

Abstract
Most drugs acting on G-protein-coupled receptors target the orthosteric binding pocket where the native hormone or neurotransmitter binds. There is much interest in finding allosteric ligands for these targets because they modulate physiologic signaling and promise to be more selective than orthosteric ligands. Here we describe a newly developed allosteric modulator of the β2-adrenergic receptor (β2AR), AS408, that binds to the membrane-facing surface of transmembrane segments 3 and 5, as revealed by X-ray crystallography. AS408 disrupts a water-mediated polar network involving E1223.41 and the backbone carbonyls of V2065.45 and S2075.46. The AS408 binding site is adjacent to a previously identified molecular switch for β2AR activation formed by I3.40, P5.50 and F6.44. The structure reveals how AS408 stabilizes the inactive conformation of this switch, thereby acting as a negative allosteric modulator for agonists and positive allosteric modulator for inverse agonists.
The orthosteric binding pockets of aminergic G-protein-coupled receptors (GPCRs) share a high degree of amino acid identity; consequently, it has been difficult to develop subtype-selective drugs. Allosteric binding sites are less conserved and may therefore offer more subtype selectivity. Additionally, allosteric ligands function by modulating responses to native hormones and neurotransmitters, and may therefore be better tolerated in clinical applications. Allosteric modulators have been described for a number of GPCRs1–5; however, only two allosteric modulators have been reported for beta adrenergic receptors, both identified in a DNA-encoded library screen6,7. Cmpd-15, a 646 Da negative allosteric modulator (NAM), was shown to bind to a pocket of the β2-adrenergic receptor (β2AR) formed by the cytoplasmic ends of transmembrane segments (TMs) 1, 2, 6 and 7 (refs.6,8). Cmpd-6 is a 611 Da positive allosteric modulator (PAM) that binds to a pocket formed by intracellular loop 2 (ICL2) and the cytoplasmic ends of TMs 3 and 4 (ref.9). Allosteric modulators for β-adrenergic receptors would have therapeutic use in several diseases including hypertension, arrhythmias and heart failure. We therefore explored the use of in silico docking to identify an allosteric modulator for the β2AR with druglike properties including a molecular weight below 500 Da. Following this strategy, we identified a compound with weak negative allosteric activity. Chemical optimization led to the development of AS408 (2), a NAM for both G-protein activation and arrestin recruitment. Using X-ray crystallography, we found that AS408 does not bind to the docking site in the extracellular vestibule, but to a pocket formed by the membrane-facing surface of TM3 and TM5. Using mutagenesis studies and molecular dynamics simulations, we provide mechanistic insights into how AS408 regulates β2AR function. Previous studies have shown that activation of the β2AR involves an inward movement of TM5 around S2075.46 leading to rearrangement of a conserved P5.50I3.40F6.44 motif resulting in the outward movement of TM6 (ref.10). By binding to residues in TM5 adjacent to S2075.46, AS408 can stabilize TM5 in an inactive conformation.
Results
Identification of the NAM pocket for AS408 on the β2AR.
We performed in silico docking using the exosite for the β2AR-selective agonist salmeterol located in the extracellular vestibule as a template11 (Fig. 1a). One of the initial docking hits, the small molecule BRAC1 (1) (236 Da), (Fig. 1b) exhibited weak, negative allosteric regulation of norepinephrine-stimulated arrestin recruitment and cAMP accumulation (Fig. 1c,d). Optimization efforts led to a brominated derivative that was more potent and efficacious (2, Fig. 1e,f), and we were able to determine the crystal structure of the NAM-bound β2AR in its inactive conformation (Fig. 1g).
Fig. 1 |. Hit-to-lead optimization, pharmacological characterization and structure of allosteric modulator AS408 bound to β2AR.
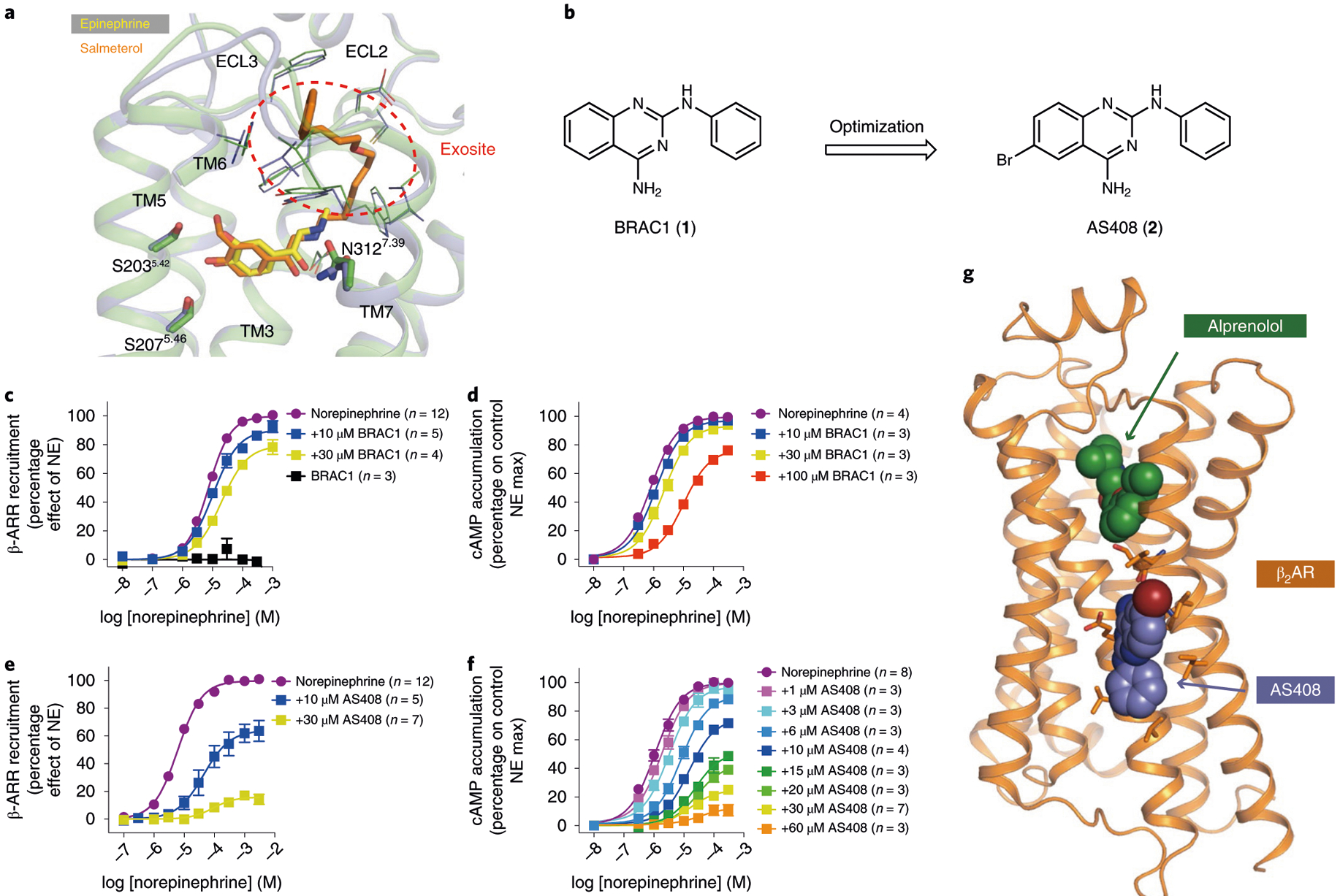
a, Salmeterol (orange) extends from the orthosteric site where epinephrine (yellow) binds to an exosite at the extracellular vestibule. The exosite was used as a docking template. b, Hit-to-lead optimization of the docking hit BRAC1. c–f, Negative allosteric effect of BRAC1 (c,d) and AS408 (e,f) on norepinephrine-stimulated β-arrestin 2 (β-ARR) recruitment (c,e) and on cAMP accumulation (d,f). Data are derived from 3–12 experiments done in duplicate and given as mean ± s.e.m.; the sample size (n) is labeled in the figure. g, Structure of AS408 bound to β2AR in the presence of antagonist alprenolol.
Crystals were obtained in lipidic cubic phase (LCP) with the β2AR bound to the neutral antagonist alprenolol and AS408. The structure was solved by molecular replacement at 3.1 Å (Supplementary Table 1). We were surprised to observe well-defined Fo–Fc electron density for AS408 at the membrane-facing surface of TM3 and TM5 (Fig. 1g and Supplementary Fig. 1a), but not in the extracellular vestibule (Fig. 1a). The position of AS408 was further confirmed by obtaining an anomalous signal for bromine (Supplementary Fig. 1b). The binding pocket is formed by predominantly hydrophobic interactions with C1253.44, V1263.45, V1293.48, V2105.49, P2115.50 and I2145.53 (superscripts indicate Ballesteros–Weinstein numbering12). The primary amine of AS408 can hydrogen bond with E1223.41 and the carbonyl of V2065.45 (Fig. 2a). L451.44 of an antiparallel symmetry mate interacts with the Br of AS408 in our crystal structure (Supplementary Fig. 1c). This interaction would not be present in cell membranes and therefore does not appear to be essential for AS408 activity. Moreover, as discussed below, the binding pose of AS408 is stable in molecular dynamics simulations without an interaction with L45.
Fig. 2 |. Structural basis of the negative allosteric activity of AS408 on agonist binding to β2AR.
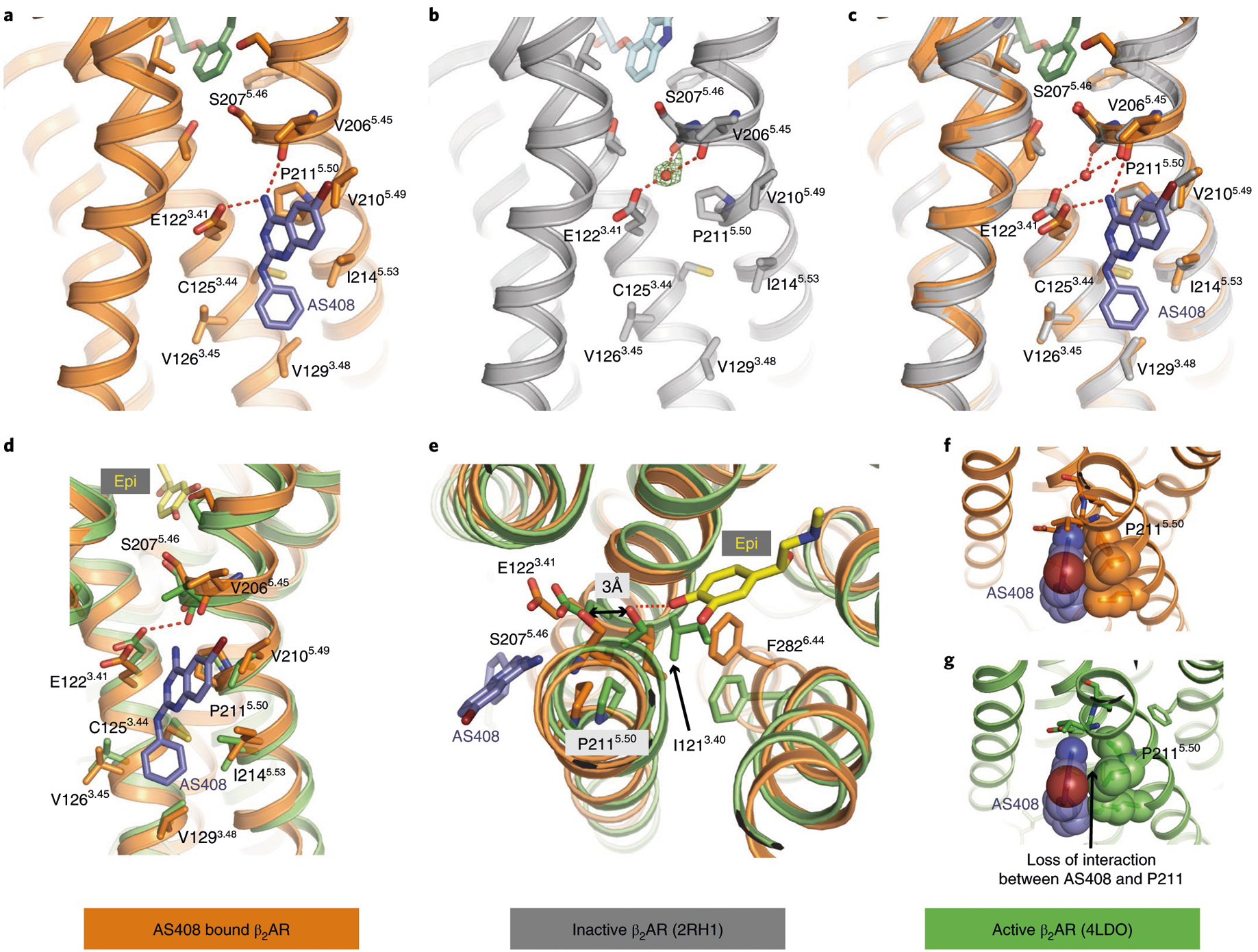
Structure of AS408 bound to β2AR in the presence of alprenolol determined by X-ray crystallography. a, The residues within 3 Å of AS408 (blue) in the presence of alprenolol (dark green). b, Inactive structure of β2AR in presence of inverse agonist carazolol (PDB 2RH1). The positive density consistent with an unmodeled water molecule is shown with green mesh. c, Superposition of the structures of the inactive form of β2AR in the presence of carazolol (PDB 2RH1, gray) on the AS408-β2AR structure and influence of AS408 on the water network formed by E1223.41, S2075.46 and V2065.45. d–g, Comparison of the AS408-bound β2AR structure with the active, agonist-bound β2AR (PDB 4LDO, green). d, Positions of the side chain of residues coordinating AS408 binding differ in the active, agonist-bound conformation. e, Illustration of the capacity of AS408 to prevent the catechol ring of epinephrine (yellow) to bind to S2075.46 (and S2035.42, not shown) in TM5 and therefore prevent transition to the active conformation. f, Interaction of P2115.50 and AS408 in the crystal structure (inactive conformation). g, Loss of interaction of P2115.50 and AS408 in the agonist-bound active conformation.
When comparing the structures of the inactive state β2AR (Protein Data Bank (PDB) 2RH1) with β2AR bound to AS408, the differences are subtle (Fig. 2a–c). However, we observe larger structural differences in residues lining the AS408 binding pocket when comparing the AS408-β2AR structure to an active state structure (Fig. 2d,e). Here, agonist binding leads to an inward movement of P2115.50 and would result in the loss of van der Waals contact with AS408 (Fig. 2f,g). Thus, the complementary interactions between AS408 and the inactive β2AR stabilize the inactive conformation. Of interest, P2115.50 was shown to be part of an allosteric hub along with I1213.40 and F2826.44. Agonist binding stabilizes an inward movement of TM5 in the binding pocket resulting in a rearrangement of packing interactions among these three amino acids to initiate the outward movement of TM610. This triad is conserved in approximately 90 nonsensory family A GPCRs.
MD simulations of the AS408 binding pocket.
To assess the stability of interactions between AS408 and the receptor, we performed 12 μs all-atom molecular dynamics simulations. AS408 adopts a binding mode that is consistent with the crystal structure (Supplementary Fig. 2). Experimental determination of basicity by titration gave a pKa of 5.2 for AS408, indicating notable but weak basicity. Therefore, in proximity to the acidic E1223.41 of the wild type receptor, proton exchange is very likely and, thus, AS408 will be protonated. We have performed MD simulations with different protonation states and positions. They show the highest stability for AS408 positively charged at the endocyclic nitrogen. The interactions of the primary amino group of the protonated resonance system of AS408 with the carboxyl group of E1223.41 and the backbone oxygen of V2065.45 were well maintained with interaction frequencies >99%. In the absence of AS408, E1223.41 may form water-mediated hydrogen bonds with the backbone carbonyls of V2065.45 and S2075.46 (Fig. 2b,c). While not modeled into the deposited inactive-state structure of the β2AR (PDB 2RH1), there is positive density consistent with a water-mediated hydrogen bond network bridging the carboxylic acid function of E1223.41 in TM3 with the backbone carbonyl oxygen of V2065.45 and S2075.46 (TM5; Fig. 2b). Activation of the β2AR involves a 2.4 Å inward movement of the alpha-carbon of S2075.46 that would disrupt this network and E1223.41 would directly hydrogen bond with the backbone carbonyl oxygen of V2065.45. On binding of AS408, the water is displaced by the amine nitrogen of the positively charged ligand enhancing stability of TM5 in an inactive conformation (Fig. 2c).
Structure–activity studies of AS408 analogs.
In the process of going from BRAC1, the initial in silico screening hit, to AS408, a number of analogs were generated and tested (3-13). Figure 3 shows how structural differences in these compounds influenced their functional properties. Except for BRAC1–5 (9), an inactive truncated version of BRAC1, all compounds show substantial lipophilicity with calculated logP values between 3 and 4.8 promoting hydrophobic interactions with the aliphatic tails of the membrane lipids. The primary amino group of the protonated AS408 forms an ionic interaction with the side chain of E1223.41 (TM3) and a hydrogen bond with the backbone oxygen of V2065.45 (TM5) (Fig. 2c). DD288 (3), missing the amino function, can no longer replace the mediating water molecule linking E1223.41 with V2065.45 and S2075.46 resulting in an attenuated, however, not abolished, negative allosteric effect. This suggests that the polar interaction between AS408 and E1223.41 is important but not crucial for NAM activity. According to the crystal structure, the heterocyclic quinazoline ring of AS408 engages in hydrophobic interactions with the aliphatic moieties of V2105.49 and P2115.50. The stronger allosteric effect of AS408, compared to the initial hit (BRAC1), can be explained by attractive interactions of the bromo substituent with the highly hydrophobic lipid–protein interface. The halogen atom fits nicely between the side chains of V2065.45 and V2105.49, when the bromine is located in position 6. A bromo substituent in position 5, 7 or 8, of the quinazoline ring confers a less complementary shape (AS436 (12), AS241 (13)) or a clash with V2065.45 (AS94 (11)). Hence, reduced allosteric modulation was observed for these regioisomers. To further probe the effect of the substituent in position 6, we replaced the bromo atom by a set of different halogens. The size of the halogen substituent and thus the extent of the hydrophobic interaction to V2065.45 and V2105.49 controls the negative allosteric modulation (F ≪ Cl < CF3 < Br < I). Further increasing the hydrophobic substituent by introduction of a phenyl group (ST240 (8)) results in partial disruption of the negative allosteric effect, as a consequence of repulsive interactions with the side chain of V2065.45. The phenyl ring of AS408 fits into a complementary hydrophobic pocket formed by C1253.44, V1263.45, V1293.48 and I2145.53. Starting from BRAC1, replacement of the phenyl group by a smaller aliphatic propyl chain reduces the hydrophobic interactions and abolishes the negative allosteric effect (BRAC1–5). Loss of the allosteric effect was also observed when we introduced a hydroxyl group to the phenyl ring (BRAC1–23 (10)). While the hydroxyl substitution may inflict repulsive interactions at the hydrophobic membrane protein interface, it is also possible that the enhanced polarity reduces partitioning of BRAC1–23 into the lipid bilayer.
Fig. 3 |. Structure–activity relationships of AS408 analogs.
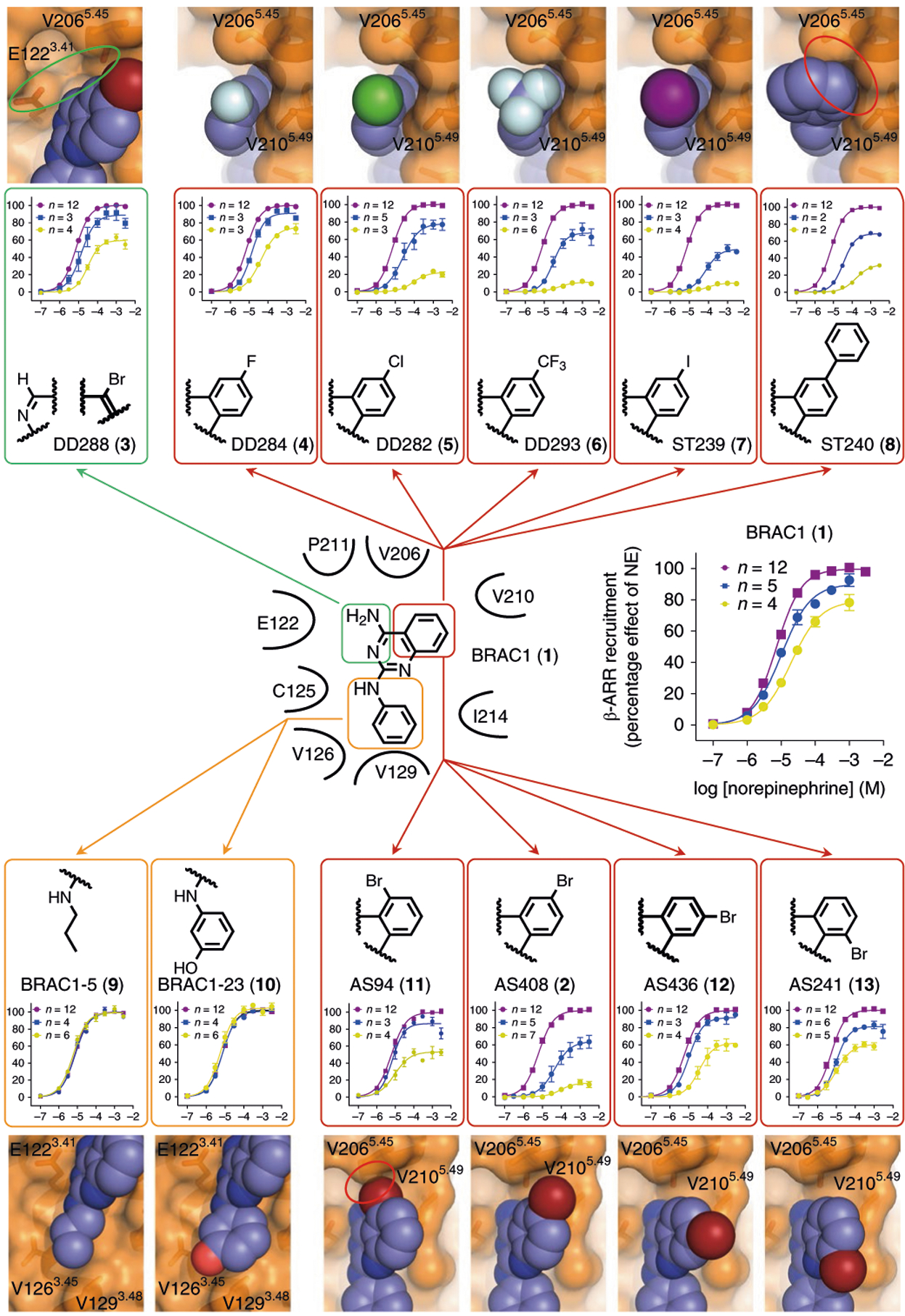
Dose–response curves indicate a negative allosteric effect of AS408 analogs on norepinephrine-stimulated β-arrestin recruitment. Modulators were tested at concentrations of 10 μM (blue) and 30 μM (yellow) compared to norepinephrine alone (purple). Data are derived from 2–12 experiments each done in duplicate and given as mean ± s.e.m. if n ≥ 3, or as mean if n = 2; the sample size (n) is labeled in the figure. Highlighted in bold is the structure of the bromo substituted phenyl ring of AS408. The receptor–ligand models show crucial parts of the crystal structure and the respective modulator aligned with the coordinates of AS408 visualizing the impact of structural modifications on receptor–ligand interactions. Red circles indicate repulsive interactions, the green circle indicates the missing polar interaction network.
Effect of AS408 on β2AR pharmacology and signaling.
As shown in Fig. 1e,f and Supplementary Fig. 3, AS408 is a nonbiased NAM, having comparable effects on norepinephrine-stimulated β-arrestin recruitment, [35S]GTPγS binding and cAMP accumulation. The data was fit using the complete operational model of efficacy13,14. Given that the effect of AS408 on agonist efficacy (β) was greater than the effect of agonist affinity (α), we fixed β near zero (β = 10−100) for the analysis using the complete operational model. Moreover, since AS408 exhibited no agonist efficacy, τB was also fixed near zero (τB = 10−100). The results (Supplementary Table 2) show that the affinity (KB) and allosteric effect on agonist affinity (α) for AS408 are similar for arrestin recruitment and cAMP accumulation, while the affinity of AS408 was approximately tenfold higher when determined by the [35S]GTPγS binding assay. We speculate that the apparent increase in affinity of AS408 might be due to better access of the modulator to the receptor in broken cell preparations (as in the [35S]GTPγS binding assays) compared to the whole cell assays (cAMP and β-arrestin recruitment assays). AS408 has the greatest effect in suppressing recruitment of arrestin by norepinephrine and the partial agonist salmeterol (Supplementary Fig. 4). The allosteric effect of AS408 on salmeterol is of particular interest because AS408 originated from a docking campaign where the salmeterol exosite was used as the docking template (Fig. 1a). This result confirms that AS408 does not bind to the intended site in the extracellular vestibule, where AS408 would be expected to act as a competitive antagonist rather than a NAM.
While AS408 is a NAM for agonist binding, G-protein activation and arrestin recruitment, AS408 is a modest PAM for binding to the inverse agonists ICI-118,551. By stabilizing the inactive state, AS408 enhances the affinity of the β2AR for ICI-118,551 by 2.3-fold (Fig. 4a) and reduces its affinity for the agonist norepinephrine (Fig. 4b). Of interest, AS408 appears to have a greater effect on the affinity of norepinephrine for uncoupled β2AR (Klow 3.6-fold reduction, P < 0.05) compared to Gs-coupled β2AR (Khigh 1.3-fold reduction, P ≈ 0.34). Likewise, AS408 has a larger effect on epinephrine binding to uncoupled β2AR (Klow 5.8-fold reduction, P < 0.001) compared to Gs-coupled β2AR (Khigh 3.8-fold reduction, P < 0.001) (Fig. 4c and Supplementary Table 3). AS408 has weak inverse agonist activity on [35S]GTPγS binding (Fig. 4d), but does not enhance the inhibition of basal activity by ICI-118,551 (Fig. 4e). The decrease in [35S]GTPγS binding observed with AS408 is not a direct effect on the Gs protein (data not shown). AS408 had no effect on the dissociation rate of [3H]formoterol in Gs-coupled β2AR (Fig. 4f) since the positive cooperativity between agonist and G protein yields formoterol affinities that are too high for AS408 to overcome. However, AS408 does accelerate the dissociation rate of [3H]formoterol from uncoupled β2AR in the presence of GTPγS. These results further confirm that AS408 does not bind above the orthosteric binding pocket, where it would be expected to slow agonist dissociation. They also suggest that AS408 does not bind to the G protein-stabilized active state of the β2AR.
Fig. 4 |. AS408 helps to stabilize the inactive conformation.
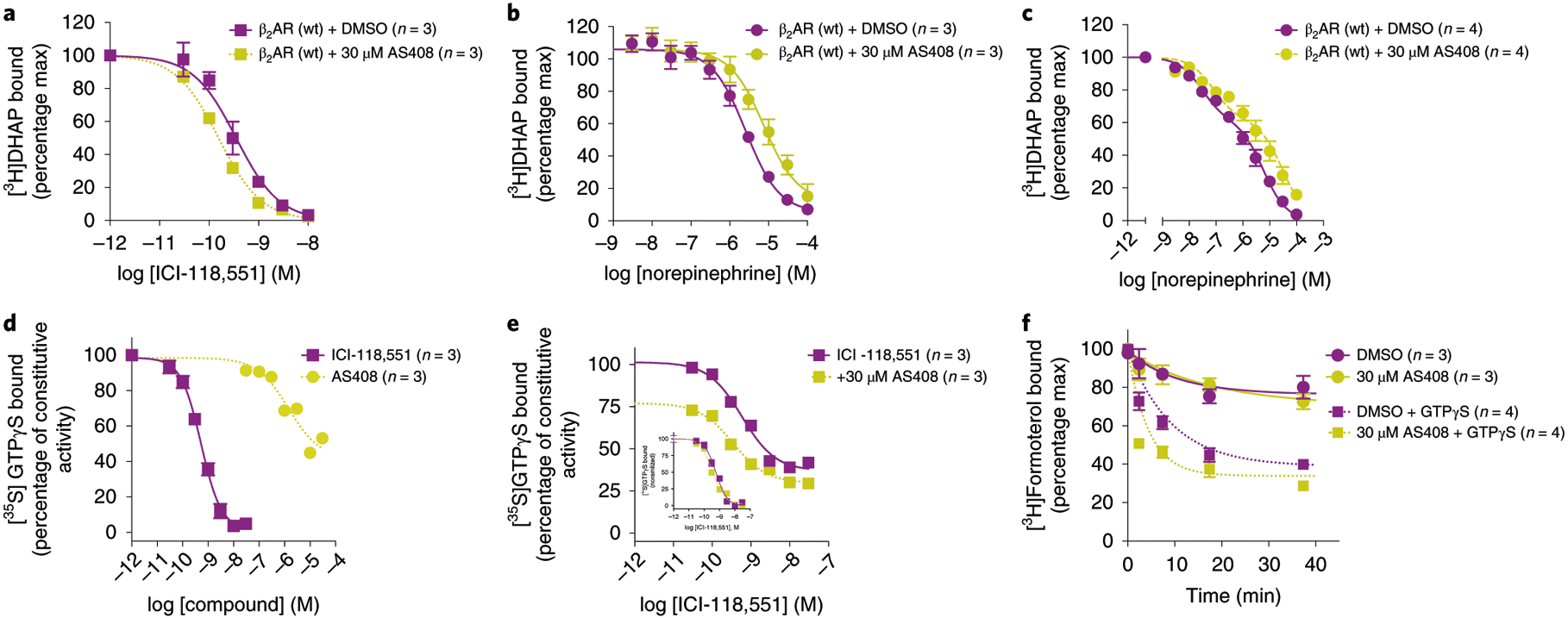
a–c, AS408 (yellow symbols) modestly enhances inverse agonist ICI-118,551 inhibition of [3H]DHAP binding (a) while diminishing full agonist norepinephrine binding to β2AR wild type (wt) in the absence (b), or presence (c) of coexpressed Gs heterotrimer. d,e, AS408 displays inverse agonist activity on constitutive G-protein activation at high concentrations (d) but does not appear to enhance ICI-118,551 inhibition of [35S]GTPγS binding (e; inset, normalized). f, AS408 accelerates the dissociation of full agonist [3H]formoterol only on G-protein uncoupling in the presence of 10 μM GTPγS. Data are given as mean ± s.e.m. of 3–4 experiments done in duplicate. The sample size (n) is labeled in the figure.
Based on the crystal structure, AS408 would be expected to interact with the aliphatic tails of membrane lipids. To assess the role of lipids on AS408 binding to the β2AR, we took advantage of the effect of AS408 on the ability of norepinephrine to compete for [3H]dihydroalprenolol (DHAP) to purified β2AR. As shown in Supplementary Fig. 5, increasing concentrations of AS408 leads to a decrease of norepinephrine’s ability to inhibit DHAP binding. The half-maximal effective concentration (EC50) for the effect of AS408 on DHAP binding was similar for β2AR in detergent, phospholipid or phospholipid with cholesterol, suggesting that lipids do not make a specific contribution to AS408 potency (Supplementary Fig. 5).
Receptor subtype selectivity.
We examined the selectivity of AS408 by performing arrestin recruitment assays on 12 family A GPCRs having different extents of sequence identity in the AS408 binding pocket (Supplementary Fig. 6). The β1AR is the only other receptor that has E at position 3.41 and differs from the β2AR only in one amino acid: V3.48 in β2AR and L3.48 in β1AR. This small conservative difference may be responsible for the approximately tenfold lower affinity of AS408 for the β1AR compared to the β2AR (Supplementary Table 4). AS408 is a weak NAM at the α1AR, however, we were unable to fit the data using the same parameters used for the β2AR and β1AR, so it was not possible to estimate the affinity of AS408 for this receptor.
We also observed effects of AS408 on arrestin recruitment by the DRD2 and μOR. In contrast to the β2AR, β1AR and α1AR, the effect of AS408 on the DRD2 and μOR was primarily on agonist potency, with little appreciable effect on agonist efficacy. As a result, we were not able to compare these receptors using the complete operational mode. We therefore compared two models to determine which best fit the data: the Allosteric Ternary Complex model and the Gaddum–Schild model15. As shown in Supplementary Table 5, we obtained a substantially better fit with the Gaddum–Schild model. The Schild slope can be used to determine whether the effect of AS408 on agonist potency is competitive (Schild slope of 1) or noncompetitive (Schild slope of <1)15. For the DRD2, which shares six out of the eight AS408 binding pocket residues with the β2AR, we obtained a Schild slope of 1.1, most consistent with competitive inhibition with an affinity approximately 100-fold lower than for the β2AR. For the μOR, the Schild slope was 1.7, a result that is not consistent with a NAM15. Although substantially different from unity, the analysis suggests a more complex pharmacology than simple competitive inhibition.
E122 mutations affect β2AR function and AS408 activity.
The location of the AS408 binding pocket is of interest given the recent reports of a PAM of GPR40 and NAMs of complement C5a receptor binding to the membrane-facing surface of TMs 2, 3, 4 and 5 (refs.16–18) (Supplementary Fig. 7), and previous mutagenesis studies revealing that several mutations of E1223.41 lead to enhanced β2AR expression and thermostability19. According to our structure, ionic interactions between AS408 in its protonated form and E1223.41 are important. To further characterize the role of E1223.41 on AS408 binding, we examined the effect of mutating E1223.41 to leucine, glutamine and arginine on agonist, antagonist and inverse agonist binding affinity (Supplementary Table 3), and on arrestin recruitment and G-protein activation (Fig. 5). E122Q and E122L expressed at levels comparable to the wild type β2AR, while expression of E122R was greatly reduced (Supplementary Fig. 8a,b). The effect of AS408 on agonist binding affinity for all of the mutants was reduced relative to the wild type receptor, with E122L being most similar to wild type for binding to epinephrine. Both E122Q and E122L exhibited substantial reduction in the allosteric response to AS408 in the arrestin recruitment assay, [35S]GTPγS binding and cAMP accumulation (Fig. 5a–k). Of interest, the effect of removing the nitrogen on AS408 (DD288 in Fig. 3) had an effect on wild type β2AR, similar to the effect of AS408 on E122L (Fig. 5c). We were unable to detect any agonist-stimulated arrestin recruitment and only weak agonist-stimulated [35S]GTPγS binding and cAMP accumulation for E122R. When we examined basal cAMP in cells expressing different levels of E122R, we were able to observe high levels of basal activity relative to wild type that could not be suppressed by the inverse agonist ICI-118,551 (Fig. 5l). We confirmed E122R was expressed on the cell membrane using immunofluorescence confocal microscopy (Supplementary Fig. 8c). Thus, in E122R the G-protein coupling interface is in a constitutively active conformation, but is largely uncoupled from the orthosteric binding pocket, except for high concentrations of norepinephrine. Similar phenomenon was reported to other mutations that affect the PIF motif20. These results reflect the importance of this allosteric hub in β2AR signaling.
Fig. 5 |. AS408 uses E1223.41 of β2AR, a residue that participates in an allosteric network.
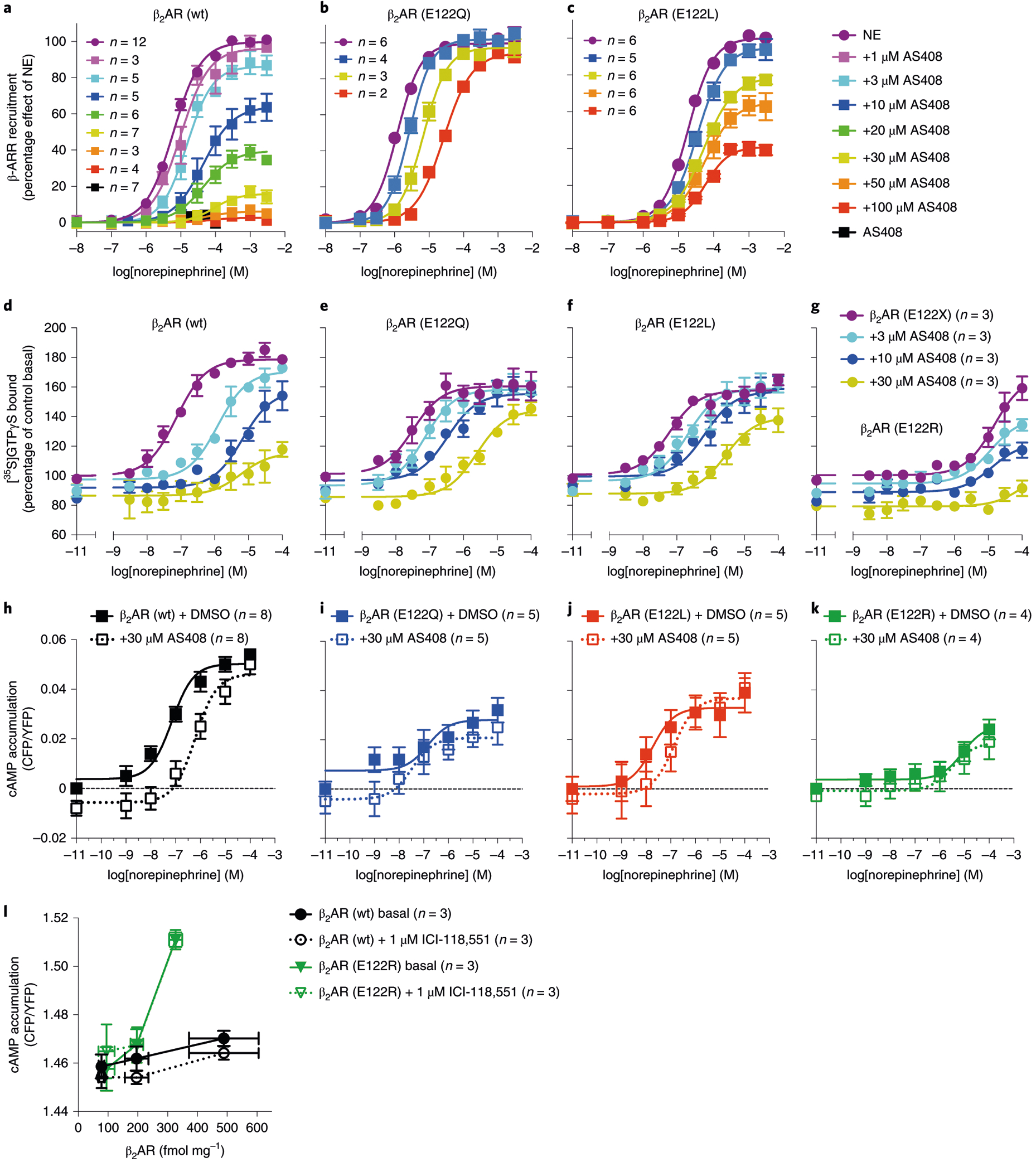
a–k, NAM activity of AS408: β2AR (wt) (a,d,h), is diminished in E122Q (b,e,i), E122L (c,f,j) and in E122R (g,k), in norepinephrine-stimulated β-arrestin 2 recruitment (HEK293 cells), [35S]GTPγS binding (Sf9 cells) (a–c) and cAMP accumulation (HEK293 cells) (d–g), compared to β2AR (wt) (h–k). l, β2AR (E122R) displayed a higher basal activity but was unresponsive to inverse agonist ICI-118,551 (CHO cells). Expression of β2AR (wt) and E122R in l were measured by [3H]DHAP binding assays. [35S]GTPγS binding assays were performed on membranes prepared from Sf9 cells coexpressing β2AR (or mutant) and Gsαβγ. Data are derived from 2–12 experiments done in duplicate and are given as mean ± s.e.m. if n ≥ 3, or as mean if n = 2. The sample size (n) is labeled in the figure.
To understand the structural basis for the functional properties of the mutants E122Q and E122R, we performed 16 μs all-atom molecular dynamics simulations of the mutants and wild type (E1223.41) β2AR. As noted above, there is evidence for a water-mediated hydrogen bond network bridging the carboxylic acid function of E1223.41 in TM3 with the backbone carbonyl oxygen of V2065.45 and S2075.46 in TM5. The E122Q and E122R mutants were modeled based on this structure. For β2AR wild type (E1223.41), the mediating water shows a very low r.m.s.d. value and the previously described interactions were maintained throughout the whole simulation (Supplementary Fig. 9a,d). The water-mediated hydrogen bond network was also observed for E122Q. However, higher r.m.s.d. values of the mediating water molecule and a reduced interaction frequency with the carbonyl oxygen of S2075.46 (97% at E122 and 85% at Q122) indicate a less stable inactive state (Supplementary Fig. 9b,e), which might explain the higher agonist binding affinity for E122Q. The loss of an allosteric effect of AS408 in the E122Q mutant can be explained by the absence of proton-donating properties of the amide group of glutamine. This results in a less stable hydrogen bond network, because an interaction of E122Q with AS408 in both its neutral and cationic form is energetically less favorable than the ionic interaction with the carboxylate anion of E1223.41 in wild type β2AR.
The E122R mutant has dramatically reduced agonist-stimulated arrestin recruitment and G-protein activation, but has high basal activity in a cAMP assay (Fig. 5g,k,l). The longer cationic side chain of E122R is expected to directly interact with the V2065.45 backbone oxygen (Supplementary Fig. 9c,f). Our MD simulations displayed a conformation of E122R that confers a stable ion-dipole interaction with the backbone oxygen of V2065.45. On the course of the simulations the arginine head group loses the contact to the backbone oxygen of S2075.46 potentially destabilizing the inactive state. As a consequence, the side chain of S2075.46 may contribute to an active-like conformation of TM5 explaining the increased basal activity of the β2AR-E122R mutant and its inability to respond to the inverse agonist ICI-118,551.
MD simulations were performed to investigate the capacity of AS408 to negatively modulate norepinephrine activation of the E122L and E122R mutants. According to its low basicity (pKa 5.2), AS408 will primarily exist as a free base in the neutral or even basic environment of the E122L and E122R mutations, respectively. In the simulations of AS408 at the E122R and E122L mutants (3.5 and 4 μs, respectively), AS408 still binds to the allosteric site indicating that negative allosteric modulation is possible. Most observed states have a hydrogen bond of AS408 to the backbone oxygen of V2065.45 in common (Supplementary Fig. 10). Three distinct conformations and a higher fluctuation between the structural states are observed. This indicates a less stable interaction with the mutants compared to β2AR wild type and explains a less pronounced allosteric modulation of norepinephrine binding.
Discussion
We present the structure of the β2AR bound to AS408, a newly discovered NAM. The crystal structure together with molecular dynamics simulations provides insights into the mechanism by which AS408 acts as a NAM for the β2AR. The AS408 binding site is composed of lipid bilayer facing residues in TM3 and TM5. This site is located adjacent to a conformation hub composed of P2115.50, I1213.40 and F2826.44, which undergo packing rearrangements on receptor activation. The AS408 binding pocket includes only one polar amino acid, E1223.41, which forms a hydrogen bond with the primary amine of AS408. Mutations of E1223.41 affect the activity of AS408 and the function of the β2AR. Notably, the E122R mutation uncouples the extracellular orthosteric pocket from the intracellular G-protein coupling interface, highlighting the importance of the conformational hub in regulating GPCR function.
The binding site of AS408 is distinct from binding sites of two other allosteric modulators recently described for the β2AR. Cmpd-15, another NAM for G-protein activation and arrestin recruitment, binds to a pocket formed by the cytoplasmic ends of TMs 1, 2, 6 and 7 (refs.6,8). By binding to this pocket Cmpd-15 directly blocks access to Gs, and likely blocks access to arrestins. Moreover, through polar interactions with TM6, Cmpd-15 stabilizes TM6 in an inactive conformation resulting in reduced affinity for agonists and enhanced affinity for inverse agonists. Cmpd-6 is a PAM for both G-protein activation and arrestin recruitment that binds to a pocket formed by ICL2 and the cytoplasmic ends of TMs 3 and 4 (ref.9). In the inactive structures of the β2AR, ICL2 is an unstructured loop, while in the active state structures, ICL2 is a helix that positions F139 for engagement with Gs. This interaction has been shown to be essential for initiating guanosine diphosphate release21. Cmpd-6 stabilizes ICL2 as a helix.
A key feature of these three allosteric modulators for the β2AR is the ability to penetrate the cell membrane. Allosteric modulators that bind intracellularly or within the membrane have recently been reported for several other Family A and Family B receptors22,23. Cmpd-15 and Cmpd-6 must cross the plasma membrane to gain access to their binding pockets. When bound, Cmpd-15 has no direct interactions with the lipid bilayer, Cmpd-6 binds at the interface of the cytosol and the inner leaflet of the bilayer, and AS408 is completely buried within the bilayer. While, allosteric modulators that bind to cytosolic or membrane imbedded sites have the potential for enhanced selectivity and the ability to modulate responses to natural hormones and neurotransmitters, they will likely have more complex pharmacodynamic and pharmacokinetic profiles compared to polar drugs that can only access the extracellular surface.
Methods
Molecular dynamics simulations.
Simulations of AS408 at β2AR wild type and the mutants E122L and E122R were based on the crystal structure reported in this manuscript. Coordinates were prepared by removing the T4L fragment and crystal water associated with T4L. The two cholesterol molecules, alprenolol, AS408, and crystal water close to the receptor were retained. Mutations were introduced using UCSF Chimera24 applying the rotamer with the highest probability and not producing clashed to the receptor or AS408. Simulations of β2AR wild type (E1223.41) and the mutants E122Q and E122R without AS408 were based on the β2AR crystal structure in complex with alprenolol (PDB 3NYA) and were prepared in the same manner. The E122Q mutant was introduced that its side chain enables the formation of the water network observed for β2AR wild type. The E122R mutation was prepared that the arginine side chain interacts with V2065.45 and S2075.46. UCSF Chimera24 was used to model missing side chains. Hydrogens were added and the protein chain termini were capped with acetyl and methylamide. Except for the neutral E1223.41 in the β2AR wild type simulation with a mediating water molecule or AS408 in its neutral state, all titratable residues were left in their dominant protonation state at pH 7.0.
Alprenolol has been used in its protonated form. AS408 was used in its neutral state, protonated at the primary amine or protonated at either of its endocyclic nitrogens. The protonation of AS408 at the endocyclic nitrogen were supported by pKa calculations using Jaguar pKa (Schrödinger release v.2018–1, Jaguar pKa)25–27 indicating significantly higher basicity for the endocyclic nitrogens.
The protein structures were aligned to orientation of proteins in membranes28 structure of β2AR (PDB 4GBR). Each complex was inserted into a pre-equilibrated membrane of dioleoyl-phosphatidylcholine lipids by means of the GROMACS tool g_membed29. Subsequently, water molecules were replaced by sodium and chlorine ions to give a neutral system with a concentration of 0.15 M of NaCl. The system dimensions were roughly 80 × 80 × 100 Å3, containing 156 lipids 58 sodium ions, 66 chlorine ions (67 in E122R systems) and about 13,000 water molecules.
Parameter topology and coordinate files were built up using the tleap (AMBER 2017, Case et al.)30 and subsequently converted into GROMACS input files. For all simulations, the general AMBER force field31 was used for alprenolol, AS408 and cholesterol, the lipid14 force field32 for dioleoyl-phosphatidylcholine molecules and ff14SB33 for protein residues. The SPC/E water model34 was applied. Parameters for ligands were assigned using antechamber (AMBER 2017, Case et al.)30. Structures of the ligands were optimized (Gaussian v.09, revision B.01, Frisch et al.)35 at the B3LYP/6–31G(d) level and charges were calculated at HF/6–31G(d) level and partial charges assigned by the RESP procedure according to the literature36. A formal charge of +1 was defined for alprenolol. AS408 exhibited a formal charge of 0 or +1 depending on the selected protonation state.
Simulations were performed using GROMACS v.5.1.3 (refs.37,38). The simulation systems were energy minimized and equilibrated in the NVT ensemble at 310 K for 1 ns followed by the NPT ensemble for 1 ns with harmonic restraints of 10.0 kcal mol−1 on protein and ligands. In the NVT ensemble the V-rescale thermostat was used. In the NPT ensemble the Berendsen barostat and a surface tension of 22 dyn cm−1 and a compressibility of 4.5 × 10−5 bar−1 was applied. The system was further equilibrated for 2 ns with restraints on protein backbone and ligands and additional 16 ns without restraints. Multiple simulations were started from the final snapshot of the equilibration resulting in productive molecular dynamics simulation runs of 2–4 μs. Simulations were performed using periodic boundary conditions and time step of 2 fs with bonds involving hydrogen constrained using LINCS39. Long-range electrostatic interactions were computed using particle mesh Ewald (PME)40 method with interpolation of order 4 and fast Fourier transform grid spacing of 1.6 Å. Nonbonded interactions were cut off at 12.0 Å.
The analysis of the trajectories was performed using the CPPTRAJ module of AMBER16 (AMBER 2017, Case et al.)30 and visualization was performed using the PyMOL Molecular Graphics System, v.2.1.1 (Schrödinger, LLC). Distance and r.m.s.d. were plotted using Matplotlib, v.2.2.2 (ref.41).
Determination of pKa.
Acid–base potentiometric titration was performed by stepwise addition of the titrant with an automatic burette. The titration was conducted in KCl (0.15 M) at 23 °C using Titrisol HCl (0.1 M) as a titrant. AS408 solution was prepared by dissolving AS408 (0.084 mmol) in 7.0 ml of 72% aqueous dimethylsulfoxide solution, at I = 0.15 M ionic strength using KCl. The acid dissociation constant was determined by titrating AS408 (7.0 ml in 72% aqueous dimethylsulfoxide solution) with HCl solution (0.1 M) to pH 2.81. A pKa value of 5.2 for AS408 was determined using a simplified Henderson–Hasselbalch equation.
Determination of logP.
The logP values for BRAC1 and its analogs were calculated using Maestro software (Schrödinger Release v.2018–1: Maestro, Schrödinger)42.
Protein expression and purification.
A previously reported β2AR-T4L43 construct was cloned into pFastbac vector and fusion protein was expressed in Sf9 cells using the Bac-to-Bac baculovirus expression system. Cells were infected with high dose baculovirus at density of around 4 × 106 cells per ml and gathered at 48 h after infection. 10 μM alprenolol was added to enhance expression. β2AR-T4L was extracted from cell membrane with DDM buffer and was purified in the same way as previously described44, using a first M1 Flag affinity column, followed by alprenolol-Sepharose chromatography44 and a second M1-Flag affinity column. Then, 100 μM alprenolol was added to the all the buffers used in the second M1 chromatography, during which detergent was exchanged from 0.1% DDM to 0.01% MNG. The purified β2AR-T4L was dialyzed against dialysis buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 0.003% MNG, 0.0003% CHS, 100 μM alprenolol) overnight at 4 °C. PNGase F was added to remove N-linked sugars. The protein was concentrated to ~50 mg ml−1 with a 50 kDa cutoff Amicon centrifugal filters (Millipore). If not used immediately, the protein was flash frozen with liquid nitrogen and stored at −80 °C.
Crystallization.
LCP crystallizations of β2AR-T4L in complex with alprenolol and AS408 were performed using a LCP crystallization robot (Gryphon, Art Robbins Instruments). In brief, protein solution was mixed with 9:1 (w/w) monoolein:cholesterol (Sigma) with protein to lipid ratio of 2:3 (w/w) and reconstituted into LCP using two-syringe method45. Then, 96-well glass sandwich plates were filled with 30 nl LCP overlaid with 1 μl precipitant solution and incubated at 20 °C. The best crystals were grown in conditions containing 0.1 M Tris-HCl, pH 8.0, 30–40% PEG400, 300–400 mM sodium formate, 6% 1,4-butanediol, 1 mM alprenolol, 1 mM AS408 and 1% DMSO.
Data collection and structure determination.
X-ray diffraction data were collected at beamline BL32XU at Spring-8, Japan. Typically wedges of 5–10° were collected for each crystal using a 10 × 10 μm2 beam. Diffraction data were processed using XDS46. A full 3.1 Å native dataset was obtained by merging data from 81 crystals collected at a 1.0 Å wavelength. While a full 4.0 Å anomalous dataset was obtained by merging data from 284 crystals collected at 0.91 Å wavelength. Crystal structure was solved by molecular replacement using high-resolution β2AR-T4L structure (PDB 2RH1) as searching model (Supplementary Table 1). The allosteric modulator AS408 was manually fit into the Fo–Fc electron density maps in Coot47. Structure refinement was performed with phenix.refine48. The occupancy of AS408 was refined as a single group, with a final value of 79%. The binding mode of AS408 was confirmed by a Fo–Fc simulated annealing omit map with starting temperature of 3,000 K. The anomalous peak for bromine was calculated using fast Fourier transform49 in ccp4 (ref.50). The final model was validated using Molprobity51. The overall Molprobity score was 1.30. In Ramachadran analysis, 98.9% of atoms were in favored regions while 1.1% of atoms were in allowed region. Data processing statistics and structural refinement statistics were shown in Supplementary Table 1. Structure figures were prepared using PyMOL Molecular Graphics System (Schrödinger, LLC).
β-arrestin-2 recruitment assay.
Determination of β-arrestin-2 recruitment was performed applying the PathHunter assay (DiscoverX), which is based on fragment complementation of β-galactosidase in human embryonic kidney 293 (HEK293) cells stably expressing (EA)-β-arrestin-2 and being transiently transfected with a receptor tagged to the PK fragment as described52. In general, cells were transfected employing Mirus TransIT-293 (peqlab) and incubated in DMEM/F12 medium (Life Technologies) at 37 °C and 5% of CO2. After 24 h, cells were detached with Versene (Life Technologies) and transferred into 384-well plates (white plate, transparent bottom, Greiner Bio-One) at a density of 5,000 cells per well using the medium CP7 Reagent (DiscoverX). After further 24 h of incubation, test compounds dissolved in PBS were added to the cells at a final volume of 25 μl and incubated at 37 °C for a distinct time that was optimized for each receptor. Determination of β-arrestin-2 recruitment was started by adding detection mix, incubation at room temperature for 60 min and measuring chemoluminescence with a Clariostar plate reader (BMG). For measuring allosteric effects, the modulator was preincubated with the cells at a distinct concentration for 30 min followed by the addition of reference agonist. Data analysis of functional experiments were performed by normalizing the raw data relative to basal activity (0%) and the maximum effect of the reference agonist (100%). Normalized curves of 3–7 individual experiments, each done as duplicate, were analyzed by nonlinear regression applying the algorithms in GraphPad Prism 6.0 (GraphPad) to get dose–response curves representing the average EC50 and Emax values.
Radioligand binding assays.
To determine the allosteric effect of AS408 on orthosteric ligand binding membranes prepared from Sf9 cells expressing β2AR or mutants alone or coinfected with Gsαβγ, were tested for their capacity to modulate [3H]DHAP binding, as described53. Typically, β2AR membranes (1–10 μg) were incubated for 3 h in binding buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 1 mM ascorbic acid) with 0.2 nM [3H]DHAP along with varying concentrations of orthosteric ligand in the absence or presence of varying concentration of AS408 (or with 50 μM propranolol to determine nonspecific binding). To test the capacity of AS408 to accelerate the dissociation of agonist [3H]formoterol, β2AR membranes (with Gsαβγ) were preincubated in binding buffer with 2 nM [3H]formoterol, 10 mM MgCl2, 10 μM GTPγS for 60 min at room temperature. Dissociation was initiated by dilution with the assay buffer containing 200 μM propranolol in the absence or presence of AS408 marking t = 0 min. Samples were taken at varying time points, filtered and washed as described below to remove free [3H]formoterol. For [3H]DHAP saturation isotherms of β2AR and mutants, membranes were incubated with varying concentrations of [3H]DHAP and filtered using rapid filtration through GF/B membranes and rinsed with ice-cold binding buffer to remove free [3H]probe. Filter plates were dried before adding Microscint 0 and counting bound [3H]probe using a Packard TopCount. All data were analyzed using GraphPad Prism 6.0 (GraphPad). Determination of Ki for antagonists and agonists in uncoupled preparations of β2AR were determined using nonlinear regression analysis using single-site fits whereas G protein-coupled Khigh and Klow estimates were determine using two-site models.
Purified β2AR in DDM or phospholipid.
Purified β2AR was reconstituted into high-density lipoprotein particles (rHDL or nanodiscs) comprising apolipoprotein A1 and a 3:2 (mol:mol) mixture of POPC:POPG lipid or a 3:2:1.25 (mol:mol:mol) mixture of POPC:POPG:cholesterol lipid54. Another sample was prepared by incubating M1-FLAG affinity resin (Sigma) with purified β2AR in DDM buffer. In total, three samples were prepared, which were β2AR in POPC/POPG high-density lipoprotein particles, β2AR in POPC/POPG/cholesterol high-density lipoprotein particles and M1 resin bound β2AR in DDM buffer. Radioligand binding assays were performed to all these three samples. Binding reactions were 500 μl in volume, containing 100 fmol functional receptor, 2 nM [3H]DHAP, 100 mM NaCl, 20 mM Tris pH 7.5, 1 mM CaCl2, 0.2% bovine serum albumin and various concentration of AS408 as indicated. Then, 0.02% DDM was added in reactions for M1 resin bound β2AR samples. Reactions were mixed and incubated for 2 h at room temperature before collection using a Brandel 48-well harvester by filtering onto a filter paper pretreated with 0.3% polyethylenimine. Radioactivity was measured by liquid scintillation counting. All experiments were triplicated and presented as means ± s.e.m.
[35S]GTPγS binding assay.
Membranes were prepared from High Five (Invitrogen) or Sf9 cells expressing β2AR or mutants and Gsαβγ. Typically, membranes (2–5 μg) were pretreated with guanosine diphosphate (final assay concentration of 10 μM) in assay buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2 and 1 mM ascorbic acid) and different concentrations of AS408 for 20 min at room temperature before adding [35S]GTPγS (for a final concentration of 0.1 nM) with a range of concentrations of agonist (epinephrine or norepinephrine). For most cases the assays were incubated at room temperature for a period of 1 h before stopping by rapid filtration through GF/B membranes and washing with ice-cold assay buffer. To determine the KB for AS408 on β2AR and mutants, assay times were reduced to 10 min at 30 °C, to avoid saturating [35S]GTPγS binding to Gs. Assays were performed in a 96-well microplate format and radioactivity was measured using a TopCount (Packard). All data were analyzed using GraphPad Prism 6.0 (GraphPad).
cAMP accumulation assays.
Intact cell cAMP accumulation was measured using the FRET-epac sensor55 in stable HEK293-Epac cells endogenously expressing β2AR or in Chinese hamster ovary (CHO) cells cotransfected with Epac and β2AR or the mutants (HEK293-Epac cells were generously provided by J. Mathiesen). Cells were collected with lifting buffer (20 mM HEPES, pH 7.4, 150 mM NaCl and 0.68 mM EDTA), centrifuged and resuspended in HBSS–HEPES (Hank’s Balanced Salt Solution plus 20 mM HEPES, pH 7.4) containing 0–150 μM modulator or vehicle (for a final assay concentration of 0–100 μM). This cell suspension (100 μl) was pipetted into the wells of a 96-well plate (black with clear bottom). After 20 min in the dark at 37 °C, 50 μl of HBSS–HEPES buffer at 37 °C containing IBMX (1 mM final), ascorbic acid (1 mM final) and norepinephrine or epinephrine (0–100 μM final) was added. Measurement of the basal activity and the effects of inverse agonist ICI-118,551 on β2AR (wt) and E122R on cAMP accumulation and binding assays were performed on CHO cells, where endogenous β2AR signaling is undetectable. The CFP/YFP ratio of the Epac-cAMP FRET sensor was immediately measured for 15 min using wavelengths of 435 nm for excitation with 485 and 530 nm for emission using a SpectraMax M5 (Molecular Devices). The CFP/YFP ratio area under the curve for 10 min was used to determine maximal agonist-stimulated cAMP accumulation and EC50 using GraphPad Prism 6.0 (GraphPad).
Assessment of cooperativity.
To assess the allosteric properties of AS408 on norepinephrine-stimulated [35S]GTPγS binding, cAMP accumulation and β-arrestin recruitment, we used the complete operational model of allostery13,14 using GraphPad Prism 6.0. In these assays dose–response curves for norepinephrine in the presence of at least three different concentrations of AS408 (1, 3, 10 and 30 μM) were generated. For the analysis we held the basal and maximal activities constant within each signaling assay and β values and τB were held constant at near zero (the value of 10−100 was used). Norepinephrine dose–response curves suggest that β values and τB should be near zero because AS408 is a NAM for agonists and an inverse agonist in cAMP and GTPγS binding assays. When screening a panel of GPCRs to determine AS408 specificity for the β2AR, we encountered results that could not be fit using the complete operational model of allostery. To do determine the mode of inhibition, the data were analyzed comparing the Gaddum–Schild model versus the allosteric ternary complex model15 using GraphPad Prism 6.0 (GraphPad). Variable were not fixed and were shared among data sets within each receptor analyzed. An F-test was performed (P < 0.05) to ascertain the preferred model. Using the preferred model (Gaddum–Schild) the EC50 values for the agonist, KB for AS408 and the Schild slope were calculated.
Immunofluorescence confocal microscopy.
HEK293T cells were plated on polylysine coat coverslips in six-well tissue culture plate and transfected with 1.5 μg β2AR-wt or β2AR-E122R DNA using the transporter 5 transfection reagent (Polysciences, Inc.). After 24 h of cell culture, the cells with high confluence were washed three times with PBS, fixed in 1% PFA in PBS for 15 min at room temperature, and blocked with the blocking solution (3% BSA/3% normal goat serum) for 30 min at room temperature. Then the cells were incubated with primary antibody (M1 mouse anti-FLAG antibody, 1:500 in blocking solution) overnight at 4 °C. After three times washing with PBS, the secondary antibody (Alexa 488 goat anti-mouse antibody, 1:500 dilution in blocking solution) was added and incubated for 1 h at room temperature. After washing again three times with PBS, the coverslips were mounted on slides with Prolong Gold Anti-fade Mountant (ThermoFisher) and allowed to dry before imaging with the confocal microscope.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Atomic coordinates and structure factors have been deposited in the PDB under accession code 6OBA.
Supplementary Material
Acknowledgements
We acknowledge support from the US NIH grant nos. GM106990 (B.K.S., P.G. and R.K.S.), and the DFG Grants Gm 13/10 and GRK 1910 (P.G.), and Beijing Advanced Innovation Center for Structural Biology, Tsinghua University (X.L.), as well as the compute resources provided by the RRZE. B.K.K. is a Chan Zuckerberg Biohub Investigator. We thank D. Steffen and M. Gilardi for help with confocal microscopy. We also thank A. Christopoulos for his critical insight with the analysis of cooperativity.
Footnotes
Competing interests
B.K.K. is a cofounder of and consultant for ConfometRx, Inc.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-020-0549-2.
Supplementary information is available for this paper at https://doi.org/10.1038/s41589-020-0549-2.
References
- 1.Kruse AC et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Digby GJ, Shirey JK & Conn PJ Allosteric activators of muscarinic receptors as novel approaches for treatment of CNS disorders. Mol. Biosyst 6, 1345–1354 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohr K, Trankle C & Holzgrabe U Structure/activity relationships of M2 muscarinic allosteric modulators. Recept. Channels 9, 229–240 (2003). [PubMed] [Google Scholar]
- 4.Wootten D, Christopoulos A & Sexton PM Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug Discov 12, 630–644 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Thal DM, Glukhova A, Sexton PM & Christopoulos A Structural insights into G-protein-coupled receptor allostery. Nature 559, 45–53 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Ahn S et al. Allosteric ‘beta-blocker’ isolated from a DNA-encoded small molecule library. Proc. Natl Acad. Sci. USA 114, 1708–1713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn S et al. Small-molecule positive allosteric modulators of the beta2-adrenoceptor isolated from DNA-encoded libraries. Mol. Pharmacol 94, 850–861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X et al. Mechanism of intracellular allosteric beta2AR antagonist revealed by X-ray crystal structure. Nature 548, 480–484 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu X et al. Mechanism of beta2AR regulation by an intracellular positive allosteric modulator. Science 364, 1283–1287 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasmussen SG et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masureel M et al. Structural insights into binding specificity, efficacy and bias of a beta2AR partial agonist. Nat. Chem. Biol 14, 1059–1066 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ballesteros JA & Weinstein H in Methods in Neurosciences Vol. 25 (ed. Sealfon SC) 366–428 (Academic Press, 1995). [Google Scholar]
- 13.Leach K, Sexton PM & Christopoulos A Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci 28, 382–389 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Aurelio L et al. Allosteric modulators of the adenosine A1 receptor: synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes. J. Med. Chem 52, 4543–4547 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Christopoulos A & Kenakin T G protein-coupled receptor allosterism and complexing. Pharmacol. Rev 54, 323–374 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Srivastava A et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 513, 124–127 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Robertson N et al. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 553, 111–114 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Liu H et al. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol 25, 472–481 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Roth CB, Hanson MA & Stevens RC Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu122(3.41), a critical residue in GPCR structure. J. Mol. Biol 376, 1305–1319 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schonegge AM et al. Evolutionary action and structural basis of the allosteric switch controlling beta2AR functional selectivity. Nat. Commun 8, 2169(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du Y et al. Assembly of a GPCR-G protein complex. Cell 177, e1211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szlenk CT, Gc JB & Natesan S Does the lipid bilayer orchestrate access and binding of ligands to transmembrane orthosteric/allosteric sites of G protein-coupled receptors? Mol. Pharmacol 96, 527–541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu S & Zhang J Small molecule allosteric modulators of G-protein-coupled receptors: drug-target interactions. J. Med. Chem 62, 24–45 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Pettersen EF et al. UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Klicić JJ, Friesner RA, Liu S-Y & Guida WC Accurate prediction of acidity constants in aqueous solution via density functional theory and self-consistent reaction field methods. J. Phys. Chem. A 106, 1327–1335 (2002). [Google Scholar]
- 26.Bochevarov AD, Watson MA, Greenwood JR & Philipp DM Multiconformation, density functional theory-based pKa prediction in application to large, flexible organic molecules with diverse functional groups. J. Chem. Theory Comput 12, 6001–6019 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Yu HS, Watson MA & Bochevarov AD Weighted averaging scheme and local atomic descriptor for pKa prediction based on density functional theory. J. Chem. Inf. Model 58, 271–286 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Lomize MA, Lomize AL, Pogozheva ID & Mosberg HI OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Wolf MG, Hoefling M, Aponte-Santamaria C, Grubmuller H & Groenhof G g_membed: efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem 31, 2169–2174 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Case DA et al. AMBER 2017 (University of California, San Francisco, 2017). [Google Scholar]
- 31.Wang J, Wolf RM, Caldwell JW, Kollman PA & Case DA Development and testing of a general amber force field. J. Comput. Chem 25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Dickson CJ et al. Lipid14: the Amber lipid force field. J. Chem. Theory Comput 10, 865–879 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maier JA et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berendsen HJC, Grigera JR & Straatsma TP The missing term in effective pair potentials. J. Phys. Chem 91, 6269–6271 (1987). [Google Scholar]
- 35.Frisch MJ et al. Gaussian 09, revision B.01 (Gaussian, Inc, Wallingford, CT, 2009). [Google Scholar]
- 36.Bayly CI, Cieplak P, Cornell WD & Kollman PA A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—the Resp model. J. Phys. Chem 97, 10269–10280 (1993). [Google Scholar]
- 37.Van Der Spoel D et al. GROMACS: fast, flexible, and free. J. Comput. Chem 26, 1701–1718 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Abraham MJ et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015). [Google Scholar]
- 39.Hess B, Bekker H, Berendsen HJC & Fraaije JGEM LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem 18, 1463–1472 (1997). [Google Scholar]
- 40.Darden T, York D & Pedersen L Particle mesh Ewald—an n.log(n) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092 (1993). [Google Scholar]
- 41.Hunter JD Matplotlib: a 2D graphics environment. Comput. Sci. Eng 9, 90–95 (2007). [Google Scholar]
- 42.Schrödinger Release 2018–1: Maestro, Schrödinger, LLC, New York, NY, 2018. [Google Scholar]
- 43.Rosenbaum DM et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266–1273 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Kobilka BK Amino and carboxyl terminal modifications to facilitate the production and purification of a G protein-coupled receptor. Anal. Biochem 231, 269–271(1995). [DOI] [PubMed] [Google Scholar]
- 45.Caffrey M & Cherezov V Crystallizing membrane proteins using lipidic mesophases. Nat. Protocol 4, 706–731 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kabsch W XDS. Acta Crystallogr. Sect. D 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. Sect. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ten Eyck LF Fast Fourier transform calculation of electron density maps. Methods Enzymol. 115, 324–337 (1985). [DOI] [PubMed] [Google Scholar]
- 50.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hubner H et al. Structure-guided development of heterodimer-selective GPCR ligands. Nat. Commun 7, 12298 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeVree BT et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 535, 182–186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whorton MR et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proceed. Natl Acad. Sci. USA 104, 7682–7687 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vedel L, Brauner-Osborne H & Mathiesen JM A cAMP biosensor-based high-throughput screening assay for identification of Gs-coupled GPCR ligands and phosphodiesterase inhibitors. J. Biomol. Screen 20, 849–857 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors have been deposited in the PDB under accession code 6OBA.