121.8, 117.1, 114.6, 65.9, 47.2, 31.4. HRMS (HESI) calcd for C18H17N3O3S2 [M+H]+ 388.0790, found 388.0773.
2-((6-(6-Acetylpyridin-3-yl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11q).
Prepared using general procedure A, with (6-acetylpyridin-3-yl)phenylboronic acid. After purification, a pale orange solid was obtained (22 mg, 30%): Melting point 181.0 – 184.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.98 (s, 1H), 9.36 (d, J = 2.4 Hz, 1H), 9.01 (s, 1H), 8.55 (dd, J = 8.2, 2.4 Hz, 1H), 8.41 (s, 1H), 8.08 (d, J = 8.2 Hz, 1H), 4.29 (s, 2H), 2.69 (s, 3H); 13C NMR (176 MHz, DMSO) δ 198.6, 169.2, 161.8, 158.9, 154.2, 153.2, 147.0, 146.8, 135.3, 131.2, 127.7, 123.1, 121.5, 31.6, 25.7. HRMS (HESI) calcd for C15H11N3O3S2 [M+H]+ 346.0320, found 346.0310.
2-((6-(Thiophen-2-yl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11r).
Prepared using general procedure A, with 2-thienylboronic acid. After purification, a pale orange solid was obtained (29 mg, 45%): Melting point 179.0 – 182.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.95 (s, 1H), 8.93 (s, 1H), 7.85 (s, 1H), 7.83 (dd, J = 5.0, 1.2 Hz, 1H), 7.80 (dd, J = 3.7, 1.2 Hz, 1H), 7.26 – 7.23 (m, 1H), 4.25 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.4, 160.9, 159.3, 154.3, 144.7, 134.7, 129.8, 129.0, 128.3, 126.3, 119.3, 31.5. HRMS (HESI) calcd for C12H8N2O2S3 [M+H]+ 308.9826, found 308.9811.
2-((6-(Benzo[b]thiophen-2-yl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11s).
Prepared using general procedure A, with benzo[b]thien-2-ylboronic acid. After purification, a pale orange solid was obtained (30 mg, 40%): Melting point >250 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.97 (s, 1H), 8.98 (s, 1H), 8.16 (s, 1H), 8.08 – 8.06 (m, 1H), 7.98 (s, 1H), 7.96 – 7.93 (m, 1H), 7.48 – 7.46 (m, 2H), 4.27 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.3, 161.2, 159.0, 154.3, 144.4, 139.6, 134.4, 126.9, 126.3, 125.4, 124.8, 124.7, 122.7, 121.2, 31.6. HRMS (HESI) calcd for C16H10N2O2S3 [M+H]+ 358.9982, found 358.9971.
2-((6-(4-Cyanophenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11t).
Prepared using general procedure A, with 4-cyanophenylboronic acid. After purification, a pale brown solid was obtained (34 mg, 50%): Melting point 226.0 – 230.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.97 (s, 1H), 8.99 (s, 1H), 8.33 (s, 1H), 8.19 – 8.17 (m, 2H), 8.03 – 7.99 (m, 2H), 4.27 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.6, 162.1, 159.1, 154.4, 149.6, 136.4, 133.5, 128.0, 127.8, 122.6, 118.6, 112.6, 31.8. HRMS (HESI) calcd for C15H9N3O2S2 [M+H]+ 328.0214, found 328.0190.
2-((6-(4-(Ethoxycarbonyl)phenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11u).
Prepared using general procedure A, with 4-ethoxycarbonylphenylboronic acid. After purification, a pale orange solid was obtained (39 mg, 50%): Melting point 182.0 – 185.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.96 (s, 1H), 8.97 (s, 1H), 8.25 (s, 1H), 8.12 (d, J = 8.4 Hz, 2H), 8.06 (d, J = 8.4 Hz, 2H), 4.33 (q, J = 7.1 Hz, 2H), 4.25 (s, 2H), 1.34 (t, J = 7.1 Hz, 3H); 13C NMR (176 MHz, DMSO) δ 169.3, 165.0, 161.6, 159.1, 154.1, 149.8, 136.0, 130.9, 130.0, 127.3, 127.0, 121.9, 61.0, 31.6, 14.1. HRMS (HESI) calcd for C17H14N2O4S2 [M+H]+ 375.0473, found 375.0445.
2-((6-(3-Fluorophenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11v).
Prepared using general procedure A, with 3-fluorophenylboronic acid. After purification, a white solid was obtained (39 mg, 58%): Melting point 194.0 –196.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.96 (s, 1H), 8.97 (s, 1H), 8.22 (s, 1H), 7.91 (dt, J = 10.2, 2.2 Hz, 1H), 7.80 (dd, J = 7.7, 1.6 Hz, 1H), 7.60 (td, J = 8.0, 6.0 Hz, 1H), 7.38 (dd, J = 8.6, 2.6 Hz, 1H), 4.27 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 169.2, 162.5 (d, J = 246.3 Hz), 161.5, 159.1, 154.1, 149.9 (d, J = 2.8 Hz), 134.1 (d, J = 8.4 Hz), 131.5 (d, J = 8.5 Hz), 127.1, 122.9 (d, J = 2.5 Hz), 121.3, 117.1 (d, J = 21.3 Hz), 113.6 (d, J = 23.7 Hz), 31.6; 19F NMR (376 MHz, DMSO-d6) δ −111.82 – −111.92 (m, 1F). HRMS (HESI) calcd for C14H9FN2O2S2 [M+H]+ 321.0167, found 321.0151.
2-((6-(2-Fluorophenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11w).
Prepared using general procedure A, with 2-fluorophenylboronic acid. After purification, a white solid was obtained (40 mg, 60%): Melting point 184.0 – 187.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.96 (s, 1H), 8.99 (s, 1H), 8.13 – 8.07 (m, 2H), 7.58 (s, 1H), 7.49 (dd, J = 11.7, 1.2 Hz, 1H), 7.43 – 7.39 (m, 1H), 4.27 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 169.3, 161.5, 158.9 (d, J = 149.4 Hz), 158.4, 154.1, 144.3 (d, J = 3.5 Hz), 132.3 (d, J = 8.8 Hz), 129.8, 127.2 (d, J = 5.3 Hz), 125.6 (d, J = 3.2 Hz), 122.9 (d, J = 4.8 Hz), 119.7 (d, J = 11.7 Hz), 116.8 (d, J = 22.0 Hz); 19F NMR (376 MHz, DMSO-d6) δ −112.35 – −112.50 (m, 1F). HRMS (HESI) calcd for C14H9FN2O2S2 [M+H]+ 321.0167, found 321.0142.
2-((6-(3,4-Difluorophenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11x).
Prepared using general procedure A, with 3,4-difluorophenylboronic acid. After purification, a white solid was obtained (32 mg, 45%): Melting point 206.0 – 209.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.96 (s, 1H), 8.97 (s, 1H), 8.20 – 8.16 (m, 2H), 7.83 (dd, J = 8.2, 3.9 Hz, 1H), 7.66 – 7.60 (m, 1H), 4.27 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.3, 161.3, 159.1, 154.1, 150.9 (dd, J = 120.7, 12.8 Hz), 149.5 (dd, J = 117.2, 12.8 Hz), 149.0, 129.6 – 129.5 (m), 127.1, 124.1 – 123.9 (m), 121.3, 118.6 (d, J = 17.7 Hz), 116.1 (d, J = 18.8 Hz), 31.5; 19F NMR (376 MHz, DMSO-d6) δ −135.61 – −135.88 (m, 1F), −136.89 – −137.13 (m, 1F). HRMS (HESI) calcd for C14H8F2N2O2S2 [M+H]+ 339.0073, found 339.0058.
2-((6-(3-Fluoro-4-(methylthio)phenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11y).
Prepared using general procedure A, with 3-fluoro-4-(methylthio)phenylboronic acid. After purification, a pale yellow solid was obtained (32 mg, 42%): Melting point 203.0 – 206.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.95 (s, 1H), 8.95 (s, 1H), 8.17 (s, 1H), 7.92 (dd, J = 11.4, 2.0 Hz, 1H), 7.80 (dd, J = 8.1, 2.0 Hz, 1H), 7.51-7.44 (m, 1H), 4.26 (s, 2H), 2.57 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.3, 161.1, 159.3, 158.6 (d, J = 242.1 Hz), 154.1, 149.8 (d, J = 2.5 Hz), 129.9 (d, J = 8.1 Hz), 128.7 (d, J = 17.1 Hz), 127.3 (d, J = 3.3 Hz), 126.8, 123.3 (d, J = 2.8 Hz), 120.6, 112.9 (d, J = 23.4 Hz), 31.5, 13.5; 19F NMR (376 MHz, DMSO-d6) δ −112.48 (dd, J = 11.4, 8.1 Hz). HRMS (HESI) calcd for C15H11FN2O2S3 [M+H]+ 367.0044, found 367.0027.
2-((6-(3-Fluoro-4-methoxyphenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11z).
Prepared using general procedure A, with 3-fluoro-4-methoxyphenylboronic acid. After purification, a pale yellow solid was obtained (29 mg, 40%): Melting point 209.0 – 211.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.93 (s, 1H), 8.06 (s, 1H), 7.95 (dd, J = 12.5, 2.4 Hz, 1H), 7.77 – 7.74 (m, 1H), 7.35 – 7.28 (m, 1H), 4.25 (s, 2H), 3.93 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.4, 160.9, 159.5, 154.1, 151.6 (d, J = 245.4 Hz), 150.4 (d, J = 2.4 Hz), 148.9 (d, J = 10.6 Hz), 126.6, 124.8 (d, J = 7.1 Hz), 123.6 (d, J = 3.1 Hz), 119.6, 114.3, 114.3 (d, J = 17.8 Hz), 56.2, 31.5; 19F NMR (376 MHz, DMSO-d6) δ −134.17 (dd, J = 12.5, 9.2 Hz). HRMS (HESI) calcd for C15H11FN2O3S2 [M+H]+ 351.0273, found 351.0260.
2-((6-(4-(Trifluoromethyl)phenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11aa).
Prepared using general procedure A, with 4-(trifluoromethyl)phenylboronic acid. After purification, a pale orange solid was obtained (39 mg, 50%): Melting point 173.0 – 176.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.00 (s, 1H), 8.30 (s, 1H), 8.21 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H), 4.28 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.2, 161.6, 159.1, 154.2, 149.4, 135.7, 130.0 (q, J = 32.0 Hz), 127.6, 127.4, 126.3 (q, J = 3.7 Hz), 123.9 (q, J = 272.0 Hz), 122.2, 31.6; 19F NMR (376 MHz, DMSO-d6) δ −61.35. HRMS (HESI) calcd for C15H9F3N2O2S2 [M+H]+ 371.0135, found 371.0118.
2-((6-(3-(Trifluoromethoxy)phenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetic acid (11ab).
Prepared using general procedure A, with 3-(trifluoromethoxy)phenylboronic acid. After purification, a whitish solid was obtained (41 mg, 50%): Melting point 210.0 – 213.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.96 (s, 1H), 8.98 (s, 1H), 8.27 (s, 1H), 8.04 – 7.98 (m, 2H), 7.71 – 7.66 (m, 1H), 7.53 (d, J = 8.2 Hz, 1H), 4.27 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.3, 161.5, 159.1, 154.1, 149.4, 149.0 (d, J = 1.8 Hz), 134.1, 131.5, 127.2, 125.9, 122.5, 121.7, 120.0 (q, J = 258.4 Hz), 119.4, 31.5; 19F NMR (376 MHz, DMSO-d6) δ −56.74. HRMS (HESI) calcd for C15H9F3N2O3S2 [M+H]+ 387.0085, found 387.0071.
2-((6-(4-(Methylthio)phenyl)thieno[3,2-d]pyrimidin-4-yl)thio)acetamide (12).
Into a 20 mL vial provided with a magnetic stir bar, reagents were loaded in the following order: compound (4) (1.0 equiv, 0.06 mmol, 20 mg), DMF (1.0 mL), N,N-diisopropylethylamine (2.2 equiv, 0.13 mmol, 20 mg), HATU (1.0 equiv, 0.06 mmol, 23 mg), and ammonia (excess, 0.1 mL of an 28% aqueous solution). The vial was sealed and the reaction mixture stirred at room temperature for 12 h. It was quenched with water, solid slowly precipitated, filtered, thoroughly rinsed with water, air and vacuum dried. No further purification was needed, yielding a pale brown solid (5 mg, 25%): Melting point 231.0 – 234.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.06 (s, 1H), 7.92 – 7.89 (m, 2H), 7.69 (s, 1H), 7.42 – 7.39 (m, 2H), 7.24 (s, 1H), 4.16 (s, 2H), 2.55 (s, 3H); 13C NMR (176 MHz, DMSO) δ 168.4, 161.4, 159.4, 154.0, 151.2, 141.7, 128.1, 127.0, 126.6, 126.0, 119.3, 32.9, 14.2. HRMS (HESI) calcd for C15H13N3OS3 [M+H]+ 348.0299, found 348.0279.
General procedure B. Synthesis of 4-chloro-6-arylthieno[3,2-d]pyrimidines (13a-b).
In a 50 mL round bottom flask provided with a reflux condenser, a rubber septum, a magnetic stir bar, and under positive N2 pressure the following reagent were added: 4-chloro-6-iodothieno[3,2-d]pyrimidine 10 (1.0 equiv, 1.20 mmol, 355 mg), NaHCO3 (1.0 equiv, 1.20 mmol, 100 mg), CsF (1.0 equiv, 1.20 mmol, 185 mg), Pd(Ph3)4 (3 mol%, 40 mg) and the corresponding arylboronic acid (1.0 equiv, 1.20 mmol). A previously N2 degassed solution of 1,4-dioxane/water (3:1, 7.0 mL) was added and the reaction mixture was stirred under mild reflux for 3 h. After this period, solvent was removed under reduced pressure and the resulting solid was purified over silica gel with hexanes/EtOAc in a gradient up to 30% of the polar solvent.
7-Chloro-2-(4-chlorophenyl)benzo[b]thiophene (13a).
Prepared using general procedure B, with (4-chlorophenyl)boronic acid. After purification, a white solid was obtained (185 mg, 55%): 1H NMR (400 MHz, CDCl3) δ 8.97 (s, 1H), 7.74 (s, 1H), 7.73 – 7.69 (m, 2H), 7.53 – 7.47 (m, 2H).
7-Chloro-2-(4-(methylthio)phenyl)benzo[b]thiophene (13b).
Prepared using general procedure B, with (4-(methylthio)phenyl)boronic acid. After purification, a pale yellow solid was obtained (300 mg, 85%): 1H NMR (400 MHz, DMSO-d6) δ 9.01 (s, 1H), 8.21 (s, 1H), 7.97 – 7.90 (m, 2H), 7.44 – 7.38 (m, 2H), 2.56 (s, 3H).
Ethyl 2-((6-(4-chlorophenyl)thieno[3,2-d]pyrimidin-4-yl)oxy)acetate (14).
Into a 20 mL microwave vial provided with a magnetic stir bar, reagents were loaded in the following order: 7-chloro-2-(4-chlorophenyl)benzo[b]thiophene 13a (1.0 equiv, 0.23 mmol, 65 mg), anhydrous DMF (3.0 mL), ethyl 2-hydroxyacetate (2.0 equiv, 0.46 mmol, 48 mg), and NaH (2.6 equiv, 0.60 mmol, 25 mg, 60% dispersion in mineral oil). The vial was sealed and the reaction mixture stirred at 100 °C for 30 min in a microwave reactor. The crude was quenched over ice/water, solvents were evaporated in vacuo, and purified over silica gel with hexanes/EtOAc in a gradient up to 50% of EtOAc. After purification, a white solid was recovered (37 mg, 45%): Melting point 145.0 – 148.0 °C . 1H NMR (700 MHz, DMSO-d6) δ 8.75 (s, 1H), 8.12 (s, 1H), 7.97 (dd, J = 8.6, 2.4 Hz, 2H), 7.63 – 7.59 (m, 2H), 5.21 (s, 2H), 4.18 (q, J = 8.2 Hz, 2H), 1.20 (t, J = 8.2 Hz, 3H). 13C NMR (176 MHz, DMSO) δ 167.6, 162.8, 162.3, 154.3, 150.5, 134.8, 131.0, 129.3, 128.3, 120.8, 115.9, 62.8, 60.8, 13.9. HRMS (HESI) calcd for C16H14N2O3SCl [M+H]+ 349.0335, found 349.0400.
2-((6-(4-Chlorophenyl)thieno[3,2-d]pyrimidin-4-yl)oxy)acetic acid (15).
In a 10 mL round bottom flask provided with a reflux condenser and a magnetic stir bar, compound 14 (1.0 equiv, 0.06 mmol, 21 mg) was dissolved in THF (2 mL), followed by the addition of LiOH (2.2 equiv, 0.12 mmol, 3 mg) pre-dissolved in water. This mixture was refluxed at 70 °C for 4–5 h. Solvents were removed under reduced pressure and the white solid obtained was taken into ice/water and the pH adjusted to 4–5 with 2M HCl(aq). The resulting solid was filtrated, thoroughly rinsed with water, and air/vacuum dried yielding a white solid (16 mg, 82%): Melting point 234.0 – 236.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.20 (s, 1H), 8.76 (s, 1H), 8.13 (s, 1H), 7.98 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 5.12 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.1, 162.8, 162.5, 154.4, 150.4, 134.8, 131.1, 129.4, 128.3, 120.8, 116.1, 62.8. HRMS (HESI) calcd for C14H9ClN2O3S [M+H]+ 321.0100, found 321.0080.
Ethyl (6-(4-(methylthio)phenyl)thieno[3,2-d]pyrimidin-4-yl)glycinate (16a).
Into a 20 mL vial provided with a magnetic stir bar, reagents were loaded in the following order: compound 13b (1.0 equiv, 0.27 mmol, 80 mg), EtOH (5.0 mL), ethyl glycinate hydrochloride (2.5 equiv, 0.68 mmol, 95 mg), and triethylamine (10 equiv, 2.7 mmol, 0.4 mL). The vial was sealed and the reaction mixture stirred at 90 °C for 18 h. After this period, solvent was evaporated in vacuo, and purified over silica gel with hexanes/EtOAc in a gradient up to 60% of EtOAc. After purification, a pale yellow solid was recovered (85 mg, 78%): Melting point 160.5 – 164.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.43 (s, 1H), 8.33 (s, 1H), 7.82 (s, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 4.21 (d, J = 5.9 Hz, 2H), 4.13 (q, J = 7.1 Hz, 2H), 2.54 (s, 3H), 1.20 (t, J = 7.1 Hz, 3H). HRMS (HESI) calcd for C17H18N3O2S2 [M+H]+ 360.0762, found 360.0844.
(6-(4-(Methylthio)phenyl)thieno[3,2-d]pyrimidin-4-yl)glycine (16b).
In a 10 mL round bottom flask provided with a reflux condenser and a magnetic stir bar, compound 16a (1.0 equiv, 0.06 mmol, 20 mg) was dissolved in THF (2 mL), followed by the addition of LiOH (2.2 equiv, 0.12 mmol, 3 mg) pre-dissolved in water. This mixture was refluxed at 70 °C for 4–5 h. Solvents were removed under reduced pressure and the white solid obtained was taken into ice/water and the pH adjusted to 4–5 with 2M HCl(aq). The resulting solid was filtrated, thoroughly rinsed with water, and air/vacuum dried yielding a pale orange solid (17 mg, 92 %): Melting point, decomposition. 1H NMR (400 MHz, DMSO-d6) δ 12.77 (s, 1H), 8.84 (s, 1H), 8.58 (s, 1H), 7.84 – 7.78 (m, 3H), 7.41 (d, J = 8.1 Hz, 2H), 4.20 (d, J = 5.9 Hz, 2H), 2.55 (s, 3H).
2-((6-Bromothieno[2,3-d]pyrimidin-4-yl)thio)acetic acid (18).
In a 50 mL round bottom flask provided with a rubber septum, a magnetic stir bar, and under positive N2 pressure, 6-bromo-4-chlorothieno[2,3-d]pyrimidine (1.0 equiv, 4.0 mmol, 1.0 g) 17 was suspended in CH3CN or EtOH (15 mL) at 0 °C (ice bath), followed by the dropwise addition of 2-mercaptoacetic acid (1.25 equiv, 5.0 mmol, 0.35 mL) and triethylamine (2.5 equiv, 10.0 mmol, 1.4 mL). Ice bath was removed and the reaction mixture was stirred overnight at room temperature. After this period, solvent was removed under reduced pressure, solid taken into ice/water, and the pH adjusted to 2–3 with 2M aq. HCl. The resulting solid was filtrated, thoroughly rinsed with water, air and vacuum dried, yielding a white solid. No further purification was needed (0.91 g, 75%). Melting point 194.5 – 198.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.90 (s, 1H), 8.83 (s, 1H), 7.83 (s, 1H), 4.18 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.3, 165.9, 161.6, 152.6, 128.0, 122.1, 116.2, 31.4. HRMS (HESI) calcd for C8H6N2O2S2Br [M+H]+ 304.8976, found 304.9059.
General procedure C. Synthesis of 6-Arylthieno[2,3-d]pyrimidines (19a-g).
In a 25 mL round bottom flask provided with a reflux condenser, a rubber septum, a magnetic stir bar, and under positive N2 pressure the following reagent were added: 2-((6-bromothieno[2,3-d]pyrimidin-4-yl)thio)acetic acid (18) (1.0 equiv, 0.25 mmol, 75 mg), K2CO3 (4.0 equiv, 1.0 mmol, 140 mg), Pd(Ph3)4 (5 mol%, 15 mg) and the corresponding arylboronic acid (1.0 equiv, 0.25 mmol). A previously N2 degassed solution of 1,4-dioxane/water (3:1, 2.5 mL) was added and the reaction mixture was stirred under mild reflux for 3 h. After this period, solvent was removed under reduced pressure, solids taken into ice/water, and the pH adjusted to 2-3 with 2M HCl(aq). The resulting solid was filtrated, thoroughly rinsed with water, air and vacuum dried, and purified over silica gel using CH2Cl2/MeOH/HOAc (95/4/1) in a gradient up 5-10% of the polar solvent.
2-((6-(4-(Methylthio)phenyl)thieno[2,3-d]pyrimidin-4-yl)thio)acetic acid (19a).
Prepared using general procedure C, with 4-(methylthio)phenylboronic acid. After purification, a pale yellow solid was obtained (45 mg, 53%): Melting point 194.0 – 196.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.89 (s, 1H), 8.81 (s, 1H), 7.91 (s, 1H), 7.85 (d, J = 8.6 Hz, 2H), 7.38 (d, J = 8.6 Hz, 2H), 4.20 (s, 2H), 2.54 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.5, 164.3, 161.9, 152.2, 143.1, 140.5, 128.8, 128.5, 126.9, 126.1, 113.7, 31.4, 14.3. HRMS (HESI) calcd for C15H12N2O2S3 [M+H]+ 349.0139, found 349.0112.
2-((6-(4-Methoxyphenyl)thieno[2,3-d]pyrimidin-4-yl)thio)acetic acid (19b).
Prepared using general procedure C, with 4-methoxyphenylboronic acid. After purification, a pale yellow solid was obtained (39 mg, 48%): Melting point 175.0 – 178.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.88 (s, 1H), 8.79 (s, 1H), 7.87 – 7.83 (m, 2H), 7.79 (s, 1H), 7.09 – 7.05 (m, 2H), 4.20 (s, 2H), 3.83 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.5, 164.2, 161.5, 160.3, 151.9, 143.6, 128.9, 128.0, 124.7, 114.7, 112.6, 55.4, 31.4. HRMS (HESI) calcd for C15H12N2O3S2 [M+H]+ 333.0367, found 333.0345.
2-((6-(3-(Methylthio)phenyl)thieno[2,3-d]pyrimidin-4-yl)thio)acetic acid (19c).
Prepared using general procedure C, with 3-(methylthio)phenylboronic acid. After purification, a pale yellow solid was obtained (57 mg, 65%): Melting point 160.0-163.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.90 (s, 1H), 8.83 (s, 1H), 8.04 (s, 1H), 8.05 – 8.03 (m, 1H), 7.66 – 7.59 (m, 1H), 7.45 (dd, J = 8.7, 7.8 Hz, 1H), 7.37 – 7.33 (m, 1H), 4.21 (s, 2H), 2.58 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.5, 164.5, 162.4, 152.41, 142.8, 139.9, 132.9, 129.8, 128.6, 126.7, 123.3, 123.2, 115.1, 31.4, 14.5. HRMS (HESI) calcd for C15H12N2O2S3 [M+H]+ 349.0139, found 349.0122.
2-((6-(3-Methoxyphenyl)thieno[2,3-d]pyrimidin-4-yl)thio)acetic acid (19d).
Prepared using general procedure C, with 3-methoxyphenylboronic acid. After purification, a pale yellow solid was obtained (42 mg, 50%): Melting point 185.0 – 188.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.89 (s, 1H), 8.82 (s, 1H), 8.01 (s, 1H), 7.50 – 7.47 (m, 1H), 7.45 – 7.40 (m, 2H), 7.09 – 6.99 (m, 1H), 4.22 (s, 2H), 3.87 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.5, 164.4, 162.3, 159.8, 152.3, 143.3, 133.5, 130.5, 128.6, 119.0, 115.6, 114.8, 111.5, 55.4, 31.4. HRMS (HESI) calcd for C15H12N2O3S2 [M+H]+ 333.0367, found 333.0370.
2-((6-(2-Methoxyphenyl)thieno[2,3-d]pyrimidin-4-yl)thio)acetic acid (19e).
Prepared using general procedure C, with 2-methoxyphenylboronic acid. After purification, a pale yellow solid was obtained (46 mg, 55%): Melting point 176.5 – 180.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.89 (s, 1H), 8.81 (s, 1H), 7.99 (dd, J = 7.8, 1.7 Hz, 1H), 7.91 (s, 1H), 7.46 (ddd, J = 8.7, 7.3, 1.7 Hz, 1H), 7.24 (dd, J = 8.7, 1.1 Hz, 1H), 7.12 (ddd, J = 7.8, 7.3, 1.1 Hz, 1H), 4.21 (s, 2H), 3.98 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.6, 164.9, 161.6, 155.8, 152.0, 139.3, 130.9, 128.8, 127.2, 121.2, 120.6, 115.3, 112.5, 55.9, 31.3. HRMS (HESI) calcd for C15H12N2O3S2 [M+H]+ 333.0368, found 333.0370.
2-((6-(3,4-Dichlorophenyl)thieno[2,3-d]pyrimidin-4-yl)thio)acetic acid (19f).
Prepared using general procedure C, with 3,4-dichlorophenylboronic acid. After purification, a white solid was obtained (35 mg, 38%): Melting point >250 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.91 (s, 1H), 8.84 (s, 1H), 8.31 (d, J = 2.2 Hz, 1H), 8.17 (s, 1H), 7.86 (dd, J = 8.4, 2.2 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 4.22 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.5, 164.7, 162.9, 152.6, 140.4, 132.8, 132.2, 131.9, 131.3, 128.5, 128.0, 126.7, 116.5, 31.5. HRMS (HESI) calcd for C14H8Cl2N2O2S2 [M+H]+ 370.9482, found 370.9459.
2-((6-(Benzo[b]thiophen-2-yl)thieno[2,3-d]pyrimidin-4-yl)thio)acetic acid (19g).
Melting point 225.0 – 228.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.92 (s, 1H), 8.85 (s, 1H), 8.06 – 8.02 (m, 2H), 7.95 – 7.90 (m, 1H), 7.80 (s, 1H), 7.46 – 7.43 (m, 2H), 4.23 (s, 2H); 13C NMR (176 MHz, DMSO) δ 169.4, 164.7, 162.4, 152.7, 139.7, 139.1, 136.6, 134.7, 128.2, 125.9, 125.3, 124.4, 123.9, 122.6, 115.5, 31.5. HRMS (HESI) calcd for C16H10N2O2S3 [M+H]+ 358.9983, found 358.9988.
Protein purification and crystallization.
The kinase domain of STK17B (aa 25–329) was expressed as an N-terminal His-tagged fusion in E. coli. The recombinant protein was purified by Ni2+-affinity chromatography, and subsequently size exclusion chromatography. The protein in 25 mM HEPES, pH 7.5, 500 mM NaCl and 0.5 mM TCEP was mixed with 1 mM inhibitor, and the complex was crystallized using sitting drop vapor diffusion method at 4 °C with the crystallization condition listed in SI Table 2. Crystals were cryo-protected with mother liquor supplemented with 20% ethylene glycol. Diffraction data collected at Diamond Light Source, BESSY II and DESY were processed with iMosflm44 and scaled with Scala or Aimless from CCP4 suite.45 The structures were solved by molecular replacement using Phaser46 and the coordinates of DAPK1 (PDB: 3EHA). Manual model rebuilding was performed in COOT,47 alternated with refinement using REFMAC5.48 Data collection and refinement statistics are summarized in SI Table 2.
Biological assays.
Kinase selectivity:
The KINOMEscan scanMAX assay panel was performed at DiscoverX Corporation as previously described49 across 403 wild type human kinases and 65 mutant human kinases at a compound concentration 1 μM. IC50 was determined on kinases where an inhibitor displayed >75% inhibition at 1 μM. False positives in the scanMAX assay were identified as those kinases where an inhibitor produced an IC50 > 10 μM.
KinaseSeeker split luciferase assay:
10 mM stock solutions of the compounds were serially diluted in DMSO to make assay stocks. Prior to initiating IC50 determinations, the test compounds were evaluated for false positives against split-luciferase. Each test compound was screened in duplicate at 1 μM against STK17A and STK17B. Cfluc-kinase was translated along with Fos-Nfluc using a cell-free system (rabbit reticulocyte lysate) at 30 °C for 90 min. 24 μL aliquot of this lysate containing either 1 μL of DMSO (for no-inhibitor control) or compound solution in DMSO was incubated for 30 min at room temperature followed by 1 h in presence of a kinase specific probe. 80 μL of luciferin assay reagent was added to each solution and luminescence was immediately measured on a luminometer. The % Inhibition was calculated using the following equation: % Inhibition = [ALU (Control) – ALU (Sample)]/ALU (Control) x 100. For IC50 determinations, each compound was tested at 8 different concentrations. The % Inhibition was plotted against compound concentration and the IC50 was determined for each compound using an 8-point curve.
NanoBRET assays:
NanoBRET assays were performed as previously described.24 For STK17B the C-terminal NanoLuc fusion (STK17B-NL) was encoded in pFN32K expression vector, including flexible Gly-Ser-Ser-Gly linkers between NanoLuc and STK17B. A 10 μg/mL solution of DNA in Opti-MEM without serum was made containing 9 μg/mL of Carrier DNA (Promega) and 1 μg/mL of STK17B-NL for a total volume of 1.05 mL. To this solution was then added 31.5 μL of FuGENE HD (Promega) to form a lipid:DNA complex. The solution was then mixed by inversion 8 times and incubated at room temperature for 20 min. The resulting transfection complex (1.082 mL) was then gently mixed with 21 mL of HEK-293 cells (ATCC) suspended at a density of 2 x 105 cells/mL in DMEM (Gibco) + 10% FBS (Corning). 100 μL of this solution was dispensed into 96-well tissue culture treated plates (Corning #3917) followed by incubation (37 °C / 5 % CO2) for 24 h. The media was removed and replaced with 85 μL of room temperature Opti-MEM without phenol red. NanoBRET Tracer K9 (Promega) was used at a final concentration of 0.66 μM. A total of 5 μL per well (20x working stock of NanoBRET Tracer K9 [13.2 μM] in Tracer Dilution Buffer (Promega N291B) was added to all wells, except the “no tracer” control wells. All test compounds were prepared initially as concentrated (10 mM) stock solutions in 100% DMSO (Sigma), then diluted in Opti-MEM media (99%) to prepare 1 % DMSO working stock solutions. A total of 10 μL per well of the 10-fold test compound stock solutions (final assay concentration 0.1 % DMSO) were added. For “no compound” and “no tracer” control wells, a total of 10 μL per well of Opti-MEM plus DMSO (9 μL Opti-MEM with 1 μL DMSO) was added, final concentration 1 % DMSO). 96-well plates containing cells with NanoBRET Tracer K9 and test compounds at eleven concentrations (100 μL total volume per well) were equilibrated (37 °C / 5 % CO2) for 2 h. The plates were cooled to room temperature for 15 min. To measure NanoBRET signal, NanoGlo substrate (Promega) at a ratio of 1:166 to Opti-MEM media in combination with extracellular NanoLuc Inhibitor (Promega) diluted 1:500 (10 μL [30 mM stock] per 5 mL Opti-MEM plus substrate) were combined to create a 3X stock solution. A total of 50 μL of the 3X substrate/extracellular NanoLuc inhibitor were added to each well. The plates were read within 10 min on a GloMax Discover luminometer (Promega, Madison, WI, USA) equipped with 450 nm BP filter (donor) and 600 nm LP filter (acceptor), using 0.3 s integration time utilizing the “NanoBRET 618” protocol. Raw milliBRET (mBRET) values were obtained by dividing the acceptor emission values (600 nm) by the donor emission values (450 nm) then multiplying by 1000. Averaged control values were used to represent complete inhibition (no tracer control: Opti-MEM + DMSO only) and no inhibition (tracer only control: no compound, Opti-MEM + DMSO + Tracer K9 only) and were plotted alongside the raw mBRET values. Biological replicates were normalized and fit using Sigmoidal, 4PL binding curve in Prism Software (version 8, GraphPad, La Jolla, CA, USA). The experiment was performed n = 3. Average IC50 and standard error (SE) were calculated in Prism Software.
Kinetic solubility:
Assays were performed at Analiza, Inc. 50-fold dilutions of each DMSO stock solution were prepared in singleton by combining 6 μL of DMSO stock with 294 μL of the appropriate media in a Millipore solubility filter plate with 0.45 μM polycarbonate filter membrane using Hamilton Starlet liquid handling. The final DMSO Concentration was 2.0% and maximum theoretical compound concentration was 200 μM (assuming stock concentration of 10 mM). The filter plate was heat sealed for the duration of the 24-hour incubation period. Phosphate Buffered Saline solution 10X, PBS (Fisher Bioreagent part number BP399-500) 50 mL was added to approximately 450 mL HPLC grade H2O. The volume of the solution was then adjusted to 500 mL for a total dilution factor of 1:10 and a final PBS concentration of 1X. The samples were place on a rotary shaker (200 RPM) at ambient temperature (22.4 – 24.0 °C) then vacuum filtered. All filtrates were injected into the nitrogen detector for quantification on Analiza’s Automated Discovery Workstation. The solubility was calculated as μg/mL and corrected for background nitrogen.
Microsomal stability:
Assays were performed at Analiza, Inc. 10 mM DMSO stock solutions were diluted to 0.5 mM with DMSO and again to 0.1 mM with acetonitrile. The resulting solution contained 0.1 mM test article in 20% DMSO /80% acetonitrile. A reaction plate was prepared by adding 691.25 μL, pre-warmed (37°C) microsomal solution (0.63 mg/mL protein in 100mM K3PO4 with 1.3mM EDTA) to an empty well of a 96 well plate and maintained at 37 °C. The diluted 0.1 mM compound (8.75 μL) was added to the microsomal solution in the reaction plate and mixed thoroughly by repeated pipetting. The resulting solutions were pre-incubated for 5 min at 37 °C before preparing the T = 0 plates. An aliquot (160 μL) of each reaction solution was added to an empty well of a 96 well plate, as an exact replicate of the Reaction Plate. Cold (4 °C) MeOH (400 μL) was added to each well and mixed thoroughly by repeated pipetting. NADPH Regeneration solution (40 μL) was added to each well and mixed thoroughly by repeated pipetting. For the T = 30-minute Incubation Plates (−NADPH), an aliquot (160 μL) of each reaction solution was added to an empty well of a 96 well plate, as an exact replicate of the Reaction Plate. LC-MS grade H2O (40 μL) was added to each well and mixed thoroughly by repeated pipetting. The plate was sealed and incubated at 37 °C for the incubation period. After incubation, cold (4 °C) MeOH (400 μL) was added to each well and mixed thoroughly by repeated pipetting. For the T= 30-minute Incubation Plate (+NADPH), NADPH (95 μL) was added to the remaining solution (microsomes + test compound) in each well in the previously-prepared reaction plate to initiate the reaction. The plate was sealed and incubated at 37 °C for the incubation period. An aliquot (100 μL) was removed from each well at the desired time point and dispensed into a well of a 96-well plate. 200μL of cold (4 °C) MeOH was added to quench the reaction. All plates were sealed, vortex and centrifuged at 3000 RPM, 4 °C for 15 min, and the supernatants are transferred for analysis by LC-TOFMS. The supernatant (20 μL) was injected onto an AQUASIL C18 column and eluted using a fast-generic gradient program. TOFMS Data was acquired using Agilent 6538 Ultra High Accuracy TOF MS in extended dynamic range (m/z 100-1000) using generic MS conditions in positive mode. Following data acquisition, exact mass extraction and peak integration was performed using MassHunter Software (Agilent Technologies). The stability of the compound is calculated as the percent remaining of the unchanged parent compound at each time point (T= 30 min) relative to the peak area at T=0 minute.
Lipophilicity:
LogD at pH 7.0 was calculated using the ChemAxon web interface https://disco.chemaxon.com/calculators/demo/plugins/logd/
Enzyme assays:
Assays were performed at Eurofins discovery services as previously described.50 Compounds were provided as 10 mM stock solutions and screened in 10 point dose-response at the Km of ATP using the full-length recombinant protein. For STK17B, the enzyme was incubated with 8 mM MOPS pH 7.0, 0.2 mM EDTA, 0.05% BSA, 10 mM sodium chloride, 250 μM KKRPQRRYSNVF, 10 mM Magnesium acetate and [γ-33P-ATP]. The reaction was initiated by the addition of the Mg/ATP mix. After incubation for 2 h at room temperature, the reaction was stopped by the addition of phosphoric acid to a concentration of 0.5%. 10 μl of the stopped reaction was spotted onto a P30 filtermat and washed four times for 4 min in 0.425% phosphoric acid and once in methanol prior to drying and scintillation counting.
Molecular modeling.
Homology model of STK17A:
A homology model for the kinase domain of human STK17A (Uniprot: Q9UEE5) was generated using MODELLER 9.24.31 The crystal structure of human STK17B bound to 11s (PDB: 6Y6H) with R41 in folded conformation A was used as the template. 11s was docked into the STK17A model using AutoDock Vina 1.1.2.51
Molecular dynamics simulation of STK17B in apo form:
The cocrystal structure of human STK17B with 11s (PDB ID: 6Y6H) was downloaded. All the hetero atoms including 11s and water molecules were removed. PDB files corresponding to the P-loop (residues 37–46) with R41 in conformations A and B were generated. Approximately 1.25 μS unbiased full atom MD simulation was performed using GROMACS 2018.152 starting from the two conformations. The hydrogen atoms were converted to virtual sites to remove the fastest vibrational freedom. The protein was parameterized with the amber99sb-ildn and then solvated with TIP3P water model in a dodecahedron box. In order to neutralize the charge on the protein, sodium and chloride ions were added to the system. Verlet cutoff was used to define neighbor list for nonbonded interactions.53 Particle Mesh Ewald (PME) was used to calculate long-range interactions. Energy minimization was performed with steepest-descent algorithm and then with conjugate descent with Fmax < 500 kJmol−1nm−1. The canonical ensemble was carried out by heating the system from 0 K to 310 K, using velocity rescaling for 100 ps.54 The isothermal-isobaric ensemble (P = 1 bar, T = 310 K) was carried out using the Berendsen barostat for 100 ps.55 The unrestrained MD productions were collected using a time step of 5 fs after the isothermal-isobaric ensemble. The trajectories were processed and analyzed using the built-in tools of Gromacs and then visualized with PyMOL 2.3.2.
Molecular dynamics simulation of STK17B in inhibitor bound form:
RosettaLoop56 was used to model the missing residues (187-295) in STK17B bound to 4 (PDB ID: 6Y6F). Unbiased full atom MD simulation was performed using GROMACS 2018.1.52 4 and 11s were parameterized with GAFF (General AMBER Force Field)57 using ACPYPE (AnteChamber PYthon Parser interfacE).58 The protein was parameterized with the amber99sb-ildn and then solvated with TIP3P water model in a dodecahedron box. The charge was neutralized by adding sodium and chloride ions to the system. Verlet cutoff was used to define neighbor list for nonbonded interactions.53 Protein parameterization and simulation protocols were identical to those used in the STK18B apo simulation described above. VMD59 was used to calculate RMSD and H-bond frequency. Xmgrace was used to plot the data. Trajectory was visualized with PyMOL 2.3.2.
Sequence alignment of STK17A and STK17B orthologs:
Kinase domains of STK17A and STK17B orthologs were identified using previously curated hierarchical eukaryotic Protein Kinase (ePK) profiles.60 The sequences were then aligned with MAFFT version 7.450.61 The alignment was then visualized using BOXSHADE version 3.2.1.
Basicity calculation.
pKa was obtained from density functional theory (DFT) calculations using the molecular electrostatic potential as the descriptor. All DFT calculations were performed with the Gaussian 16 package version A0362 with tight self-consistent field convergence criterion and ultrafine integration grids. A full structure optimization was first carried out at the B3LYP/6-311+G(d,p) level of theory. After the structure optimization, single point calculations were performed to obtain the molecular electrostatic potential using the pop=chelpg keyword63 for each of the nuclei followed by a full NBO analysis.64
Supplementary Material
SI Table 1: Experimental and calculated pKa values for thienopyridine.
SI Table 2: X-ray data collection and refinement statistics.
SI Figure 1: SMARTCyp prediction of most liable points to metabolism in 4.
SI Figure 2: X-ray structure of the catalytic domain of STK17B/11h complex.
SI Figure 3: Multiple sequence alignment of the kinase domain of STK17A and STK17B orthologs.
SI Figure 4: Binding pocket comparison of STK17A/11s with STK17B/11s.
SI Movie 1: MD of apo-STK17B conformation A. (SI Movie 1.mp4)
SI Movie 2: MD of apo-STK17B conformation B. (SI Movie 2.mp4)
SI Movie 3: MD of STK17B/4. (SI Movie 3.mp4)
SI Movie 4: MD of STK17B/11s conformation A. (SI Movie 4.mp4)
SI Movie 5: MD of STK17B/11s conformation B. (SI Movie 5.mp4)
SI File 1: DiscoverX KINOMEscan data. (SI File 1.xlsx)
SI File 5: Coordinates of human STK17A homology model (SI File 5.pdb)
SI File 2: NMR spectra. (SI File 2.pdf)
SI File 3: Purity traces for lead compounds (SI File 3.pdf)
SI File 4: Molecular formula strings (SI File 4.cvs)
Acknowledgements
The SGC, a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative EUbOPEN (No 875510), Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome. Funding for this project was provided by National Institutes of Health (1R44TR001916-02) to Luceome Biotechnologies and the SGC-UNC. Additional funding for the SGC-UNC was provided by UNC Lineberger Comprehensive Cancer Center, PharmAlliance, and National Institutes of Health (1R01CA218442-01 and 1U24DK116204-01). S.K. is grateful for support by the German cancer network (DKTK) and the Frankfurt cancer center (FCI). Funding for N.K. from the National Institutes of Health (1U01CA239106-01) is acknowledged. The authors thank staff at Diamond Light Source, BESSY II and DESY for their assistance during data collection, and the UNC Department of Chemistry Mass Spectrometry Core Laboratory for their assistance with compound characterization. NanoBRET assays were aided by the provision of a CAMKK2-NLuc clone by Carol C. Preta and Priscila Zonzini (SGC-UNICAMP, Brazil). Other NanoBRET constructs and tracer K9 were kindly provided by Promega Corporation (Madison WI). The authors thank Alexander Tropsha (UNC Chapel Hill) for insights and discussion of computational models and electrostatics calculations.
Abbreviations
- NanoBRET
bioluminescense resonace energy transfer assay, trademark of Promega Corporation
- STK17A
serine/threonine-protein kinase 17A
- STK17B
serine/threonine-protein kinase 17B
- DRAK1
DAP kinase-related apoptosis-inducing protein kinase (DRAK)1
- DRAK2
DAP kinase-related apoptosis-inducing protein kinase (DRAK)2
- TGFβ1
Transforming Growth Factor Beta 1
- DFG
“Asp-Phe-Gly”
- MAP4K4
Mitogen-activated protein kinase kinase kinase kinase 4
- PKIS2
Published Kinase Inhibitor Set 2
- TPL2
tumor progression locus 2 kinase
- MAPK3K8
mitogen-activated protein kinase 3K8
- COT
cancer Osaka thyroid kinase
- KD
dissociation constant
- SNAr
nucleophilic aromatic substitution
- LogD
logarithm of distribution coefficient
- CAMKK1
Calcium/Calmodulin Dependent Protein Kinase Kinase 1
- CAMKK2
Calcium/Calmodulin Dependent Protein Kinase Kinase 2
- AURKB
Aurora kinase B
- PIM2
Serine/threonine-protein kinase PIM-2
- MET
Mesenchymal epithelial transition kinase
- NEK6
Serine/threonine-protein kinase Nek6
- WEE1
Wee1-like protein kinase
- STK38L
Serine/threonine-protein kinase 38-like
- HIPK4
Homeodomain-interacting protein kinase 4
- pka
-logarithm of acid dissociation constant Ka
- CSNK2
casein kinase II
- CDCl3
deuterated chloroform
- EtOAc
ethyl acetate
- EtOH
ethanol
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- TCEP
tris(2-carboxyethyl)phosphine
- Fluc
split-firefly luciferase pairs
- ALU
Arithmetic logic unit
- NanoLuc
luciferase trademark of Promega Corporation
- FuGene HD
transfections reagent trademark of Promega Corporation
- DMEM
Dulbecco's Modified Eagle Medium
- FBS
fetal bovine serum
- LC-TOFMS
Liquid chromatography-time-of-flight-mass spectrometry
- MOPS
3-(N-morpholino)propanesulfonic acid
Footnotes
PDB ID of New Crystal (X-ray) Structures
6Y6F STK17B/4
6Y6H STK17B/11h
6ZJF STK17B/11s
3LM5 STK17B/1
7AKG STK17B/2
Authors will release the atomic coordinates upon article publication.
References
- 1.Taylor SS; Radzio-Andzelm E, Three protein kinase structures define a common motif. Structure 1994, 2 (5), 345–355. [DOI] [PubMed] [Google Scholar]
- 2.Manning G; Whyte DB; Martinez R; Hunter T; Sudarsanam S, The protein kinase complement of the human genome. Science 2002, 298 (5600), 1912–1934. [DOI] [PubMed] [Google Scholar]
- 3.Shiloh R; Bialik S; Kimchi A, The DAPK family: a structure-function analysis. Apoptosis 2014, 19 (2), 286–297. [DOI] [PubMed] [Google Scholar]
- 4.Farag AK; Roh EJ, Death-associated protein kinase (DAPK) family modulators: Current and future therapeutic outcomes. Med Res Rev 2019, 39 (1), 349–385. [DOI] [PubMed] [Google Scholar]
- 5.McGargill MA; Choy C; Wen BG; Hedrick SM, Drak2 regulates the survival of activated T cells and is required for organ-specific autoimmune disease. J Immunol 2008, 181 (11), 7593–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramos SJ; Hernandez JB; Gatzka M; Walsh CM, Enhanced T cell apoptosis within Drak2-deficient mice promotes resistance to autoimmunity. J Immunol 2008, 181 (11), 7606–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boly R; Gras T; Lamkami T; Guissou P; Serteyn D; Kiss R; Dubois J, Quercetin inhibits a large panel of kinases implicated in cancer cell biology. Int J Oncol 2011, 38 (3), 833–842. [DOI] [PubMed] [Google Scholar]
- 8.Hsu KC; Sung TY; Lin CT; Chiu YY; Hsu JT; Hung HC; Sun CM; Barve I; Chen WL; Huang WC; Huang CT; Chen CH; Yang JM, Anchor-based classification and type-C inhibitors for tyrosine kinases. Sci Rep 2015, 5, 10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Library of Integrated Network-based Cellular Signatures: TKI-258 KINOMEscan - Dataset (ID:20168). http://lincs.hms.harvard.edu/db/datasets/20168/results?proteins=200117&proteins=200118. [Google Scholar]
- 10.Leonczak P; Gao LJ; Ramadori AT; Lescrinier E; Rozenski J; De Jonghe S; Herdewijn P, Synthesis and structure-activity relationship studies of 2-(1,3,4-oxadiazole-2(3H)-thione)-3-amino-5-arylthieno[2,3-b]pyridines as inhibitors of DRAK2. ChemMedChem 2014, 9 (11), 2587–2601. [DOI] [PubMed] [Google Scholar]
- 11.Jung ME; Byun BJ; Kim HM; Lee JY; Park JH; Lee N; Son YH; Choi SU; Yang KM; Kim SJ; Lee K; Kim YC; Choi G, Discovery of indirubin derivatives as new class of DRAK2 inhibitors from high throughput screening. Bioorg Med Chem Lett 2016, 26 (11), 2719–2723. [DOI] [PubMed] [Google Scholar]
- 12.Ali I; Park S; Jung ME; Lee N; Bibi M; Chae CH; Yang K-M; Kim S-J; Choi G; Lee K, Identification of TRD-35 as potent and selective DRAK2 inhibitor. Bull. Korean Chem. Soc. 2020, 41 (5), 567–569. [Google Scholar]
- 13.Wang S; Xu L; Lu YT; Liu YF; Han B; Liu T; Tang J; Li J; Wu J; Li JY; Yu LF; Yang F, Discovery of benzofuran-3(2H)-one derivatives as novel DRAK2 inhibitors that protect islet beta-cells from apoptosis. Eur J Med Chem 2017, 130, 195–208. [DOI] [PubMed] [Google Scholar]
- 14.Huang D; Zhou T; Lafleur K; Nevado C; Caflisch A, Kinase selectivity potential for inhibitors targeting the ATP binding site: a network analysis. Bioinformatics 2010, 26 (2), 198–204. [DOI] [PubMed] [Google Scholar]
- 15.Muller S; Chaikuad A; Gray NS; Knapp S, The ins and outs of selective kinase inhibitor development. Nat Chem Biol 2015, 11 (11), 818–821. [DOI] [PubMed] [Google Scholar]
- 16.Hari SB; Perera BG; Ranjitkar P; Seeliger MA; Maly DJ, Conformation-selective inhibitors reveal differences in the activation and phosphate-binding loops of the tyrosine kinases Abl and Src. ACS Chem Biol 2013, 8 (12), 2734–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guimaraes CR; Rai BK; Munchhof MJ; Liu S; Wang J; Bhattacharya SK; Buckbinder L, Understanding the impact of the P-loop conformation on kinase selectivity. J Chem Inf Model 2011, 51 (6), 1199–1204. [DOI] [PubMed] [Google Scholar]
- 18.Drewry DH; Wells CI; Andrews DM; Angell R; Al-Ali H; Axtman AD; Capuzzi SJ; Elkins JM; Ettmayer P; Frederiksen M; Gileadi O; Gray N; Hooper A; Knapp S; Laufer S; Luecking U; Michaelides M; Muller S; Muratov E; Denny RA; Saikatendu KS; Treiber DK; Zuercher WJ; Willson TM, Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS One 2017, 12 (8), e0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ni Y; Gopalsamy A; Cole D; Hu Y; Denny R; Ipek M; Liu J; Lee J; Hall JP; Luong M; Telliez JB; Lin LL, Identification and SAR of a new series of thieno[3,2-d]pyrimidines as Tpl2 kinase inhibitors. Bioorg Med Chem Lett 2011, 21 (19), 5952–5956. [DOI] [PubMed] [Google Scholar]
- 20.Drewry DH; Wells CI; Zuercher WJ; Willson TM, A Perspective on Extreme Open Science: Companies Sharing Compounds without Restriction. SLAS Discov 2019, 24 (5), 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieske J; Cerv M; Kreida S; Komadina D; Fischer J; Barthelmess M; Fischer P; Pakendorf T; Yefanov O; Mariani V; Seine T; Ross BH; Crosas E; Lorbeer O; Burkhardt A; Lane TJ; Guenther S; Bergtholdt J; Schoen S; Tornroth-Horsefield S; Chapman HN; Meents A, On-chip crystallization for serial crystallography experiments and on-chip ligand-binding studies. IUCrJ 2019, 6 (Pt 4), 714–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rydberg P; Gloriam DE; Zaretzki J; Breneman C; Olsen L, SMARTCyp: A 2D method for prediction of cytochrome P450-mediated drug metabolism. ACS Med Chem Lett 2010, 1 (3), 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jester BW; Cox KJ; Gaj A; Shomin CD; Porter JR; Ghosh I, A coiled-coil enabled split-luciferase three-hybrid system: applied toward profiling inhibitors of protein kinases. J Am Chem Soc 2010, 132 (33), 11727–11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vasta JD; Corona CR; Wilkinson J; Zimprich CA; Hartnett JR; Ingold MR; Zimmerman K; Machleidt T; Kirkland TA; Huwiler KG; Ohana RF; Slater M; Otto P; Cong M; Wells CI; Berger BT; Hanke T; Glas C; Ding K; Drewry DH; Huber KVM; Willson TM; Knapp S; Müller S; Meisenheimer PL; Fan F; Wood KV; Robers MB, Quantitative, wide-spectrum kinase profiling in live cells for assessing the effect of cellular ATP on target engagement. Cell Chem Biol 2018, 25 (2), 206–214.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roskoski R Jr., Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol Res 2020, 152, 104609. [DOI] [PubMed] [Google Scholar]
- 26.Arrowsmith CH; Audia JE; Austin C; Baell J; Bennett J; Blagg J; Bountra C; Brennan PE; Brown PJ; Bunnage ME; Buser-Doepner C; Campbell RM; Carter AJ; Cohen P; Copeland RA; Cravatt B; Dahlin JL; Dhanak D; Edwards AM; Frederiksen M; Frye SV; Gray N; Grimshaw CE; Hepworth D; Howe T; Huber KV; Jin J; Knapp S; Kotz JD; Kruger RG; Lowe D; Mader MM; Marsden B; Mueller-Fahrnow A; Muller S; O'Hagan RC; Overington JP; Owen DR; Rosenberg SH; Roth B; Ross R; Schapira M; Schreiber SL; Shoichet B; Sundstrom M; Superti-Furga G; Taunton J; Toledo-Sherman L; Walpole C; Walters MA; Willson TM; Workman P; Young RN; Zuercher WJ, The promise and peril of chemical probes. Nat Chem Biol 2015, 11 (8), 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao X; Rong C; Zhong A; Lu T; Liu S, Molecular acidity: An accurate description with information-theoretic approach in density functional reactivity theory. J Comput Chem 2018, 39 (2), 117–129. [DOI] [PubMed] [Google Scholar]
- 28.Liu S; Pedersen LG, Estimation of molecular acidity via electrostatic potential at the nucleus and valence natural atomic orbitals. J Phys Chem A 2009, 113 (15), 3648–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klemm LH; Jacquot RD, Basicities of thienopyridines. Potentiometric determination and use in effecting chemical separations. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry 1973, 45 (2), 181–186. [Google Scholar]
- 30.Kenny PW, Hydrogen bonding, electrostatic potential, and molecular design. J Chem Inf Model 2009, 49 (5), 1234–1244. [DOI] [PubMed] [Google Scholar]
- 31.Webb B; Sali A, Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics 2016, 54, 5.6.1–5.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knapp S; Arruda P; Blagg J; Burley S; Drewry DH; Edwards A; Fabbro D; Gillespie P; Gray NS; Kuster B; Lackey KE; Mazzafera P; Tomkinson NC; Willson TM; Workman P; Zuercher WJ, A public-private partnership to unlock the untargeted kinome. Nat Chem Biol 2013, 9 (1), 3–6. [DOI] [PubMed] [Google Scholar]
- 33.Fedorov O; Muller S; Knapp S, The (un)targeted cancer kinome. Nat Chem Biol 2010, 6 (3), 166–169. [DOI] [PubMed] [Google Scholar]
- 34.Gatzka M; Newton RH; Walsh CM, Altered thymic selection and increased autoimmunity caused by ectopic expression of DRAK2 during T cell development. J Immunol 2009, 183 (1), 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGargill MA; Wen BG; Walsh CM; Hedrick SM, A deficiency in Drak2 results in a T cell hypersensitivity and an unexpected resistance to autoimmunity. Immunity 2004, 21 (6), 781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miduturu CV; Deng X; Kwiatkowski N; Yang W; Brault L; Filippakopoulos P; Chung E; Yang Q; Schwaller J; Knapp S; King RW; Lee JD; Herrgard S; Zarrinkar P; Gray NS, High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem Biol 2011, 18 (7), 868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falke H; Chaikuad A; Becker A; Loaec N; Lozach O; Abu Jhaisha S; Becker W; Jones PG; Preu L; Baumann K; Knapp S; Meijer L; Kunick C, 10-iodo-11H-indolo[3,2-c]quinoline-6-carboxylic acids are selective inhibitors of DYRK1A. J Med Chem 2015, 58 (7), 3131–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parker LJ; Watanabe H; Tsuganezawa K; Tomabechi Y; Handa N; Shirouzu M; Yuki H; Honma T; Ogawa N; Nagano T; Yokoyama S; Tanaka A, Flexibility of the P-loop of Pim-1 kinase: observation of a novel conformation induced by interaction with an inhibitor. Acta Crystallogr Sect F Struct Biol Cryst Commun 2012, 68 (Pt 8), 860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xing L; Rai B; Lunney EA, Scaffold mining of kinase hinge binders in crystal structure database. J Comput Aided Mol Des 2014, 28 (1), 13–23. [DOI] [PubMed] [Google Scholar]
- 40.Xing L; Klug-Mcleod J; Rai B; Lunney EA, Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg Med Chem 2015, 23 (19), 6520–6527. [DOI] [PubMed] [Google Scholar]
- 41.Attia MH; Elrazaz EZ; El-Emam SZ; Taher AT; Abdel-Aziz HA; Abouzid KAM, Synthesis and in-vitro anti-proliferative evaluation of some pyrazolo[1,5-a]pyrimidines as novel larotrectinib analogs. Bioorg Chem 2020, 94, 103458. [DOI] [PubMed] [Google Scholar]
- 42.Golub AG; Bdzhola VG; Briukhovetska NV; Balanda AO; Kukharenko OP; Kotey IM; Ostrynska OV; Yarmoluk SM, Synthesis and biological evaluation of substituted (thieno[2,3-d]pyrimidin-4-ylthio)carboxylic acids as inhibitors of human protein kinase CK2. Eur J Med Chem 2011, 46 (3), 870–876. [DOI] [PubMed] [Google Scholar]
- 43.O'Byrne SN; Scott JW; Pilotte JR; Santiago ADS; Langendorf CG; Oakhill JS; Eduful BJ; Counago RM; Wells CI; Zuercher WJ; Willson TM; Drewry DH, In depth analysis of kinase cross screening data to identify CAMKK2 inhibitory scaffolds. Molecules 2020, 25 (2), 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Powell HR; Battye TGG; Kontogiannis L; Johnson O; Leslie AGW, Integrating macromolecular X-ray diffraction data with the graphical user interface iMosflm. Nat Protoc 2017, 12 (7), 1310–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Potterton L; Agirre J; Ballard C; Cowtan K; Dodson E; Evans PR; Jenkins HT; Keegan R; Krissinel E; Stevenson K; Lebedev A; McNicholas SJ; Nicholls RA; Noble M; Pannu NS; Roth C; Sheldrick G; Skubak P; Turkenburg J; Uski V; von Delft F; Waterman D; Wilson K; Winn M; Wojdyr M, CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr D Struct Biol 2018, 74 (Pt 2), 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCoy AJ, Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr 2007, 63 (Pt 1), 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murshudov GN; Skubak P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA, REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 2011, 67 (Pt 4), 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis MI; Hunt JP; Herrgard S; Ciceri P; Wodicka LM; Pallares G; Hocker M; Treiber DK; Zarrinkar PP, Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 2011, 29 (11), 1046–1051. [DOI] [PubMed] [Google Scholar]
- 50.Gao Y; Davies SP; Augustin M; Woodward A; Patel UA; Kovelman R; Harvey KJ, A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem J 2013, 451 (2), 313–328. [DOI] [PubMed] [Google Scholar]
- 51.Trott O; Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010, 31 (2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abraham MJ; Murtola T; Schulz R; Pall S; Smith JC; Hess B; Lindahl E, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19-25. [Google Scholar]
- 53.Pall S; Hess B, A flexible algorithm for calculating pair interactions on SIMD architectures. Computer Physics Communications 2013, 184 (12), 2641–2650. [Google Scholar]
- 54.Bussi G; Donadio D; Parrinello M, Canonical sampling through velocity rescaling. J Chem Phys 2007, 126 (1), 014101. [DOI] [PubMed] [Google Scholar]
- 55.Berendsen HJC; Postma JPM; van Gunsteren WF; DiNola A; Haak JR, Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81 (8), 3684–3690. [Google Scholar]
- 56.Wang C; Bradley P; Baker D, Protein-protein docking with backbone flexibility. J Mol Biol 2007, 373 (2), 503–519. [DOI] [PubMed] [Google Scholar]
- 57.Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA, Development and testing of a general amber force field. J Comput Chem 2004, 25 (9), 1157–1174. [DOI] [PubMed] [Google Scholar]
- 58.Sousa da Silva AW; Vranken WF, ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res Notes 2012, 5, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Humphrey W; Dalke A; Schulten K, VMD: visual molecular dynamics. J Mol Graph 1996, 14 (1), 33–38. [DOI] [PubMed] [Google Scholar]
- 60.McSkimming DI; Dastgheib S; Baffi TR; Byrne DP; Ferries S; Scott ST; Newton AC; Eyers CE; Kochut KJ; Eyers PA; Kannan N, KinView: a visual comparative sequence analysis tool for integrated kinome research. Mol Biosyst 2016, 12 (12), 3651–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katoh K; Standley DM, MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013, 30 (4), 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JJA; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- 63.Chirlian LE; Francl MM, Atomic charges derived from electrostatic potentials – a detailed study. J. Comp. Chem. 1987, 8, 894–905. [Google Scholar]
- 64.Weinhold F; Carpenter JE, In The Structure of Small Molecules and Ions, Naaman R; Vager Z, Eds. Plenum: 1988; pp 227–236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SI Table 1: Experimental and calculated pKa values for thienopyridine.
SI Table 2: X-ray data collection and refinement statistics.
SI Figure 1: SMARTCyp prediction of most liable points to metabolism in 4.
SI Figure 2: X-ray structure of the catalytic domain of STK17B/11h complex.
SI Figure 3: Multiple sequence alignment of the kinase domain of STK17A and STK17B orthologs.
SI Figure 4: Binding pocket comparison of STK17A/11s with STK17B/11s.
SI Movie 1: MD of apo-STK17B conformation A. (SI Movie 1.mp4)
SI Movie 2: MD of apo-STK17B conformation B. (SI Movie 2.mp4)
SI Movie 3: MD of STK17B/4. (SI Movie 3.mp4)
SI Movie 4: MD of STK17B/11s conformation A. (SI Movie 4.mp4)
SI Movie 5: MD of STK17B/11s conformation B. (SI Movie 5.mp4)
SI File 1: DiscoverX KINOMEscan data. (SI File 1.xlsx)
SI File 5: Coordinates of human STK17A homology model (SI File 5.pdb)
SI File 2: NMR spectra. (SI File 2.pdf)
SI File 3: Purity traces for lead compounds (SI File 3.pdf)
SI File 4: Molecular formula strings (SI File 4.cvs)