

Abstract
Helicobacter pylori, a Gram‐negative bacterium, is associated with a wide range of gastric diseases such as gastritis, duodenal ulcer, and gastric cancer. The prevalence of H pylori and risk of disease vary in different parts of the world based on the prevailing bacterial lineage. Here, we present a contextual and comparative genomics analysis of 20 clinical isolates of H pylori from patients in Bangladesh. Despite a uniform host ethnicity (Bengali), isolates were classified as being part of the HpAsia2 (50%) or HpEurope (50%) population. Out of twenty isolates, eighteen isolates were cagA positive, with two HpEurope isolates being cagA negative, three EPIYA motif patterns (AB, AB‐C, and ABC‐C) were observed among the cagA‐positive isolates. Three vacA genotypes were observed with the s1m1i1dic1 genotype observed in 75% of isolates; the s1m2i1d1c2 and s2m2i2d2c2 genotypes were found to be 15% and 10% of isolates, respectively. The non‐virulent genotypes s2m2i2d2c2 was only observed in HpEurope population isolates. Genotypic analysis of oipA gene, present in all isolates, revealed five different patterns of the CT repeat; all HpAsia2 isolates were in “ON” while 20% of HpEurope isolates were genotypically “OFF.” The three blood group antigen binding adhesins encoded genes (bab genes) examined and we observed that the most common genotype was (babA/babB/‐) found in eight isolates, notably six were HpAsia2 isolates. The babA gene was found in all HpAsia2 isolates but present in only half of the HpEurope isolates. In silico antibiotic susceptibility analysis revealed that 40% of the strains were multi‐drug resistant. Mutations associated with resistance to metronidazole, fluoroquinolone, and clarithromycin were detected 90%, 45%, and 5%, respectively, in H pylori strain. In conclusion, it is evident that two populations of H pylori with similar antibiotic profiles are predominant in Bangladesh, and it appears that genotypically the HpAisa2 isolates are potentially more virulent than the HpEurope isolates.
Keywords: genome sequencing, H pylori, outer membrane proteins, phylogeny, virulence
1. INTRODUCTION
Helicobacter pylori is a highly successful human pathogen that colonizes the human gastric mucosa of over half of the world's population. The prevalence of H pylori infection appears to be higher in developing countries than developed countries and its prevalence vary between populations and between groups within the same population. 1 , 2 The prevalence in Asia, Africa, varies from 54.7% to 79.1%, in North and South America the prevalence is 37.1% and 63.4% and in Europe, the prevalence is averages 47.0%. 3 Bangladesh, one of the low socioeconomic country of South Asia with more than 160 million inhabitants, is the eight most populous country in the world. In a study, a high prevalence of H pylori (92%) was reported in Bangladeshi population. 4 , 5 Furthermore, H pylori infection in Bangladesh was reported to be significantly higher in smokers as compared to non‐smokers or subjects below 15 years of age. 6 Despite high occurrence rate of H pylori infection in Bangladesh, the incidence of gastric cancer is low as in other developing countries. 7
Helicobacter pylori has been co‐evolving with humans for more than 60 000 years. 8 This long intimate symbiotic association of H pylori with human has been led to the emergence of different genotypes by accumulation of host specific adaptive changes over the period 9 and is characterized by distinct genotypes that predominate in different geographical regions around the world. 10 Moreover, the occurrence frequency and severity of gastric illnesses associated with H pylori are found to be strongly associated with the dominant genotype in that region. For example, the incidences of gastric cancer is higher in East Asian countries such as Japan and Korea when compared to Western countries. 11
Helicobacter pylori can colonize a host during infancy establishing a chronic infection that can persist for decades, if not eradicated. 12 The bacterium has numerous mechanisms to manipulate and evade host defenses to ensure its stomach survival. To counter the acidic pH in the stomach, H pylori utilizes urease enzyme that forms a cloud of neutral micro‐environment around the bacterium. 13 CagPAI (cytotoxin‐associated gene pathogenicity island) containing several virulence genes that trigger abnormal cellular signals is considered to be the most important risk factor for H pylori‐associated gastric cancer. CagA, the most important virulence factor of CagPAI, plays a crucial role in H pylori pathogenesis. Comparative analysis revealed a significant functional difference in East Asian cagA which showed to induce higher pro‐inflammatory secretions as compared to Western cagA. Furthermore, cagA activates a number of signal transduction pathways that bind and disrupt the function of epithelial junctions, leading to aberrations in functioning of tight junction, cell polarity, and cell differentiation. 14 H pylori also produces vacuolating cytotoxin A (vacA), which after entering in host cells by endocytosis, induces various cellular activities, including membrane channel formation, cytochorome c release leading to cell death, and cell membrane receptor binding, which initiates a proinflammotory response. 15 Similar differences in its functionality like cagA have also been associated with vacA which is believed to be the result of host specific adaptive changes. 16 The gene encoding vacA shows allelic diversity in its signal (s) regions (alleles: s1 and s2) and middle regions (alleles: m1 and m2). In vitro experiments showed s1m1 strains induce cell vacuolation more frequently than s1m2 and s2m2, from which it was inferred that the s1m1 was more cytotoxic. 17 Allelic differences in the intermediate (i) region (alleles: i1 and i2) are suggested to be correlated with severity of disease as compared to the “s” and “m” regions. 18 The more frequently observed deletions in the VacA have been classified and it has been observed that the deletion of 69‐81 bp (d2) type is less virulent than the no deletion (d1) type. 19
The outer membrane proteins of H pylori are considered to be possible virulence factors. OipA (outer inflammatory protein), one member of this large protein family, is involved in bacterial adherence to the gastric epithelial cells and in mucosal inflammation. 20 Additionally OipA is associated with interleukin (IL)‐8 induction, mucosal damage and with duodenal ulcer. 20 In addition to this, the bab genes have been shown to be positively correlated with gastric cancer and duodenal ulcers. 21 , 22 Similar to other H pylori OMPs, BabA has two closely related paralogues, BabB (also known as HopT) and BabC (also known as HopU), although these paralogues are not well characterized.
Inappropriate and irrational use of antibiotics against infectious diseases has already resulted in the emergence of multi‐drug‐resistant bacteria globally. Currently, triple therapy comprising two antibiotics and a proton pump inhibitor is used as eradication regime for H pylori. However, increasing antibiotic resistance trend to metronidazole, clarithromycin, and fluoroquinolones in H pylori has decreased the success rate of its eradication. Inactivation mutations such as frameshift and nonsense mutations, insertions, and deletions of the rdxA and frxA genes suggested to confer resistance to metronidazole in H pylori. 23 In addition, amino acid substitutions in rdxA are also suggested to confer resistance to metronidazole in H pylori. 24 , 25 , 26 , 27 On the other hand, the mechanism of fluoroquinolone resistance in H pylori has been associated to the mutations in quinolone resistance determining regions (QRDR) of the gyrA. 28 Moreover, the strategy employed by H pylori for clarithromycin resistance has been elucidated mainly due to mutations at nucleotide positions (A2142G, A2143G, A2142C, A2146, A2147G, and G2224A) in the 23S rRNA. 29 , 30 , 31
Despite high prevalence of H pylori in Bangladesh, little is known about the local lineages or the local prevalence of particular genotypes for cagA or vacA and babA genotypes and any associations with disease severity. 32 Therefore, this study was directed to evaluate the prevalence of distinct genotypes of cagA, vacA, and babA/B and other virulence factors together with its genotype‐based antibiotic resistance profiles in prevailing Bangladeshi H pylori lineages and also to find the association with clinical outcome using a genomics‐based approach to characterize genotypes and infer lineage classification. The findings would also provide a better understanding of the prevalence of antibiotic‐resistant H pylori strains and its genotypic molecular mechanisms to facilitate the designing of more rational and effective combinatorial antibiotics therapy for eradication of H pylori infection. These data enable contextualization and comparison of drug‐resistant H pylori‐associated disease in Bangladesh with disease in other parts of the world.
2. MATERIALS AND METHODS
The genome sequences for each of 20 randomly selected H pylori isolates collected from a cohort of 174 (125 adult and 49 children) H pylori‐positive symptomatic or asymptomatic patients were determined (File S1). Out of 2010 H pylori isolates, 1% were selected in a two‐step randomization process. In the first step, 20 patients (out of 174) were selected randomly and in the second step one isolate from each patient (from ten single colony isolates stored for each patient) was selected. The selected isolates were sub‐cultured in selective medium (BHI‐7.5% sheep blood plate, 0.4% isovitalex, 0.4% DENT supplement) under microaerophilic conditions (5% O2; 15% CO2; 80% N2) at 37°C for 3‐5 days. 32 Identity of the isolates was confirmed by mass spectrometry using MALDI TOF (Bruker, Germany). Genomic DNA was prepared from confluent growth using a commercial DNA extraction kit (Qiagen DNA Mini kit, Germany). Genomic library was prepared using Nextera DNA sample preparation kit (Illumina, San Diego, CA, USA) and sequenced on an Illumina MiSeq platform at the Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam, using a V3 600 cycles, paired‐end kit (Illumina).
2.1. Bacterial genome assembly and annotation
Paired Fastq files obtained for each isolate were processed as follows. Low‐quality bases were removed and trimmed using the NGS QC toolkit 33 and FAST‐X Toolkit (http://hannonlab.cshl.edu/fastx_toolkit), respectively. Read sets were then assembled using SPAdes to produce a draft genome sequence for each isolate. 34 Contigs were reordered to be consistent with the genome of H pylori 26695 using Contig Layout Authenticator (CLA). 35 Gene prediction and annotation of the assembled draft genomes were carried out by using Prokka. 36 The Artemis genome viewer was used to access specific annotated features in each of the draft genome sequences. 37 tRNA and rRNA were identified in the draft genomes using tRNAScan and RNAmmer, respectively. 38 , 39 The identification of phage‐related regions was carried out using PHASTER. 40 In addition, sequences of CagPAI‐positive and CagPAI‐negative isolates were aligned against H pylori 26695 (Typical HpEurope) and H pylori F57 (Typical HpEastAsia) reference strains using Blast Ring Image Generator (BRIG). 41
2.2. Phylogenetic analysis
Thirty‐one 31 reference H pylori genome sequences representing different population/lineages were downloaded from the National Center for Biotechnology Information (NCBI) (Listed in Table S1) database. All sequences from the present study and the reference sequences were used to construct whole genome‐based phylogenetic tree using Harvest. 42 The H pylori 26695 genome sequence was used as a reference for the core genome alignment. The tool was run to build a core genome‐based phylogenetic tree that excluded recombination regions as previously done by Kumar et al 43 The phylogenetic tree was visualized in interactive tree of life (iTOL). 44
2.3. Core genome and pan genome analysis
OrthoMCL was used to identify orthologous gene clusters using predicted protein sequences of all studied isolates (minimum threshold of 50 amino acid in length with identity and e‐value parameters were at 70% and 0.00001, respectively). 45 The identified genes were aligned against the EggNOG database to define their functional categories. Genes contained more than one domain of distinct categories were classified as multiple class genes. The functional categories were graphically represented using R (http://www.R-project.org). Genes without appropriate hit against the database were classified as unknown genes. Furthermore, H pylori isolate‐specific genes were also identified using OrthMCL followed by an in‐house perl script. Similarly, the functionally categorized strain‐specific genes by EggNOG were depicted graphically using R. In addition, a floral Venn diagram depicted the number of core genes and isolate‐specific genes was produced using R. In addition to this, the core and specific gene content of HpAsia2 and HpEurope strains were also analyzed separately using OrthoMCL with the above‐mentioned parameters.
2.4. Identification of virulence‐related and outer membrane proteins
The amino acid sequences of the predicted genes were compared with the H pylori virulence genes listed in VFDB. 46 Similarly, major OMPs from H pylori J99 were used to detect the genes encoding these protein in each of the isolates as described previously 47 using BLASTp. Identity and coverage 80% and 70%, respectively, was used as the detection threshold. 48 Among the virulence genes, we mainly focused on the presence and absence of BabA, BabB, and BabC gene and translation potential of a major OMP, OipA in the isolates.
2.5. Phylogenetic analysis of CagA and VacA genotypes
The nucleotide sequences of the cagA and vacA genes from the genomes of the isolates and from the genomes of ten representative H pylori genomes from a range of genotypes from NCBI (Table S1) were extracted and aligned using CLUSTALW as implemented in MEGA 5.2. 49 These alignments were then utilized to construct cagA and vacA gene‐based phylogenetic tree using neighbor‐joining algorithm with 1000 bootstraps values. The output tree was visualized in iTOL in each instance. The multiple sequence alignments were also used for the allelic/genotypic classification of the cagA and vacA gene present in each isolate.
2.6. In silico analysis of antibiotic susceptibility
Blastn search was performed to extract rdxA, frxA, gyrA, and 23S rRNA nucleotide sequences from each strain using reference genes hp0954, hp0642, hp0701, and hpr01, respectively, of H pylori 26695. Extracted rdxA, frxA, and gyrA gene sequences were then translated into amino acid sequences prior to alignment. Sequence alignment of extracted 23S rRNA gene of each strain was performed at nucleotide level. Aligned sequences were then compared with reference sequence of H pylori 26695 to examine for reported and novel mutations.
3. RESULTS
3.1. Patient information and strains isolation
Twenty H pylori isolates, one each from 20 patients were selected through 2‐step randomization procedure and were analyzed. The average age of the patients was 29 years; 7‐59 years range. Seventy percent (14/20) of the patients were male. None of the patients had any other concomitant illness. Socioeconomically, 15% (3/20), 25% (5/20), and 60% (12/20) of the patients were from a high (>5000 taka/month), medium (2500‐5000 taka/month), or low income group (<2500 taka/month), respectively. More than half, 65% (13/20), of the patients had basic literacy skills (5‐10 years schooling) and 35% were illiterate. Twenty‐five‐percent (5/20) of the patients were smokers and none reported raw tobacco chewing or alcohol intake. Thirty percent (6/20) of the patients reported dyspepsia, heartburn, and occasional H2 blocker use. Among the isolates 12 (60%) were from gastric antrum, seven (35%) from gastric body and one (5%) were from gastric juice.
3.2. Genome characteristics
Table 1 presents the general characteristics for each of the 20 genome sequences. There were 28‐70 contigs with a high (40×–1070×) genome coverage. The mean genome size was 1.6 Mb and the mean G + C content was 38.9%. Each genome contains between 1586 and 1710 annotated CDS with an average ~92% of the genome is used for the coding region.
Table 1.
Genome statistics of the whole genome sequences of the 20 H pylori isolates in this study
| Strains designation | Genome coverage | No. of contigs | Genome size | No. of CDS | Coding percentage | G + C percentage | rRNA | tRNA | EPIYA Motif |
|---|---|---|---|---|---|---|---|---|---|
| 17A6 | 92 | 61 | 1643694 | 1710 | 91.9 | 38.85 | 3 | 36 | AB‐C |
| 19B6 | 84 | 38 | 1600763 | 1601 | 92.2 | 39.01 | 4 | 37 | AB‐C |
| 25b2 | 140 | 41 | 1641783 | 1695 | 92.5 | 38.79 | 4 | 36 | AB‐C |
| 20A8 | 77 | 40 | 1659973 | 1692 | 92.4 | 38.9 | 3 | 37 | AB‐C |
| 28B4 | 82 | 22 | 1616466 | 1635 | 92.5 | 39.02 | 4 | 37 | AB‐C |
| 37A5 | 111 | 70 | 1662458 | 1700 | 91.7 | 38.85 | 3 | 36 | AB‐C |
| 40A6 | 139 | 53 | 1640998 | 1660 | 92.1 | 38.84 | 3 | 37 | AB‐C |
| 43a2 | 40 | 39 | 1635561 | 1648 | 92.2 | 38.86 | 3 | 37 | AB‐C |
| 44A4 | 92 | 35 | 1591152 | 1608 | 92.5 | 39.04 | 4 | 36 | AB‐C |
| 59a9 | 80 | 42 | 1641723 | 1694 | 92.6 | 38.8 | 4 | 36 | AB‐C |
| 60A7 | 61 | 46 | 1642721 | 1654 | 92.1 | 38.85 | 4 | 37 | AB |
| 61A5 | 78 | 36 | 1602458 | 1600 | 91.3 | 38.92 | 6 | 36 | AB‐C |
| 149A3 | 1070 | 49 | 1634281 | 1672 | 92.5 | 38.9 | 3 | 36 | AB‐CC |
| 86A5 | 103 | 28 | 1616047 | 1640 | 92.3 | 38.92 | 4 | 37 | AB‐C |
| 88A4 | 107 | 43 | 1617524 | 1651 | 92.3 | 38.9 | 4 | 37 | AB‐C |
| 89B9 | 80 | 33 | 1619489 | 1614 | 92.2 | 38.91 | 4 | 37 | AB‐C |
| GJ906 | 79 | 30 | 1612378 | 1639 | 92.3 | 38.9 | 4 | 37 | AB‐C |
| S106A3 | 51 | 40 | 1609865 | 1624 | 92.4 | 39.03 | 4 | 37 | AB‐C |
| S108A3 | 68 | 41 | 1645721 | 1668 | 91.9 | 38.84 | 5 | 36 | AB‐CC |
| 152B5 | 89 | 28 | 1579779 | 1586 | 92.3 | 38.99 | 4 | 36 | AB‐C |
Fifty‐five percent (11/20) of the isolate genomes harbored one/two either intact, incomplete phage sequence (4.8‐52.6 kb). The phage sequences consisting of between 9 and 39 CDSs, encode helicase, terminase, integrase, transposase, methylase, and other hypothetical proteins in addition to phage‐related genes (Table S2).
The OrthoMCL analysis identified 30 267 proteins forming 1765 orthologus gene clusters. The core genome consisted of 1256 orthologous gene clusters. Out of 1256 core gene clusters, 1094 clusters could be assigned to different functional classes by EggNOG database (Figure S1A). A high proportion (~11%, 117/1094) of genes belong to J (translation, ribosomal structure and biogenesis) and M (cell membrane/envelope biogenesis) functional classes. Approximately 15% (168/1094) of genes were classified as unknown where the function could not be predicted. In addition, 212 unique genes were identified in the 20 isolates and only 99 of these were assigned to various classes by EggNOG database. The majority were classified the S (hypothetical) or L (replication, recombination, and repair) functional classes (Figure S1B). A small but variable number of unique genes were identified for each isolate (Figure S1C). Moreover, an additional performance of OrthoMCL analysis predicted 1712 and 1669 orthologous gene clusters in HpAsia2 and HpEurope strains, respectively. The core of HpAsia2 contained 1308 genes while 1295 genes were present in the core of HpEurope. In addition to this, 142 and 210 genes were identified as unique in HpAsia2 and HpEurope strains, respectively. In both lineages, majority of the unique genes were classified to L (replication, recombination and repair) and S (function unknown) classes.
3.3. Core genome phylogenetic analysis
We used whole genome SNP‐based phylogenetic analysis to infer the lineage of each of the isolates. All isolates from the present study were separated into two distinct population; 50% (10/20) of the isolates belonged to HpAsia2 (50%) and 50% (10/20) to HpEurope cluster despite being from one ethnicity (Figure 1).
Figure 1.
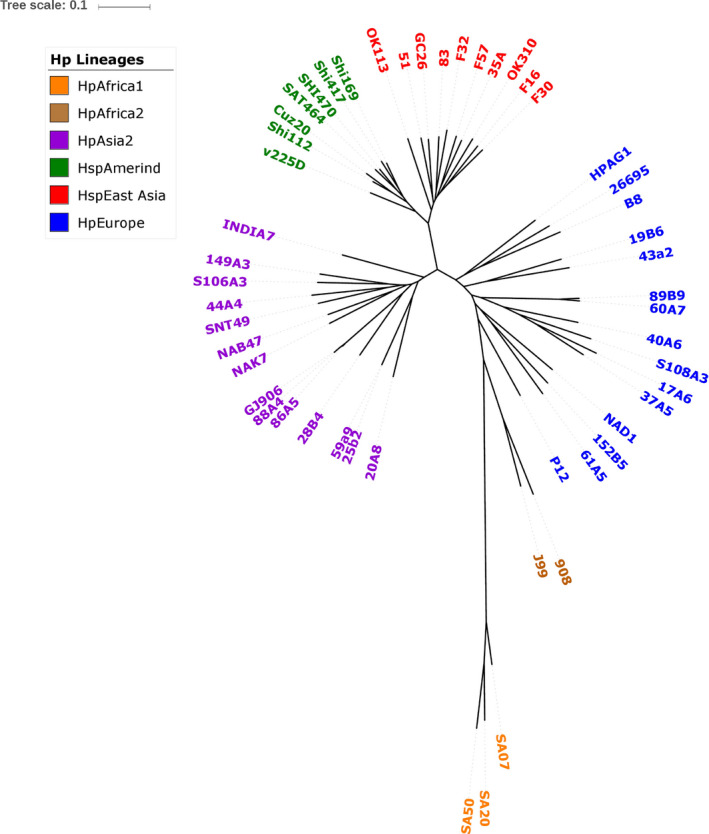
Whole genome core SNP‐based phylogenetic tree of the 20 Bangladeshi H pylori isolates and 31 H pylori reference genomes. The analysis was done using Harvest tool and the output of Harvest was visualized using iTOL
3.4. Identification of virulence genes
3.4.1. Cag Pathogenicity Island and CagA genotype
Ninety percent (18/20) of the isolate genomes harbored a complete CagPAI. Of these, ten CagPAI were associated with HpAsia2 lineage isolates and eight belonged to HpEurope lineage isolates. All CagPAI of HpAsia2 lineage isolates were lacking both DNA and DNA‐RNA helicases coding sequences while five of the HpEurope lineage isolates were missing only DNA helicase when compared with Western type CagPAI sequence of 26695 strain (Figure S2A). Comparison of CagPAI sequences with East Asian strain (F57) showed that six isolates (five HpAsia2 and one HpEurope) manifested a major gap of 2093 kb which encodes for a hypothetical protein in F57 (Figure S2B).
Virulence factor mapping indicates most of the Bangladesh isolates harbored 118 to 131 virulence genes with the exception of two (61A5, 152B5; CagPAI‐negative strain lacking cag1 to cag5 and cagA to cagZ) (Figure S3). Moreover, all urease enzymes, most of the flagellar associated proteins, endotoxins, and most of the Lewis antigens such as FutA, FutB, and NeuA/FlmD were found in most of the isolates (Figure S3).
The phylogenetic analysis of cagA gene of 18 Bangladeshi H pylori isolates together with five Western and five East Asian cagA gene sequences, revealed a close clustering with the Western strains and not with East Asian strains as shown in Figure 2. In all Western type cagA, 55.5% (10/18) of the population were clustered with HpAsia2 while 44.45% with HpEurope (Figure 1). A closer examination of these gene sequences revealed AB‐C EPIYA motif in 15 (75%) isolates which is more reflective of HpEurope lineage. Two (10%) isolates had one extra EPIYA‐C motif (ABC‐C type) as compared to normal AB‐C EPIYA motif in which one was associated with HpAsia2 and other with HpEurope. Moreover, one isolate belonged to HpEurope was lacking EPIYA‐(C) motif (AB type) (Table 2). Seven cagA‐encoded strains (six AB‐C type and one ABC‐C) possessed an EPIY (T) B‐type motif while remaining isolates (9 AB‐C, 1 ABC‐C, and 1 AB types) contained an EPIY (A) B‐type motif at C‐terminal. In addition to this, we found that 5 out of 7 EPIY (T) B‐type motif harboring isolates were associated to HpAsia2 (25b2, 59a9, 86A5, 88A4, and GJ906) while only 2 belonged to HpEurope (17A6 and S108A3) (Figure 1).
Figure 2.
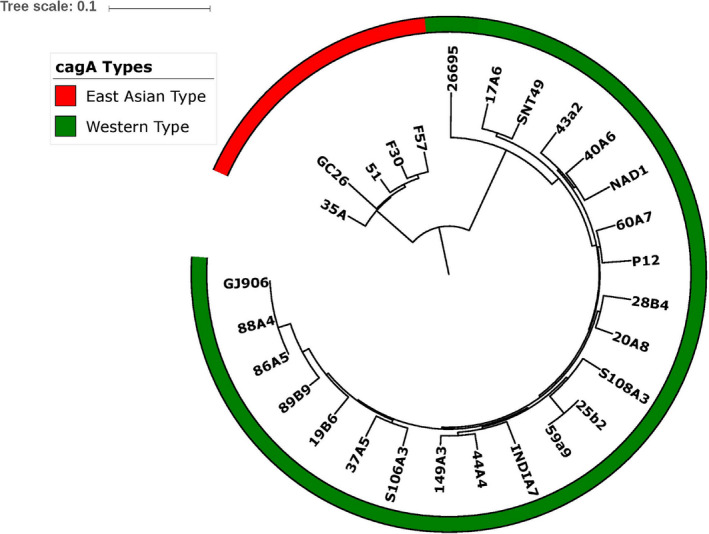
The cagA gene‐based phylogenetic tree of 20 BD H pylori isolates generated using Mega 5.2 with a bootstrap replicates of 1000 employing neighbor‐joining (NJ) algorithm and visualized in iTOL (A)
Table 2.
Prevalence of Helicobacter pylori virulence factors (cagA and vacA) among different lineage of isolates
| Genotypes description | Total, no. (%) | Lineage wise distribution | |
|---|---|---|---|
| HpAsia2 | HpEurope | ||
| Total studied | 20 | 10 (50%) | 10 (50%) |
| cagA | |||
| cagA positive | 18 (90) | 10 (55.5%) | 8 (44.45%) |
| AB | 1 (5) | 0 (0%) | 1 (100%) |
| ABC | 15 (75) | 9 (60%) | 6 (40%) |
| ABCC | 2 (10) | 1 (50%) | 1 (50%) |
| cagA negative | 2 (10) | 0 (0%) | 2 (100%) |
| Pre‐EPIYA type (no deletion) | 18 (100) | 10 (55.5%) | 8 (44.45%) |
| vacA | |||
| s1 | 18 (90) | 10 (55.5%) | 8 (44.45%) |
| s2 | 2 (10) | 0 (0%) | 2 (100%) |
| m1 | 15 (75) | 8 (53.3%) | 7 (46.6%) |
| m2 | 5 (25) | 2 (40%) | 3 (60%) |
| i1 | 18 (90) | 10 (55.5%) | 8 (44.45%) |
| i2 | 2 (10) | 2 (100%) | 0 (0%) |
| d1 | 18 (90) | 10 (55.5%) | 8 (44.45%) |
| d2 | 2 (10) | 2 (100%) | 0 (0%) |
| c1 | 15 (75) | 8 (53.3%) | 7 (46.6%) |
| c2 | 5 (25) | 2 (40%) | 3 (60%) |
| vacA s1m1i1d1c1 | 15 (75) | 8 (53.3%) | 7 (46.6%) |
| s1m2i1d1c2 | 3 (15) | 2 (66.6%) | 1 (33.3%) |
| s2m2i2d2c2 | 2 (10) | 0 (0%) | 2(100%) |
| CagA positive/vacA s1m1i1d1c1 | 15 (75) | 8 (53.3%) | 7 (46.6%) |
| s1m2i1d1c2 | 3 (15) | 2 (66.6%) | 1 (33.3%) |
| CagA negative/vacA s2m2i2d2c2 | 2 (10) | 0 (0%) | 2(100%) |
Moreover, sequence analysis of the pre‐EPIYA revealed that none of the isolates had a 39 bp deletion (approximately 300‐bps upstream of the first EPIYA motif) that is typically observed in strains from Western countries. A total 55 EPIYA motifs were identified in 18 cagA‐positive isolates. We identified two types of motifs: EPIY (A) (48/55, 87.2%) and EPIY (T) (7/55, 12.7%). The EPIYA‐B contained two types of motifs (EPIYA, EPIYT), and EPIY (T) was found in this motif (Table S3).
3.4.2. VacA Genotype
The predominant vacA “s” allele type was s1 (18/20, 90%) which is a typical of HpAsian and Western strains. The remaining isolates were s2 genotype and were present in HpEurope lineage isolates. The occurrence rate of the vacA m1 allele type was 75% (15/20) while the m2 allele type accounted for 25% (5/20). VacA gene‐based phylogenetic analysis of all 20 Bangladeshi isolates together with ten Western and East Asian harboring vacA gene revealed a clear distinction between m1 and m2 sequences (Figure 3). The prevalence of s1m1 genotype was found to be 75% (15/20), while 15% was s1m2 and 10% s2m2 in all 20 isolates. An examination of allelic diversity of different regions of vacA genotype found that all isolates that possessed s1 and m1, also retained i1, d1, and c1 (75%, 15/20). In contrast, all isolates that contained less‐virulent genotype s2 and m2 harbored i2, d2, and c2 genotype (10%, 2/20). Moreover, both cagA‐ and vacA‐positive isolates were predominant with genotype s1m1i1d1c1 while other group of less‐virulent isolates contained s1m2i1d1c1 genotype. A third cagA‐negative and non‐virulent group was identified as s2m2i2d2c2 genotype (Table 2).
Figure 3.
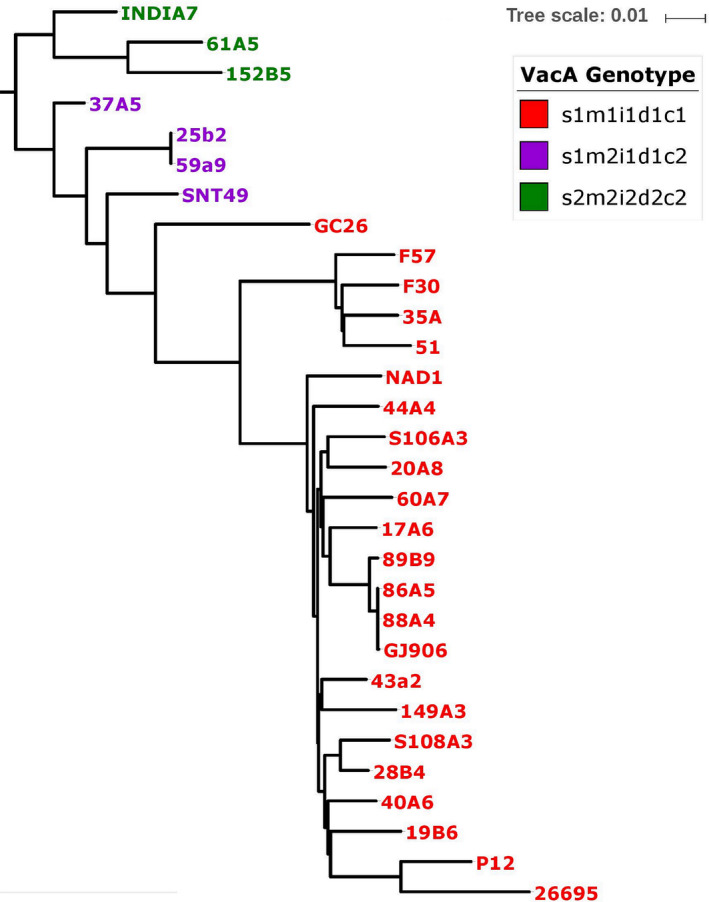
The vacA gene‐based phylogenetic tree of 20 studied H pylori strains generated using Mega 5.2 with a bootstrap replicates of 1000 employing neighbor‐joining (NJ) algorithm and visualized in iTOL (A)
3.5. Outer membrane proteins analysis
3.5.1. OipA gene
We found that oipA gene is present in all 20 isolates and showed five different CT repeat patterns (Table 3). Furthermore, we found that nine isolates out of 20 contained six CT repeat sequence displaying the “ON” status of OipA, while five strains that also had similar status (“ON”) contained (2 + 3) pattern of CT repeats. Two strains were found to harbor a CT repeat architecture of (5 + 2) and another two with nine direct repeat of CT dinucleotide and in both cases the gene status was “ON.” In addition to this, 61A5 and 89B9 isolates were having 10 direct CT dinucleotide repeat and in that case gene status was found to be “OFF” and these two were belonged to HpEurope lineage.
Table 3.
Number and types of repeats and “On and Off” status of OipA gene in 20 H pylori strains
| Strains | Number and types of repeats | Status | Hp Lineage |
|---|---|---|---|
| 149A3 | 6 | On | HpAsia2 |
| 20A8 | 6 | On | HpAsia2 |
| 25b2 | 6 | On | HpAsia2 |
| 28B4 | 6 | On | HpAsia2 |
| 37A5 | 6 | On | HpEurope |
| 43a2 | 6 | On | HpEurope |
| 59a9 | 6 | On | HpAsia2 |
| 60A7 | 6 | On | HpEurope |
| S108A3 | 6 | On | HpEurope |
| 17A6 | 2 + 3 | On | HpEurope |
| 86A5 | 2 + 3 | On | HpAsia2 |
| 88A4 | 2 + 3 | On | HpAsia2 |
| 152B5 | 2 + 3 | On | HpEurope |
| GJ906 | 2 + 3 | On | HpAsia2 |
| 44A4 | 5 + 2 | On | HpAsia2 |
| 19B6 | 5 + 2 | On | HpEurope |
| 40A6 | 9 | On | HpEurope |
| S106A3 | 9 | On | HpAsia2 |
| 89B9 | 10 | Off | HpEurope |
| 61A5 | 10 | Off | HpEurope |
3.5.2. Bab genes
The prevalence of bab genes among the isolates were studied. The babA gene was detected in 75% (15/20) of the isolates, while babB gene was identified in 55% (11/20) strains. The babA gene was found in all HpAsia2 population isolates, but was present in only 50% of HpEurope isolates. The genotype babA/babB/‐ (babA and babB present and babC absent) was detected in 40% (8/20) isolates, and six of these eight isolates were HpAsia2 isolates. We also observed that babA was absent in both s2m2i2d2c2 and one of the three s1m2i1d1c1 vacA genotype isolates (Figure S4). Clinical outcome data showed that isolates with (babA/babB/‐) or (babA/‐/‐), cagA positive and the s1m1i1d1c1 vacA genotypes were associated with either more or less severe clinical gastric outcome, while normal clinical outcomes were observed for (‐/babB/‐) or (‐/‐/‐) isolates except in 37A5 (Table S4).
3.6. In silico antibiotics susceptibility analysis
In silico antimicrobial susceptibility analysis revealed that 40% of the strains were multi‐drug resistant. We also detected that 90% (18/20) of the strains were resistant to metronidazole. Of those metronidazole‐resistant strains, nine were expected to express non‐functional or altered RdxA and/or FrxA proteins resulted from truncation, frameshift mutations, insertion of extra bases, and partial gene deletions. The remaining nine genotype‐based predicted metronidazole‐resistant strains resulted from amino acid exchanges (R16C, K64N, and P106S) that merely occur in the rdxA gene of metronidazole‐resistant H pylori strains. Of those nine strains, two exhibited double mutations (K64N and P106S) while one contained triple mutations (R16C, K64N, and P106S) (Table 4).
Table 4.
Truncations, frameshift mutations, insertion, deletions, and amino acid exchanges identified in the rdxA, frxA, gyrA, and 23S rRNA genes of sequenced H pylori isolates
| Genes | Mutation type (Position) | No. of H pylori strain (s) | References |
|---|---|---|---|
| rdxA | Nonsense (50) | 4 | Truncations in rdxA suggested to lead to metronidazole résistance 23 |
| Nonsense (86) | 1 | ||
| Fragmented | 1 | Fragmentation in rdxA suggested to be responsible for metronidazole résistance 31 | |
| Frameshift (146) | 1 | These frameshift, deletions have not been previously reported in the literature | |
| deletion of 35 bps N‐terminal region | 1 | ||
| deletion of 18 bps between codon 91 to 97 of N‐terminal region | 1 | ||
| R16C | 1 | R16C occurs only in metronidazole‐resistant H pylori strains 24 , 25 , 26 | |
| K64N | 7 | K64N occurs solely in metronidazole‐resistant H pylori strains 27 | |
| P106S | 5 | P106S occurs solely in metronidazole‐resistant H pylori strains 27 | |
| frxA | Frameshift (18) | 5 | Frameshifts in frxA suggested to confer metronidazole resistance 31 |
| Frameshift (106) | 1 | ||
| Nonsense (73) | 2 | This nonsense mutation has not been previously reported in the literature | |
| Insertion (45 543 bp) | 1 | This insertion in frxA has not been previously reported in the literature | |
| gyrA | 5‐ amino acid N‐terminal extension (MQDNS) | 5 | 5‐amino acid N‐terminal extension suggested to confer fluoroquinolone resistance 31 |
| D91G | 2 | Amino‐ acid exchanges in QRDR of GyrA suggested to confer resistance to fluoroquinolone 31 | |
| D91N | 1 | ||
| R295C | 1 | Amino acid exchange at 295 suggested to be responsible for fluoroquinolone resistant in H pylori 31 | |
| 23S rRNA | G2224A | 1 | G2224A mutation in 23S rRNA occurs solely in clarithromycin‐resistant H pylori strain 31 |
An extraordinary N‐terminal extension of GyrA by five amino acid residues (QDNSV) and amino acid exchanges in QRDR (N87, D91, and R295) occurs solely in fluoroquinolone‐resistant H pylori. 31 Our genotypic analysis also revealed that 45% (9/20) of the H pylori strains were resistant to fluoroquinolone antibiotics. Out of these nine fluoroquinolone‐resistant strains, five had an unprecedented N‐terminal extension of GyrA by five amino acid residues, immediately after starting codon, three exhibited amino acid exchanges in QRDR (D91) of H pylori and one strain showed a mutation at R295. Point mutation at G2224 in 23S rRNA was detected in only one strain which is suggested to confer resistance to clarithromycin (Table 4).
4. DISCUSSION
Helicobacter pylori has a complex and long‐standing coexistence with humans, so much so that particular bacterial lineages are strongly associated with regionally associated human lineages. 8 The plasticity of the H pylori genome, in particular recombination leads to situations in a modern world, where human populations are more mobile, where the long‐standing patterns of co‐evolution are uncoupled with instances of adverse clinical consequences for the host. 50 Hence, identifying the demographic composition and understanding the virulence potential of the region's dominant strains becomes essential. Here, we sequenced 20 Bangladeshi H pylori isolates and performed whole genome–based comparative analysis to investigate and understand the genetic architecture and virulence gene profile of these isolates.
Helicobacter pylori can be categorized into seven distinct lineages with names that are associated with geographic regions but are really representative of the human lineages that have been long‐term inhabitants of these regions: HpAfrica1, HpAfrica2, HpSahul, HpEurope, HpAsia2, HpAmerind, and HpEastAsia. 8 , 10 In this study, the 20 isolates from one ethnic group, namely Bengali, were not uniform in their H pylori lineage. The observation of two distinct lineages has been also reported previously. 51 The maintenance of independent lineages of H pylori within a stable ethnic group seems counterintuitive given the plasticity of the H pylori genome and an expectation that these co‐existent lineages would converge over time. The H pylori lineages present are consistent being at a point of intersection between humans co‐evolving with HpAsia2 lineage H pylori on the Indian subcontinent and humans from Central Asia with ancestral links to Europe. The mobility of ancient human populations is highlighted by H pylori genome from a European Copper age glacier mummy found in the European Alps belonged to HpAsia2 lineage. 52 Other studies, however, investigated that all Indian strains sequenced from indigenous Indians, who are predominantly Aryan and Dravidian ancestry, have significant homology to the HpEurope population suggested that introduction of H pylori took place with Indo‐Aryan migration. 53
The accessory genome content provides an overview of differences in gene content between Bangladeshi isolates, while the core genome comparison has allowed to observe the generalized preservation of lineage‐specific differences. Higher percentage of core genes belonging to J (translation, ribosomal structure, and biogenesis) and M (cell membrane/biogenesis envelope) functional classes also identified in our analysis may potentially indicate the adaptive stress imposed by the dynamic micro‐environment of the stomach to survive on this gastric pathogen as also previously reported. 43 Analysis of this study also revealed that a majority of specific genes belonging to L (replication, recombination, and repair) functional classes also indicating the requirement of H pylori to maintain a robust recombination and repair mechanism.
Based on the incidence rate of gastric cancer in people from Europe and South‐Central Asia, HpEurope population is thought to be associated with higher gastric cancer risk than HpAsia2 (http://globocan.iarc.fr). An unexpected result from our study which revealed that all HpAsia2 strains had cagA gene while two HpEurope isolates were completely lacking cagA gene, even the whole CagPAI, suggesting that HpAsia2 strains may be more virulent than HpEurope isolates. This study also corroborate with a similar type of comparative genomic study of H pylori isolates from Bangladesh, which showed that subjects infected with HpAsia2 had greater activity and inflammation in the antrum than with patients where HpEurope isolates were obtained which might be due to higher proportion of less‐virulent cagA and vacA genotype in HpEurope isolates. 51 In corroboration with this, oipA gene analysis result of this study identified that expression status of oipA gene of all the isolates (100%) of HpAsia2 was “ON,” while 20% of the HpEurope isolates were “OFF.” This result also suggested that the inducing potential of inflammation in HpAsia2 strains is high as compared to HpEurope.
There are several reports about the association of BabA‐positive status of H pylori with increased risk for the development of peptic ulcer disease. 21 In this study, we looked into the prevalence of different bab genes in various combination among the isolates. The study revealed that all HpAsia2 isolates were harboring babA and also most of the isolates had both babA and babB and no babC gene (babA/babB/‐). Clinical outcomes of the patients carrying HpAsia2 lineage H pylori showed more gastric severity as compared to HpEurope carriers. This could be one of the important explanation because of absence of babA or babA/babB/‐ genes in most of the Bangladeshi isolates of HpEurope lineage. This result is also in concordance with the others which explains that the pathogenic potential of HpAsia2 is more as compared to HpEurope as described above.
Prevalence of metronidazole‐resistant H pylori in Bangladesh is quite high, with resistance rate more than 90%. The resistance rates of levofloxacin and clarithromycin have been also increasing in H pylori isolates of Bangladesh. 32 , 54 Although the main mechanism of acquiring metronidazole resistance involves RdxA and/or FrxA inactivation mutations, a considerable number of missense mutations in both rdxA and frxA genes cannot rule out their role in metronidazole resistance by inducing conformational changes of RdxA and FrxA proteins. In tandem with remarkably high occurrence of metronidazole, our study also showed that about half of the strains would be resistant to fluoroquinolones. This acquired resistance is primarily due to amino acid substitution mutations in QRDR of GyrA protein that we also observed in some genotype predicted fluoroquinolone‐resistant strains. Interestingly, more than half of the predicted fluoroquinolone‐resistant strains had an insertion of QDNSV residues immediate to the start codon at N‐terminal region. This insertion of five amino acid residues would likely cause a conformational change in the GyrA protein and thus reducing its binding affinity to fluoroquinolone antibiotics. Importantly, genotype‐based analysis of antibiotic susceptibility of our study showed a high occurrence of metronidazole‐ and fluoroquinolone‐resistant H pylori strains in Bangladesh. This suggests that triple therapy based on metronidazole and fluoroquinolone may not be useful to treat H pylori infection in Bangladesh.
In conclusion, we showed that Bangladeshi strains with similar antibiotic resistance pattern, separated into two major population with distinct cagA and vacA genotypes. The comparative analysis of genomes provides important clues to the virulence potential of two segregated population (HpAsia2 and HpEurope). The study revealed that HpAsia2 strains are more virulent as compared to HpEurope in Bangladesh. Finally, as the pathology of H pylori is multifactorial, it would be important to understand even host and environmental factors in addition to bacterial genotype to better understand the disease outcome.
Our study had several limitations; a two‐step randomization procedure used in this study may not represent all variations which might be available in a population. In silico analysis of antimicrobial resistance may not identify novel or yet to be determined mutations associated with antimicrobial resistance.
DISCLOSURE
The authors have no competing interests.
Supporting information
Fig S1A
Fig S1B
Fig S1C
Fig S2A
Fig S2B
Fig S3
Supplementary Material
Table S1
Table S2
Table S3
Table S4
Supplementary Material
ACKNOWLEDGEMENTS
Authors from icddr,b are grateful to the Governments of Canada, Sweden, Bangladesh, and the UK for providing core/unrestricted support.
Qumar S, Nguyen TH, Nahar S, et al. A comparative whole genome analysis of Helicobacter pylori from a human dense South Asian setting. Helicobacter.2021;26:e12766 10.1111/hel.12766
REFERENCES
- 1. Frenck RW Jr, Clemens J. Helicobacter in the developing world. Microbes Infect. 2003;5(8):705‐713. [DOI] [PubMed] [Google Scholar]
- 2. Eusebi LH, Zagari RM, Bazzoli F. Epidemiology of Helicobacter pylori infection. Helicobacter. 2014;19(Suppl 1):1‐5. [DOI] [PubMed] [Google Scholar]
- 3. Hooi JKY, Lai WY, Ng WK, et al. Global prevalence of Helicobacter pylori infection: systematic review and meta‐analysis. Gastroenterology. 2017;153(2):420‐429. [DOI] [PubMed] [Google Scholar]
- 4. Ahmad MM, Rahman M, Rumi AK, et al. Prevalence of Helicobacter pylori in asymptomatic population–a pilot serological study in Bangladesh. J Epidemiol. 1997;7(4):251‐254. [DOI] [PubMed] [Google Scholar]
- 5. Bardhan PK. Epidemiological features of Helicobacter pylori infection in developing countries. Clin Infect Dis. 1997;25(5):973‐978. [DOI] [PubMed] [Google Scholar]
- 6. Kibria KM, Nahar S, Hossain M, et al. Epidemiology of H pylori and its relation with gastrointestinal disorders, a community‐based study in Dhaka, Bangladesh. J Gastroenterol Hepatol Res. 2018;7:2709‐2716. [Google Scholar]
- 7. Fock KM, Ang TL. Epidemiology of Helicobacter pylori infection and gastric cancer in Asia. J Gastroenterol Hepatol. 2010;25(3):479‐486. [DOI] [PubMed] [Google Scholar]
- 8. Linz B, Balloux F, Moodley Y, et al. An African origin for the intimate association between humans and Helicobacter pylori . Nature. 2007;445(7130):915‐918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamaoka Y Helicobacter pylori typing as a tool for tracking human migration. Clin Microbiol Infect. 2009;15(9):829‐834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Falush D, Wirth T, Linz B, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299(5612):1582‐1585. [DOI] [PubMed] [Google Scholar]
- 11. Bickenbach K, Strong VE. Comparisons of gastric cancer treatments: east vs. west. J Gastric Cancer. 2012;12(2):55‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuipers EJ, Israel DA, Kusters JG, et al. Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart from the same host. J Infect Dis. 2000;181(1):273‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mobley HL. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment Pharmacol Ther. 1996;10(Suppl 1):57‐64. [DOI] [PubMed] [Google Scholar]
- 14. Backert S, Tegtmeyer N, Selbach M. The versatility of Helicobacter pylori CagA effector protein functions: the master key hypothesis. Helicobacter. 2010;15(3):163‐176. [DOI] [PubMed] [Google Scholar]
- 15. Roesler BM, Rabelo‐Goncalves EM, Zeitune JM. Virulence factors of Helicobacter pylori: a review. Clin Med Insights Gastroenterol. 2014;7:9‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19(3):449‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamaoka Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat Rev Gastroenterol Hepatol. 2010;7(11):629‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Basso D, Zambon C, Letley DP, et al. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology. 2008;135(1):91‐99. [DOI] [PubMed] [Google Scholar]
- 19. Ogiwara H, Sugimoto M, Ohno T, et al. Role of deletion located between the intermediate and middle regions of the Helicobacter pylori vacA gene in cases of gastroduodenal diseases. J Clin Microbiol. 2009;47(11):3493‐3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamaoka Y, Kikuchi S, El–Zimaity HMT, Gutierrez O, Osato MS, Graham DY. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology. 2002;123(2):414‐424. [DOI] [PubMed] [Google Scholar]
- 21. Gerhard M, Lehn N, Neumayer N, et al. Clinical relevance of the Helicobacter pylori gene for blood‐group antigen‐binding adhesin. Proc Natl Acad Sci USA. 1999;96(22):12778‐12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sheu S‐M, Sheu B‐S, Chiang W‐C, et al. H pylori clinical isolates have diverse babAB genotype distributions over different topographic sites of stomach with correlation to clinical disease outcomes. BMC Microbiol. 2012;12(1):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gerrits MM, van der Wouden E‐J, Bax DA, et al. Role of the rdxA and frxA genes in oxygen‐dependent metronidazole resistance of Helicobacter pylori . J Med Microbiol. 2004;53(Pt 11):1123‐1128. [DOI] [PubMed] [Google Scholar]
- 24. Marais A, Bilardi C, Cantet F, Mendz GL, Megraud F. Characterization of the genes rdxA and frxA involved in metronidazole resistance in Helicobacter pylori . Res Microbiol. 2003;154(2):137‐144. [DOI] [PubMed] [Google Scholar]
- 25. Jeong J‐Y, Mukhopadhyay AK, Dailidiene D, et al. Sequential inactivation of rdxA (HP0954) and frxA (HP0642) nitroreductase genes causes moderate and high‐level metronidazole resistance in Helicobacter pylori . J Bacteriol. 2000;182(18):5082‐5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Binh TT, Suzuki R, Trang TT, Kwon DH, Yamaoka Y. Search for novel candidate mutations for metronidazole resistance in Helicobacter pylori using next‐generation sequencing. Antimicrob Agents Chemother. 2015;59(4):2343‐2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goodwin A, Kersulyte D, Sisson G, Veldhuyzen van Zanten SJ, Berg DE, Hoffman PS. Metronidazole resistance in Helicobacter pylori is due to null mutations in a gene (rdxA) that encodes an oxygen‐insensitive NADPH nitroreductase. Mol Microbiol. 1998;28(2):383‐393. [DOI] [PubMed] [Google Scholar]
- 28. Trespalacios‐Rangel AA, Otero W, Arevalo‐Galvis A, Poutou‐Pinales RA, Rimbara E, Graham DY. Surveillance of levofloxacin resistance in Helicobacter pylori isolates in Bogota‐Colombia (2009–2014). PLoS One. 2016;11(7):e0160007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stone GG, Shortridge D, Flamm RK, et al. Identification of a 23S rRNA gene mutation in clarithromycin‐resistant Helicobacter pylori . Helicobacter. 1996;1(4):227‐228. [DOI] [PubMed] [Google Scholar]
- 30. Lauener F, Imkamp F, Lehours P, et al. Genetic determinants and prediction of antibiotic resistance phenotypes in Helicobacter pylori . J Clin Med. 2019;8(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shetty V, Lamichhane B, Tay CY, et al. High primary resistance to metronidazole and levofloxacin, and a moderate resistance to clarithromycin in Helicobacter pylori isolated from Karnataka patients. Gut Pathogens. 2019;11:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nahar S, Mukhopadhyay AK, Khan R, et al. Antimicrobial susceptibility of Helicobacter pylori strains isolated in Bangladesh. J Clin Microbiol. 2004;42(10):4856‐4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2):e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. J Comput Biol. 2012;19(5):455‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shaik S, Kumar N, Lankapalli AK, Tiwari SK, Baddam R, Ahmed N. Contig‐layout‐authenticator (CLA): a combinatorial approach to ordering and scaffolding of bacterial contigs for comparative genomics and molecular epidemiology. PLoS One. 2016;11(6):e0155459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068‐2069. [DOI] [PubMed] [Google Scholar]
- 37. Mural RJ. ARTEMIS: a tool for displaying and annotating DNA sequence. Brief Bioinform. 2000;1(2):199‐200. [DOI] [PubMed] [Google Scholar]
- 38. Lowe TM, Eddy SR. tRNAscan‐SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100‐3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arndt D, Grant JR, Marcu A, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44(W1):W16‐W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genom. 2011;12:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core‐genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15(11):524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kumar N, Albert MJ, Al Abkal H, Siddique I, Ahmed N. What constitutes an Arabian Helicobacter pylori? Lessons from comparative genomics. Helicobacter. 2017;22(1). [DOI] [PubMed] [Google Scholar]
- 44. Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44(W1):W242‐W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178‐2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen L, Yang J, Yu J, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2004;33(Database issue):D325‐D328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Alm RA, Bina J, Andrews BM, Doig P, Hancock RE, Trust TJ. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect Immun. 2000;68(7):4155‐4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qumar S, Majid M, Kumar N, et al. Genome dynamics and molecular infection epidemiology of multidrug‐resistant Helicobacter pullorum isolates obtained from broiler and free‐range chickens in India. Appl Environ Microbiol. 2017;83(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870‐1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suerbaum S, Achtman M Helicobacter pylori: recombination, population structure and human migrations. Int J Med Microbiol. 2004;294(2–3):133‐139. [DOI] [PubMed] [Google Scholar]
- 51. Aftab H, Miftahussurur M, Subsomwong P, et al. Two populations of less‐virulent Helicobacter pylori genotypes in Bangladesh. PLoS One. 2017;12(8):e0182947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maixner F, Krause‐Kyora B, Turaev D, et al. The 5300‐year‐old Helicobacter pylori genome of the Iceman. Science. 2016;351(6269):162‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Devi SM, Ahmed I, Francalacci P, et al. Ancestral European roots of Helicobacter pylori in India. BMC Genom. 2007;8:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aftab H, Miftahussurur M, Subsomwong P, Ahmed F, Khan AK, Yamaoka Y Helicobacter pylori antibiotic susceptibility patterns in Bangladesh: emerging levofloxacin resistance. J Infect Dev Count. 2016;10(3):245‐253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1A
Fig S1B
Fig S1C
Fig S2A
Fig S2B
Fig S3
Supplementary Material
Table S1
Table S2
Table S3
Table S4
Supplementary Material