

Abstract
The search for solar‐driven photocatalysts for overall water splitting has been actively pursued. Although metal oxynitrides with metal d0/d10‐closed shell configuration are very promising candidates in terms of their visible light absorption, they usually suffer from serious photo‐generated charge recombination and thus, little photoactivity. Here, by forming their solid solutions of LaTaON2 and CaTaO2N, which are traditionally considered to be inorganic yellow‐red pigments but have poor photocatalytic activity, a class of promising solar‐driven photocatalysts La1‐ xCaxTaO1+yN2‐ y (0 ≤ x, y ≤ 1) are explored. In particular, the optimal photocatalyst with x = 0.9 has the ability of realizing overall water splitting with stoichiometric H2/O2 ratio under the illumination of both AM1.5 simulated solar light and visible light. The modulated key parameters including band structure, Ta bonding environment, defects concentration, and band edge alignments revealed in La0.1Ca0.9TaO1+ yN2‐ y have substantially promoted the separation of photogenerated charge carriers with sufficient energetics for water oxidation and reduction reactions. The results obtained in this study provide an important candidate for designing efficient solar‐driven photocatalysts for overall water splitting.
Keywords: oxynitrides, perovskites, solid solution, visible light, water splitting
Forming solid solutions between LaTaON2 and CaTaO2N leads to a series of promising solar‐driven photocatalysts La1‐ xCaxTaO1+ yN2‐ y (0 ≤ x, y ≤ 1). Overall water splitting with stoichiometric H2/O2 ratio can be achieved at optimal composition of x = 0.9 under the illumination of both AM1.5 simulated solar light and visible light.

Water splitting into H2 and O2 on particulate photocatalysts stands as one of the simplest and most cost‐effective techniques for storing solar energy into chemical fuels.[ 1 ] With easy access to both reactant (water) and energy input (solar insolation), this technique signifies a promising means to setup a clean and sustainable energy infrastructure that can eventually decarbonize our fossil‐fuel based energy economy.[ 2 ] For instance, large water splitting panels fabricated from particulate photocatalyst has shown to be a viable means for large‐scale production of low‐cost renewable solar hydrogen.[ 3 ] Although efficient overall water splitting has been realized on wide bandgap semiconductors using ultra‐violet (UV) photons,[ 4 , 5 ] visible‐light‐driven overall water splitting has been rather difficult, occurring to be the bottleneck problem for efficient solar energy conversion.[ 6 , 7 ] Thereby, exploring photocatalysts sensitive to visible light photons for overall water splitting becomes a premier target.[ 8 ]
Previous reports on single‐absorber type photocatalyst for overall water splitting using visible light photons are almost exclusively referred to metal oxynitrides with d 10 or d° closed‐shell electronic structure, such as (Ga1‐ xZnx)(N1‐ xOx),[ 9 ] (Zn1+ xGe)(N2Ox),[ 10 ] TaON,[ 11 ] Ta3N5,[ 12 ] LaMgxTa1‐ xO1+3 xN2‐3 x [ 13 ] etc. Fine composition/structure tuning as well as peculiar surface modifications is often indispensable to realize stoichiometric H2 and O2 evolution.[ 10 , 14 ] This can be rationalized by a need to minimize energy barriers for charge migration and transfer so as to compensate the reduced energetics of charges generated by visible light photons.[ 15 ] This imposes stringent standard on the type of crystal structures upon which various modifications can be performed. Perovskite oxynitrides are emerging groups of functional materials that show potential applications in a wealth of fields including colossal magnetoresistivity, dielectrics, pigments, phosphors, etc.[ 16 ] Their perovskite type structures are extremely flexible and tolerant to diverse cation/anion displacements,[ 17 , 18 ] opening ample playgrounds to tailor the desired physicochemical properties.
Perovskite oxynitrides with chemical formula AM(O,N)3 (A = La, Ca, Sr, Ba; M = Ti, Nb, Ta) have bandgap ranging from 1.7 to 2.6 eV, showing strong absorption in the visible light region.[ 19 ] However, their photocatalytic activity for water splitting is normally very poor even with an aid of sacrificial agents.[ 20 , 21 , 22 , 23 , 24 ] This has been generally ascribed to several key factors that are strongly linked to charge separation, migration, and transfer:[ 12 , 20 , 25 , 26 ] i) Defects generated during synthetic procedures (high temperature ammonolysis) as their presence induces rapid charge recombination events; ii) structural distortions (tilting of M(O,N)6 octahedrons) which prevent fast charge migrations; iii) improper band edge alignments which result in incommensurate energetics for water reduction and oxidation half‐reactions. How to properly tackle these problems is a prerequisite to achieve visible‐light water splitting on these compounds and still remains a great challenge.
In this work, solid solutions between LaTaON2 and CaTaO2N were managed to finely tune their optical absorption, Ta bonding environment, defects concentration, and band edge alignments. The successful manipulation of these important parameters offers us flexible modifications on their photocatalytic activities. At optimal conditions, visible light driven overall water splitting has been achieved on this material system for the first time, to our best knowledge.
Typical synthesized sample powders with a variation of Ca content are shown in Figure S1a, Supporting Information, detailed synthetic and analytic information can be found in the Supporting Information. The formation of solid solutions La1‐ xCaxTaO1+ yN2‐ y (0 ≤ x, y ≤ 1) is confirmed by their X‐ray powder diffraction (XRD) patterns that closely resemble standard ones of LaTaON2 and CaTaO2N. All reflections consecutively shift toward high angles along with Ca uptake (Figure S1a, Supporting Information), indicating continuous shrinkage of LaTaON2 unit cell by dissolving CaTaO2N. This is further verified by Rietveld refinements of their XRD data (Figure 1a, Table S1, Supporting Information) and is in a good agreement with vegard's law as La3+/N3− ions are gradually replaced by smaller Ca2+/O2− ions in the structure.[ 27 ] La/Ca probably has random distributions at the A sites of perovskite as super‐lattice reflections from cation ordering are not observed. Nevertheless, there is a watershed point around x = 0.7 whereby space group changes from I12/m to Pnma (Figure S1b, Supporting Information). Such a symmetry change from monoclinic to orthorhombic likely stems from the tilt of Ta(O, N)6 octahedrons in a manner that is gradually increased from LaTaON2 (Glazer notion: a−b−c0) to CaTaO2N (Glazer notion: a−b−c+).[ 17 , 22 ] This can be seen from the refined crystal structures of LaTaON2, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) and CaTaO2N (Figure 1a). For these Ta‐based perovskite oxynitrides, smaller unit cell generally means shorter Ta—O/N bonds whilst increased tilt of Ta(O, N)6 octahedrons often indicates decreased Ta—O/N—Ta bond angles that is more deviated from 180° (see Table S1, Supporting Information). These two factors are of critical importance to their electronic structures since the hybridizations between Ta 5d and O/N 2p orbitals contribute mostly to the conduction and valence bands.[ 26 , 28 , 29 ] For instance, shorter Ta—O/N bonds favor p‐d σ bonds while deviation of Ta—O/N—Ta bond angles away from 180° increases the difficulties for p‐d π bonds formation.[ 28 , 30 ] Thereby, forming solid solutions between LaTaON2 and CaTaO2N is a useful tool to finely tune their electronic structures which govern the behavior of photo‐generated charges under light illuminations.
Figure 1.
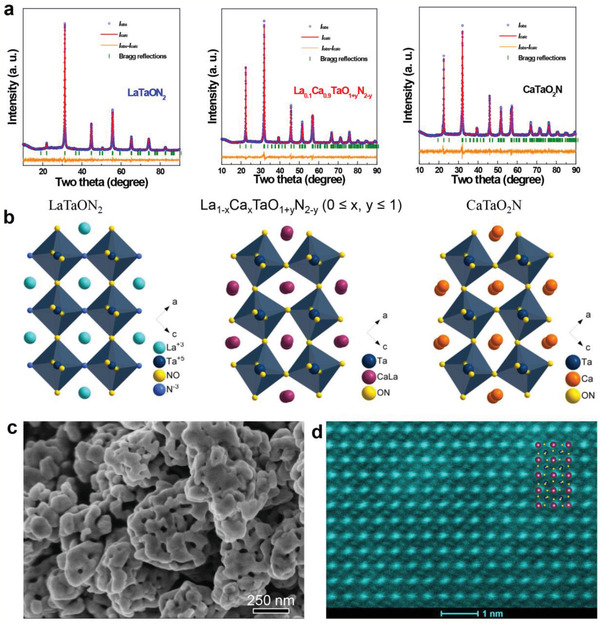
a) Observed and calculated X‐ray powder diffraction patterns of LaTaON2, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) and CaTaO2N; the refinements all converge with reasonable R factors and χ2 factor (LaTaON2: R p = 3.76%, R wp = 4.91%, χ2 = 1.311; La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9): R p = 4.94%, R wp = 6.37%, χ2 = 2.762; CaTaO2N: R p = 5.58%, R wp = 6.91%, χ2 = 2.835). b) Schematic illustration of crystal structures of LaTaON2 (left panel), La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) (middle panel) and CaTaO2N (right panel), these structures are projected along [010] direction. c) Field emission scanning electron microscopy images of La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9). d) High‐angle annular dark‐field scanning transmission electron microscopy (HAADF‐STEM) image of La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) along [010] direction. Inset: Depicted crystal structure of La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) projected along [010] direction.
All sample powders are composed of particles with particle size around 500 nm (Figure S2a–c, Supporting Information). These particles contain interconnected granules of 40–100 nm in size according to their particle size distribution (Figure S3a, Supporting Information) and own highly porous microstructures according to their SEM and TEM images (Figure 1c, Figure S2, Supporting Information). Such porosity is responsible for their large surface area (≈13 m2·g−1) and originates the commonly observed structural shrinkage occurred in the transition from metal oxides to metal oxynitrides or metal nitrides during the high temperature ammonolysis.[ 31 ] Porous microstructures have been very useful to improve photocatalytic activity of oxynitrides and are generally introduced by altering preparation methods.[ 32 , 33 ] The pore size of all samples mostly falls into mesoporous range (2–50 nm) according to BET analysis (Figure S3b, Supporting Information), which is beneficial for mass transportations in water splitting reactions. Further analysis under HAADF‐STEM conditions suggests the high crystallinity of the oxynitride particles. A typical HAADF‐STEM image of La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) in Figure 1d illustrates the random occupation of La/Ca at A sites which matches well with crystal structures from Rietveld refinement (see inset in Figure 1d).
Although sharing similar crystal structures and microstructures, these sample powders have distinct colors that are strongly linked to the content of Ca at A sites. The color of sample powders varies sequentially from red, orange, yellow to light yellow as the Ca content increases from 0 (LaTaON2) to 100 at% (CaTaO2N), being potential applications as non‐toxic pigments.[ 34 ] This is further verified by their UV–vis diffuse reflectance spectroscopy where the absorption edge of solid solutions has a continuous blue‐shift from 650 nm for LaTaON2 to 500 nm for CaTaO2N (Figure 2a). Correspondingly, the direct band gap of solid solutions can be finely tuned between 1.95 and 2.47 eV simply by controlling the Ca content at A sites according to Tauc plot (Figures S4 and S5, Table S2, Supporting Information). This phenomenon most probably arises from a change on O/N ratio and gradual increase on the level of structure distortions.[ 26 , 34 ] This peculiar property is quite useful as it offers us freedoms in manipulating the energetics of photo‐generated charges if compared with conventional semiconductors whose band gap is fixed. The band edge positions of solid solutions La1‐ xCaxTaO1+ yN2‐ y (0 ≤ x, y ≤ 1) were revealed by a combination of Mott–Schottky (MS) analysis in conjunction with X‐ray photoelectron spectroscopy (XPS) valence band scan. Their MS plots illustrated in Figure S6, Supporting Information, suggest the flat band potential changes from −0.49 to −0.92 V versus NHE along with Ca content. XPS valence band scan informs that the energy gap between valence band edge and Fermi level for these samples varies from 1.45 to 2.02 eV (Figure 2b). Considering their band gap, a picture of their band‐edge positions is schematically illustrated in Figure 2c. It can be seen from the figure that continuous band edge alignment can be realized simply by adjusting the composition of solid solutions. Compared with LaTaON2 and CaTaO2N, the band edge positions of La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) have been adjusted in a manner that a high driving force of ≥ 0.5 V is available for both water reduction and oxidation reactions. This is of critical importance for overall water splitting on photocatalysts of single‐light semiconducting absorber which has both water reduction and oxidation reactions occurring on single particle.
Figure 2.

a) Ultraviolet‐visible diffuse reflectance spectroscopy (UV–vis DRS) of La1‐ xCaxTaO1+ yN2‐ y (0 ≤ x, y ≤ 1). b) XPS valence band scan of La1‐ xCaxTaO1+ yN2‐ y (0 ≤ x, y ≤ 1). c) Schematic illustration of the band structures of La1‐ xCaxTaO1+ yN2‐ y (0 ≤ x, y ≤ 1) referring to water redox potentials.
In addition, Raman spectra (Figure S7, Supporting Information) reveal interesting features on local bonding environment in solid solutions. Raman peak assigned to the breathing mode of Ta(O, N)6 octahedrons shifts from 795 to 828 cm−1 as Ca content increases in solid solutions.[ 35 ] This implies the enhanced Ta—O/N bond strength or increased crystal field in Ta(O, N)6 octahedrons along with Ca uptake. A stronger Ta—O/N bond or crystal field in Ta(O, N)6 octahedrons helps stabilize Ta at its high oxidation state which in turn inhibits defects such as Ta4+ species.[ 36 ] This deduction is confirmed by XPS analysis whereby Ta4+ 4f signals centered at around 24.2 and 26.3 eV[ 37 ] decrease clearly as Ca content increases in solid solutions (Figure S9, Supporting Information). Ta4+ species is generally detrimental to photocatalytic activity as they may trap photo‐generated charges and accelerate charge recombination.[ 26 , 38 ] As anticipated, the Ca uptake is accompanied by the decrement of N content in solid solutions, which is affirmed by XPS (Figure S9, Supporting Information) as well as thermogravimetric analysis (Figure S8, Table S2, Supporting Information). All other constituent elements such as O, La, and Ca have reasonable XPS bonding states (Figure S9, Supporting Information) and are consistent with literatures.[ 5 , 39 , 40 ]
As the key parameters including the bandgap, Ta—O/N bonding environment and defects content vary much among solid solutions, their photocatalytic activity was evaluated first by investigating two half‐reactions of water splitting before optimal conditions were screened out for subsequent overall water splitting reactions. Control experiments that lack one of the following components such as sample powders, water, and light illumination did not produce detectable H2/O2, thereby ruling out possible spontaneous water reduction/oxidation reactions. Figure 3a,b illustrate temporal photocatalytic H2 and O2 evolution half‐reactions of LaTaON2, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) and CaTaO2N under visible light illumination (λ ≥ 420 nm). Complete results for all solid solutions can be found in Figure S11, Supporting Information. Here, sodium sulfite and silver nitrate aqueous solution are used as sacrificial agents to promote H2 and O2 evolution half‐reactions, respectively. In both cases, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) shows the highest activity with striking improvements over its parent compounds LaTaON2 and CaTaO2N for H2 and O2 evolutions. In particular, LaTaON2 and CaTaO2N show very low photocatalytic O2 evolution rate which is accelerated by more than one order of magnitude in La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9). Water oxidation into O2 is four‐proton‐four‐electron reaction that is considered as the rate‐limited step for overall water splitting reactions.[ 41 ] The realization of both O2 and H2 evolution half‐reactions with La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) opens the avenue toward overall water splitting reaction.
Figure 3.

a) Photocatalytic H2 evolution for LaTaON2, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) and CaTaO2N under visible light illumination (λ ≥ 420 nm). Sodium sulfite aqueous solution (0.05 m) was used as sacrificial agent and 1 wt% Pt as reductive cocatalyst was loaded onto sample powders. b) Photocatalytic O2 evolution for LaTaON2, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) and CaTaO2N under visible light illumination (λ ≥ 420 nm). Silver nitrate aqueous solution (0.05 m) was applied as sacrificial agent and 2 wt% CoOx as oxidative cocatalyst was loaded onto sample powders. 0.2 g La2O3 was added to maintain the pH at ≈8.5. c) Direct water splitting reactions for La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) under AM 1.5 illumination, evacuation was performed every 6 h and 0.5 wt% RhCrOx as a cocatalyst was loaded onto sample powders. d) Action spectra of La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) for direct water splitting.
Prior to overall water splitting testing, the activity of La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) was further investigated by varying the amount of cocatalysts loaded. 1 wt% Pt and 3 wt% CoOx were found to be the optimal loading amount for H2 and O2 evolution half‐reactions (Figure S12a,b, Supporting Information), respectively. An apparent quantum efficiency (AQE) as high as 9.4% at 420 ± 20 nm has been achieved for O2 evolution half‐reactions which stands as the highest value reported to date on LaTaON2 and CaTaO2N. Detailed comparison of our materials with other metal oxynitrides in the literatures is given in Table S3, Supporting Information. La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) is one of few compounds active for both photocatalytic water reduction and oxidation reactions under visible light illumination. Moreover, its activity is apparently better than or at least comparable to other metal oxynitrides reported.
Nevertheless, our attempts for overall water splitting using both Pt and CoOx loaded La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) did not produce stoichiometric H2 and O2. This is probably due to the drawback of Pt cocatalyst that promotes back reactions on the products.[ 42 ] Thereby, RhCrOx was loaded onto La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) as a cocatalyst which is known to be resistant to the back reactions.[ 43 , 44 ] In this case, successful overall water splitting with a H2/O2 molar ratio of 2:1 was achieved both under visible light (λ ≥ 420 nm) and simulated AM 1.5 illuminations (Figure 3d, Figure S12c,d, Supporting Information) while no activity was observed for LaTaON2 and CaTaO2N. Although CaTaO2N has been reported to be active for overall water splitting,[ 45 ] as far as comparing the samples with La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) and CaTaO2N prepared in the present experiments, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) showed much higher activity. The activity for overall water splitting was further investigated for other compositions particularly those near x = 0.9. The activity shows a clear dependence on the composition and reaches the highest value at x = 0.9 (Figure S12c, Supporting Information). As all samples were prepared under the same conditions, such strong correlations between composition and activity suggest that some intrinsic factors probably play an important role here. One possible factor is the Ta—O/N—Ta bond angle that changes along with composition and is critical in shaping the electronic structure. This can be inferred from their conduction band edge positions which are very sensitive to Ta—O/N—Ta bond angle. As already seen previously, Ta—O/N—Ta bond angle likely reaches an optimal point at x = 0.9 whereby band edge positions are properly adjusted so that a high driving force of ≥ 0.5 V is available for both water reduction and oxidation reactions. The overall water splitting of La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) was further investigated by changing the wavelength of incident photons. The action spectra for overall water splitting at x = 0.9 are illustrated in Figure 3d. TheAQE shows a clear dependence on incident photon wavelength and matches well with the absorption spectra, confirming real photon‐driven processes. More importantly, the activity of La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) is maintained for extended cycles of usage, indicating a good stability. The sample powders after these experiments show identical XRD patterns to those before the photocatalytic measurement experiments and STEM‐EDX mapping suggests a homogeneous distribution of all constituent elements (Figure S13, Supporting Information), suggesting the good stability nature of the samples under water splitting conditions.
To gain a deep insight into the origin of the improved photocatalytic activity, we have performed photoelectrochemical (PEC) analysis on LaTaON2, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) and CaTaO2N. La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) exhibits a higher anodic photocurrent at the bias of over 0.5 V (Figure 4a,b) and smaller interfacial charge transfer resistance than LaTaON2 and CaTaO2N under both visible light (λ ≥ 420 nm) and simulated AM 1.5 illuminations (Figure S14, Supporting Information), indicating a better charge separation capability in the solid solution. These PEC results are consistent with the photocatalytic results. This deduction is also supported by time‐resolved photoluminescence (PL) decay spectra (Figure 4c). La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) gives a much slower PL decay compared with LaTaON2 and CaTaO2N. Specifically, its average PL lifetime is extended up to 50.41 ns while LaTaON2 and CaTaO2N have the much shorter lifetime of 32.44 and 5.53 ns, respectively (Table S4, Supporting Information). This confirms that behavior of photo‐generated charges can be modified by forming solid solutions. The behavior of photo‐generated electrons was further investigated by open‐circuit voltage (V oc) decay (OCVD) measurements in which O2 was used as an electron scavenger and was compared with control experiment using inert Ar (Figure 4d). OCVD records the dissipation processes of photo‐generated electrons which are stored in semiconductors during light illumination.[ 46 ] O2 is a useful prober to those electrons that reach surface and are of chemical reactivity.[ 33 ] For La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9), O2 considerably accelerates OCVD decay with respect to Ar, indicating that a large amount of photo‐generated electrons are accessible to sample surface for chemical reactions. On the contrary, LaTaON2 and CaTaO2N show little changes on OCVD decay profiles in O2 and Ar, implying that most photo‐generated electrons are consumed by recombining with holes or trapping at surface rather than participate in chemical reactions.
Figure 4.

Linear sweep voltammetry of LaTaON2, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) and CaTaO2N under chopped a) visible light illumination (λ ≥ 420 nm) and b) simulated AM 1.5 illumination. c) Time‐resolved photoluminescence decay spectra of LaTaON2, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) and CaTaO2N monitored at 625 nm by time‐correlated single‐photon counting. 471.8 nm light generated by a picosecond pulsed light‐emitting diode was used to excite the samples. Average lifetime is tabulated in inset table. d) Open‐circuit voltage (V oc) decay (OCVD) profiles of photoelectrodes fabricated from LaTaON2, La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) and CaTaO2N powders in either Ar or O2 atmosphere. The illumination initiated once V oc was stabilized in the dark and was terminated at 100 s. V oc in the dark was marked by dotted line.
In summary, overall water splitting with stoichiometric H2/O2 ratio under visible light (λ ≥ 420 nm) has been achieved in solid solutions between LaTaON2 and CaTaO2N, both of which are inactive photocatalysts under the same conditions. The optimal constituent compositions of La0.1Ca0.9TaO1+ yN2‐ y (x = 0.9) shows the highest activity with a good durability and stability. The modulated key parameters including Ta—O/N bond length, Ta—O/N—Ta bond angle, defects concentration, bandgap, and band edge alignments greatly promote the separation of photogenerated charges as evidenced by the combination of several characterizations. Moreover, the appropriate band edges guarantee the sufficient driving force of the charge carriers for both water reduction and oxidation half‐reactions. The global merits of the developed solid solution make it a promising solar‐driven photocatalyst for overall water splitting.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
The authors would like to thank the National Natural Science Foundation of China (Grant No. 51972233, 51825204, 21633009) and Natural Science Foundation of Shanghai (Grant No. 19ZR1459200) for funding. The work was supported by Shanghai Science and Technology Commission (14DZ2261100) and the Fundamental Research Funds for the Central Universities. G.L. thanks Newton Advanced Fellowship.
Wang Y., Kang Y., Zhu H., Liu G., Irvine J. T. S., Xu X., Perovskite Oxynitride Solid Solutions of LaTaON2‐CaTaO2N with Greatly Enhanced Photogenerated Charge Separation for Solar‐Driven Overall Water Splitting. Adv. Sci. 2021, 8, 2003343 10.1002/advs.202003343
Contributor Information
Gang Liu, Email: gangliu@imr.ac.cn.
John T. S. Irvine, Email: jtsi@st-andrews.ac.uk.
Xiaoxiang Xu, Email: xxxu@tongji.edu.cn.
References
- 1.a) Chen S. S., Takata T., Domen K., Nat. Rev. Mater. 2017, 2, 17050; [Google Scholar]; b) Tachibana Y., Vayssieres L., Durrant J. R., Nat. Photonics 2012, 6, 511; [Google Scholar]; c) Xu X. X., Randorn C., Efstathiou P., Irvine J. T. S., Nat. Mater. 2012, 11, 595; [DOI] [PubMed] [Google Scholar]; d) Wang Q., Hisatomi T., Jia Q. X., Tokudome H., Zhong M., Wang C. Z., Pan Z. H., Takata T., Nakabayashi M., Shibata N., Li Y. B., Sharp I. D., Kudo A., Yamada T., Domen K., Nat. Mater. 2016, 15, 611; [DOI] [PubMed] [Google Scholar]; e) Listorti A., Durrant J., Barber J., Nat. Mater. 2009, 8, 929; [DOI] [PubMed] [Google Scholar]; f) Zou Z. G., Ye J. H., Sayama K., Arakawa H., Nature 2001, 414, 625; [DOI] [PubMed] [Google Scholar]; g) Yu Z. B., Chen X. Q., Kang X. D., Xie Y. P., Zhu H. Z., Wang S. L., Ullah S., Ma H., Wang L. Z., Liu G., Ma X. L., Cheng H. M., Adv. Mater. 2018, 30, 1706259. [DOI] [PubMed] [Google Scholar]
- 2.a) Turner J. A., Science 2004, 305, 972; [DOI] [PubMed] [Google Scholar]; b) Turner J. A., Science 1999, 285, 687; [DOI] [PubMed] [Google Scholar]; c) Hejazi S., Mohajernia S., Osuagwu B., Zoppellaro G., Andryskova P., Tomanec O., Kment S., Zboril R., Schmuki P., Adv. Mater. 2020, 1902069;32(16), [DOI] [PubMed] [Google Scholar]; d) Wang Y., Du P. P., Pan H. Z., Fu L., Zhang Y., Chen J., Du Y. W., Tang N. J., Liu G., Adv. Mater. 2019, 31, 1807540. [DOI] [PubMed] [Google Scholar]
- 3. Goto Y., Hisatomi T., Wang Q., Higashi T., Ishikiriyama K., Maeda T., Sakata Y., Okunaka S., Tokudome H., Katayama M., Akiyama S., Nishiyama H., Inoue Y., Takewaki T., Setoyama T., Minegishi T., Takata T., Yamada T., Domen K., Joule 2018, 2, 509. [Google Scholar]
- 4.a) Kato H., Asakura K., Kudo A., J. Am. Chem. Soc. 2003, 125, 3082; [DOI] [PubMed] [Google Scholar]; b) Miseki Y., Kato H., Kudo A., Energy Environ. Sci. 2009, 2, 306; [Google Scholar]; c) Chiang T. H., Lyu H., Hisatomi T., Goto Y., Takata T., Katayama M., Minegishi T., Domen K., ACS Catal. 2018, 8, 2782; [Google Scholar]; d) Takata T., Jiang J. Z., Sakata Y., Nakabayashi M., Shibata N., Nandal V., Seki K., Hisatomi T., Domen K., Nature 2020, 581, 411. [DOI] [PubMed] [Google Scholar]
- 5. Zhao Z. Q., Goncalves R. V., Barman S. K., Willard E. J., Byle E., Perry R., Wu Z. K., Huda M. N., Moule A. J., Osterloh F. E., Energy Environ. Sci. 2019, 12, 1385. [Google Scholar]
- 6. Bolton J. R., Strickler S. J., Connolly J. S., Nature 1985, 316, 495. [Google Scholar]
- 7. Chen Z. B., Jaramillo T. F., Deutsch T. G., Kleiman‐Shwarsctein A., Forman A. J., Gaillard N., Garland R., Takanabe K., Heske C., Sunkara M., McFarland E. W., Domen K., Miller E. L., Turner J. A., Dinh H. N., J. Mater. Res. 2010, 25, 3. [Google Scholar]
- 8. Yerga R. M. N., Galvan M. C. A., del Valle F., de la Mano J. A. V., Fierro J. L. G., ChemSusChem 2009, 2, 471. [DOI] [PubMed] [Google Scholar]
- 9. Ohno T., Bai L., Hisatomi T., Maeda K., Domen K., J. Am. Chem. Soc. 2012, 134, 8254. [DOI] [PubMed] [Google Scholar]
- 10. Lee Y. G., Teramura K., Hara M., Domen K., Chem. Mater. 2007, 19, 2120. [Google Scholar]
- 11. Maeda K., Lu D. L., Domen K., Chem. ‐ Eur. J. 2013, 19, 4986. [DOI] [PubMed] [Google Scholar]
- 12. Wang Z., Inoue Y., Hisatomi T., Ishikawa R., Wang Q., Takata T., Chen S. S., Shibata N., Ikuhara Y., Domen K., Nat. Catal. 2018, 1, 756. [Google Scholar]
- 13. Pan C. S., Takata T., Nakabayashi M., Matsumoto T., Shibata N., Ikuhara Y., Domen K., Angew. Chem., Int. Ed. 2015, 54, 2955. [DOI] [PubMed] [Google Scholar]
- 14.a) Maeda K., Domen K., Chem. Mater. 2010, 22, 612; [Google Scholar]; b) Nurlaela E., Ould‐Chikh S., Harb M., del Gobbo S., Aouine M., Puzenat E., Sautet P., Domen K., Basset J. M., Takanabe K., Chem. Mater. 2014, 26, 4812; [Google Scholar]; c) Wang X. C., Maeda K., Lee Y., Domen K., Chem. Phys. Lett. 2008, 457, 134; [Google Scholar]; d) Maeda K., Teramura K., Domen K., J. Catal. 2008, 254, 198; [Google Scholar]; e) Maeda K., Teramura K., Takata T., Hara M., Saito N., Toda K., Inoue Y., Kobayashi H., Domen K., J. Phys. Chem. B 2005, 109, 20504; [DOI] [PubMed] [Google Scholar]; f) Tessier F., Maillard P., Lee Y., Bleugat C., Domen K., J. Phys. Chem. C 2009, 113, 8526. [Google Scholar]
- 15. Maeda K., Domen K., J. Phys. Chem. Lett. 2010, 1, 2655. [Google Scholar]
- 16.a) Fuertes A., J. Mater. Chem. 2012, 22, 3293; [Google Scholar]; b) Fuertes A., Dalton Trans. 2010, 39, 5942; [DOI] [PubMed] [Google Scholar]; c) Fuertes A., Mater. Horiz. 2015, 2, 453; [Google Scholar]; d) Liu Y., Ye S., Xie H. C., Zhu J., Shi Q., Ta N., Chen R. T., Gao Y. Y., An H. Y., Nie W., Jing H. W., Fan F. T., Li C., Adv. Mater. 2020, 32, 1906513; [DOI] [PubMed] [Google Scholar]; e) Xiao M., Hao M. M., Lyu M. Q., Moore E. G., Zhang C., Luo B., Hou J. W., Lipton‐Duffin J., Wang L. Z., Adv. Funct. Mater. 2019, 29, 1905683. [Google Scholar]
- 17. Mitchell R. H., Perovskites: Modern and Ancient, Almaz Press Inc., Ontario, Canada: 2002. [Google Scholar]
- 18. Dong B. B., Cui J. Y., Liu T. F., Gao Y. Y., Qi Y., Li D., Xiong F. Q., Zhang F. X., Li C., Adv. Energy Mater. 2018, 8, 1801660. [Google Scholar]
- 19.a) Kim Y. I., Ceram. Int. 2014, 40, 5275; [Google Scholar]; b) Kim Y. I., Woodward P. M., Baba‐Kishi K. Z., Tai C. W., Chem. Mater. 2004, 16, 1267; [Google Scholar]; c) Jacobs J., Oehler F., Schettlock J., Ebbinghaus S. G., Z. Anorg. Allg. Chem. 2018, 644, 1832; [Google Scholar]; d) Oehler F., Ebbinghaus S. G., Solid State Sci. 2016, 54, 43; [Google Scholar]; e) Seo J., Hisatomi T., Nakabayashi M., Shibata N., Minegishi T., Katayama M., Domen K., Adv. Energy Mater. 2018, 8, 1800094. [Google Scholar]
- 20. Siritanaratkul B., Maeda K., Hisatomi T., Domen K., ChemSusChem 2011, 4, 74. [DOI] [PubMed] [Google Scholar]
- 21.a) Higashi M., Abe R., Teramura K., Takata T., Ohtani B., Domen K., Chem. Phys. Lett. 2008, 452, 120; [Google Scholar]; b) Higashi M., Abe R., Takata T., Domen K., Chem. Mater. 2009, 21, 1543. [Google Scholar]
- 22. Clark L., Oro‐Sole J., Knight K. S., Fuertes A., Attfield J. P., Chem. Mater. 2013, 25, 5004. [Google Scholar]
- 23. Kato H., Ueda K., Kobayashi M., Kakihana M., J. Mater. Chem. A 2015, 3, 11824. [Google Scholar]
- 24. Zhang L., Song Y., Feng J. Y., Fang T., Zhong Y. J., Li Z. S., Zou Z. G., Int. J. Hydrogen Energy 2014, 39, 7697. [Google Scholar]
- 25.a) Matsukawa M., Ishikawa R., Hisatomi T., Moriya Y., Shibata N., Kubota J., Ikuhara Y., Domen K., Nano Lett. 2014, 14, 1038; [DOI] [PubMed] [Google Scholar]; b) Luo Y., Wang Z., Suzuki S., Yubuta K., Kariya N., Hisatomi T., Domen K., Teshima K., Cryst. Growth Des. 2020, 20, 255; [Google Scholar]; c) Sun X. Q., Mi Y. L., Jiao F., Xu X. X., ACS Catal. 2018, 8, 3209; [Google Scholar]; d) Feng J. Y., Huang H. T., Fang T., Wang X., Yan S. C., Luo W. J., Yu T., Zhao Y. X., Li Z. S., Zou Z. G., Adv. Funct. Mater. 2019, 29, 1808389. [Google Scholar]
- 26. Balaz S., Porter S. H., Woodward P. M., Brinson L. J., Chem. Mater. 2013, 25, 3337. [Google Scholar]
- 27.a) Shannon R. D., Prewitt C. T., Acta Crystallogr. 1969, 25, 925; [Google Scholar]; b) Shannon R. D., Prewitt C. T., Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 1970, 26, 1046. [Google Scholar]
- 28. Kubo A., Giorgi G., Yamashita K., Chem. Mater. 2017, 29, 539. [Google Scholar]
- 29. Garcia‐Esparza A. T., Tyminska N., Al Orabi R. A., Le Bahers T., J. Mater. Chem. C 2019, 7, 1612. [Google Scholar]
- 30.a) Cox P. A., The Electronic Structure and Chemistry of Solids, Oxford University Press, Oxford, UK: 1987; [Google Scholar]; b) Cox P. A., Transition Metal Oxides: An Introduction to their Electronic Structure and Properties, Oxford University Press, Oxford, UK: 1992. [Google Scholar]
- 31. Dong B. B., Cui J. Y., Gao Y. Y., Qi Y., Zhang F. X., Li C., Adv. Mater. 2019, 31, 1808185. [DOI] [PubMed] [Google Scholar]
- 32. Yang M. H., MacLeod M. J., Tessier F., DiSalvo F. J., J. Am. Ceram. Soc. 2012, 95, 3084. [Google Scholar]
- 33. Xie Y. H., Wang Y. W., Chen Z. F., Xu X. X., ChemSusChem 2016, 9, 1403. [DOI] [PubMed] [Google Scholar]
- 34. Jansen M., Letschert H. P., Nature 2000, 404, 980. [DOI] [PubMed] [Google Scholar]
- 35. Sun X. Q., Wu F. F., Liu G., Xu X. X., J. Mater. Chem. A 2018, 6, 20760. [Google Scholar]
- 36. Etourneau J., Portier J., Menil F., J. Alloys Compd. 1992, 188, 1. [Google Scholar]
- 37.a) Wang Y. W., Jin S., Pan G. X., Li Z. X., Chen L., Liu G., Xu X. X., J. Mater. Chem. A 2019, 7, 5702; [Google Scholar]; b) Ettema A. R. H. F., Haas C., J. Phys.: Condens. Matter 1993, 5, 3817. [Google Scholar]
- 38. Jadhav S., Hasegawa S., Hisatomi T., Wang Z., Seo J., Higashi T., Katayama M., Minegishi T., Takata T., Peralta‐Hernandez J. M., Torres O. S., Domen K., J. Mater. Chem. A 2020, 8, 1127. [Google Scholar]
- 39. Wang Y. W., Wei S. H., Xu X. X., Appl. Catal., B 2020, 263, 118315. [Google Scholar]
- 40. Wu F. F., Liu G., Xu X. X., J. Catal. 2017, 346, 10. [Google Scholar]
- 41. Du J. L., Chen Z. F., Chen C. C., Meyer T. J., J. Am. Chem. Soc. 2015, 137, 3193. [DOI] [PubMed] [Google Scholar]
- 42. Yoshida H., Yamada R., Yoshida T., ChemSusChem 2019, 12, 1958. [DOI] [PubMed] [Google Scholar]
- 43. Maeda K., Teramura K., Lu D. L., Saito N., Inoue Y., Domen K., Angew. Chem., Int. Ed. 2006, 45, 7806. [DOI] [PubMed] [Google Scholar]
- 44. Maeda K., Sakamoto N., Ikeda T., Ohtsuka H., Xiong A. K., Lu D. L., Kanehara M., Teranishi T., Domen K., Chem. ‐ Eur. J. 2010, 16, 7750. [DOI] [PubMed] [Google Scholar]
- 45. Xu J. S., Pan C. S., Takata T., Domen K., Chem. Commun. 2015, 51, 7191. [DOI] [PubMed] [Google Scholar]
- 46.a) Zaban A., Greenshtein M., Bisquert J., ChemPhysChem 2003, 4, 859; [DOI] [PubMed] [Google Scholar]; b) Meekins B. H., Kamat P. V., ACS Nano 2009, 3, 3437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information