

Abstract
Helicobacter pylori is a highly potential pathogen to colonize in the human stomach. This bacterial strain is now alarming serious health concern all over the world. Combating through available drugs is a difficult task due to lack of appropriate common targets against genetically diverse strains. Therefore, the developments of effective targets vaccines require alternative strategies to eliminate the H. pylori infection. In this study, we developed a novel vaccine construct using B-cell derived T-cell epitopes from four target antigenic proteins (HpaA, FlaA, FlaB and Omp18), and found the induction of possible immune response using advanced immunoinformatics approaches. In order to boost immune system, we tagged adjuvant (50S ribosomal protein L7/L12) with a suitable linker at the N-terminus side of vaccine sequence. Protein–protein docking between human Toll like receptor 5 (TLR5) and vaccine construct help to predict the way of inductive signaling that leads to immune-response. The calculated negative score (− 151.4, + / − 8.7) of molecular docking complex signify the best binding interface. Molecular dynamics simulation studies confirmed the proper docking between TLR5 and vaccine candidate. Moreover, Normal mode analysis (NMA) calculates the molecular motion of the docking complex. The low eigenvalue (2.935e−05) indicates the stable and flexible molecular motion in the binding interaction side. Finally, in-silico cloning of vaccine candidate was performed using expression vector pET28b (+) with the optimized restriction sites.
Supplementary Information
The online version of this article (10.1007/s10989-020-10157-w) contains supplementary material, which is available to authorized users.
Keywords: Helicobacter pylori, B-cell, T-cell, In-silico, Novel vaccine, Molecular dynamics simulation
Introduction
The flagellated gram-negative bacterium H. pylori is a spiral-shaped, microaerophilic bacteria which colonizes in human gastric epithelial cell surface region (Amieva and El–Omar 2008; Meza et al. 2017). Approximately half of the global human population is suffering from chronic gastritis, among which maximum cases remains asymptomatic. However, 10% of those infection leads to severe peptic ulcer (gastric and duodenal), gastric adenocarcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma (Cover and Blaser 2009; Salama et al. 2013; Yamaoka 2018). As per report of Testerman and Morris (2014) gastric cancer kills over 700,000 people per year globally (Testerman and Morris 2014). The virulent surface proteins of this extracellular pathogen play a pivotal role in host–pathogen interactions and pathogenicity including flagella-driven motility in the gastric epithelial layer (Viala et al. 2004). Virulent factors of H. pylori cause chronic and progressive gastric mucosal inflammation, resulting in tissues necrosis, atrophy, lesions, intestinal metaplasia and subsequently stomach cancer (Khan et al. 2019). It was observed that the infection of H. pylori is increasing day by day globally (Fig. 1) (Hooi et al. 2017; Khoder et al. 2019; Zamani et al. 2018). Motility, adherence, colonization and several virulence factors are the key requirements of the gastrointestinal disease by H. pylori. This phenomenon is mainly facilitated by bacterial outer membrane proteins (OMPs) and flagellar proteins. HpaA and Omp18 are well characterized outer membrane proteins of H. pylori (Lundström et al. 2001; Voland et al. 2003). On the other hand, FlaA and FlaB are mostly studied flagellar proteins (Allan et al. 2000; Josenhans et al. 1995).
Fig. 1.

The mapping of H. pylori infection from previous published data (Hooi et al. 2017; Khoder et al. 2019; Zamani et al. 2018)
Toll like receptors (TLRs) plays a crucial role in pathogen recognition, host–pathogen interaction and downstream signal regulation (Bhattacharya et al. 2020b; Netea et al. 2004). TLRs are most studied pathogen recognition receptors (PRRs), which recognizes specific antigenic epitope of surface proteins of the pathogen, known as a pathogen associated molecular patterns (PAMPs). However, in our study bacterial flagellin protein is most proper antagonist of TLR5 for pathogen recognition.
Previously, there is no effective antibiotic or drug-resistant medication available to eradicate H. pylori strain (Meza et al. 2017; Testerman and Morris 2014). So, designing of potential vaccine is an urgent need to combat H. pylori (Del Giudice et al. 2009). Among the large number of virulent proteins, identification of potential virulent proteins is the most challenging task for the development of effective vaccine candidate. Researchers are seeking an effective virulent protein to breakdown the virulence of H. pylori through vaccination. But, classical or conventional approach of vaccine candidate formulation is a time consume process and there are some limitations and drawbacks of conventional vaccinology, like in case of live attenuated or inactivated vaccines, which expose an individual with whole pathogens, is very nonspecific and there is also a risk of the attenuated pathogen reverting to full virulence. Therefore, scientists are now using modern immunoinformatics approach to design a vaccine against pathogenic infection (Bhattacharya et al. 2020a; Dhal et al. 2019; Pandey et al. 2018).
In this current study we selected four such antigenic proteins (HpaA, FlaA, FlaB and Omp18) for in-silico design of multi-epitope-based vaccine model. A comprehensive computational framework was used to identify antigenic common B -cell and T- cell epitope to construct a vaccine construct against deleterious H. pylori. HpaA and Omp18 proteins are surface localizing protein and a promising candidate for a vaccine against H. pylori infection (Carlsohn et al. 2006). Similarly, FlaA and FlaB protein of H. pylori flagellin also having virulence property (Gu 2017). This two protein are promising peptide based vaccine candidate against H. pylori. According to Song et al. flagellin is recognized by human toll-like receptor 5 (TLR5) as a pathogen invasion signal and subsequently evokes the immune response (Song et al. 2017). So FlaA and FlaB protein, a part of the flagellin protein, can recognized by TLR5 and also induces immune response a part of activity response against human TLR5.
In this work, we selected four antigenic proteins (HpaA, FlaA, FlaB and Omp18) from H. pylori. Using these antigenic protein common B -cell and T-cell epitopes was predicted and vaccine construct was developed. We have also characterized different properties of vaccine candidate such as secondary structure, antigenicity, allergenicity and physiochemical properties. We have also performed tertiary structure modeling and model validation of vaccine construct. The vaccine construct was docked with human TLR5 to understand the binding affinity. We profiled the thermodynamics stability, in-silico cloning and expression was performed using vaccine candidate. The present research aims to achieve potent multi epitopic peptide vaccine component against H. pylori to control its infection.
Materials and Methods
Collection of Proteins
Four proteins were selected for the development of computer aided multi-epitope peptide vaccine designing against H. pylori, including HpaA, FlaA, FlaB and Omp18, based on their role in H. pylori infection. The target protein sequences of H. pylori were retrieved as FASTA format from the NCBI database for further computational analyses to detect the antigenic peptide sequence for vaccine designing (Pruitt et al. 2007).
Prediction of B-Cell Epitopes
B lymphocytes cells are generating antibody and memory cells; herewith it turns active long-term immune responses against pathogens. For prediction of sequence-based linear B- cell epitope of the four target proteins, we employed Bepipred Linear Epitope Prediction 2.0 algorithm under the Immune Epitope Database (IEDB) online server (Jespersen et al. 2017; Kim et al. 2012). FASTA sequences of those targeted proteins were provided as input and selected all default parameters.
Identification of MHC-I and MHC-II Epitopes Within B-Cell Epitopes
T-cells play a pivotal role in stimulating both cytotoxic and helper T-cell mediated immune response (Saha et al. 2017). MHC-I and MHC-II class epitopes were selected from the previously retrieved. The Bcell epitope of target proteins were predicted using T-cell epitope prediction tool under the IEDB server (Patra et al. 2020). Here, we opted IEDB recommended 2.22 method for identifying MHC-I & MHC-II epitopes and chosen all repository. After successful identification of T-cell epitopes, we selected common epitope for B-cells, T-cells and both MHC molecules, which might serve as potential vaccine as it can induce both humoral and cell mediated immunity.
Construction of Vaccine Sequence
Common B-cell and T- cell epitopes are selected for final vaccine construction on the basis of high antigenicity and non-allergenicity nature. In order to establish final multi-epitopes-based vaccine construct, the selected MHC-I and MHC-II epitopes were linked together by AAY and GPGPG linkers, respectively. Finally, 50S ribosomal protein L7/L12 adjuvant is linked with N-terminus of the vaccine model with the help of EAAAK peptide linker to accelerate immune response.
Secondary Structure Prediction of Vaccine
PSIPRED 4.0 web server was used to predict the secondary structure of vaccine construct, as it delivered highly accurate result (Buchan and Jones 2019). This server has two feed-forward neural networks-based algorithms, which executes a study on output generated from PSI-BLAST and refine the secondary structure.
Evaluation of Antigenicity, Allergenicity and Physiochemical Properties of the Designed Vaccine
Analysis and evaluation of allergenic property is very much crucial for modulating any vaccine. Antigenic propensity of vaccine model was evaluation by using VaxiJen v2.0 server and further validated by AntigenPro server (Doytchinova and Flower 2007; Magnan et al. 2010). For additional analysis, allergenicity was evaluated by performing AllergenFP v.1.0 server (Dimitrov et al. 2014). Protein-Sol webserver was used to determine the solubility of vaccine construct (Hebditch et al. 2017). Finally, various physicochemical properties of each designed vaccine constructs were determined by ExPASy ProtParam server (Gasteiger et al. 2005).
Tertiary Structure Modelling
Three-dimensional modelling of the vaccine construct is a critical step in epitope-based vaccine development (Roy et al. 2012; Willard et al. 2003). SPARKS-X; protein fold recognition web server was used to develop 3D structure of vaccine construct (Yang et al. 2011). The FASTA format of vaccine construct (amino acid sequence) was an input for the SPARKS-X. This server implements BLASTp for searching template for 3D structure prediction.
Model Validation
Predicted tertiary structure validation of amino acids chain is a crucial step in model building process. Subsequently, SPARKS-X provided protein model has to go through some validation servers such as (a) ProSA-web: calculates energy plot as well as Z score value that indicate overall quality score of target vaccine construct model (Wiederstein and Sippl 2007). (b) PROCHECK-server predicts the residue-by-residue stereochemical qualities validation of the vaccine model through Ramachandran plot (Laskowski et al. 1993).
Discontinuous B-Cell Epitope Prediction
Discontinuous B-cell epitopes of the vaccine construct was predicted through Ellipro tool of IEDB server (Ponomarenko et al. 2008). If the score appears as 0.8, it indicates 80% of the protein residues are inside the ellipsoids and 20% being outside. The clustering of discontinuous epitopes is subjected by distance R, which is measured in Å and more R indicates larger discontinuous epitope.
Molecular Docking Analysis and Binding Affinity Analysis
Molecular docking is very promising and most fundamental process to characterize the interaction and binding affinity between the vaccine constructs and the human Toll-like receptor 5 (TLR5). The crystal structure of human TLR5, downloaded from RCSB protein data bank (PDB ID: 3J0A) was used for molecular docking. Molecular docking was carried out with HADDOCK web server at the easy interface (De Vries et al. 2010). Prior of molecular docking, the active residues (directly involved in the interaction) and passive residues (surrounding residues) for both TLR5 and vaccine construct were determined using CPORT server (de Vries and Bonvin 2011).
The binding affinity of the docking complex or Gibbs free energy (ΔG) determinate the actual form of interaction pattern and dissociation constant (KD) values of docked complex was predicted at 37 °C temperature by PRODIGY web server (Xue et al. 2016).
Molecular Dynamics Simulation of Vaccine Construct
The design vaccine component were completely energy-minimized by the default energy minimizer process of the NAMD (Nanoscale Molecular Dynamics). Software which uses conjugate with gradient and line search algorithms based on NVT ensemble at the pressure of 1.025 atm (Phillips et al. 2005). The molecular simulations were passed out by the CHARMM27 coordinate protein-based topology, and all atom force field with map correction with a time-step of 1 fs and periodic boundary conditions applied in all three dimensions (MacKerell et al. 2000). RMSF and RMSD plot of the docking complex showed its residue by residue fluctuation graph.
Normal Mode Analysis
The Normal Mode Analysis study was performed on the docking complex (TLR5-vaccine construct). Here, we opted iMODS online server to complete the NMA study (López-Blanco et al. 2014). iMODS was employed for investigation of the structural dynamics of the docking complex as well as determined the molecular motion. iMODs server is a customable, easy and user-friendly online server. This web server also generates complex deformability, B-factor, eigenvalues, variance, co-variance map and elastic network. The docked PDB files act as an input file and uploaded to the iMODS server and the results were displayed keeping all the parameters as default.
Codon Optimization and In-Silico Cloning
Codon optimization of vaccine construct was performed through online web server namely Java Codon Adaptation Tool (JCAT) for high-level expression of the vaccine sequence in E. coli K12 strain (Grote et al. 2005). We used pET28b( +) expression vector to execute in silico cloning of vaccine sequence(Bhattacharya et al. 2020c). This expression vector map and sequences was downloaded from “addgene” vector database (Kamens 2015). Here, SnapGene 5.1.7 restriction cloning tool was employed for finalizing in silico cloning (GSL Biotech LLC 2015).
Result
Collection of Target Proteins
In this study, four proteins of Helicobacter pylori were screening for the possible vaccine construction. These proteins were: HpaA (accession number: ANH47002.1), FlaA (accession number: AAU21201.1), FlaB (accession number: AAU21202.1) and Omp18 (accession number: WP_171924268.1) were retrieved as FASTA format from NCBI database.
Prediction of B-Cell Epitopes
B-cell epitopes of four target protein of Helicobacter pylori were predicted different algorithms from Bepipred Linear Epitope Prediction 2.0 algorithm of IEDB server. IEDB predicted B-cell epitopes of varying length of each target proteins were identified and tabulated along with epitopes with their start position, sequence and length (Supplementary Table 1). The predicted B-cell epitopic regions of each target protein shown in supplementary Fig. 1.
Identification of MHC-I and MHC-II Epitopes Within B-Cell Epitopes
Common B-cell and T-cell epitopes are more applicable for vaccine designing, here each four-target protein of H. pylori have same number of MHC-I and MHC-II binding epitopes. In order to identification of MHC class I epitopes, we retrieved 9mer epitopic sequence. Similarly, for MHC-II epitopes identification we collected 15mer epitopic sequence. These two types of epitopic sequences were further used vaccine construction which is illustrated in Table 1 respectively.
Table 1.
A Selection of MHC-I epitopes (derived from B-cell epitopes) based on VaxiJen score > 1
| Protein name | B-cell epitope sequence | MHC-I epitope sequence | MHC-I binding alleles | VaxiJen score |
|---|---|---|---|---|
| A | ||||
| HpaA | NPSVETKTQNDAKNQQPVQTHERMKTSSEHVTPLDFNYPIHIVQAPQN | HVTPLDFNYP |
HLA-A*0201 HLA-A*0205 HLA-B*2702 HLA-B*3501 HLA-B*3701 HLA-B*3801 HLA-B*0702 |
1.1099 |
| GYQVLRFQDEKALSAQDKR | QDEKALSAQ |
HLA-A*1101 HLA-A*3101 HLA-B*2702 HLA-B*5801 |
1.1087 | |
| PESNRVVHDFAVEVG | VVHDFAVEV |
HLA-A*0201 HLA-A*0205 HLA-B*3701 HLA-B*4403 HLA-B*5301 |
1.3684 | |
| YTYKHSNSGGLDSSNNIIHEALEKNKEDA | KHSNSGGLD |
HLA-B*3902 HLA-B*5301 HLA-B*5401 |
2.2506 | |
| FlaA | NALKTSLERLSSGLRINKAAD | RLSSGLRIN |
HLA-B*2702 HLA-B*2705 HLA-B*3901 HLA-B*5801 |
1.5169 |
| AAQDGQTTESRKAIQSD | QDGQTTESR |
HLA-B*2702 HLA-B*5101 HLA-B*5801 |
2.9528 | |
| VTGNFNANVKSASGANYNAVIASGNQSLGSGVTTLRG | NANVKSASG | HLA-B*5401 | 2.2390 | |
| VNNISITQVNVKAAESQIRDV | VNVKAAESQ | HLA-B*5801 | 1.9766 | |
| FlaB | QNNRDLSSSLEKLSSGLRINKAADD | KLSSGLRIN |
HLA-A*0205 HLA-B*3901 HLA-B*5801 |
1.5519 |
| AQDGQTLESRRALQSDIQRLLEE | QDGQTLESR |
HLA-A*3302 HLA-B*2702 HLA-B*3501 HLA-B*3902 HLA-B*5801 |
2.2153 | |
| VRGIFDANVASAAGANANGAQAETNSQGIGAGVTSLKGAM | NANGAQAET |
HLA-B*5101 HLA-B*5103 |
2.4424 | |
| ISVTQVNVKAAESQIRDVDF | VNVKAAESQ | HLA-B*5801 | 1.9766 | |
| Omp18 | HKMDNKTVAGDVSAKTVQTAPVTTEPAPEKEEPKQEPAPVVEEKPAVESGT | GDVSAKTVQ |
HLA-A*3302 HLA-B*5101 HLA-B*5102 HLA-B*5103 HLA-B*5201 HLA-B*5801 |
1.9177 |
| HKMDNKTVAGDVSAKTVQTAPVTTEPAPEKEEPKQEPAPVVEEKPAVESGT | EKEEPKQEP |
HLA-B*4403 HLA-B*0702 |
1.3706 | |
| FDFDKYEIKESDQET | EIKESDQET |
HLA-A*3302 HLA-B*3701 HLA-B*3901 HLA-B*3902 HLA-B*4403 HLA-B*5301 |
1.8008 | |
| ISFGETKPKCTQKTRECYKEN | GETKPKCTQ |
HLA-A*1101 HLA-A*3101 HLA-B*5401 HLA-B*5801 |
1.7641 |
| Sl. No | B-cell epitope sequence | MHC-II epitope sequence | MHC-II binding alleles | VaxiJen score |
|---|---|---|---|---|
| B | ||||
| HpaA | NPSVETKTQNDAKNQQPVQTHERMKTSSEHVTPLDFNYPIHIVQAPQN | VQTHERMKTSSEHVT |
HLA-DRB1*1302 HLA-DRB1*0901 HLA-DRB1*0701 HLA-DRB1*0301 HLA-DRB1*0405 |
1.2231 |
| NPSVETKTQNDAKNQQPVQTHERMKTSSEHVTPLDFNYPIHIVQAPQN | PSVETKTQNDAKNQQ |
HLA-DRB1*0802 HLA-DRB1*1302 HLA-DRB4*0101 HLA-DRB3*0101 |
1.6689 | |
| GYQVLRFQDEKALSAQDKR | LRFQDEKALSAQDKR |
HLA-DRB1*0401 HLA-DQA1*0401 HLA-DPA1*0103 HLA-DRB5*0101 |
1.1177 | |
| YTYKHSNSGGLDSSNNIIHEALEKNKEDA | YTYKHSNSGGLDSSN |
HLA-DRB1*0101 HLA-DQA1*0501 HLA-DRB1*0401 HLA-DRB3*0101 HLA-DRB1*0405 |
1.3437 | |
| FlaA | AAQDGQTTESRKAIQSD | QDGQTTESRKAIQSD |
HLA-DQA1*0301 HLA-DQA1*0102 HLA-DQA1*0401 HLA-DRB1*0701 HLA-DQA1*0501 |
1.7622 |
| VTGNFNANVKSASGANYNAVIASGNQSLGSGVTTLRG | ASGNQSLGSGVTTLR |
HLA-DQA1*0301 HLA-DPA1*0301 HLA-DRB1*0405 HLA-DQA1*0501 HLA-DPA1*0201 |
1.3656 | |
| VTGNFNANVKSASGANYNAVIASGNQSLGSGVTTLRG | NANVKSASGANYNAV |
HLA-DRB1*1101 HLA-DRB3*0101 HLA-DQA1*0301 HLA-DQA1*0501 HLA-DPA1*0103 |
1.6352 | |
| VNNISITQVNVKAAESQIRDV | NISITQVNVKAAESQ |
HLA-DQA1*0101 HLA-DRB3*0101 |
1.6160 | |
| FlaB | AQDGQTLESRRALQSDIQRLLEE | QDGQTLESRRALQSD |
HLA-DRB1*1101 HLA-DQA1*0102 HLA-DRB1*0802 HLA-DRB1*0301 HLA-DRB1*0901 |
1.2748 |
| VRGIFDANVASAAGANANGAQAETNSQGIGAGVTSLKGAM | ETNSQGIGAGVTSLK |
HLA-DQA1*0102 HLA-DQA1*0301 HLA-DRB1*0101 HLA-DQA1*0401 HLA-DRB1*0301 |
1.7236 | |
| VRGIFDANVASAAGANANGAQAETNSQGIGAGVTSLKGAM | GANANGAQAETNSQG |
HLA-DRB1*0401 HLA-DQA1*0501 HLA-DRB1*0405 HLA-DRB4*0101 HLA-DPA1*0103 |
2.3765 | |
| ISVTQVNVKAAESQIRDVDF | QVNVKAAESQIRDVD |
HLA-DPA1*0103 HLA-DQA1*0101 HLA-DRB1*1201 HLA-DRB1*1501 HLA-DPA1*0301 |
1.6015 | |
| Omp18 | HKMDNKTVAGDVSAKTVQTAPVTTEPAPEKEEPKQEPAPVVEEKPAVESGT | KMDNKTVAGDVSAKT |
HLA-DQA1*0501 HLA-DRB1*0401 HLA-DRB1*0802 HLA-DRB3*0101 HLA-DRB1*0301 |
1.7244 |
| HKMDNKTVAGDVSAKTVQTAPVTTEPAPEKEEPKQEPAPVVEEKPAVESGT | NKTVAGDVSAKTVQT |
HLA-DQA1*0401 HLA-DRB1*0802 HLA-DQA1*0301 HLA-DRB1*0901 HLA-DRB1*1302 |
1.4458 | |
| FDFDKYEIKESDQET | FDFDKYEIKESDQET |
HLA-DPA1*0201 HLA-DPA1*0103 HLA-DRB1*0401 HLA-DRB1*0301 HLA-DRB4*0101 |
1.3271 | |
| ISFGETKPKCTQKTRECYKEN | GETKPKCTQKTRECY |
HLA-DRB1*0301 HLA-DRB1*1201 HLA-DRB1*1101 HLA-DRB1*0701 HLA-DRB5*0101 |
1.3419 |
Designing of Vaccine Construct
Common MHC-I and MHC-II epitopes were used to design vaccine constructs; here total 15 MHC class I and 16 MHC class II epitopes were filtered and linked them with AAY and GPGPG linker respectively. 50S ribosomal protein L7/L12 with (NCBI accession no. P0A7K2) as an adjuvant, was added to the amino terminus of the vaccine construct with linker EAAAK, graphical representation shown in Fig. 2. Lastly, all of the linked MHC-I, MHC-II and adjuvant considered as a final vaccine construct had a total of 632 amino acids making it ~ 64.367 kDa peptide and an Extinction coefficient of 25960 M−1 cm−1.
Fig. 2.

Schematic diagram of final multi epitopic peptide vaccine construct
Secondary Structure Prediction of Vaccine
PSIPRED v4.o assessed the secondary structure of the vaccine component, where server calculates the number of the amino acids in the alpha helix, beta-sheet and coil formation within the vaccine model. Vaccine model had a total of 207 (32.75%) amino acids in the alpha-helix formation, 62 (9.81%) amino acids in the beta-sheet formation, and 363 (57.43%) amino acids in the coil structure formation represented in Table 2 and PSIPRED generated cartoon structure shown in Fig. 3.
Table 2.
Percentage and number of amino acids of secondary structure (Alpha helix, beta sheet and coil) present in vaccine construct
| Candidate name | Alpha helix (percentage and number of amino acids) | Beta sheet (percentage and number of amino acids) | Coil structure (percentage and number of amino acids) |
|---|---|---|---|
| Vaccine construct (632 amino acids) | 207(32.75%) amino acids | 62 (9.81%) amino acids | 363 (57.43%) amino acids |
Fig. 3.

Schematic representation of secondary structure prediction of the multi-epitope vaccine, secondary structure prediction result represents the arrangement of α-helix (32.75%) β-strands (9.81%) and coils (57.43%)
Evaluation of Antigenicity, Allergenicity and Physiochemical Properties of the Designed Vaccine
Antigenic propensity is an important parameter for designing vaccine construct that typically induce the humoral as well as cell mediated immune responses against the H. pylori. The predicted data recommend that our vaccine is antigenic with an antigenic probability score of 1.2317 and 0.918291predicted by VaxiJen v2.0 and ANTIGENPro servers, individually (Table 3).
Table 3.
Antigenicity, allergenicity, solubility, and physicochemical property assesment of the primary sequence of multi-epitope-based vaccine construct
| Sl. No | Features | Assessment |
|---|---|---|
| 1 | Antigenicity |
1.2317 (Probable ANTIGEN) by VaxiJen v2.0 0.918291 (Probable ANTIGEN) by ANTIGENPro |
| 2 | Allergenicity |
Probable non-allergen (AllerTOP v.2.0) Probable non-allergen (AllergenFP v.1.0) |
| 3 | Solubility |
0.668 (Soluble) |
| 4 | Number of amino acids | 632 |
| 5 | Molecular weight |
64,366.48 Dalton |
| 6 | Theoretical Isoelectric point (pI) | 5.12 |
| 7 | Total number of atoms | 8920 |
| 8 | Formula | C2764H4392N800O957S7 |
| 9 | Estimated half-life |
30 h (mammalian reticulocytes, in vitro) > 20 h (yeast, in vivo) > 10 h (Escherichia coli, in vivo) |
| 10 | Instability index |
27.70 (Stable) |
| 11 | Aliphatic index | 59.45 |
| 12 | Grand average of hydropathicity (GRAVY) | − 0.646 |
Bold denotes that antigenic score is higher than 0.45 (threshold value)
AllergenFP v.1.0 server, concluded that the vaccine constructs as a non-allergenic sequence although the AllerTOP v.2.0 supported the vaccine non-allergenic nature. Therefore, the vaccine construct might proceed for further investigation. As per the Protein-Sol server, predicted scaled solubility of the vaccine construct is 0.668 (Fig. 4). The ExPASy protparam result of all other physiochemical property of vaccine construct listed within the Table 3.
Fig. 4.
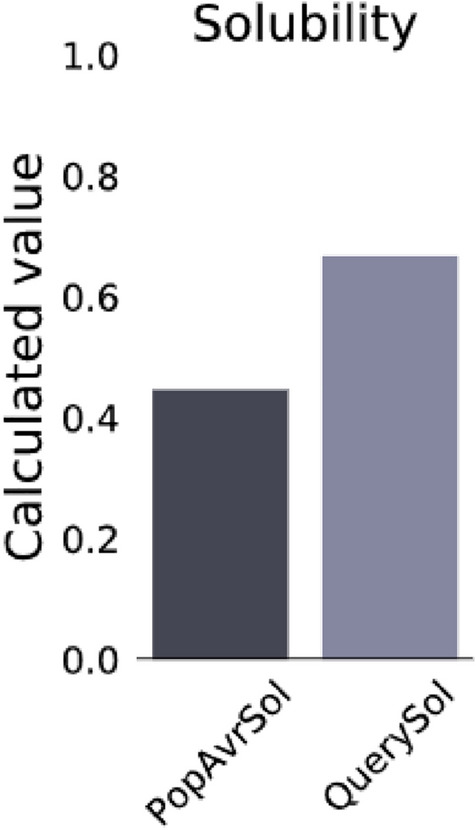
The diagram shows vaccine solubility plot
Tertiary Structure Modelling
SPARKS-X sever predicted 3D protein architecture from amino acid sequences represents one of the most crucial steps in computational structural biology. The 3D structure of vaccine construct was visualized by PyMol software. The 3D structure of vaccine construct is represented in Fig. 5, where red color depicts helix, blue color depicts coiled structure and cyan color depicts beta strand.
Fig. 5.
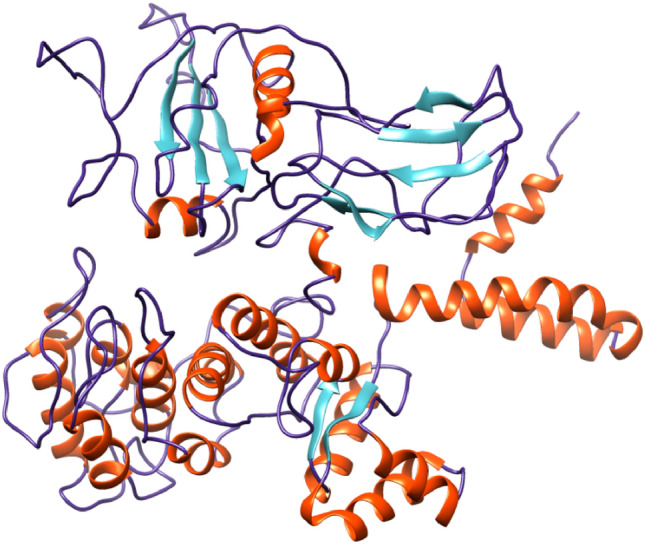
3-dimensional (3D) structure of final vaccine construct (orange color depicts helix, blue color depicts coiled structure and cyan color depicts beta strand) (Color figure online)
Model Validation
In this section, we used two online web servers regarding validation of vaccine tertiary structure. ProSA and PROCHECK web servers simultaneously revealed the model quality as well as analysis of vaccine stereo-chemical properties. ProSA ‘Z’ score of vaccine construct is − 3.74 and energy plot represented in Fig. 6a and b, respectively. PROCHECK server analyse residue-by-residue localization in Ramachandran plot, as shown in Fig. 6c. According to PROCHECK, 80.0% of amino acids residues are placed in most favored region of Ramachandran plot, moreover, as all amino acid residues are tabulated in the Table 4.
Fig. 6.

Vaccine model validation a ProSA validation of predicted structure by Z score; b energy plot for structure validation; c Ramachandran plot
Table 4.
Distribution of amino acid residues showing in Ramachandran plot
| Type of amino acid residues | No. of amino acid residues | Percentage |
|---|---|---|
| Most favoured region [A, B, L] | 404 | 80.0% |
| Additional allowed region [a, b, l, p] | 69 | 13.7% |
| Generously allowed region [~ a, ~ b, ~ l, ~ p] | 21 | 4.2% |
| Disallowed region | 11 | 2.2% |
| Total Number of non-glycine and non-proline residues | 505 | 100% |
| End-residues except glycine and proline | 2 | – |
| Glycine residues | 83 | – |
| Proline residues | 42 | – |
| Total number of residues | 632 | |
Discontinuous B-Cell Epitope Prediction
ElliPro online web server was predicted discontinuous B-cell epitopes based on tertiary structure of vaccine construct. Five epitopes were predicted from the ElliPro server from which epitope with maximum prediction score 0.841 was nominated as a discontinuous epitope. 3D structure of predicted discontinuous epitopes were shown in Fig. 7.
Fig. 7.
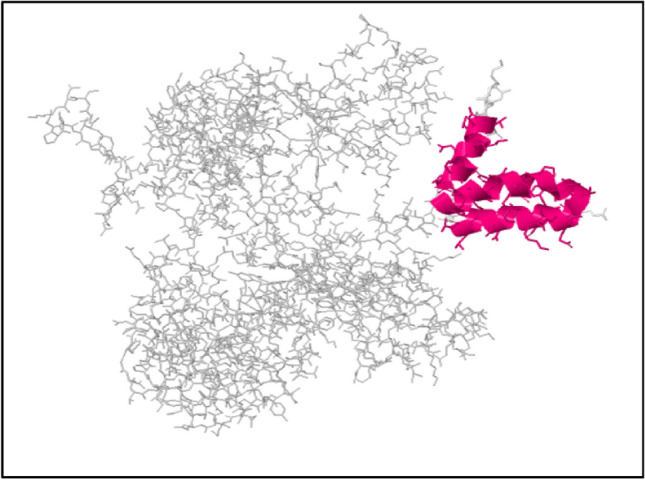
Magenta colour helix structure showed discontinuous B-cell epitopes in the 3D model of multi-epitope vaccine (Color figure online)
Molecular Docking Analysis and Binding Affinity Analysis
HADDOCK clustered 163 structures in 7 cluster(s), which represents 81% of the water refined HADDOCK generated models. The top ranked cluster with the lowest HADDOCK score is the most significant one for docking analysis. The HADDOCK score of − 151.4 + / − 8.7; indicates proper interaction between the vaccine construct and TLR5. The score of buried surface area (BSA) is 2692.6 + / − 99.3 Å2. It reflected close proximity and a less water-exposed on protein binding surface. Subsequently, RMSD plots determined the docking interaction which is represented in Fig. 9. The small RMSD value of the docked complex specifies the formation of good quality docked. The HADDOCK calculated electrostatic energy, desolvation energy, restraints violation energy, van der waals energy and Z-Score values listed in the Table 5. The Fig. 8 show molecular docking between Human TLR5 and vaccine construct, and Table 6 represented the hydrogen bond between both protein molecules.
Fig. 9.

a Molecular dynamics simulation of vaccine construct (through Cα fluctuations, atom distances, and dihedral angles). b B-factor plot analysis along with RMSD graphical plot
Table 5.
The statistical analysis of docking complex (TLR5 and vaccine construct)
| Vaccine construct-TLR5 | |
|---|---|
| HADDOCK score (a.u) | − 151.4 + / − 8.7 |
| Cluster size | 8 |
| RMSD from the overall lowest-energy structure | 1.4 + / − 0.8 |
| Van der Waals energy (kcal mol−1) | − 78.3 + / − 1.3 |
| Electrostatic energy (kcal mol−1) | − 306.4 + / − 34.1 |
| Desolvation energy (kcal mol−1) | − 12.2 + / − 3.8 |
| Restraints violation energy (kcal mol−1) | 3.8 + / − 2.1 |
| Buried surface area (Å2) | 2692.6 + / − 99.3 |
| Z-score | − 1.8 |
| PRODIGY binding score | Gibbs free energy (ΔG) − 11.7 kcal mol−1 and dissociation constant (Kd) 5.5E-09 M |
Negative HADDOCK score signifies strong protein interaction which is expressed in arbitrary units a.u) and binding affinity score
Fig. 8.
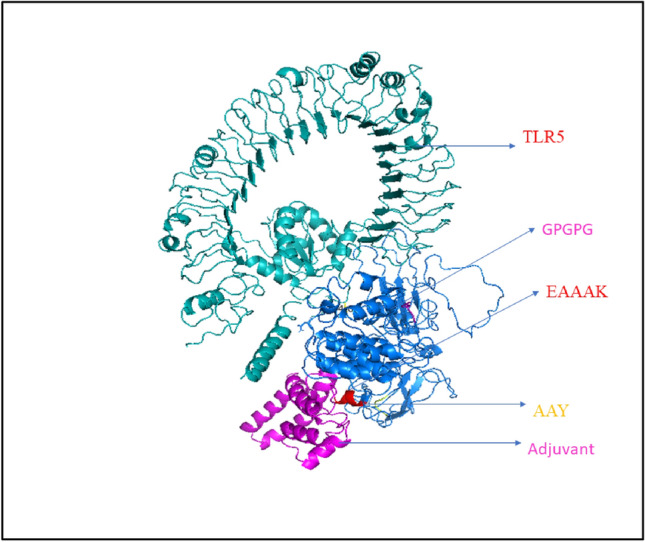
The interaction pattern of designed vaccine construct with human TLR5
Table 6.
Hydrogen Bond (distance, Å) between TLR5 and vaccine construct
| Vaccine construct (Atom) | TLR5 (Atom) | Distance (Å) |
|---|---|---|
| O:12,705 | H:16,357 | 3.28 |
| H:15,306 | H:12,133 | 4.46 |
| H:15,363 | H:20,751 | 3.42 |
| H:15,997 | H:20,775 | 4.46 |
| H:16,342 | C:12,415 | 4.94 |
| H:4343 | O:11,787 | 2.49 |
| O:4896 | H:21,124 | 3.91 |
PRODIGY provided binding affinity of Gibbs free energy (ΔG) and dissociation constant (Kd) values of docked complex, which was − 11.7 kcal mol−1 and 5.5E-09 M, respectively (Table 5). The negative value of docking complex has indicated the thermodynamically stable docking structure.
Molecular Dynamics Simulation of Vaccine Construct
Molecular dynamics simulation is a computer-based simulation method for analyzing the physical movements of atoms and molecules. For the visualisation of the molecular structures and the trajectories, we used VMD1.9.3 software package (Humphrey et al. 1996). The trajectories were analysed by means of backbone carbon atom Cα fluctuations, atom distances, and dihedral angles through VMD program, as represented in Fig. 9a. Analyses of essential dynamics (ED), NAMD software based CHARMM topology file had been involve for structure fluctuation determination. In the Fig. 9b represented our calculation of 1000 time step (TS) molecular dynamic simulation and analysed the overall vaccine structure was stable, it also flexible for binding properties accessed via B-factor and RMSD plot.
Normal Mode Analysis
Flexibility in biological macromolecules is a crucial property for interacting with substrate or protein–protein interactions machinery. Therefore, iMODs calculates the molecular motion as well as structural flexibility through NMA study that is incorporated with coordinates of the docked complex. The output of server directed the domains mobility represented by two coloured affine-arrows that indicates two clusters showing the NMA mobility (Fig. 10). The complex deformability mainly depends on the individual distortion of each residues (Cα atom), presented through coloured hinges in the chain (Fig. 11b). The server calculated eigenvalue as 2.935e−05, as represented in Fig. 11d. Moreover, eigenvalue and the variance are inversely related associated to each normal mode (Fig. 11c). The B-factor graph shows the average RMS, however B-factor plot represents the stable structure docked molecules shown in Fig. 11a. The covariance matrix represents with colours graph where, correlated, uncorrelated or anti-correlated motions, are represented by red, white and blue colors, respectively (Fig. 11e). An elastic network model shows docked protein molecule (Cα) atoms are interconnected with “springs” of certain strengths (the darker greys representation of the stiffer springs).
Fig. 10.
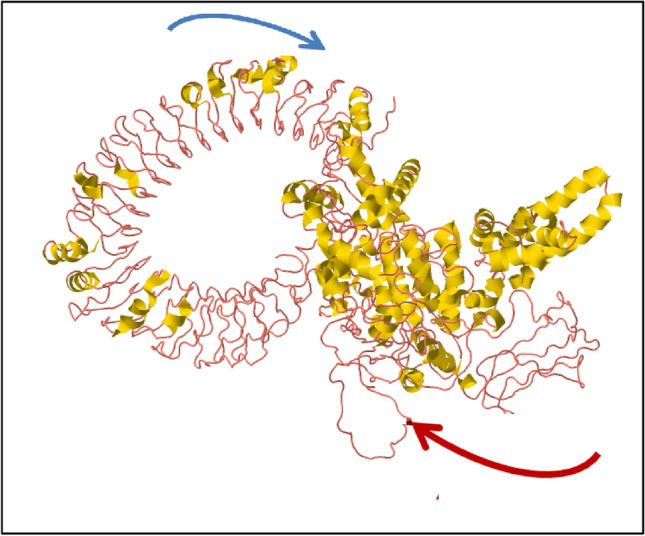
Molecular mobility assessed through NMA of docked complex. (Two coloured affine-arrows showing the domains mobility)
Fig. 11.

Results of iMODS a B-factor; b deformability plot; c variance plot; d eigenvalue; e covariance matrix analysis; f elastic network model
Codon Optimization and In-Silico Cloning
In silico cloning was performed to understand the expression of our developed multi epitopic vaccine candidate on E. coli hosts. Java Codon Adaptation (JCat) tool optimized our construct for the expression in E. coli (K12 strain). Additionally, we selected two filter parameters to evade unnecessary error, those are (a) avoiding rho-independent transcription terminators, (b) avoid prokaryotic ribosome binding sites. The CAI value of optimized vaccine construct is 0.971788060 (range between 0.8 and 1.0), and an optimal range of GC content is 53.1118 (range between 30 and 70%), representing the strong probability of protein expression. In the following step, we incorporate two restricted endonucleases (XhoI and XbaI) in the both ends of vaccine construct to assist the cloning process. Lastly, the vaccine construct was cloned within the pET28b ( +) vector with the help of SnapGene software (Fig. 12). The clone was 7.094 kb long.
Fig. 12.

In silico cloning of vaccine construct expressed in the E. coli host
Discussion
H. pylori infection is increasing day by day in developed as well as developing countries. The different studies indicate chronic infection may cause of gastric carcinoma (Correa 1995; Leung et al. 2004; Nomura et al. 1991). Additional infection with H. pylori leads to chronic gastritis, multifocal atrophic gastritis, occasionally gastric dysplasia, lesions and rarely intestinal metaplasia (Correa 1995). Till now, no safe and effective vaccine candidate is not available for treating H. pylori infection.
The current study is based on the development of a multi-epitope-based peptide vaccine against H. pylori. The main advantage of the epitope-based vaccine is that it can generate specific immune responses against epitopes and enhance binding affinity with the target receptor molecules and in turn avoid unfavorable non-epitopes selection (Bhattacharya et al. 2020d; Moise et al. 2015; Terry et al. 2015). The selected common epitopes elicits the immune-response with the target molecules for the next step of cellular cascades (Zhou et al. 2009). However, in the past Meza et al. reported multi epitope-based peptide (MEBP) vaccine candidates against H. pylori using reverse vaccinology approach (Meza et al. 2017). They predicted both B and T-cell epitopes of four virulent protein (FliD, Urease B, VacA and CagA) for designing a MEBP vaccine candidate against H. pylori. Lately, Khan et al. did the same and predicted B-cell and T-cell epitopes from different virulent protein (CagA, OipA, GroEL and VacA) and planned a MEBP vaccine against H. pylori (Khan et al. 2019).
Now, in this work we selected different four antigenic proteins (HpaA, FlaA, FlaB and Omp18) to design a MEBP vaccine through modern immunoinformatics approach. For the development of both humoral and cytotoxic immune response, T-cell epitopes derived from B-cell epitopes were identified in this study. Afterwards, by using several web tools, both MHC-I and MHC-II epitopes were predicted successfully. Subsequently, we have predicted common B-cell and T-cell epitope and joined with appropriate peptide linkers for formulation of this vaccine construct. An adjuvant 50S ribosomal protein L7/L12 was added at N terminal of the vaccine to boost up immunogenicity at cellular level which was used by other researcher for vaccine development (Shey et al. 2019).
In this study the 3D structure of vaccine candidate developed through SPARKS-X web server, and its validated through ProSA and PROCHECK web-server. Subsequently the Z score, energy plot was evaluated to understand its proper folding. Ramachandran plot of the amino acid residues of vaccine candidate was studied. Whereas, we predicted negative value through Z score analysis which indicates its reliable structure quality.
HADDOCK server demonstrated the binding interaction of the design vaccine candidate and TLR5, and calculated HADDOCK score is noted as − 151.4 + / − 8.7. It indicates proper interaction between the protein–protein complexes. Subsequently, the binding affinity of the designed vaccine with the TLR5 we passed through the PRODIGY server for calculating exact binding affinity score (Gibbs free energy (ΔG) is − 11.7 kcal mol−1 and dissociation constant (KD) is 5.5E-09 M). The stability of the vaccine structure was accessed through molecular dynamics simulation with the help of NAMD software package, it generated NAMD plot to show the molecular flexibility and stability. Whereas, NMA study performed to analyzed the molecular mobility, comparative deformability. The eigenvalue was noted as 2.935e−05 which indicates the better flexibility of the docking complex. Finally, the SnapGene cloning tool was employed to amplify the desired vaccine sequence within the pET28b ( +) expression vector.
Conclusion
In the summary, we demonstrated a reverse vaccinology approach to design and developed a MEBP vaccine which might be elicited simultaneous cellular and humoral immune responses in humans. The constructed vaccine was gone through the antigenicity and allergenicity assessment server, and concluded that our vaccine construct showed high antigenicity and non-allergenic nature. 3D structures of vaccine construct show its binding affinity with human TLR5 receptor and elicit the cellular responses. Though we found immunogenic response in our study through in-silico approaches but there is a strong need of in-vitro and in-vivo studies to validate the effectively, immunogenicity and most vital host safety to vaccine structure before human administration.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Information 1 (DOCX 23 kb)
Supplementary figure 1: The predicted B-cell epitopic region of each target protein shown in two color gradients where yellow color peaks represents epitopic region as well as green represents non epitopic region. (DOCX 359 kb)
Authors Contributions
PG, SB, MB and ARS conceptualized the methodology and data retrieval. GS and ARS did the analysis and validation. PG, SB, MB and CC wrote, review and edited the manuscript. CC and SSL supervised the whole study. All the authors finalized and approved the manuscript.
Funding
None.
Data Availability
All data includes within the manuscript.
Compliance with Ethical Standards
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Pratik Ghosh, Swarnav Bhakta and Manojit Bhattacharya have contributed equally.
Contributor Information
Sang-Soo Lee, Email: 123sslee@gmail.com.
Chiranjib Chakraborty, Email: drchiranjib@yahoo.com.
References
- Allan E, Dorrell N, Foynes S, Anyim M, Wren BWJ. Mutational analysis of genes encoding the early flagellar components of Helicobacter pylori: evidence for transcriptional regulation of flagellin A biosynthesis. J Bacteriol. 2000;182:5274–5277. doi: 10.1128/jb.182.18.5274-5277.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieva MR, El-Omar EMG. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology. 2008;134:306–323. doi: 10.1053/j.gastro.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Bhattacharya M, Malick RC, Mondal N, Patra P, Pal BB, Patra BC, Kumar Das B. Computational characterization of epitopic region within the outer membrane protein candidate in Flavobacterium columnare for vaccine development. J Biomol Struct Dyn. 2020;38:450–459. doi: 10.1080/07391102.2019.1580222. [DOI] [PubMed] [Google Scholar]
- Bhattacharya M, Sharma AR, Mallick B, Sharma G, Lee S-S, Chakraborty C. Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex. Infect Genet Evol. 2020;85:104587. doi: 10.1016/j.meegid.2020.104587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya M, et al. A SARS-CoV-2 vaccine candidate: in-silico cloning and validation. Inf Med Unlocked. 2020;20:100394. doi: 10.1016/j.imu.2020.100394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya M, et al. Computer aided novel antigenic epitopes selection from the outer membrane protein sequences of Aeromonas hydrophila and its analyses. Infect Genet Evol. 2020;82:104320. doi: 10.1016/j.meegid.2020.104320. [DOI] [PubMed] [Google Scholar]
- Buchan DW, Jones DT. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019;47:W402–W407. doi: 10.1093/nar/gkz297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsohn E, Nyström J, Bölin I, Nilsson CL, Svennerholm AM. HpaA is essential for Helicobacter pylori colonization in mice. Infect Immunity. 2006;74:920–926. doi: 10.1128/IAI.74.2.920-926.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol. 1995;19:S37–43. [PubMed] [Google Scholar]
- Cover TL, Blaser MJJG. Helicobacter pylori in health and disease. Gastroenterology. 2009;136:1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries SJ, Bonvin AM. CPORT: a consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS ONE. 2011;6:e17695. doi: 10.1371/journal.pone.0017695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries SJ, Van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010;5:883. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- Del Giudice G, Malfertheiner P, Rappuoli RJ. Development of vaccines against Helicobacter pylori. Expert Rev Vaccines. 2009;8:1037–1049. doi: 10.1586/erv.09.62. [DOI] [PubMed] [Google Scholar]
- Dhal AK, Pani A, Mahapatra RK, Yun S-IJI. An immunoinformatics approach for design and validation of multi-subunit vaccine against Cryptosporidium parvum. Immunobiology. 2019;224:747–757. doi: 10.1016/j.imbio.2019.09.001. [DOI] [PubMed] [Google Scholar]
- Dimitrov I, Naneva L, Doytchinova I, Bangov IJB. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30:846–851. doi: 10.1093/bioinformatics/btt619. [DOI] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The proteomics protocols handbook. Berlin: Springer; 2005. pp. 571–607. [Google Scholar]
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn DJ. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33:W526–W531. doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GSL Biotech LLC (2015) SnapGene software [web]. www.snapgene.com
- Gu H. Role of flagella in the pathogenesis of Helicobacter pylori. Curr Microbiol. 2017;74:863–869. doi: 10.1007/s00284-017-1256-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker JJB. Protein–Sol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33:3098–3100. doi: 10.1093/bioinformatics/btx345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooi JKY, et al. Global prevalence of Helicobacter pylori Infection: systematic review and meta-analysis. Gastroenterology. 2017;153:420–429. doi: 10.1053/j.gastro.2017.04.022. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten KJ. VMD: visual molecular dynamics. J Mol Graph Model. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jespersen MC, Peters B, Nielsen M, Marcatili PJ. BepiPred-20: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45:W24–W29. doi: 10.1093/nar/gkx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josenhans C, Labigne A, Suerbaum SJ. Comparative ultrastructural and functional studies of Helicobacter pylori and Helicobacter mustelae flagellin mutants: both flagellin subunits, FlaA and FlaB, are necessary for full motility in Helicobacter species. J Bacteriol. 1995;177:3010–3020. doi: 10.1128/jb.177.11.3010-3020.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamens JJNAR. The Addgene repository: an international nonprofit plasmid and data resource. Nucleic Acids Res. 2015;43:D1152–D1157. doi: 10.1093/nar/gku893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Khan S, Ali A, Akbar H, Sayaf AM, Khan A, Wei D-QJ. Immunoinformatics approaches to explore Helicobacter pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci Rep. 2019;9:1–13. doi: 10.1038/s41598-019-49354-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoder G, Muhammad JS, Mahmoud I, Soliman SSM, Burucoa C. Prevalence of Helicobacter pylori and its associated factors among healthy asymptomatic residents in the United Arab Emirates. Pathogens. 2019;8:44. doi: 10.3390/pathogens8020044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, et al. Immune epitope database analysis resource. Nucleic Acids Res. 2012;40:W525–W530. doi: 10.1093/nar/gks438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Leung W, et al. Factors predicting progression of gastric intestinal metaplasia: results of a randomised trial on Helicobacter pylori eradication. Gut. 2004;53:1244–1249. doi: 10.1136/gut.2003.034629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Blanco JR, Aliaga JI, Quintana-Ortí ES, Chacón PJ. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res. 2014;42:W271–W276. doi: 10.1093/nar/gku339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundström AM, Blom K, Sundaeus V, Bölin IJ. HpaA shows variable surface localization but the gene expression is similar in different Helicobacter pylori strains. Microbial Pathog. 2001;31:243–253. doi: 10.1006/mpat.2001.0466. [DOI] [PubMed] [Google Scholar]
- MacKerell AD, Jr, Banavali N, Foloppe NJ. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, Baldi PJB. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26:2936–2943. doi: 10.1093/bioinformatics/btq551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza B, Ascencio F, Sierra-Beltrán AP, Torres J, Angulo CJI, Genetics E. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: an in silico approach. Infect Genet Evol. 2017;49:309–317. doi: 10.1016/j.meegid.2017.02.007. [DOI] [PubMed] [Google Scholar]
- Moise L, et al. iVAX: An integrated toolkit for the selection and optimization of antigens and the design of epitope-driven vaccines. Human Vaccines Immunother. 2015;11:2312–2321. doi: 10.1080/21645515.2015.1061159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Van der Graaf C, Van der Meer JW, BJJ Kullberg JW. Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. J Leukoc Biol. 2004;75:749–755. doi: 10.1189/jlb.1103543. [DOI] [PubMed] [Google Scholar]
- Nomura A, Stemmermann GN, Chyou P-H, Kato I, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med. 1991;325:1132–1136. doi: 10.1056/NEJM199110173251604. [DOI] [PubMed] [Google Scholar]
- Pandey RK, Bhatt TK, Prajapati VK. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci Rep. 2018;8:1–11. doi: 10.1038/s41598-018-19456-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra P, et al. Identification and design of a next-generation multi epitopes bases peptide vaccine candidate against prostate cancer: an in silico approach. Cell Biochem Biophys. 2020;4:495–509. doi: 10.1007/s12013-020-00912-7. [DOI] [PubMed] [Google Scholar]
- Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, Peters BJ. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinf. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Yang J, Zhang YJ. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012;40:W471–W477. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha CK, Hasan MM, Hossain MS, Jahan MA, Azad AK. In silico identification and characterization of common epitope-based peptide vaccine for Nipah and Hendra viruses Asian Pacific. J Trop Med. 2017;10:529–538. doi: 10.1016/j.apjtm.2017.06.016. [DOI] [PubMed] [Google Scholar]
- Salama NR, Hartung ML, Müller AJNRM. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol. 2013;11:385–399. doi: 10.1038/nrmicro3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shey RA, et al. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci Rep. 2019;9:1–18. doi: 10.1038/s41598-019-40833-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WS, Jeon YJ, Namgung B, Hong M, S-iJSr Y. A conserved TLR5 binding and activation hot spot on flagellin. Sci Rep. 2017;7:1–11. doi: 10.1038/srep40878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry FE, Moise L, Martin RF, Torres M, Pilotte N, Williams SA, De Groot AS. Time for T? Immunoinformatics addresses vaccine design for neglected tropical and emerging infectious diseases. Expert Rev Vaccines. 2015;14:21–35. doi: 10.1586/14760584.2015.955478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testerman TL, Morris JJ. Beyond the stomach: an updated view of Helicobacter pylori pathogenesis, diagnosis, and treatment. World J Gastroenterol. 2014;20:12781. doi: 10.3748/wjg.v20.i36.12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viala J, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- Voland P, Hafsi N, Zeitner M, Laforsch S, Wagner H, Prinz CJI. Antigenic properties of HpaA and Omp18, two outer membrane proteins of Helicobacter pylori. Infect Immunity. 2003;71:3837–3843. doi: 10.1128/IAI.71.7.3837-3843.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, Wishart DS. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003;31:3316–3319. doi: 10.1093/nar/gkg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone AJB. PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics. 2016;32:3676–3678. doi: 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- Yamaoka YJNRCO. How to eliminate gastric cancer-related death worldwide? Nat Rev Clin Oncol. 2018;15:407–408. doi: 10.1038/s41571-018-0029-8. [DOI] [PubMed] [Google Scholar]
- Yang Y, Faraggi E, Zhao H, Zhou YJB. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics. 2011;27:2076–2082. doi: 10.1093/bioinformatics/btr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamani M, Ebrahimtabar F, Zamani V, Miller WH, Alizadeh-Navaei R, Shokri-Shirvani J, Derakhshan MH. Systematic review with meta-analysis: the worldwide prevalence of Helicobacter pylori infection. Aliment Pharmacol Ther. 2018;47:868–876. doi: 10.1111/apt.14561. [DOI] [PubMed] [Google Scholar]
- Zhou W-Y, et al. Therapeutic efficacy of a multi-epitope vaccine against Helicobacter pylori infection in BALB/c mice model. Vaccine. 2009;27:5013–5019. doi: 10.1016/j.vaccine.2009.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information 1 (DOCX 23 kb)
Supplementary figure 1: The predicted B-cell epitopic region of each target protein shown in two color gradients where yellow color peaks represents epitopic region as well as green represents non epitopic region. (DOCX 359 kb)
Data Availability Statement
All data includes within the manuscript.