

Abstract
The islet of Langerhans produces endocrine hormones to regulate glucose homeostasis. The normal function of the islet relies on the homeostatic regulations of cellular composition and cell–cell interactions within the islet microenvironment. Immune cells populate the islet during embryonic development and participate in islet organogenesis and function. In obesity, a low-grade inflammation manifests in multiple organs, including pancreatic islets. Obesity-associated islet inflammation is evident in both animal models and humans, characterized by the accumulation of immune cells and elevated production of inflammatory cytokines/chemokines and metabolic mediators. Myeloid lineage cells (monocytes and macrophages) are the dominant types of immune cells in islet inflammation during the development of obesity and type 2 diabetes mellitus (T2DM). In this review, we will discuss the role of the immune system in islet homeostasis and inflammation and summarize recent findings of the cellular and molecular factors that alter islet microenvironment and β cell function in obesity and T2DM.
Introduction
Obesity has become a global epidemic and is the major cause of insulin resistance. Obesity is associated with a systemic low-grade inflammation, which manifests in multiple organs. In pancreatic islet, obesity dampens β cell homeostasis. One major detrimental consequence of obesity-associated islet inflammation is the impairment of β cell function, causing insufficient insulin release. This leads to hyperglycemia and the onset of type 2 diabetes mellitus (T2DM).
Normal islet function, in particular insulin secretion, relies on homeostatic regulations within the islet microenvironment. Endocrine cells, immune cells, neurons, and vascular endothelial cells are all involved in the maintenance of islet homeostasis and β cell function. Obesity-associated metabolic factors (e.g. free fatty acids, FFAs) can directly impinge β cells or indirectly via various ‘accessory’ cells in the islet. Numerous studies have directly focused on β cells to determine how obesity affects insulin secretion and the underlying mechanisms in these cells (Hudish et al., 2019). Meanwhile, increasing body of evidence has supported the important roles of non-β cells in islet homeostasis and β cell activities. For instance, obesity dampens the abundancy of pericytes, which controls the diameter of islet capillaries and the release of insulin from β cells (Almaca et al., 2018). In many cases, islet macrophages act as sensors to sense metabolic and inflammatory cues and subsequently regulate β cell proliferation and function (Weitz et al., 2018; Zinselmeyer et al., 2018; Ying et al., 2019). These findings indicate a multifaceted cellular crosstalk in the regulation of islet homeostasis and the dysregulation of β cell function in obesity and T2DM.
During the development of obesity, the islets undertake adaptive responses evidenced by the compensatory proliferation and increased insulin production. These adaptation mechanisms may temporarily compensate the need for more insulin to lower blood glucose. With the gradually aggravated insulin resistance, β cell adaptation will fail. It remains to be determined what factors trigger β cell adaptation and why it fails eventually. Emerging studies suggest that elevated degree of inflammation might be responsible for the transition from β cell adaptation to β cell functional failure (Donath et al., 2009; Ying et al., 2019).
Understanding the cellular and molecular regulators for both islet physiology and pathophysiology will provide important knowledge for developing better therapeutic regimens for all forms of diabetes. While most of the knowledge of islet homeostasis and inflammation we have learned so far is from studies in rodent models, human studies and clinical trials are rapidly growing in numbers. Among these efforts, strategies targeting inflammatory pathways (e.g. IL-1β blockade) emerge as a promising lead for resorting β cell mass and function. In this review, we will first summarize how a homeostatic islet microenvironment at steady state is established, with a focus on the resident immune cells and their functions. Then we will provide an updated review covering the cellular and molecular mediators in immune system that are involved in obesity-associated islet inflammation. We will conclude by summarizing recent progresses in human data and clinical trials and raising key questions for future studies.
Islet homeostasis
Islet histology
The pancreas is composed of exocrine (acinar and ductal tissues) and endocrine tissues that control the body’s digestive activities and glucose homeostasis, respectively. The endocrine tissues (i.e. the islets of Langerhans) only comprise ~2% of the total mass of the pancreas. The islet harbors a panel of endocrine cells bearing distinct functions (Figure 1). Alpha (α) cells and β cells are the two cell populations representing the primary functions of the islet. The proportions of each endocrine cell types vary among species. In mouse, ~80% of the islet cells are β cells that produce insulin, and glucagon-producing α cells are <10%. In contrast, in humans, the proportion of α cells is remarkably higher (~30%–40%), with a relatively lower β cell percentage (~50%–60%) (Dolensek et al., 2015). Other islet endocrine cell types, to much smaller proportions, are delta (δ), pancreatic polypeptide (PP or γ), and  cells that produce somatostatin, pancreatic polypeptide, and ghrelin, respectively. The reasons for the species-specific ratios among endocrine cells within the islets remain unknown.
cells that produce somatostatin, pancreatic polypeptide, and ghrelin, respectively. The reasons for the species-specific ratios among endocrine cells within the islets remain unknown.
Figure 1.
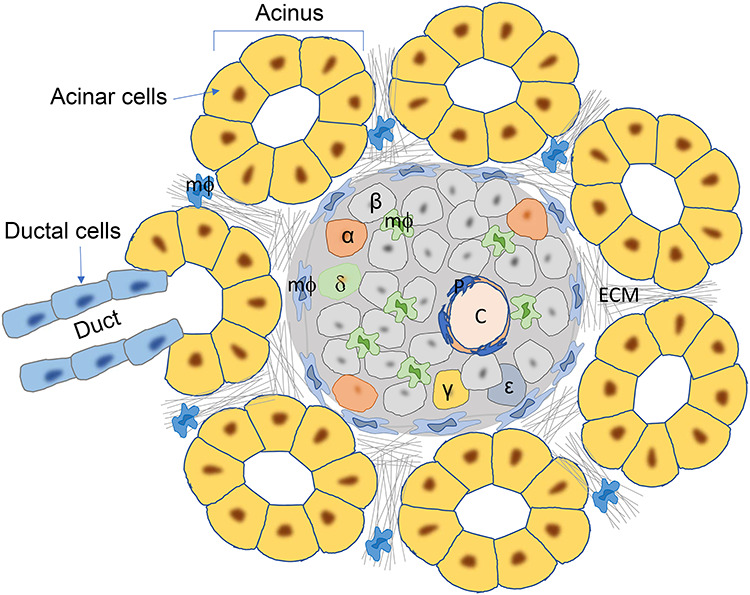
Pancreas histology at steady state, shown by micrographic view of the mouse pancreas. The islet of Langerhans is surrounded by pancreatic acini, which are composed of exocrine acinar cells. The acinus is also connected to exocrine duct, which is formed by ductal cells. The islet contains various types of endocrine cells, depicted by different colors and labeled (α, β, δ, γ, and  ). The islet capillaries (C) are coated by pericytes (P). Heterogeneous populations of macrophages (mФ) are seen in both exocrine pancreas and the islet. ECM is found in all area of the pancreas, including the exocrine–endocrine interfaces.
). The islet capillaries (C) are coated by pericytes (P). Heterogeneous populations of macrophages (mФ) are seen in both exocrine pancreas and the islet. ECM is found in all area of the pancreas, including the exocrine–endocrine interfaces.
In addition to endocrine cells, various types of non-endocrine ‘accessory’ cells are present in both exocrine and endocrine pancreas (Tang et al., 2018a). These cells form connective tissues and vascular and neural networks (Aamodt and Powers, 2017; Tang et al., 2018b). Functionally, these cells are important for islet morphogenesis, maintenance, and function. Resident immune cells can be found in both exocrine and endocrine pancreas (see ‘Immune cells in the islet and exocrine pancreas and their physiological role’ below for details).
The exocrine–endocrine boundary and the capsule of the islet
The islets are embedded in a much larger exocrine pancreas and the total mass of the islets only accounts for 2% of the pancreas. The physical interface between exocrine and endocrine pancreas is a critical site permissive for exocrine–endocrine communications (Figure 1). The interface is enriched for extracellular matrix (ECM) that is essential for the physiological structure and function of the islets (Wang and Rosenberg, 1999; Pinkse et al., 2006; Miao et al., 2013). Collagen type IV and VI and laminins are the most abundant molecules in the islet ECM. Interesting questions include whether and how exocrine and endocrine pancreases communicate with each other and how these communications play a role in pancreatic diseases. Emerging studies from various settings have suggested that exocrine and endocrine pancreas indeed affect each other (Wang and Rosenberg, 1999; Cobelli and Vella, 2017; Dirice et al., 2019). For instance, it has been demonstrated that pancreatic exocrine cells in adult mice can be reprogrammed into cells that resemble β cells (Xu et al., 2008; Zhou et al., 2008).
The capsule of the islet is another interesting topic with recently increasing numbers of studies to explore its structure and function (Korpos et al., 2013; Tang et al., 2018a). In islet transplantation, optimizing human islet isolation is a critical step to ensure the quality and survival of the transplanted islets. However, enzymatic digestion methods that are commonly used may dampen the ECM, especially the capsule of the islets (Irving-Rodgers et al., 2014). Preventing or restoring the integrity of the islet basal membrane would have a great impact on the success of islet transplantation-based therapy. Pre-exposure of islets to matrix proteins may improve the survival and therefore function of transplanted islets (Wang and Rosenberg, 1999). Macrophages are enriched in the peri-islet capsular area. Ying et al. (2019) found a population of F4/80+ tissue-resident macrophages that line along this peri-islet capsular zone (Figure 1). The role of these macrophages under steady state remains to be defined. However, under diseased conditions, these peri-islet macrophages may act as a barrier to block the infiltration by other types of immune cells (Yuan et al., 2017; Ying et al., 2019) or to promote β cell proliferation under obese conditions (Ying et al., 2019).
Schwann cells, the glial cells in the periphery, are also highly enriched in the peripheral area of the islets (Sunami et al., 2001; Tang et al., 2014; Juang et al., 2015). These cells release neurotrophic factors and the glial cell line-derived neurotrophic factor (Teitelman et al., 1998; Sunami et al., 2001; Mwangi et al., 2008; Thorens, 2014). Together with other parasympathetic and sympathetic nerves, these innervations form peri-islet neural network support islet function (Thorens, 2014). Defects in these neural regulations impair β cell mass and function.
Cell–cell communications within the islet
An increasing number of studies have revealed the cellular crosstalk in the islets. The communications among individual β cells contribute to glucose homeostasis during fasting and feeding. Konstantinova et al. (2007) provided data showing that β–β interaction inhibited basal insulin secretion but enhanced glucose-stimulated insulin secretion (GSIS). They proposed a bidirectional signaling pathway connecting neighboring β cells via the EphA–ephrinA axis (Figure 2; Konstantinova et al., 2007). In addition, the crosstalk between β cells and other endocrine cells has also been reported (Figure 2). For instance, human β cells release serotonin to inhibit glucagon secretion from α cells (Almaca et al., 2016). In this loop interaction, serotonin secreted from β cells decreases cyclic AMP levels in neighboring α cells, leading to suppressed secretion of glucagon. Islet δ cells are also important paracrine regulators of β cell and α cell secretory activity. Glucagon secreted from α cells stimulates β cell insulin secretion (Svendsen et al., 2018; Zhu et al., 2019). δ cells can balance the secretion of insulin by β cells and glucagon by α cells (Rorsman and Huising, 2018). By experimentally expressing a light-sensitive ion channel in β cells, Briant et al. (2018) examined the interactions among α, β, and δ cells. They found that optogenetic activation of the β cells propagated to neighboring δ cells via gap junctions and the consequential stimulation of somatostatin secretion inhibited α cell electrical activity in a paracrine manner (Briant et al., 2018). Interestingly, δ cells can interact with β cells through filopodia-like structure and regulate β cell insulin secretion in an IGF-1/VEGF signaling-dependent manner (Arrojo et al., 2019). An interesting crosstalk between β and δ cells was revealed by van der Meulen et al. (2015). In this study, they found that mature β cells expressed urocortin3 that can promote glucose-stimulated somatostatin secretion via cognate receptors on δ cells (van der Meulen et al., 2015). In summary, all these studies support a notion that endocrine cells in the islets actively interact among each other, via various mechanisms. This intra-organ crosstalk is essential for a normal function of the islet.
Figure 2.
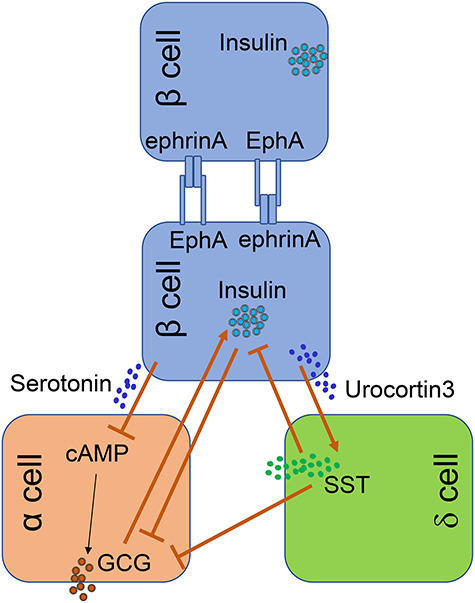
Cellular crosstalk in the islet. The crosstalk among different types of endocrine cells has been increasingly revealed. These cell–cell interactions (as depicted herein) are important for establishing an equilibrium of islet hormone production. These data reflect a critical aspect of islet homeostasis occurring within islet microenvironment. GCG, glucagon; SST, somatostatin.
The islet is one of the most vascularized tissues. The dilation of islet vasculature is critical for the secretion of insulin and other islet hormones. Pericytes, the smooth muscle-like cells that line the outside of blood vessels throughout the body, are involved in insulin secretion and islet homeostasis (Figure 1). Through the involvement of sympathetic neurons and endogenous adenosine, islet pericytes control local blood flow through regulating the diameter of the islet capillaries (Almaca et al., 2018). In addition, the interactions between β cells and pericytes play a key role in the regulation of islet blood flow, capillary diameter, and insulin secretion. This was evidenced by that β cell-derived angiopoietin-1 regulated GSIS. Knockout of angiopoietin-1 in β cells resulted in less pericyte coverage, disorganized endothelial cell ultrastructure, and enhanced infiltration of inflammatory cells (Park et al., 2019).
Immune cells in the islet and exocrine pancreas and their physiological role
Pancreas-resident immune cell
Immune cells are frequently present in all nonlymphoid tissues where they participate in host defense against infection and tissue repair after injury (Burzyn et al., 2013; Davies et al., 2013; Heath and Carbone, 2013; Jenne and Kubes, 2013). In the pancreas, yolk sac-derived primitive macrophages populate the pancreas during embryogenesis (Schulz et al., 2012). Calderon et al. (2015) analyzed the origin of pancreatic macrophages and found that macrophages residing in the islets at steady state are derived from adult hematopoiesis not yolk sac. In contrast, the origins of adult exocrine macrophages are mixed, with some of them derived from yolk sac and others from adult definitive hematopoiesis. Another study reported a transient appearance of CCR2+F4/80+ myeloid lineage cells during a narrow perinatal window (between birth and 2 weeks of age) in B6 mice (Mussar et al., 2017). This population was not found in adult B6 mice, suggesting a time-restricted condition permissive for the recruitment and retention of CCR2+ cells in the pancreas. These cells populated the newborn pancreas within the mesenchyme surrounding acinar and endocrine epithelial clusters. While they express CCR2, morphological analysis revealed that these cells are of macrophage, not monocyte identity. In addition to CCR2 and F4/80, these cells also express other macrophage markers, such as CD115, and scavenger receptors CD206 and CD93 (Mussar et al., 2017). With the combination of in situ immunostaining of pancreatic cryosections and flow cytometric analysis, Ying et al. (2019) identified two major subsets of macrophages distinguished by their anatomical locations and surface markers. The CD11c+F4/80+ cells are restricted to the intra-islet distribution and the CD11c−F4/80hi subset is located at the peri-islet capsular zone. Transcriptome profiling and gene signature analysis of sorted subsets showed that both populations are of macrophage not dendritic cell identity (Ying et al., 2019).
Therefore, macrophages with different origins populate both the exocrine and endocrine pancreas perhaps at different stages of development (Table 1). Phenotypically distinct myeloid subsets are selectively enriched in discrete compartments of the pancreas and at distinct developmental stages. After birth, both islet and exocrine macrophages undertake a further differentiation process, acquiring distinct phenotypic and functional properties. It is possible that local factors derived from either the islet or the exocrine pancreas can instruct resident macrophages to differentiate. Parabiosis and adoptive transfer studies revealed that islet macrophages have a minimal exchange with circulating monocytes (Calderon et al., 2015; Ying et al., 2019), suggesting that these macrophages are self-maintained locally. Indeed, BrdU incorporation assays revealed that at steady state, macrophages in both the islet and exocrine pancreas can proliferate (Calderon et al., 2015; Ying et al., 2019).
Table 1.
Myeloid cells in the islet and exocrine pancreas at steady state.
| Cell type | Location | Origin | Maintenance | Reference |
|---|---|---|---|---|
| F4/80hi | Embryonic pancreas | Yolk sac | Unknown | Schulz et al. (2012) |
| F4/80+CX3CR1+ | Islet | Adult hematopoiesis | Local proliferation | Calderon et al. (2015) |
| F4/80+CD206+ | Exocrine | Mixed origins | Local proliferation | Calderon et al. (2015) |
| F4/80+CD206− | Exocrine | Mixed origins | Local proliferation | Calderon et al. (2015) |
| F4/80+CCR2+ | Neonatal pancreas | Unknown | Mussar et al. (2017) | |
| CD11c+F4/80+ | Intra-islet | Unknown | Local proliferation | Ying et al. (2019) |
| CD11c−F4/80hi | Peri-islet | Unknown | Local proliferation | Ying et al. (2019) |
| Gr1+F4/80− granulocytes | E14.5 pancreas | Unknown | Unknown | Mussar et al. (2017) |
No dendritic cell has been found in mouse or human pancreas in non-T1D subjects. One study reported the presence of Gr1+F4/80− granulocytes in the E14.5 pancreas of B6 mouse (Mussar et al., 2017); however, the anatomical distribution of these Gr1+F480− cells and whether they persist through adulthood remain unclear. Under steady state, adaptive immune cells (such as T cells and B cells) are mainly found in the exocrine pancreas not the islets (Calderon et al., 2015; Ying et al., 2019).
Immune cells in islet morphogenesis and normal β cell function
Macrophages are involved in islet organogenesis. Banaei-Bouchareb et al. (2004) showed that F4/80+ macrophages can be detected in the fetal pancreatic connective tissue in Swiss mice from E12.5 and onward. These macrophages express CSF1R (CD115). The role of macrophages in islet organogenesis is evidenced by the defective islet development in osteopetrotic (op/op) mice. Due to the lack of colony-stimulating factor 1 (CSF1), very few macrophages are detected in the pancreas (and islets) of op/op mice. The islets in op/op mice are poorly developed, characterized by remarkably reduced β cell mass and cell size, and the islet exhibits a reduced rate of expansion postnatally (Banaei-Bouchareb et al., 2004; Calderon et al., 2015). Interestingly, in these mice, α cell mass is not affected by macrophage deficiency, suggesting a more profound effect of islet macrophages on β cell development pre- and postnatally (Banaei-Bouchareb et al., 2004). Moreover, Mussar et al. (2017) found that during the perinatal window of B6 mice, a population of CCR2+ myeloid cells are restrictedly required for the proliferation of the endocrine pancreatic epithelium. Using CCR2-specific depletion models, they found that the loss of this CCR2+ myeloid population caused a reduction in β cell proliferation, dysfunctional islet phenotypes, and glucose intolerance in newborns. Importantly, replenishment of pancreatic CCR2+ myeloid compartments by adoptive transfer rescued these defects (Mussar et al., 2017).
Tissue-resident macrophages sense microenvironmental cues and respond by producing effector cytokines, chemokines, and growth factors (Iwasaki and Medzhitov, 2015). Macrophages secrete numerous substances, such as cytokines (IL-6, TNF-α, and IFN-γ), growth factors (EGF, TGF-β, VEGF, HGF, NGF, and IGF-1/2), metalloproteinases, and ECM proteins, involving in pancreas organogenesis and β cell neogenesis (Bonner-Weir, 2000; Nielsen et al., 2001). These various types of cytokines and growth factors may be utilized by different types of immune cells or involved in different types of cell–cell communications. For example, Mussar et al. (2017) identified IGF-2 as a molecular mediator for CCR2+ myeloid cells to promote perinatal β cell proliferation.
Macrophages are also involved in regulating β cell function. Ying et al. (2019) found that in lean B6 mice, depletion of islet macrophages resulted in an impaired β cell GSIS. How macrophages exert a beneficial impact on β cell insulin secretion at steady state remains to be investigated. Of note, Weitz et al. (2018) provided data supporting an interesting interaction between macrophages and β cells. In this interaction loop, islet purinergic receptor-expressing macrophages sense ATP that is co-released with insulin from β cells and monitor the functional status of β cells (Weitz et al., 2018).
Obesity-associated islet inflammation
Adaptation of the islet to obesity and insulin resistance
Under stress, injury, or metabolic pressure, the host tissues undertake adaptive responses to retain their functions (Kotas and Medzhitov, 2015; De Jesus and Kulkarni, 2019). In obesity, pancreatic islets can adapt to insulin resistance, evidenced by islet hypertrophy and compensatory β cell proliferation (Burke et al., 2016). Islet hypertrophy is characterized by an increase of the number of β cells per islet. This is primarily due to increased proliferation of β cells (Hull et al., 2005; Ebato et al., 2008; Peyot et al., 2010; Stamateris et al., 2013; Mosser et al., 2015). As a consequence, the size of the islet is increased in obesity. Multiple factors, including glucose (Alonso et al., 2007; Levitt et al., 2011; Porat et al., 2011), insulin (Assmann et al., 2009), and hepatocyte growth factor (Garcia-Ocana et al., 2000; Araujo et al., 2012; Demirci et al., 2012) can stimulate β cell replication under obese conditions. In addition, peri-islet ECM also plays a critical role permissive for the growth of the islet under obese conditions. Accompanying with these islet adaptations are the vascular changes. Dai et al. (2013) investigated islet vascular alterations in response to insulin resistance in three different rodent models of obesity, namely, ob/ob mice, GLUT4+/−, and high fat diet (HFD)-induced obesity. They found that islet vessel area was increased; however, intra-islet vessel density was decreased (Dai et al., 2013). Interestingly, these changes mostly occurred in the core of the islet and independent of islet size.
Cellular and molecular components of islet inflammation
Inflammation is a key feature of obesity-induced alterations in pancreatic islet microenvironment. In pancreatic islets, obesity-associated metaflammation is characterized by the accumulation of immune cells and elevated production of inflammatory cytokines/chemokines and metabolic mediators (Table 2).
Table 2.
Immune cells and related molecules in islet inflammation in animal models.
| Model | Cell type | Main function | Molecular mediator | Reference |
|---|---|---|---|---|
| HFD B6 mice and GK rats | Macrophage (CD68+CD11b+) | Sign of inflammation | Ehses et al. (2007) | |
| Palmitate treated (B6); db/db and KKAy mice | Monocyte (CD11b+Ly-6C+) | Induce β cell dysfunction | IL-1β and TNF-α | Eguchi et al. (2012) |
| db/db mice | Macrophage (CD68+F4/80− and CD68+F4/80+) | Proinflammatory | IL-6, KC, MCP-1, M-CSF, GM-CSF | Cucak et al. (2014) |
| HFD B6 mice | Macrophage (CD11chiMHCIIhi) | Impair GSIS | Cell–cell contact | Ying et al. (2019) |
| HFD B6 mice | Macrophage (CD11chiMHCIIhi) | Promote β cell proliferation | PDGF | Ying et al. (2019) |
| HFD B6 mice | Macrophage (F4/80hiMHCIIhi) | Promote β cell proliferation | PDGF | Ying et al. (2019) |
| HFD, db/db mice | Macrophage (F4/80+CD68+) | Vascular remodeling | VEGF | Chittezhath et al. (2019) |
| HFD, Il33−/− B6, BalB/C mice | Type 2 innate lymphoid cells | Promote β cell function | IL-33, IL-13, CSF2, RA | Dalmas et al. (2017) |
Cellular mediators
The accumulation of immune cells in pancreatic islets is evidenced in both rodent models of and humans with obesity and T2DM (Ehses et al., 2007; Richardson et al., 2009; Eguchi et al., 2012; Cucak et al., 2014; Ying et al., 2019). Multiple types of immune cells have been found in the islets, especially under obese conditions. Myeloid lineage cells (primarily monocytes and macrophages) are the dominant cellular component of obesity-associated islet inflammation in both humans and animal models.
(I) Macrophages. The accumulation of myeloid lineage cells, especially macrophages, is reported in nearly all studies so far concerning the cellular components of islet inflammation in obesity in both animal models and humans (Ehses et al., 2007; Richardson et al., 2009; Eguchi et al., 2012; Cucak et al., 2014; Ying et al., 2019). We highlight below several of these studies. One of the first comprehensive analyses of islet immune profiles was performed by Ehses et al. (2007). Using CD68 and CD11b as markers, they found an increased number of macrophages in the pancreas of HFD-fed C57BL/6J (B6) mice and also GK rats (Ehses et al., 2007). This general phenomenon of myeloid cell accumulation was confirmed by Cucak et al. (2014). In a different mouse model of obesity that was leptin receptor deficient (db/db mouse). Interestingly, in this study, they identified two subsets of macrophages distinguished by the expression of CD68 and F4/80. Both CD68+F4/80− and CD68+F4/80+ subsets exhibited a proinflammatory M1-like phenotype (Cucak et al., 2014). Using both diet- and genetically induced rodent models of obesity, Ying et al. (2019) provided an in-depth assessment of obesity-associated islet metaflammation in mice. Obesity induces a massive increase of intra-islet macrophages characterized by the expression of CD11c and MHCII. Interestingly, these CD11c+MHCII+ intra-islet macrophages can engulf insulin-containing granules, and this process was dramatically enhanced under obese conditions. Another study reported that macrophage depletion compromised islet remodeling in terms of size, vascular density, and insulin secretion capacity (Chittezhath et al., 2019). Together, it becomes clear that myeloid lineage cells especially macrophages dominate the immune cell accumulation of pancreatic islets in obesity and affect islet homeostasis and β cell function via various mechanisms.
An important question is what causes the accumulation of macrophages in pancreatic islets during the development of obesity. It has been known that tissue-resident macrophages are maintained by local self-proliferation (Jenkins et al., 2011). This mechanism has also been reported in islet macrophages by different groups (Calderon et al., 2015; Mussar et al., 2017; Ying et al., 2019). Calderon et al. (2015) found that at steady state, both islet and pancreas exocrine macrophages are only minimally derived from blood cells and replicate locally at a low rate. Similarly, Ying et al. (2019) reported that in lean mice, both intra-islet and peri-islet macrophages were maintained at a very low turnover rate. However, under obese conditions, local proliferation of islet-resident macrophages was significantly enhanced. What causes the islet macrophages to replicate remains unknown. Macrophages can adapt to local environmental cues (Gosselin et al., 2014; Lavin et al., 2014). It is thus possible that elevated glucose and/or fatty acids (FAs) in obesity can trigger the release of proliferation-stimulating factors that promote the local proliferation of islet macrophages.
(II) Monocytes. Different from tissue-resident macrophages, circulating monocytes patrol the body and infiltrate tissue where there is an injury or inflammation. Using saturated fatty acid-induced obesity model, Eguchi et al. (2012) reported that CD11b+Ly-6C+ M1-type proinflammatory monocytes were recruited from the circulation into the pancreas. Ying et al. (2019) reassessed the infiltration of monocytes using HFD models. Indeed, they found that purified and track-mark labeled monocytes can be found within the pancreas of HFD mice one week after the adoptive transfer. However, surprisingly, these monocytes were trapped in the peri-islet area and failed to penetrate the basal membrane of the islets (Ying et al., 2019). More interestingly, a time-course dynamic analysis showed that the accumulation of transferred monocytes peaked at one week post-transfer and decreased after that, with a simultaneous increase of the accumulation of transferred monocytes in the draining lymph nodes. What exact role do monocytes play in the development of obesity and T2DM remains to be further investigated.
(III) Other immune cells. A special type of immune cells called type 2 innate lymphoid cells (ILC2) has been recently found in the islets, and these ILC2 cells promote insulin secretion through retinoic acid produced by myeloid cells in the islets (Dalmas et al., 2017). An interleukin 33 (IL-33)–ILC2 axis was proposed to explain the adaptation of β cells after acute stress. This axis became impaired during chronic obesity.
While it is clear that adaptive immune cells (T cells and B cells) are important players in adipose tissue in obesity, it remains controversial whether adaptive immune cells are involved in pancreatic inflammation and β cell dysfunction during the development of obesity. Recent studies found no T cells or B cells in pancreatic islets under either healthy or obese conditions (Calderon et al., 2015; Ying et al., 2019), arguing against the involvement of the adaptive immune cells in islet pathologies provoked by obesity.
Molecular mediators
The manifestation of islet inflammation in obesity and T2D is featured by the broadened spectrum and/or altered profile of secreted soluble factors. These factors contribute to both the compensatory responses of β cells and the impairment of β cell function.
(I) Interleukin 1β (IL-1β) is the most characterized cytokine in pancreatic islets of obesity and T2DM. Numerous studies and reviews have reported and discussed the multifaceted roles of IL-1β in various aspects of β cell physiology and pathophysiology (Maedler et al., 2002; Dinarello et al., 2010; Herder et al., 2015; Dror et al., 2017; Boni-Schnetzler et al., 2018). We here only highlight one critical aspect that IL-1β acts as a messenger mediating the crosstalk between macrophages and β cells.
Macrophages are the major sources of islet IL-1β production (Eguchi et al., 2012; Westwell-Roper et al., 2016). At steady condition, islet and peritoneal macrophages produce IL-1β at basal level (Calderon et al., 2015; Dror et al., 2017). This low-level IL-1β can stimulate postprandial insulin secretion (Dror et al., 2017). These findings suggest a beneficial role of low-level IL-1β in regulating glucose metabolism. IL-1β and insulin increase the uptake of glucose by macrophages. However, insulin released from β cells promotes proinflammatory status of macrophages, resulting in the production of reactive oxygen species, and the secretion of IL-1β in an NLRP3 inflammasome-dependent manner. Therefore, IL-1β-induced release of insulin occurs physiologically. In another study, deletion of IL-1 receptor (IL-1R) in β cells was reported to cause an impaired GSIS and β cell dedifferentiation (Burke et al., 2018).
High glucose can promote IL-1β production (Jourdan et al., 2013). In vitro incubation of macrophages in media containing high concentrations of glucose or FFAs induces increased level of IL-1β. With the progression of obesity and insulin resistance, the amount of IL-1β increases dramatically. β cells express the IL-1R (Dror et al., 2017). Thus, it can be speculated that a higher concentration of IL-1β within the islet microenvironment plays a crucial role mediating the interactions between islet macrophages and β cells. IL-1R-mediated intracellular signaling in β cells activates NF-κB (Donath et al., 2013) and JNK pathways (Welsh, 1996; Ammendrup et al., 2000; Bonny et al., 2001; Major and Wolf, 2001). The main pathological consequences caused by activated NF-κB and JNK pathways include increased β cell stress and apoptosis and loss-of-maintenance of β cell differentiation (Kim-Muller et al., 2014). All these pathways contribute to the impairment of β cell function.
(II) Islet amyloid polypeptide (IAPP) has a proinflammatory effect in human T2DM (Westermark et al., 2011). In the synthetic human IAPP (hIAPP) treatment or the hIAPP transgenic mouse model, hIAPP enhances the production of IL-1β in islet-resident macrophages upon HFD feeding (Westwell-Roper et al., 2014, 2016; Park et al., 2017). Amyloid formation also reduces the level of IL-1R antagonist (IL-1Ra), thus rebalancing the ratio between IL-1β and IL-1Ra (Hui et al., 2017). Through enhancing IL-1β and simultaneous repressing IL-1Ra, IAPP plays an important role in amplifying the inflammatory responses in the islet.
Amyloid deposition is associated with macrophage accumulation and differentiation. Islet amyloid formation is an important determinant for inducing islet inflammation in a long-term HFD-fed hIAPP transgenic mice (Meier et al., 2014). This was evidenced by increased accumulation of F4/80+ macrophages in the islets and simultaneously increased expression of inflammatory genes—Ccl2, Cxcl1, Emr1, Il1b, Tnf, and Il6 (Meier et al., 2014). Extracellular amyloidosis is directly related to the degree of β cell apoptosis in islets in T2DM (Raleigh et al., 2017). Kamata et al. (2014) examined >100 autopsy cases with T2DM. They found that amyloid-rich islets contained higher number of macrophages, and signs of oxidative stress-related DNA damage, and repressed insulin expression in β cells (Kamata et al., 2014). Together, these mechanisms promote a more proinflammatory islet microenvironment contributing to β cell dysfunction, reflected by reduced β cell mass and insulin release.
Human IAPP is preferentially degraded by autophagy rather than proteasomal degradation (Lee et al., 2019). Defect in the autophagy process leads to the accumulation of hIAPP and islet cell injury. On the other hand, accumulated hIAPP further impairs autophagy (Kim et al., 2014; Lee et al., 2019). Elevated expression of hIAPP induces the activation of MTORC1 and the inhibition of mitophagy, together causing damaged mitochondria and ER stress (Hernandez et al., 2018).
(III) Monocyte chemoattractant protein 1 (MCP-1, a.k.a. CCL2), the ligand for CCR2, is a key chemokine attracting proinflammatory monocytes to the tissue sites. Increased MCP-1 in the serum has been reported in the db/db mice at an early stage (8 weeks of age) (Cucak et al., 2014). Several inflammatory and metabolic factors have been reported to increase MCP-1 in the islets. IL-1β induces the production of MCP-1 and IL-6 in mouse and human islets, whereas high glucose has no such effect (Welsh et al., 2005; Jourdan et al., 2013). In addition, palmitate acid can induce MCP-1 production in MIN6 β cell line. MCP-1 is required for monocyte infiltration into the pancreas (though not islets), because monocytes with CCR2 (the receptor for MCP-1) null mutations do not appear in pancreas (Ying et al., 2019). Interestingly, in situ immunofluorescence analysis revealed that MCP-1 shows a higher gradient in the peri-islet area, especially under obese condition. F4/80+ macrophages contribute to the production of MCP-1 (Ying et al., 2019).
(IV) IL-33 is another cytokine that belongs to the IL-1 cytokine family (Dinarello, 2018). In recent years, IL-33 has been increasingly recognized in the regulation of tissue immune responses, injury, and repair (Molofsky et al., 2015). Dalmas et al. (2017) reported that islet mesenchymal cells produce IL-33. IL-33 promotes β cell function through ILC2 cells, which are present in the islet as a very low abundance. The IL-33–ILC2 axis is activated after an acute β cell stress, which results in an increase of insulin section. However, while glucose and IL-1β can increase the production of IL-33 in the islet, the IL-33–ILC2 axis is defective during chronic obesity (Dalmas et al., 2017). Myeloid cells, especially macrophages, are the dominant type of cells to produce IL-1β (Eguchi et al., 2012; Westwell-Roper et al., 2016). It is thus possible that islet macrophages act upstream of ILCs to regulate IL-33 production from these cells. These findings suggest a loop interaction between islet-derived IL-33, ILC2s, and myeloid cells to regulate insulin secretion.
Islet inflammation in human T2DM and therapeutic prospects
Fetal and neonatal human pancreas contains leukocyte infiltrates, mainly T cells, which are present in the connective tissue of septa and in the capsule (Jansen et al., 1993). Jansen et al. (1993) analyzed five fetal and six neonatal human pancreases and found infiltration of the exocrine pancreas with dendritic-like cells and T cells. They also examined immune infiltration in an 8-month-old diabetic infant and found a more pronounced immune infiltration. Islet inflammation is also revealed in human T2DM patients (Donath et al., 2008). Evidence supporting this notion includes immune cell accumulation, amyloid deposition (as described above) (Westermark et al., 2011), and the production of IL-1β in β cells of T2DM patients (Maedler et al., 2002). Increased numbers of macrophages have also been reported in the islets of human obese and T2D patients (Ehses et al., 2007; Richardson et al., 2009; Butcher et al., 2014; Kamata et al., 2014). In these studies, the accumulation of CD68+ macrophages was detected in pancreatic samples from T2D patients (Ehses et al., 2007; Richardson et al., 2009; Gerst et al., 2017), indicative of the functional relevance of macrophages in affecting human β cell insulin secretion in obesity and T2D.
Another feature indicting an inflamed islet microenvironment in human T2DM is the detection of increased concentration and broadened spectrum of inflammatory cytokines. This has been comprehensively summarized by Boni-Schnetzler and Meier (2019). Similar to what has been observed in rodent models of obesity and T2DM, IL-1β is one of the most evident cytokines augmented in the islets of human T2DM (Boni-Schnetzler and Meier, 2019).
These findings support a rationale to target islet inflammatory molecules as a strategy to restore β cell mass and function. A number of clinical trials have been undertaken mainly to test the effect of blocking IL-1β through the administration of either IL-1Ra or anti-IL-1β mAb. The outcomes vary among different trials. The administration of IL-1Ra (Anakinra) modestly reduced fasting blood glucose level (Larsen et al., 2007). Meta-analysis of a large cohort of individuals revealed that IL-1 antagonism, through either anti-IL-1 antibodies or IL-1Ra lowered hemoglobin A1c, an indicator for how well diabetes is controlled (Kataria et al., 2019).
Conclusions and future perspectives
There are variations in the reported phenotypes and functions of macrophages in different studies. This may be due to the differences of surface markers, animal models, and disease stages. It may also indicate that while macrophages are the dominant immune cells, heterogeneous subsets with different phenotypes and functions are mixed within the islets or at the peri-islet capsular area. Their origins, differentiation, and functions are probably more complicated than what we have learned so far. Further studies using more comprehensive and high-throughput approaches may provide a more complete view of the immune profiles in pancreatic islets at steady and diseased conditions. This goal becomes more approachable due to the advancements of new techniques. Single-cell RNA-sequencing can provide important insights into the heterogeneity of immune cell phenotypes and functions in each specific condition. The location of each immune cell population within the islet (intra-islet vs. peri-islet distribution) is another critical parameter for understanding the role of each of them in islet homeostasis or pathology. In this regard, light-sheet microscope (Voigt et al., 2019) and 3D reconstitution (Chien et al., 2016), for example, will provide useful information.
While β cells are at the center of the loss-of-homeostasis of the islets in obesity and T2DM, mounting evidence clearly indicate that it is not an isolated event occurred only in β cells. Instead, it is more likely a consequence of dysregulated interactions among various types of cells residing in the islets. Better understanding the distribution and function of other endocrine cells and ‘accessary’ cells will provide important clues to the impairment of β cell maintenance and function. Interorgan crosstalk has emerged as another important topic to understand β cell adaptation and dysfunction. For instance, the crosstalk between the liver and the islet has been revealed. In a genetically induced insulin resistance model, liver-derived serpinB1 promotes human and mouse β cell proliferation (El Ouaamari et al., 2013, 2016). Future studies should further explore the cellular and molecular communications among different organs, because it will provide important clues for how islet adapts to insulin resistance and how systemically elevated metabolic and inflammatory cues impinge on β cell function.
Cell-based therapy has recently been actively investigated, in part based on the notion that transfused cells can precisely deliver a targeted therapy (Lee et al., 2016). Macrophages are key cellular players in regulating islet homeostasis, adaptation, and β cell function (Banaei-Bouchareb et al., 2004; Calderon et al., 2015; Ying et al., 2019). It thus can be speculated that targeting islet macrophages would have a therapeutic potential for restoring islet homeostasis and β cell function. Macrophages can actively monitor and modulate local microenvironment. Animal studies have demonstrated that adoptive transfer of pre-conditioned macrophages (Parsa et al., 2012) or reprogramming islet-resident macrophages (Fu et al., 2014) led to the suppression of diabetes. However, challenges also exist, especially given the fact that macrophages are heterogeneous and plastic. It is difficult to identify the specific subset that can be targeted as a therapeutic. In addition, how to achieve a tissue-specific intervention via modulating macrophages is another task that warrants future investigations.
How can the knowledges we have learned from animal studies be translated into the understanding of human obesity and T2DM? It is particularly challenging given the known differences of islet structure between humans and rodents (Arrojo e Drigo et al., 2015). Little is known about how immune system regulates human islet homeostasis and affect β cell function in normal and diseased conditions. Realizing this urgent need, the NIH has initiated and continued the supports for analyzing pancreata recovered from human tissue donors with T2DM and related metabolic disorders. It can be expected that increasing number of these collaborative efforts will be invaluable for the development of more efficient therapeutic strategies.
Acknowledgements
We thank J.M. Olefsky, Y.S. Lee, W. Ying, and M.-X. Yang for insightful discussions. We apologize to those individuals whose work could not be cited here due to space limitation.
Funding
This work was funded by the US National Institutes of Health (DK114427 to W.F.).
Conflict of interest: none declared.
Author contributions: J.G. and W.F. for conceptualization, writing, and editing.
References
- Aamodt, K.I., and Powers, A.C. (2017). Signals in the pancreatic islet microenvironment influence β-cell proliferation. Diabetes Obes. Metab. 19(Suppl 1), 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almaca, J., Molina, J., Menegaz, D., et al. (2016). Human β cells produce and release serotonin to inhibit glucagon secretion from α cells. Cell Rep. 17, 3281–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almaca, J., Weitz, J., Rodriguez-Diaz, R., et al. (2018). The pericyte of the pancreatic islet regulates capillary diameter and local blood flow. Cell Metab. 27, 630–644.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso, L.C., Yokoe, T., Zhang, P., et al. (2007). Glucose infusion in mice: a new model to induce β-cell replication. Diabetes 56, 1792–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammendrup, A., Maillard, A., Nielsen, K., et al. (2000). The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic β-cells. Diabetes 49, 1468–1476. [DOI] [PubMed] [Google Scholar]
- Araujo, T.G., Oliveira, A.G., Carvalho, B.M., et al. (2012). Hepatocyte growth factor plays a key role in insulin resistance-associated compensatory mechanisms. Endocrinology 153, 5760–5769. [DOI] [PubMed] [Google Scholar]
- Arrojo e Drigo, R., Ali, Y., Diez, J., et al. (2015). New insights into the architecture of the islet of Langerhans: a focused cross-species assessment. Diabetologia 58, 2218–2228. [DOI] [PubMed] [Google Scholar]
- Arrojo, E.D.R., Jacob, S., Garcia-Prieto, C.F., et al. (2019). Structural basis for δ cell paracrine regulation in pancreatic islets. Nat. Commun. 10, 3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann, A., Ueki, K., Winnay, J.N., et al. (2009). Glucose effects on β-cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol. Cell. Biol. 29, 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaei-Bouchareb, L., Gouon-Evans, V., Samara-Boustani, D., et al. (2004). Insulin cell mass is altered in Csf1op/Csf1op macrophage-deficient mice. J. Leukoc. Biol. 76, 359–367. [DOI] [PubMed] [Google Scholar]
- Boni-Schnetzler, M., Hauselmann, S.P., Dalmas, E., et al. (2018). β cell-specific deletion of the IL-1 receptor antagonist impairs β cell proliferation and insulin secretion. Cell Rep. 22, 1774–1786. [DOI] [PubMed] [Google Scholar]
- Boni-Schnetzler, M., and Meier, D.T. (2019). Islet inflammation in type 2 diabetes. Semin. Immunopathol. 41, 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner-Weir, S. (2000). Islet growth and development in the adult. J. Mol. Endocrinol. 24, 297–302. [DOI] [PubMed] [Google Scholar]
- Bonny, C., Oberson, A., Negri, S., et al. (2001). Cell-permeable peptide inhibitors of JNK: novel blockers of β-cell death. Diabetes 50, 77–82. [DOI] [PubMed] [Google Scholar]
- Briant, L.J.B., Reinbothe, T.M., Spiliotis, I., et al. (2018). δ-cells and β-cells are electrically coupled and regulate α-cell activity via somatostatin. J. Physiol. 596, 197–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke, S.J., Batdorf, H.M., Burk, D.H., et al. (2018). Pancreatic deletion of the interleukin-1 receptor disrupts whole body glucose homeostasis and promotes islet β-cell de-differentiation. Mol. Metab. 14, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke, S.J., Karlstad, M.D., and Collier, J.J. (2016). Pancreatic islet responses to metabolic trauma. Shock 46, 230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn, D., Benoist, C., and Mathis, D. (2013). Regulatory T cells in nonlymphoid tissues. Nat. Immunol. 14, 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher, M.J., Hallinger, D., Garcia, E., et al. (2014). Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia 57, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon, B., Carrero, J.A., Ferris, S.T., et al. (2015). The pancreas anatomy conditions the origin and properties of resident macrophages. J. Exp. Med. 212, 1497–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien, H.J., Peng, S.J., Hua, T.E., et al. (2016). 3-D imaging of islets in obesity: formation of the islet-duct complex and neurovascular remodeling in young hyperphagic mice. Int. J. Obes. 40, 685–697. [DOI] [PubMed] [Google Scholar]
- Chittezhath, M., Gunaseelan, D., Zheng, X., et al. (2019). Islet macrophages are associated with islet vascular remodeling and compensatory hyperinsulinemia during diabetes. Am. J. Physiol. Endocrinol. Metab. 317, E1108–E1120. [DOI] [PubMed] [Google Scholar]
- Cobelli, C., and Vella, A. (2017). Exocrine and endocrine interactions in cystic fibrosis: a potential key to understanding insulin secretion in health and disease? Diabetes 66, 20–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucak, H., Grunnet, L.G., and Rosendahl, A. (2014). Accumulation of M1-like macrophages in type 2 diabetic islets is followed by a systemic shift in macrophage polarization. J. Leukoc. Biol. 95, 149–160. [DOI] [PubMed] [Google Scholar]
- Dai, C., Brissova, M., Reinert, R.B., et al. (2013). Pancreatic islet vasculature adapts to insulin resistance through dilation and not angiogenesis. Diabetes 62, 4144–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmas, E., Lehmann, F.M., Dror, E., et al. (2017). Interleukin-33-activated islet-resident innate lymphoid cells promote insulin secretion through myeloid cell retinoic acid production. Immunity 47, 928–942 e927. [DOI] [PubMed] [Google Scholar]
- Davies, L.C., Jenkins, S.J., Allen, J.E., et al. (2013). Tissue-resident macrophages. Nat. Immunol. 14, 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jesus, D.F., and Kulkarni, R.N. (2019). ‘Omics’ and ‘epi-omics’ underlying the β-cell adaptation to insulin resistance. Mol. Metab. 27S, S42–S48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirci, C., Ernst, S., Alvarez-Perez, J.C., et al. (2012). Loss of HGF/c-Met signaling in pancreatic β-cells leads to incomplete maternal β-cell adaptation and gestational diabetes mellitus. Diabetes 61, 1143–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello, C.A. (2018). Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 281, 8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello, C.A., Donath, M.Y., and Mandrup-Poulsen, T. (2010). Role of IL-1β in type 2 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 17, 314–321. [DOI] [PubMed] [Google Scholar]
- Dirice, E., De Jesus, D.F., Kahraman, S., et al. (2019). Human duct cells contribute to β cell compensation in insulin resistance. JCI Insight 4, pii:99576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolensek, J., Rupnik, M.S., and Stozer, A. (2015). Structural similarities and differences between the human and the mouse pancreas. Islets 7, e1024405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath, M.Y., Boni-Schnetzler, M., Ellingsgaard, H., et al. (2009). Islet inflammation impairs the pancreatic β-cell in type 2 diabetes. Phys. Ther. 24, 325–331. [DOI] [PubMed] [Google Scholar]
- Donath, M.Y., Dalmas, E., Sauter, N.S., et al. (2013). Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell Metab. 17, 860–872. [DOI] [PubMed] [Google Scholar]
- Donath, M.Y., Schumann, D.M., Faulenbach, M., et al. (2008). Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care 31(Suppl 2), S161–S164. [DOI] [PubMed] [Google Scholar]
- Dror, E., Dalmas, E., Meier, D.T., et al. (2017). Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 18, 283–292. [DOI] [PubMed] [Google Scholar]
- Ebato, C., Uchida, T., Arakawa, M., et al. (2008). Autophagy is important in islet homeostasis and compensatory increase of β cell mass in response to high-fat diet. Cell Metab. 8, 325–332. [DOI] [PubMed] [Google Scholar]
- Eguchi, K., Manabe, I., Oishi-Tanaka, Y., et al. (2012). Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 15, 518–533. [DOI] [PubMed] [Google Scholar]
- Ehses, J.A., Perren, A., Eppler, E., et al. (2007). Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56, 2356–2370. [DOI] [PubMed] [Google Scholar]
- El Ouaamari, A., Dirice, E., Gedeon, N., et al. (2016). SerpinB1 promotes pancreatic β cell proliferation. Cell Metab. 23, 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ouaamari, A., Kawamori, D., Dirice, E., et al. (2013). Liver-derived systemic factors drive β cell hyperplasia in insulin-resistant states. Cell Rep. 3, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, W., Farache, J., Clardy, S.M., et al. (2014). Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and β cells. eLife 3, e04631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ocana, A., Takane, K.K., Syed, M.A., et al. (2000). Hepatocyte growth factor overexpression in the islet of transgenic mice increases β cell proliferation, enhances islet mass, and induces mild hypoglycemia. J. Biol. Chem. 275, 1226–1232. [DOI] [PubMed] [Google Scholar]
- Gerst, F., Wagner, R., Kaiser, G., et al. (2017). Metabolic crosstalk between fatty pancreas and fatty liver: effects on local inflammation and insulin secretion. Diabetologia 60, 2240–2251. [DOI] [PubMed] [Google Scholar]
- Gosselin, D., Link, V.M., Romanoski, C.E., et al. (2014). Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath, W.R., and Carbone, F.R. (2013). The skin-resident and migratory immune system in steady state and memory: innate lymphocytes, dendritic cells and T cells. Nat. Immunol. 14, 978–985. [DOI] [PubMed] [Google Scholar]
- Herder, C., Dalmas, E., Boni-Schnetzler, M., et al. (2015). The IL-1 pathway in type 2 diabetes and cardiovascular complications. Trends Endocrinol. Metab. 26, 551–563. [DOI] [PubMed] [Google Scholar]
- Hernandez, M.G., Aguilar, A.G., Burillo, J., et al. (2018). Pancreatic β cells overexpressing hIAPP impaired mitophagy and unbalanced mitochondrial dynamics. Cell Death Dis. 9, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudish, L.I., Reusch, J.E., and Sussel, L. (2019). β cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J. Clin. Invest. 129, 4001–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui, Q., Asadi, A., Park, Y.J., et al. (2017). Amyloid formation disrupts the balance between interleukin-1β and interleukin-1 receptor antagonist in human islets. Mol. Metab. 6, 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull, R.L., Kodama, K., Utzschneider, K.M., et al. (2005). Dietary-fat-induced obesity in mice results in β cell hyperplasia but not increased insulin release: evidence for specificity of impaired β cell adaptation. Diabetologia 48, 1350–1358. [DOI] [PubMed] [Google Scholar]
- Irving-Rodgers, H.F., Choong, F.J., Hummitzsch, K., et al. (2014). Pancreatic islet basement membrane loss and remodeling after mouse islet isolation and transplantation: impact for allograft rejection. Cell Transplant. 23, 59–72. [DOI] [PubMed] [Google Scholar]
- Iwasaki, A., and Medzhitov, R. (2015). Control of adaptive immunity by the innate immune system. Nat. Immunol. 16, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, A., Voorbij, H.A., Jeucken, P.H., et al. (1993). An immunohistochemical study on organized lymphoid cell infiltrates in fetal and neonatal pancreases. A comparison with similar infiltrates found in the pancreas of a diabetic infant. Autoimmunity 15, 31–38. [DOI] [PubMed] [Google Scholar]
- Jenkins, S.J., Ruckerl, D., Cook, P.C., et al. (2011). Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332, 1284–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenne, C.N., and Kubes, P. (2013). Immune surveillance by the liver. Nat. Immunol. 14, 996–1006. [DOI] [PubMed] [Google Scholar]
- Jourdan, T., Godlewski, G., Cinar, R., et al. (2013). Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates β cell loss in type 2 diabetes. Nat. Med. 19, 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang, J.H., Kuo, C.H., Peng, S.J., et al. (2015). 3-D imaging reveals participation of donor islet Schwann cells and pericytes in islet transplantation and graft neurovascular regeneration. EBio Medicine 2, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata, K., Mizukami, H., Inaba, W., et al. (2014). Islet amyloid with macrophage migration correlates with augmented β-cell deficits in type 2 diabetic patients. Amyloid 21, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataria, Y., Ellervik, C., and Mandrup-Poulsen, T. (2019). Treatment of type 2 diabetes by targeting interleukin-1: a meta-analysis of 2921 patients. Semin. Immunopathol. 41, 413–425. [DOI] [PubMed] [Google Scholar]
- Kim, J., Cheon, H., Jeong, Y.T., et al. (2014). Amyloidogenic peptide oligomer accumulation in autophagy-deficient β cells induces diabetes. J. Clin. Invest. 124, 3311–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Muller, J.Y., Zhao, S., Srivastava, S., et al. (2014). Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab. 20, 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinova, I., Nikolova, G., Ohara-Imaizumi, M., et al. (2007). EphA–ephrin-A-mediated β cell communication regulates insulin secretion from pancreatic islets. Cell 129, 359–370. [DOI] [PubMed] [Google Scholar]
- Korpos, E., Kadri, N., Kappelhoff, R., et al. (2013). The peri-islet basement membrane, a barrier to infiltrating leukocytes in type 1 diabetes in mouse and human. Diabetes 62, 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotas, M.E., and Medzhitov, R. (2015). Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, C.M., Faulenbach, M., Vaag, A., et al. (2007). Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 356, 1517–1526. [DOI] [PubMed] [Google Scholar]
- Lavin, Y., Winter, D., Blecher-Gonen, R., et al. (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S., Kivimae, S., Dolor, A., et al. (2016). Macrophage-based cell therapies: the long and winding road. J. Control. Release 240, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y.H., Kim, J., Park, K., et al. (2019). β-cell autophagy: mechanism and role in β-cell dysfunction. Mol. Metab. 27S, S92–S103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt, H.E., Cyphert, T.J., Pascoe, J.L., et al. (2011). Glucose stimulates human β cell replication in vivo in islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia 54, 572–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler, K., Sergeev, P., Ris, F., et al. (2002). Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 110, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major, C.D., and Wolf, B.A. (2001). Interleukin-1β stimulation of c-Jun NH2-terminal kinase activity in insulin-secreting cells: evidence for cytoplasmic restriction. Diabetes 50, 2721–2728. [DOI] [PubMed] [Google Scholar]
- Meier, D.T., Morcos, M., Samarasekera, T., et al. (2014). Islet amyloid formation is an important determinant for inducing islet inflammation in high-fat-fed human IAPP transgenic mice. Diabetologia 57, 1884–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen, T., Donaldson, C.J., Caceres, E., et al. (2015). Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 21, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, G., Zhao, Y., Li, Y., et al. (2013). Basement membrane extract preserves islet viability and activity in vitro by up-regulating α3 integrin and its signal. Pancreas 42, 971–976. [DOI] [PubMed] [Google Scholar]
- Molofsky, A.B., Savage, A.K., and Locksley, R.M. (2015). Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity 42, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser, R.E., Maulis, M.F., Moulle, V.S., et al. (2015). High-fat diet-induced β-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am. J. Physiol. Endocrinol. Metab. 308, E573–E582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussar, K., Pardike, S., Hohl, T.M., et al. (2017). A CCR2+ myeloid cell niche required for pancreatic β cell growth. JCI Insight 2, e93834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mwangi, S., Anitha, M., Mallikarjun, C., et al. (2008). Glial cell line-derived neurotrophic factor increases β-cell mass and improves glucose tolerance. Gastroenterology 134, 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, J.H., Galsgaard, E.D., Moldrup, A., et al. (2001). Regulation of β-cell mass by hormones and growth factors. Diabetes 50, S25–S29. [DOI] [PubMed] [Google Scholar]
- Park, H.S., Kim, H.Z., Park, J.S., et al. (2019). β-cell-derived angiopoietin-1 regulates insulin secretion and glucose homeostasis by stabilizing the islet microenvironment. Diabetes 68, 774–786. [DOI] [PubMed] [Google Scholar]
- Park, Y.J., Warnock, G.L., Ao, Z., et al. (2017). Dual role of interleukin-1β in islet amyloid formation and its β-cell toxicity: implications for type 2 diabetes and islet transplantation. Diabetes Obes. Metab. 19, 682–694. [DOI] [PubMed] [Google Scholar]
- Parsa, R., Andresen, P., Gillett, A., et al. (2012). Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes 61, 2881–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyot, M.L., Pepin, E., Lamontagne, J., et al. (2010). β-cell failure in diet-induced obese mice stratified according to body weight gain: secretory dysfunction and altered islet lipid metabolism without steatosis or reduced β-cell mass. Diabetes 59, 2178–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkse, G.G., Bouwman, W.P., Jiawan-Lalai, R., et al. (2006). Integrin signaling via RGD peptides and anti-β1 antibodies confers resistance to apoptosis in islets of Langerhans. Diabetes 55, 312–317. [DOI] [PubMed] [Google Scholar]
- Porat, S., Weinberg-Corem, N., Tornovsky-Babaey, S., et al. (2011). Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 13, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raleigh, D., Zhang, X., Hastoy, B., et al. (2017). The β-cell assassin: IAPP cytotoxicity. J. Mol. Endocrinol. 59, R121–R140. [DOI] [PubMed] [Google Scholar]
- Richardson, S.J., Willcox, A., Bone, A.J., et al. (2009). Islet-associated macrophages in type 2 diabetes. Diabetologia 52, 1686–1688. [DOI] [PubMed] [Google Scholar]
- Rorsman, P., and Huising, M.O. (2018). The somatostatin-secreting pancreatic δ-cell in health and disease. Nat. Rev. Endocrinol. 14, 404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz, C., Gomez, P.E., Chorro, L., et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90. [DOI] [PubMed] [Google Scholar]
- Stamateris, R.E., Sharma, R.B., Hollern, D.A., et al. (2013). Adaptive β-cell proliferation increases early in high-fat feeding in mice, concurrent with metabolic changes, with induction of islet cyclin D2 expression. Am. J. Physiol. Endocrinol. Metab. 305, E149–E159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunami, E., Kanazawa, H., Hashizume, H., et al. (2001). Morphological characteristics of Schwann cells in the islets of Langerhans of the murine pancreas. Arch. Histol. Cytol. 64, 191–201. [DOI] [PubMed] [Google Scholar]
- Svendsen, B., Larsen, O., Gabe, M.B.N., et al. (2018). Insulin secretion depends on intra-islet glucagon signaling. Cell Rep. 25, 1127–1134.e2. [DOI] [PubMed] [Google Scholar]
- Tang, S.C., Jessup, C.F., and Campbell-Thompson, M. (2018a). The role of accessory cells in islet homeostasis. Curr. Diab. Rep. 18, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, S.C., Peng, S.J., and Chien, H.J. (2014). Imaging of the islet neural network. Diabetes Obes. Metab. 16(Suppl 1), 77–86. [DOI] [PubMed] [Google Scholar]
- Tang, S.C., Shen, C.N., Lin, P.Y., et al. (2018b). Pancreatic neuro-insular network in young mice revealed by 3D panoramic histology. Diabetologia 61, 158–167. [DOI] [PubMed] [Google Scholar]
- Teitelman, G., Guz, Y., Ivkovic, S., et al. (1998). Islet injury induces neurotrophin expression in pancreatic cells and reactive gliosis of peri-islet Schwann cells. J. Neurobiol. 34, 304–318. [DOI] [PubMed] [Google Scholar]
- Thorens, B. (2014). Neural regulation of pancreatic islet cell mass and function. Diabetes Obes. Metab. 16(Suppl 1), 87–95. [DOI] [PubMed] [Google Scholar]
- Voigt, F.F., Kirschenbaum, D., Platonova, E., et al. (2019). The mesoSPIM initiative: open-source light-sheet microscopes for imaging cleared tissue. Nat. Methods 16, 1105–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R.N., and Rosenberg, L. (1999). Maintenance of β-cell function and survival following islet isolation requires re-establishment of the islet-matrix relationship. J. Endocrinol. 163, 181–190. [DOI] [PubMed] [Google Scholar]
- Weitz, J.R., Makhmutova, M., Almaca, J., et al. (2018). Mouse pancreatic islet macrophages use locally released ATP to monitor β cell activity. Diabetologia 61, 182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh, N. (1996). Interleukin-1β-induced ceramide and diacylglycerol generation may lead to activation of the c-Jun NH2-terminal kinase and the transcription factor ATF2 inthe insulin-producing cell line RINm5F. J. Biol. Chem. 271, 8307–8312. [DOI] [PubMed] [Google Scholar]
- Welsh, N., Cnop, M., Kharroubi, I., et al. (2005). Is there a role for locally produced interleukin-1 in the deleterious effects of high glucose or the type 2 diabetes milieu to human pancreatic islets? Diabetes 54, 3238–3244. [DOI] [PubMed] [Google Scholar]
- Westermark, P., Andersson, A., and Westermark, G.T. (2011). Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 91, 795–826. [DOI] [PubMed] [Google Scholar]
- Westwell-Roper, C., Denroche, H.C., Ehses, J.A., et al. (2016). Differential activation of innate immune pathways by distinct islet amyloid polypeptide (IAPP) aggregates. J. Biol. Chem. 291, 8908–8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwell-Roper, C.Y., Ehses, J.A., and Verchere, C.B. (2014). Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1β production and β-cell dysfunction. Diabetes 63, 1698–1711. [DOI] [PubMed] [Google Scholar]
- Xu, X., D'Hoker, J., Stange, G., et al. (2008). β cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132, 197–207. [DOI] [PubMed] [Google Scholar]
- Ying, W., Lee, Y.S., Dong, Y., et al. (2019). Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab. 29, 457–474.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, X., Yang, B.H., Dong, Y., et al. (2017). CRIg, a tissue-resident macrophage specific immune checkpoint molecule, promotes immunological tolerance in NOD mice, via a dual role in effector and regulatory T cells. eLife 6, e29540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Q., Brown, J., Kanarek, A., et al. (2008). In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature 455, 627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, L., Dattaroy, D., Pham, J., et al. (2019). Intra-islet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight 5, e127994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinselmeyer, B.H., Vomund, A.N., Saunders, B.T., et al. (2018). The resident macrophages in murine pancreatic islets are constantly probing their local environment, capturing β cell granules and blood particles. Diabetologia 61, 1374–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]