

Abstract
Introduction
This study assessed the hypothesis that circulating human amylin (amyloid‐forming) cross‐seeds with amyloid beta (Aβ) in early Alzheimer's disease (AD).
Methods
Evidence of amylin‐AD pathology interaction was tested in brains of 31 familial AD mutation carriers and 20 cognitively unaffected individuals, in cerebrospinal fluid (CSF) (98 diseased and 117 control samples) and in genetic databases. For functional testing, we genetically manipulated amylin secretion in APP/PS1 and non‐APP/PS1 rats.
Results
Amylin‐Aβ cross‐seeding was identified in AD brains. High CSF amylin levels were associated with decreased CSF Aβ42 concentrations. AD risk and amylin gene are not correlated. Suppressed amylin secretion protected APP/PS1 rats against AD‐associated effects. In contrast, hypersecretion or intravenous injection of human amylin in APP/PS1 rats exacerbated AD‐like pathology through disruption of CSF‐brain Aβ exchange and amylin‐Aβ cross‐seeding.
Discussion
These findings strengthened the hypothesis of circulating amylin‐AD interaction and suggest that modulation of blood amylin levels may alter Aβ‐related pathology/symptoms.
Keywords: amylin, amyloid, familial Alzheimer's disease, islet amyloid polypeptide, sporadic Alzheimer's disease
1. INTRODUCTION
Disease course‐modifying therapeutic targets remain the highest unmet needs in Alzheimer's disease (AD), 1 a brain disorder that afflicts more than 5.7 million Americans of all ages, a number projected to triple by 2050. Given that deterioration of cognition and cerebral accumulation of amyloid beta (Aβ) and tau tangles are characteristics of AD dementia, in brain imaging and at autopsy, 2 current therapeutic strategies are aimed at interrupting cerebral Aβ plaque formation. Alternative approaches may involve modulation of blood levels of proteins that cross‐seed with Aβ. Recent data from multiple research teams 3 , 4 , 5 , 6 , 7 , 8 suggest that one potential modifier of AD pathology may be circulating amylin, an amyloidogenic pancreatic hormone. 9
The pancreatic hormone amylin (or islet amyloid polypeptide [IAPP]) normally participates in the regulation of satiation through signaling pathways within the hypothalamus. 10 Recent analyses of plasma data from Alzheimer's Disease Neuroimaging Initiative and in serum data from the Texas Alzheimer's Research and Care Consortium identified circulating pancreatic amylin as a mediator of age‐specific effects on dementia severity. 8 Brain parenchyma and microvasculature in individuals with late‐onset sporadic AD (sAD) have amylin deposits and mixed amylin–Aβ plaques. 3 , 4 , 5 , 6 , 7 Aggregated amylin with similar molecular weights were identified in matched plasma and brain tissues from humans with sAD 3 suggesting that toxic amylin species include circulating aggregated amylin.
Because amylin is co‐secreted with insulin, 11 conditions with insulin dysregulation, such as insulin resistance and type 2 diabetes, may increase the amount of amylin crossing from blood to the central nervous system. High circulating amylin levels in plasma and cerebrospinal fluid (CSF) 12 could increase the risk for AD. Even if amylin secretion is normal in AD, amylin may be harmful to the brain by interacting with Aβ pathology. Importantly, insulin secretion is to some extent modifiable, and therapeutic agents to block amyloidogenicity of circulating amylin 13 may be a means of reducing amylin–Aβ cross‐seeding.
Because individuals with early‐onset familial forms of AD will develop AD pathology, 14 evidence of amylin–Aβ cross‐seeding in the brains of persons with familial AD (fAD) could strengthen the hypothesis of a role of circulating amylin in AD pathology. We therefore analyzed brains of individuals inheriting mutations in the amyloid precursor protein (APP) and presenilin 1 (PS1) for immunochemical evidence of amylin interaction with areas of AD‐related histopathology (Aβ andtau deposits). To test whether circulating amylin is involved in early AD pathological processes, we analyzed the amylin–Aβ42 relationship in CSF from humans with AD pathologic change and a diagnosis of mild cognitive impairment (MCI) or dementia, and in CSF from cognitively unimpaired (CU) individuals. To directly address the potential for modulation of circulating amylin levels as means to alter AD‐related pathology/symptoms, we manipulated the pancreatic secretion of amylin through gene suppression and overexpression in APP/PS1 rats and infused human amylin intravenously in APP/PS1 rats. Our work provides a basis for additional studies to validate circulating amylin as a potential therapeutic target for AD.
2. METHODS
Detailed methods are included in Appendix A in supporting information.
2.1. Human studies
Institutional review board approval and informed consent were obtained prospectively at the institutions participating in this study. Formalin fixed and frozen temporal cortex tissue from 31 fAD mutation carriers were provided by the Queen Square Brain Bank for Neurological Disorders at UCL Queen Square Institute of Neurology (United Kingdom) and King's College London (United Kingdom). Both formalin fixed and frozen tissue was available for 14 brains. Brain tissue samples from 20 age‐matched CU individuals were obtained from the AD Center at the University of Kentucky (USA). AD Centers at the University of Kentucky, University of Washington (USA), Wake Forest University (USA), and University of Gothenburg (Sweden) provided the CSF samples for this study (98 diseased and 117 control samples). Details on patients and sample size for each analysis are summarized in Appendix B, Tables B.1‐B.3 in supporting information.
2.1.1. Immunochemical analyses
Frozen and formalin fixed brain tissues were processed as previously described. 3 , 7 Brain amylin and Aβ concentrations were measured using the enzyme‐linked immunosorbent assay (ELISA) for amylin (EZHA‐52K, Millipore) and Aβ42 (KHB3441, Thermo Fisher). Brain amylin deposition and interaction with AD‐related histopathology were assessed by immunohistochemistry (IHC) and confocal microscopy. For IHC analysis, the immunoreactivity intensity signals of amylin and Aβ were deconvoluted using ImageJ. Details on antibodies, reagents, and image analysis are included in Appendix A.
2.1.2. CSF amylin and CSFAβ42 levels
Observer‐blind measurement of CSF amylin levels was performed by using amylin ELISA (EIA‐AMY; RayBiotech). The results were communicated to the AD Centers providing the samples for assessing the relationships with CSF Aβ42 and cognitive function of the individuals providing the sample. CSF Aβ42 levels were measured with the INNO‐BIA AlzBio3 multiplex assay (FujiRebio) at the AD Centers providing the samples.
2.1.3. Genomic analysis
Databases from the International Genomics of Alzheimer's Project (IGAP; n = 17,008 AD cases and n = 13,154 controls) and Healthy Exosomes (HEX; n = 331 AD cases and n = 468 controls) were used to test the association of common and rare amylin gene variants with the risk for AD.
Research in Context
Systematic review: Although the association between dysregulation of circulating amylin (islet amyloid polypeptide [IAPP]) and Alzheimer's disease (AD) pathology is not yet as widely studied as other aspects of AD biology, accumulating evidence from multiple research teams describe amylin–amyloid beta (Aβ) cross‐seeding in brains of individuals with sporadic AD. One study reported that high blood amylin and high cerebrospinal fluid (CSF) amylin correlate in sporadic AD.
Interpretation of results: Early AD processes may affect the central regulation of pancreatic amylin leading to abnormal circulating amylin and amylin–Aβ cross‐seeding in the brain.
Future directions: This study provides a basis for additional hypothesis‐driven research and evidence‐based validation studies to help understand: the triggers of amylin–Aβ interaction and whether reducing amylin dyshomeostasis could slow the progression of AD, the impact of peripheral amylin dyshomeostasis on Aβ clearance across the blood‐brain barrier, and effects of circulating amylin levels on CSF–brain Aβ exchange in human AD.
2.2. Animal studies
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85‐23, revised 2011) and was approved by the Institutional Animal Care and Use Committees at University of Kentucky.
2.2.1. Human amylin expression in APP/PS1 and non‐APP/PS1 rats
Rats that overexpress (three‐fold) human amylin in the pancreas (HIP rats) and develop brain amylin pathology 6 , 7 , 15 were crossed with APPSwe/PS1ΔE9 rats (TgF344‐19 rats) that develop AD‐like pathology 16 to generate APP/PS1/HIP rats (amylin‐Aβ model rats). Because rodent amylin is non‐amyloidogenic, 9 APP/PS1 littermates served as controls for the amyloidogenic human amylin effects. HIP and wild‐type (WT) rats from the same litters as APP/PS1/HIP and APP/PS1 rats were used to assess the effects of brain amylin pathology, in the absence of APPSwe/PS1ΔE9 mutations. F2 litters were used in experiments. Male rats were used, because amylin dyshomeostasis develops earlier in male (≈12 months) than in female (> 18 months) rats. 7
2.2.2. Suppressed amylin expression in APP/PS1 rats
To test whether suppressing amylin secretion reduces AD‐related neurologic deficits, we crossed APP/PS1 rats with amylin knockout rats (AKO rats) to generate APP/PS1/AKO rats that lack the amylin gene. AKO rats were previously characterized. 7 Rats from the F3 generation were used in these experiments to ensure the homozygosity of AKO genotype in the APP/PS1/AKO model.
2.2.3. Immunochemical detection of human versus rat amylin
Because homozygous HIP and APP/PS1/HIP rats have increased mortality rates, we used heterozygous rats that express both human and rat amylin. ELISA and anti‐amylin antibody sensitivities/affinities are higher for rat versus human amylin (technical evaluation in Appendix A).
3. RESULTS
3.1. Circulating amylin cross‐seeds with Aβ in brains of PS1 and APP mutation carriers
To test the hypothesis (Figure 1A) that amylin secreted from the pancreas interacts with fAD pathology, brain tissue from fAD mutation carriers and CU individuals were analyzed using ELISA, IHC, and confocal microscopy. Amylin ELISA revealed that brains of PS1 mutation carriers had higher amylin levels compared to CU individuals (Figure 1B). Increased brain amylin concentrations were associated with greater Aβ42 levels in PS1 versus CU brains (ELISA; Figure 1C). A nonsignificant increased brain accumulation of amylin was also identified in APP versus CU brains (P = 0.09; Figure 1B), which is similar to the tendency for higher Aβ42 in this group (P = 0.07; Figure 1C). Overall, increased brain amylin levels correlated strongly (R2 = 0.40, P < 0.01) with increased brain Aβ42 levels in fAD (Figure 1D), whereas the amylin–Aβ relationship was variable in CU brains (Figure 1E).
FIGURE 1.
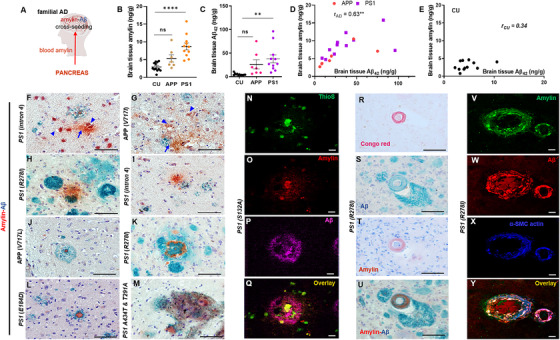
Brain amylin–amyloid beta (Aβ) relationship in PSEN1 and APP mutation carriers. A, Schematic amylin–Aβ cross‐seeding hypothesis. B‐E, Amylin and Aβ42 levels and the relationship between amylin and Aβ42 concentrations in temporal cortex specimens from PS1 and APP mutation carriers (n = 11 and n = 7, respectively) and cognitively unaffected individuals (CU; n = 12). F‐M, Representative images of immunohistochemistry analysis using anti‐amylin (brown) and anti‐Aβ (green) antibodies on temporal cortex slices from familial Alzheimer's disease (fAD) brains. Amylin deposition in neurons (F, G; arrowheads) or forming homogenous plaques (F, G; arrows; H, I) and amylin–Aβ plaques (J‐M) are shown. N‐Q, Confocal microscopic analysis of a fAD brain section triple stained with Thioflavin S (ThioS, green; N), anti‐amylin antibody (red; O), and anti‐Aβ antibody (magenta; P). R‐U, representative images of consecutive fAD brain sections stained with Congo red (R), anti‐Aβ (S), anti‐amylin (T), or a combination of anti‐amylin (brown) and anti‐Aβ (green; U) antibodies. V‐Y, Confocal microscopic analysis of a fAD brain section triple stained with anti‐amylin antibody (green; V), anti‐Aβ antibody (red; W) and anti‐α smooth muscle cell (SMC) actin antibody (blue; X). Type of PS1 or APP mutation is labeled on each image; three sections/brain, n = 27 fAD brains and n = 27 CU brains. Scale bars, 50 μm (F‐M, R‐U), 10 μm (N‐Q) and (V‐Y). Data are means ± standard error of the mean; P ≤ 0.01 **, P ≤ 0.0001 ****; one‐way analysis of variance with Dunnett's multiple comparisons test (B, C); correlation analysis (D, E)
IHC co‐staining with anti‐amylin and anti‐Aβ antibodies in fAD brain sections detected distinct amylin and Aβ plaques and mixed amylin–Aβ plaques (Figure 1F‐M). Some plaques stained positive for amylin adjacent to Aβ deposition (Figures 1H and 1I), had layered amylin–Aβ compositions with amylin‐positive cores (Figure 1J‐L) or formed intercalated amylin–Aβ deposits as shown in Figure 1M. For deconvolution and analysis of amylin immunoreactivity intensity signals on IHC images, pancreatic tissue from a patient with type 2 diabetes served as positive control for amylin deposition (Figure S1A in supporting information). The IHC analysis detected amylin in about one third of the plaques in fAD brains (Figure S1B). CU brains have sporadic amylin deposits (Figure S1C); however, amylin–Aβ cross‐seeding is lower in CU versus fAD brains (Figure 1C).
In fAD brains, amylin immunoreactivity was detected in neuronal soma (Figures 1F and 1G; arrowheads; 26 of 27 patients) and neuritic plaques (Figure 1H‐M, 16 of 27 patients). In some amylin–Aβ neuritic plaques, immunostaining showed the presence of amylin in small proteinaceous fragments (Figures 1F and 1G; arrows). Confocal microscopic analysis of areas containing amylin‐positive neurons revealed no overlap between the immunoreactivity signals for amylin and p‐tau (Figure S2A‐S2C in supporting information) suggesting distinct amylin and tau pathologies.
To test whether mixed amylin–Aβ plaques have biochemical characteristics of amyloid, brain slices were triple‐stained with Thioflavin S and anti‐amylin and anti‐Aβ antibodies. Confocal microscopy analysis identified Thioflavin S–positive areas within the Aβ deposits (Figure S2D) and in the amylin cores of mixed amylin‐Aβ plaques (Figure 1N‐Q). Amylin amyloid plaques have similar structures to those formed in sAD (Figure S2E).
In sAD brains, amylin was identified within the arteriole wall, as part of cerebral amyloid angiopathy (CAA). 3 , 4 Post mortem histological reports included comorbid CAA in many fAD brains from this cohort (Table B.4). Histological evidence of cerebral small vessel disease was reported also in CU brains (Table B.4). We tested the association of brain amylin accumulation with blood‐brain barrier (BBB) leak by assessing the brain uptake of immunoglobulins (IgG). Analysis of anti‐IgG staining in brain sections revealed variable levels of compromised BBB (Figure S3 in supporting information) in both fAD and CU brain groups. IHC analyses of serial sections for amylin, amylin–Aβ cross‐seeding, and Congo red staining (Figure 1R‐U) suggest that amylin is part of CAA. This was further confirmed by confocal microscopy analysis of brain sections triple‐stained for amylin, Aβ, and α‐smooth muscle cell actin (Figure 1V‐Y). Brain amylin pathology is summarized in Table B.5.
Analyses of the association of common and rare amylin gene variants with the risk of developing AD revealed no statistically significant differences (Table B6). Taken together, our results indicate that circulating amyloid‐forming amylin secreted from the pancreas interacts with fAD pathology through mechanisms that are not linked to a genetic predisposition and may involve amylin as a part of CAA.
3.2. High CSF amylin levels are associated with decreased CSF Aβ42 levels in early AD
The order of biological changes in fAD begins with deposition of amyloid, followed by neurodegenerative changes (assessed from CSF biomarkers) and cognitive decline. 17 To test whether amylin homeostasis in the central nervous system may change in early sAD, we investigated the relationships between CSF amylin and CSF Aβ42, a validated AD biomarker, 2 in CU, MCI, and sAD dementia CSF samples. The average CSF levels of both amylin and Aβ42 are associated with the cognitive status (Figure 2A‐B). Decreased CSF Aβ42 levels correlated with decreased CSF amylin levels in sAD (Figure 2C). In MCI, however, CSF amylin and Aβ42 levels were inversely correlated, with lower Aβ42 levels being associated with higher amylin levels (Figure 2D). Both CSF amylin and Aβ42 levels were highly variable in the CU individuals (Figure 2E).
FIGURE 2.
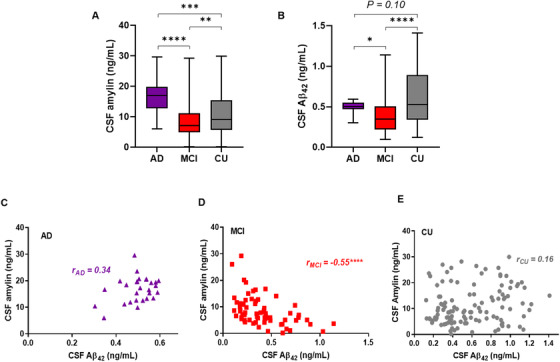
Cerebrospinal fluid (CSF) amylin–amyloid beta (Aβ)42 relationship in Alzheimer's disease (AD), mild cognitive impairment (MCI), and cognitively unaffected (CU) humans. A, B, Levels of CSF amylin (A) and CSF Aβ42 (B) in patients with sporadic AD (sAD, n = 28); individuals with MCI (n = 70), and in CU individuals (n = 117). C‐E, Correlation analysis of CSF amylin levels versus CSF Aβ42 levels in patients from (A, B). Data are presented as box and whisker plot (A, B) and as individual values (C‐E); P ≤ 0.05 *, P ≤ 0.01 **, P ≤ 0.001 ***, P ≤ 0.0001 ****; two‐tailed, unpaired t test (A, B); correlation analysis (C‐E)
These results suggest that increases in CSF amylin and decreases in CSF Aβ42 levels may be characteristic of early cognitive change in the MCI stage of sAD, which needs to be further evaluated in a larger sample with longitudinal characterization of the cohort.
3.3. Genetically suppressed amylin secretion in APP/PS1 rats protects against AD effects
To address the potential for altering AD‐related pathology/symptoms through modulation of the blood amylin level, we genetically manipulated amylin secretion in APP/PS1 and non‐APP/PS1 rats by pancreatic overexpression of human amylin (3‐fold) or amylin gene deletion. Representative genotyping results for newly generated transgenic rats are shown in Figure S4A‐S4C in supporting information. Study design and study time points are described in Figure 3A‐C.
FIGURE 3.
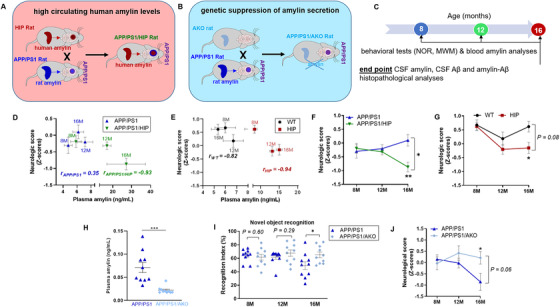
Behavior deficits in APP/PS1 and non‐APP/PS1 rats with genetically manipulated amylin secretion. A‐C, Schematic summary of rat models generated for this study and time points of behavior and molecular analyses. Genetic manipulation of circulating amylin was performed in rats with and without Alzheimer's disease (AD)‐like pathology through transgenic expression of human amylin in the pancreas (red color code, A) and by deletion of the amylin gene (light blue color code, B). The time points for behavioral studies were chosen based on previous reports that amylin dyshomeostasis develops at ≈12 months of age in human amylin expressing rats 7 , 15 and that amyloid beta (Aβ) pathology is developed in APP/PS1 rats at about 16 months of age 16 (C). D‐G, Longitudinal neurological scores of cognitive function (assessed from Novel Object Recognition and water maze performance tests) versus plasma amylin levels (both rat and human amylin; D, E) and versus age (F, G) in APP/PS1/HIP versus APP/PS1 rats, and HIP versus wild‐type (WT) rats (n = 10 rats/group). Data for individual tests are shown in Figure S6 in supporting information. H, Plasma amylin levels in APP/PS1 rats versus APP/PS1 rats with suppressed amylin expression (APP/PS1/AKO rats) at 16 months of age. I, Novel object recognition in APP/PS1 rats versus APP/PS1/AKO rats at 8, 12, and 16 months of age (n = 10 rats/group). J, Longitudinal neurological scores of cognitive function in APP/PS1 versus APP/PS1/AKO rats (n = 10 rats/group). Data are means ± standard error of the mean. P ≤ 0.05 *, P ≤ 0.01 **, *, P ≤ 0.001 ***; correlation analysis (D, E); two‐way analysis of variance with Sidak post hoc (F, G, J); two‐tailed, unpaired t test (H, I)
Although APP/PS1/HIP and HIP rats overexpress human amylin throughout their lives, their plasma levels of amylin (combined rat and human amylin) increase with aging (> 12 months of age), whereas there is no significant change of plasma levels of amylin (rat amylin only) in APP/PS1 and WT littermates (Figures 3D and 3E). Plasma from APP/PS1/HIP rats had increased amylin immunoreactivity intensity signals at high molecular weight bands (> 3.9 kDa; monomeric amylin) compared to plasma from APP/PS1 littermates (Figure S5 in supporting information). The results are consistent with previous reports in HIP versus WT rats 7 , 15 , 18 and reflect the constructive validity of the human amylin‐expressing rats. We interpret the presence of aggregated amylin in the systemic circulation as a possible sign of amylin dyshomeostasis that may be triggered by different conditions, including amylin hypersecretion (common in prediabetes). 7 APP/PS1/HIP rats with high plasma levels of amylin (combined rat and human amylin) had greater behavior deficits (Figure 3D and Figure S6A‐S6C in supporting information), which worsened at a higher rate with aging than in APP/PS1 littermates expressing only rat amylin (Figure 3F). Similarly, cognitive performance declined faster in aged HIP rats with high plasma amylin levels (combined rat and human amylin) than in WT littermates (Figures 3E and 3G and S6D‐S6F). The neurological score‐plasma amylin relationship in HIP rats versus WT littermates (Figure 3E) suggests that high blood amylin levels affect neurological score in the absence of genetic determinants of AD pathology.
The Novel Object Recognition (NOR) test in HIP rats (Figure S6D in supporting information) suggests that amylin dyshomeostasis impairs short‐term memory with age in the absence of AD‐like pathology. We therefore tested whether suppressed amylin expression in APP/PS1 rats alters the results of the NOR task. APP/PS1 with the amylin gene deleted (APP/PS1/AKO rats) had lower plasma amylin and less severe memory retention deficits at age 16 months compared to APP/PS1 littermates (Figure 3H‐J).
Our results suggest the following: (1) rats that are prone to develop mixed amylin–Aβ pathology have greater neurological deficits compared to APP/PS1 rats; (2) high blood amylin levels affect brain function in the absence of genetic determinants of AD pathology in HIP rats; and (3) suppression of amylin secretion provides a sustained protection longitudinally against recognition memory deficits in APP/PS1/AKO rats, compared to APP/PS1 rats.
3.4. High blood amylin disrupts CSF–brain Aβ exchange by amylin–Aβ cross‐seeding in brain
Next, we tested amylin–Aβ interaction in aged APP/PS1/HIP and APP/PS1 littermates using IHC co‐staining with anti‐amylin and anti‐Aβ antibodies in brain slices (Figure 4A‐E). The density of plaques (amylin, Aβ, and amylin–Aβ plaques) was higher in 16‐month‐old APP/PS1/HIP rat brains compared to controls (Figure 4F). Amylin formed the protein core of Aβ plaques in both groups (Figure 4A‐D); however, surface area covered by mixed amylin–Aβ deposits was higher in APP/PS1/HIP brains compared to that in APP/PS1 rat brains (Figures 4G and 4H). Non‐APP/PS1 rats expressing human amylin (ie, the HIP rats) had brain amylin deposition in the absence of AD mutations (Figure S7A‐S7B in supporting information). In contrast, WT rats had no cerebral amylin plaques suggesting that rat amylin does not form cerebral plaques in non‐APP/PS1 rats that express only rat amylin. In addition to amylin‐core Aβ plaques, APP/PS1/HIP rat brains had amylin accumulation in neuronal soma (Figure 4E) mirroring our findings in fAD brains (Figure 1). Cerebral vascular deposition of amylin was identified in APP/PS1/HIP and HIP rats, but not APP/PS1 and WT rats (Figure S7C‐S7F), indicating a possible relationship between high blood amylin levels and cerebral vascular amylin deposition. Pancreatic tissue from rats in all groups were used as controls for amylin immunoreactivity signals in the brains (Figure S8A‐S8E in supporting information).
FIGURE 4.
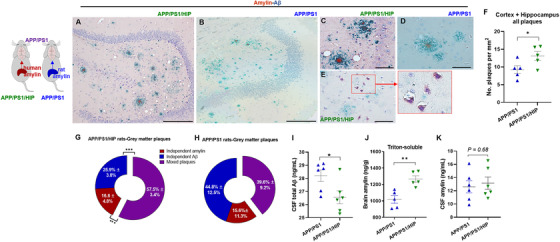
Amylin–amyloid beta (Aβ) cross‐seeding and impaired cerebrospinal fluid (CSF)–brain Aβ exchange by high blood amylin levels (both rat and human amylin) in APP/PS1 rats. A‐E, Representative immunohistological images of co‐staining with anti‐amylin (brown) and anti‐Aβ (green) antibodies in brain sections from 16‐month‐old APP/PS1/HIP rats (A, C, E) and APP/PS1 rats (B, D; n = 5 rats/group). Amylin–Aβ plaques (A‐D) and amylin depositions within neurons (E) are shown. F, Analysis of total plaques including Aβ plaque, amylin plaque, and amylin‐core Aβ plaque depositions in APP/PS1 and APP/PS1/HIP rat brains, assessed from immunohistological images (n = 5 rats/group; data were normalized to total imaging area). G, H, Relative distribution of amylin, Aβ, and amylin–Aβ in plaques in the gray matter of APP/PS1/HIP rat brains (G; n = 5) and APP/PS1 rat brains (H; n = 5). I‐K, CSF total Aβ (I; n = 6 rats/group), brain amylin (Triton‐soluble fractions of tissue homogenate; n = 5 rats/group; J), and CSF amylin (K; n = 6 rats/group) levels in APP/PS1/HIP versus APP/PS1 rats at 16 months of age. Scale bars, 200 μm (A, B), 50 μm (C‐E). Data are means ± standard error of the mean; P ≤ 0.05 *, P ≤ 0.01 **, P ≤ 0.001 ***; two‐tailed, unpaired t test
Higher plasma levels of amylin (combined human and rat amylin) in APP/PS1/HIP versus APP/PS1 rats are associated with lower CSF Aβ levels (Figure 4I) and more cerebral plaques (Figure 4F). Amylin secreted from the pancreas accumulated in the brain (Figure 4J) and cross‐seeded with Aβ (Figure 4G) without significant change of the CSF concentration of amylin (combined human and rat amylin; Figure 4K).
These results suggest that increased circulating amylin levels may alter CSF–brain Aβ exchange through amylin–Aβ cross‐seeding in the brain.
3.5. Suppressed amylin secretion decreases amyloid density in plaques
Cross‐seeding between rat amylin and Aβ in APP/PS1 rat brains was unanticipated, because rodent amylin does not form amyloid. 9 In contrast to the human brain (which lacks amylin mRNA 3 ), some rodent brain cells appear to secrete amylin 10 that may promote direct amylin–Aβ interaction in APP/PS1 rats. We therefore tested whether genetically suppressed amylin secretion in APP/PS1 rats is associated with decreased cerebral amylin–Aβ plaque formation. Essentially, APP/PS1/AKO rats with the amylin gene deleted lacked amylin–Aβ cross‐seeding in the brains, whereas APP/PS1 littermates had amylin‐core Aβ plaques (CP) in brain sections (Figure 5A‐G). Decreased amylin–Aβ cross‐seeding in APP/PS1/AKO rat brains correlated with lower amylin immunoreactivity intensity signal in pancreatic tissues (Figure S8E), plasma (Figure 3H), and brain homogenates (Figure 5H) from these rats, compared to APP/PS1 rats.
FIGURE 5.
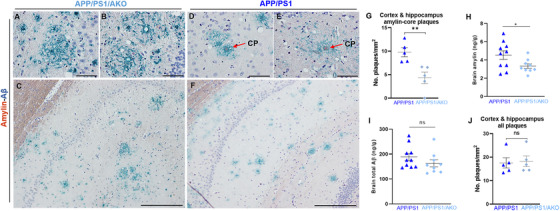
Decreased brain amylin amyloid‐core plaques in APP/PS1 rats with genetically suppressed amylin secretion. A‐F, Representative images of immunostaining for amylin (brown) and amyloid beta (Aβ) (green) in brain tissues from 16‐month‐old APP/PS1/AKO rats (A‐C) and APP/PS1 littermates (D‐F). Arrows indicate amylin‐core Aβ plaques (CP). G, Counts of amylin‐core Aβ plaques in APP/PS1 and APP/PS1/AKO rat brains assessed from immunohistological images (n = 5 rats/group; data were normalized to total imaging area). H, I, Amylin and Aβ levels in brain homogenates from 16‐month‐old APP/PS1/AKO rats and APP/PS1 littermates. J, Counts of Aβ plaques in the same APP/PS1 and APP/PS1/AKO rat brains as in (G; n = 5 rats/group). Scale bars, 50 μm (A, B, D, E), 200 μm (C, F). Data are means ± standard error of the mean. P ≤ 0.05 *; P ≤ 0.01 **; two‐tailed, unpaired t test
It is worth noting that lack of endogenous rat amylin secretion in APP/PS1/AKO rats appears to not increase brain Aβ concentration (measured by total Aβ ELISA) in the APP/PS1/AKO rat brains, compared to APP/PS1 littermates (Figure 5I). The extent of Aβ pathology was similar in APP/PS1 and APP/PS1/AKO rats (Figure 5J). Furthermore, western blot analysis of brain homogenates from APP/PS1 versus APP/PS1/AKO rat brains suggests that suppressed circulating amylin does not increase soluble Aβ levels in the brain (Figure S9 in supporting information).
3.6. Circulating aggregated amylin accelerates amylin–Aβ cross‐seeding in the brain
Greater pathological processes in brains of aged APP/PS1/HIP versus APP/PS1 rats were associated with circulating aggregated amylin (Figure S5), and this was also the case in plasma from aged HIP versus WT rats. 7 , 15 , 18 To assess the impact of circulating aggregated human amylin (amyloid forming) on early AD pathological processes, young APP/PS1 rats (age 7 months), studied before the development of Aβ pathology (which occurs at age ≈16 months), were injected intravenously with pre‐aggregated human amylin (Figure 6A; the schematic schedule of the treatment). Amylin was infused for 2 months at a dose that only slightly elevated the plasma amylin level (Figure 6B). The aggregation status/size distribution of injected amylin is shown in Figure S10 in supporting information. At the conclusion of the infusion regimen, amylin‐core Aβ plaques were detected in treated (Figure 6C) but not in untreated rats. Brains of APP/PS1 rats intravenously infused with human amylin also showed neuronal amylin deposition (Figure 6D), consistent with our findings in APP/PS1/HIP rats (Figure 4E) and human fAD brains (Figure 1F‐G), and higher soluble amylin and Aβ levels by western blot analysis of brain homogenates (Figure 6E and Figure S11 in supporting information).
FIGURE 6.
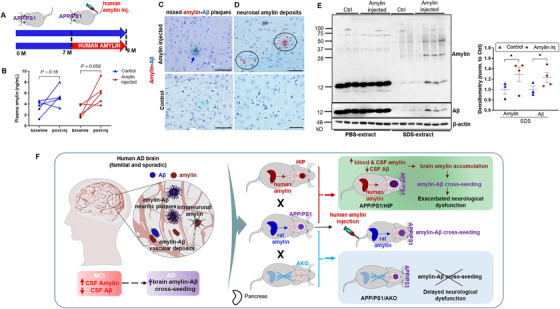
Circulating aggregated amylin cross‐seeds with brain amyloid beta (Aβ) in young APP/PS1 rats. A, Experimental protocol for intravenous amylin infusion in rats. B, Plasma amylin levels in APP/PS1 rats intravenously infused with human amylin and age matched APP/PS1 control rats at baseline and after injections (n = 5 rats/group). C, D, Immunohistological analysis with anti‐amylin (brown) and anti‐Aβ (green) antibodies showing amylin‐Aβ plaques (C; arrow) and amylin accumulation in neurons (D; circles) of APP/PS1 rats intravenously infused with human amylin and age‐matched APP/PS1 control rats (n = 3 rats/group). E, Western blot analysis of amylin and Aβ levels in the brain homogenate extracted using phosphate‐buffered saline or sodium dodecyl sulfate from APP/PS1 rats intravenously infused with human amylin and age‐matched APP/PS1 control rats (n = 3‐4 rats/group). F, Schematic summary of the results. Circulating aggregated amylin affects brain and cerebrospinal fluid (CSF) Aβ levels via brain amylin accumulation and amylin‐Aβ cross‐seeding. Amylin gene deletion in APP/PS1 rats blocks amylin accumulation in the brain and amylin–Aβ cross‐seeding, providing protection against AD‐associated effects. Scale bars, 50 μm (C, D). Data are means ± standard error of the mean; two‐tailed, paired t test (B), P ≤ 0.05 *; two‐tailed, unpaired t test (E)
The results support the hypothesis that circulating aggregated amylin (amyloid‐forming) has a great effect on the cerebral plaque structure by promoting amylin–Aβ cross‐seeding.
4. DISCUSSION
This study shows that circulating amyloid‐forming amylin secreted by the pancreas cross‐seeded with Aβ in brain parenchyma and blood vessel walls in PSEN1 and APP mutation carriers. Increases in CSF amylin and decreases in CSF Aβ42 levels appeared in the setting of early cognitive change in the MCI stage of sAD. In APP/PS1 rats, genetically suppressed amylin secretion protected against AD effects, whereas hypersecretion of human amylin accelerated AD‐like pathology through (at least in part) circulating aggregated amylin. At the molecular level, circulating aggregated amylin correlated with disrupted CSF–brain Aβ exchange and amylin–Aβ cross‐seeding in the brain. Intravenously infused aggregated human amylin induced amylin–Aβ cross‐seeding in young APP/PS1 rats before the development of Aβ pathology. These results (summarized in Figure 6F) demonstrate a role of circulating aggregated amylin in early AD and suggest that restoring amylin homeostasis may reduce AD pathology.
Amylin dysregulation contributes to type 2 diabetes. 9 , 19 Aβ cross‐seeds with amylin in the pancreatic islets 5 suggesting that AD‐related pathological processes may affect β‐cell function. Because the hypothalamus regulates pancreatic β‐cell function 20 and is affected by AD pathology, it was hypothesized that AD‐related pathological processes may impair central signaling pathways that regulate amylin and insulin secretion. 21 Our results emphasize the need to elucidate the temporal sequence of amylin‐ and early AD–mediated pathological events in the brain and peripheral tissues, as well as the impact of AD pathology on central function of amylin as a satiation hormone. 10
In sAD, brain amylin pathology was explained by hyperamylinemia, 22 as it coincides with insulin resistance and type 2 diabetes, which are risk factors for sAD. 23 , 24 We did not anticipate amylin dyshomeostasis in fAD, given the earlier onset of disease with reduced age‐dependence of amyloid pathology. The triggers of amylin dyshomeostasis in fAD mutation carriers has yet to be elucidated. It is intriguing that the brain amylin levels are higher in PSEN1 versus APP mutation carriers. Presenilins are present in pancreatic β‐cells; 25 whether mutant presinilin is a link between amylin dyshomeostasis and fAD awaits further investigation.
In non‐APP/PS1 rats, hypersecretion of human amylin impaired brain function in the absence of AD pathology. In APP/PS1 rat brains, rat amylin cross‐seeded with Aβ (an unanticipated result), whereas suppressed amylin secretion protected against AD effects. These results directly address the potential for modulation of circulating amylin as a means to alter AD pathology. It has been reported that injection of the pancreatic peptide amylin potently reduces behavioral impairment and brain amyloid pathology in murine AD models 26 and proposed that amylin protects against AD effects in humans. 27 If amylin lowers Aβ pathology, we then should expect that genetically suppressed amylin gene in APP/PS1 rats will exacerbate AD pathology and behavior deficits, whereas overexpressing amylin will slow disease progression. Our results demonstrated the opposite and are consistent with data showing that intracerebroventricular human amylin administration in murine AD models promotes robust amylin–Aβ cross‐seeding, accelerating AD pathology; 28 and pancreatic expression of amyloid‐forming human amylin in murine AD mice exacerbate cerebral amyloid burden through cumulative diabetogenic effects of the coexpressed human amylin and Aβ. 29
There are limitations to our study of fAD brain specimens and human CSF. We were not able to track possible glucose dysregulation in patients. Neither effects of anti‐AD treatments nor specific forms of anti‐depressant drugs given to individual patients, which may affect pancreatic function including amylin and insulin secretion, were available. The CSF amylin–Aβ42 relationship needs to be further tested in a larger sample with longitudinal characterization of the cohort.
In conclusion, finding amylin–Aβ cross‐seeding in fAD mutation carriers is of interest because: (1) a role of pancreatic amylin in familial AD was previously unknown, (2) there is accumulating evidence showing brain amylin dyshomeostasis in the much more prevalent sporadic form of AD, and (3) development of anti‐amylin drugs may improve the lives of fAD mutation carriers and slow the progression of sporadic AD.
AUTHOR CONTRIBUTIONS
Conceptualization: F.D.; methodology: J.H., H.Z., T.L., G.J.B., L.B.G., R.G., J.B., AESG, A.J.M., E.L.A., S.D., and F.D.; formal analysis: H.L., N.V., S.S., D.K., E.L.A., S.D., R.G., J.B., and F.D.; resources: J.H., H.Z., P.T.N., G.A.J., D.M.W., A.J.H., S.C., C.T., and F.D.; writing: F.D. drafted the manuscript, all other authors contributed to the final form of the manuscript.
CONFLICT OF INTEREST
The authors declare no competing interests.
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
AppendixA
AppendixB
ACKNOWLEDGMENTS
Funding in part by:
● National Institutes of Health AG057290, AG053999, NS116058, UK ADC P30 AG028383;
● University of Kentucky Research Alliance to Reduce Diabetes‐Associated Microvascular Dysfunction;
● UK Dementia Research Institute, which receives its funding from DRI Ltd, UK Medical Research Council, Alzheimer's Society and Alzheimer's Research UK;
● Medical Research Council (award number MR/N026004/1);
● Wellcome Trust Hardy (award number 202903/Z/16/Z);
● Dolby Family Fund;
● National Institute for Health Research University College London Hospitals Biomedical Research Centre;
● BRCNIHR Biomedical Research Centre at University College London Hospitals NHS Foundation Trust and University College London;
● Wake Forest Alzheimer's Disease Research Center P30 AG049638 ;
● H.L. is supported by an American Heart Association fellowship (18PRE33990154);
● Alzheimer's Association VMF‐15‐363458 ;
● T.L. is supported by an Alzheimer's Research UK Senior Fellowship;
● R.G. and J.B. received fellowships from the Alzheimer's Society.
● Resources from the University of Kentucky COVD Pathology Core were used in this study.
Ly H, Verma N, Sharma S, et al. The association of circulating amylin with β‐amyloid in familial Alzheimer's disease. Alzheimer's Dement. 2021;7:e12130. 10.1002/trc2.12130
REFERENCES
- 1. Khachaturian AS, Hayden KM, Mielke MM, et al. Future prospects and challenges for Alzheimer's disease drug development in the era of the NIA‐AA Research Framework. Alzheimers Dement. 2018;14(4):532‐534. [DOI] [PubMed] [Google Scholar]
- 2. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jackson K, Barisone GA, Diaz E, Lw Jin, DeCarli C, Despa F. Amylin deposition in the brain: a second amyloid in Alzheimer disease?. Ann Neurol. 2013;74(4):517‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oskarsson ME, Paulsson JF, Schultz SW, Ingelsson M, Westermark P, Westermark GT. In Vivo Seeding and Cross‐Seeding of Localized Amyloidosis A Molecular Link between Type 2 Diabetes and Alzheimer Disease. Am J Pathol. 2015;185(3):834‐846. [DOI] [PubMed] [Google Scholar]
- 5. Martinez‐Valbuena I, Valenti‐Azcarate R, Amat‐Villegas I, et al. Amylin as a potential link between type 2 diabetes and alzheimer disease. Ann Neurol. 2019;86(4):539‐551. [DOI] [PubMed] [Google Scholar]
- 6. Verma N, Ly H, Liu M, et al. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL‐1beta Synthesis in Brains of Alzheimer's Disease Patients with Type‐2 Diabetes and in Diabetic HIP Rats. J Alzheimers Dis. 2016;53(1):259‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ly H, Verma N, Wu F, et al. Brain microvascular injury and white matter disease provoked by diabetes‐associated hyperamylinemia. Ann Neurol. 2017;82(2):208‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Royall DR. Palmer RF and The Alzheimer's Disease Neuroimaging Initiative, Blood‐based protein mediators of senility with replications across biofluids and cohorts. Brain Commun. 2020;2(1):fcz036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci U S A. 1990;87(13):5036‐5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lutz TA. Control of energy homeostasis by amylin. Cell Mol Life Sci. 2012;69(12):1947‐1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kahn SE, D'Alessio DA, Schwartz MW, et al. Evidence of cosecretion of islet amyloid polypeptide and insulin by beta‐cells. Diabetes. 1990;39(5):634‐638. [DOI] [PubMed] [Google Scholar]
- 12. Schultz N, Janelidze S, Byman E, et al. Levels of islet amyloid polypeptide in cerebrospinal fluid and plasma from patients with Alzheimer's disease. PLoS One. 2019;14(6):e0218561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akter R, Bower RL, Abedini A, Schmidt AM, Hay DL, Raleigh DP. Amyloidogenicity, Cytotoxicity, and Receptor Activity of Bovine Amylin: implications for Xenobiotic Transplantation and the Design of Nontoxic Amylin Variants. ACS Chem Biol. 2018;13(9):2747‐2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8(6):595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Srodulski S, Sharma S, Bachstetter AB, et al. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Molecular Neurodegeneration. 2014;9(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cohen RM, Rezai‐Zadeh K, Weitz TM, et al. A Transgenic Alzheimer Rat with Plaques, Tau Pathology, Behavioral Impairment, Oligomeric Aβ, and Frank Neuronal Loss. The Journal of Neuroscience. 2013;33(15):6245‐6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367(9):795‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verma N, Liu M, Ly H, et al. Diabetic microcirculatory disturbances and pathologic erythropoiesis are provoked by deposition of amyloid‐forming amylin in red blood cells and capillaries. Kidney Int. 2020;97(1):143‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jurgens CA, Toukatly MN, Fligner CL, et al. beta‐cell loss and beta‐cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am J Pathol. 2011;178(6):2632‐2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosario W, Singh I, Wautlet A, et al. The Brain‐to‐Pancreatic Islet Neuronal Map Reveals Differential Glucose Regulation From Distinct Hypothalamic Regions. Diabetes. 2016;65(9):2711‐2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Despa F, Goldstein LB, Biessels GJ. Amylin as a Potential Link between Type 2 Diabetes and Alzheimer Disease. Ann Neurol. 2020;87(3):486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Biessels G, Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol. 2018;14(10):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stanley M, Macauley SL, Holtzman DM. Changes in insulin and insulin signaling in Alzheimer's disease: cause or consequence?. J Exp Med. 2016;213(8):1375‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arnold SE, Arvanitakis Z, Macauley‐Rambach SL, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14(3):168‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dror V, Kalynyak TB, Bychkivska Y, et al. Glucose and endoplasmic reticulum calcium channels regulate HIF‐1beta via presenilin in pancreatic beta‐cells. J Biol Chem. 2008;283(15):9909‐9916. [DOI] [PubMed] [Google Scholar]
- 26. Zhu H, Wang X, Wallack M, et al. Intraperitoneal injection of the pancreatic peptide amylin potently reduces behavioral impairment and brain amyloid pathology in murine models of Alzheimer's disease. Mol Psychiatry. 2015;20(2):252‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu H, Tao Q, Ang TFA, et al. Association of Plasma Amylin Concentration With Alzheimer Disease and Brain Structure in Older Adults. JAMA Netw Open. 2019;2(8):e199826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moreno‐Gonzalez I, Iii EG, Salvadores N, Shahnawaz M, Diaz‐Espinoza R, Soto C. Molecular interaction between type 2 diabetes and Alzheimer's disease through cross‐seeding of protein misfolding. Mol Psychiatry. 2017;22(9):1327‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wijesekara N, Ahrens R, Sabale M, et al. Amyloid‐beta and islet amyloid pathologies link Alzheimer's disease and type 2 diabetes in a transgenic model. FASEB J. 2017;31(12):5409‐5418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
AppendixA
AppendixB