

Abstract
Therapeutic plasma exchange is an effective empiric treatment for thrombotic thrombocytopenic purpura (TTP), but how therapy affects the level of a disintegrin and metalloprotease with thrombospondin type 1 motif 13 (ADAMTS13) or inhibitor has not been reported in many patients. We prospectively analyzed ADAMTS13 activity and inhibitor levels in 37 adults with TTP. ADAMTS13 level at presentation was lower than 5% in 16 of 20 patients with idiopathic TTP and in none of 17 patients with TTP associated with hematopoietic stem cell transplantation, cancer, drugs, or pregnancy (P < .00001). Seven of the 16 patients with ADAMTS13 activity lower than 5% (≈ 44%) had inhibitors. For 8 patients followed serially with ADAMTS13 activity lower than 5% but no inhibitor at presentation, plasma exchange led to complete clinical remission and a rise in ADAMTS13 level. In contrast, 4 patients with low ADAMTS13 activity but high-titer inhibitor (> 5 units/mL) had neither a rise in ADAMTS13 activity nor a reduction in the inhibitor titer: 3 had recurrent disease and 1 died. Among 17 patients with ADAMTS13 activity at presentation higher than 25%, 10 died. Mortality rate for idiopathic TTP was 15%, whereas mortality for nonidiopathic TTP was 59% (P < .02). We conclude that assays of ADAMTS13 activity and inhibitors in addition to the clinical categories (idiopathic TTP and nonidiopathic TTP) are predictive of outcome and may be useful to tailor patient treatment.
Introduction
Thrombotic thrombocytopenic purpura (TTP), a life-threatening disseminated thrombotic microangiopathy, occurs mainly in adults and is characterized by thrombocytopenia, hemolytic anemia, neurologic disturbances, renal abnormalities, and fever.1,2 The incidence for idiopathic TTP is estimated to be approximately 3.7 annually per million persons.3 If untreated, the mortality rate is approximately 90%. Plasma exchange therapy reduces the mortality rate to approximately 20%.4 Although many cases are idiopathic, TTP sometimes occurs in association with pregnancy5; autoimmune diseases6; infection7; drugs such as cyclosporine A, FK506, mitomycin, or ticlopidine8; and bone marrow transplantation.9 This clinical heterogeneity poses a challenge for understanding the pathogenesis of TTP and selecting appropriate therapies.
For many patients with idiopathic TTP, the underlying defect is deficiency of ADAMTS13, a member of the a disintegrin and metalloprotease with thrombospondin type 1 motifs (ADAMTS) family of metalloproteases.10–13 ADAMTS13 has recently been purified14–16 and the complete cDNA sequence has been analyzed.16–18 ADAMTS13 cleaves the Tyr1605-Met1606 bond in the central A2 subunit of the von Willebrand factor (VWF)19,20 and prevents accumulation of “unusually large” VWF multimers that are very adhesive and can lead to platelet aggregation and microvascular thrombosis.21–23 The cleavage can be stimulated by shear forces like those occurring at sites of arterial thrombosis or by low ionic strength and 1.5 M urea or guanidine.19,20 Congenital ADAMTS13 deficiency is rare and associated with missense or frame-shift mutations of the ADAMTS13 gene,18,24–29 whereas acquired ADAMTS13 deficiency is more common and often caused by immunoglobulin G (IgG) inhibitors.13,30–33 Therefore, the efficacy of plasma infusion or therapeutic plasma exchange probably depends on replenishing the missing ADAMTS13 metalloprotease or removing the inhibitory antibodies.
Despite these advances in understanding the pathogenesis of TTP, the clinical utility of ADAMTS13 assays has not been established. ADAMTS13 activity was essentially undetectable in 80% to 100% of selected patients with idiopathic TTP,13,30 but a prospective study of unselected patients with thrombotic microangiopathy concluded that ADAMTS13 deficiency correlates poorly with the pattern of organ involvement at presentation or the subsequent response to plasma exchange.34 However, 18 patients in that study with severe ADAMTS13 deficiency had a mortality of 17%, whereas 124 patients without severe ADAMTS13 deficiency had a mortality of 65%,34 suggesting that ADAMTS13 assays may have prognostic value. In addition, the effect of plasma exchange on plasma levels of ADAMTS13 and inhibitors has not been systemically investigated. To address these issues, we have investigated ADAMTS13 activity and inhibitor levels in 37 patients with TTP receiving plasma exchange therapy.
Patients, materials, and methods
Patients
Consecutive adult patients with a clinical diagnosis of TTP admitted to Barnes-Jewish-Hospital, Washington University were prospectively enrolled between October 1999 and June 2003. Diagnostic criteria were as follows: (1) microangiopathic hemolytic anemia (hemoglobin level < 125 g/L [<12.5 g/dL], direct antiglobulin test negative, 3 or more fragmented red cells or helmet cells per high-power field in the peripheral blood smear); (2) thrombocytopenia (platelet count < 150 × 109/L [< 150 × 103/μL] or a 50% drop from the previous count); (3) no requirement for neurologic symptoms, renal function abnormalities, or fever. Each participating patient or legal guardian (if a patient had neorologic symptoms on admission) signed informed consent. The patient data were collected at presentation and during plasma exchange therapy, according to the protocol approved by the institutional review board of the Washington University School of Medicine.
Plasma exchange
Administration of plasma exchange therapy was at the discretion of the attending physician, but 1.5 volumes of plasma exchange daily with plasma replacement was typically ordered until the platelet count and lactate dehydrogenase (LDH) level had been normal for 3 days, after which plasma exchange was reduced in frequency or terminated. Either fresh frozen plasma or cryosupernatant was used as the replacement fluid.
Sample collection
Before plasma exchange was initiated, venous blood was collected into 5-mL blue-top tubes (3.2% sodium citrate) and kept at 4°C for less than 24 hours. Anticoagulated blood was centrifuged at 1100g for 10 minutes, and plasma was stored at −70°C until assay. Plasma samples were collected at presentation in all patients and twice weekly immediately before plasma exchange sessions in 30 patients.
Clinical definition
Idiopathic TTP was defined as TTP that occurred in patients with no apparent pre-existing illness. Hematopoietic stem cell transplantation (HSCT)–associated TTP occurred after HSCT in patients who received cyclosporine prophylaxis for graft-versus-host disease. Cancer-, drug-, and pregnancy-associated TTPs were self-evident. Three patients were in the “Other” category: one had idiopathic pulmonary fibrosis and was treated with tacrolimus (FK506), the others had sickle cell disease or lupus nephritis. Response to treatment was defined as the achievement of a platelet count higher than 150 × 109/L (150 × 103/μL) during plasma exchange treatment or within 1 week after stopping treatment.34 Remission was defined as normal platelet count and no plasma exchange treatment for 30 days or more. Relapse was defined as the recurrence of TTP following a remission.34
ADAMTS13 activity assay
The ADAMTS13 activity was assayed with a collagen-binding enzyme-linked immunoassay (ELISA) method as described previously35 with some modifications.33,36 Samples were assayed in batch to minimize the interassay variation. The intra-assay variation was less than 10%.
ADAMTS13 inhibitor assay
Patient plasma was heat-treated at 56°C for 30 minutes to inactivate endogenous ADAMTS13 protease activity. The heat-treated plasma was diluted with phosphate-buffered saline (PBS; 1:1, 1:2, 1:4, 1:8, 1:16). Five microliters of the undiluted or diluted patient plasma was incubated with 5 μL normal human plasma at 37°C for 30 minutes. The residual ADAMTS13 activity was measured as described in “ADAMTS13 activity assay.”33,36 One unit of inhibitor per milliliter was defined as the concentration able to reduce the ADAMTS13 activity of an equal volume of normal human plasma by 50%. If an inhibitor was not detected in the initial sample prior to plasma exchange, subsequent assays for inhibitor were not performed during therapy.
Statistics
Association of categoric variables was evaluated by the chi-square analysis with the Yates continuity correction for 2 × 2 contingency tables. Continuous variables were compared by the Student t test.
Results
The clinical and laboratory data from our patients with TTP are summarized in Tables 1 and 2. The female-to-male ratio was more than 4:1, and African American females accounted for 40% of the patients. The median age was 47 years with a range of 16 to 79 years. Of 37 patients, 26 presented with their first episode of TTP and 11 either presented with a recurrence or achieved a remission and later had a recurrence. All patients had thrombocytopenia, antiglobulin test–negative hemolytic anemia, and schistocytosis in the peripheral blood smear. The median platelet count was 18 × 109/L (18 × 103/μL) and the median hematocrit was 0.28 (28%). All patients received plasma exchange therapy and many patients received corticosteroids, vincristine, cyclophosphamide, or splenectomy as indicated in Tables 1 and 2.
Table 1.
Demographic data and clinical presentation of each TTP patient
| No. | Age, y | Race and sex | Etiology | Episodes | Plt, × 109/L | Hct | LDH, U/L | Dialysis | Cr, μM | Fever | CNS symptoms and signs | % ADAMTS13 | Inhibitor, U/mL | Other treatments | Outcome | F/U, mo |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 38 | AAF | Idiopathic | Initial | 10 | 0.314 | 1836 | No | 123.76 | No | None | < 5 | Neg | None | Res-Rem | 13 |
| 2 | 62 | AAF | Idiopathic | Initial | 8 | 0.165 | 2090 | No | 88.4 | Yes | Fatigue and dizziness | < 5 | Neg | P, V, S | Res-Rem | 13 |
| 3 | 39 | WF | Idiopathic | Initial | 20 | 0.156 | 1818 | No | 88.4 | No | None | < 5 | Neg | P | Res-Rem | 33 |
| 4 | 47 | AAF | Idiopathic | Initial | 23 | 0.18 | 769 | No | 79.56 | No | None | < 5 | Neg | None | Res-Rem | 21 |
| 5 | 77 | AAF | Idiopathic | Multi | 11 | 0.266 | 408 | No | 132.6 | Yes | Briefly aphasic | 16 | Neg | IVIg, P | Res-Rem | 32 |
| 6 | 79 | AAF | Idiopathic | Initial | 7 | 0.274 | 1660 | No | 123.76 | No | AMS | < 5 | Neg | P, CVP | Res-Rem | 25 |
| 7 | 56 | AAF | Idiopathic | Initial | 19 | 0.29 | 654 | No | 97.24 | No | AMS | < 5 | Neg | P, V | Res-Rem | 22 |
| 8 | 50 | AAF | Idiopathic | Initial | 6 | 0.326 | 627 | No | 88.4 | No | Mild headache | < 5 | Neg | None | Res-Rem | 17 |
| 9 | 62 | AAF | Idiopathic | Initial | 8 | 0.213 | 1062 | No | 150.28 | Yes | AMS | < 5 | Neg | P, V, S | Res-Rem | 8 |
| 10 | 62 | WF | Idiopathic | Multi | 12 | 0.304 | 1084 | No | 97.24 | Yes | None | < 5 | 6.5* | CVP, S | Rem-Rel-Rem | 35 |
| 11 | 52 | AAF | Idiopathic | Multi | 20 | 0.20 | 866 | No | 97.24 | No | None | < 5 | 8* | P, S, CVP | Rem-Rel-Died | 2† |
| 12 | 44 | AAF | Idiopathic | Multi | 15 | 0.23 | 695 | No | 97.24 | Yes | Stroke, blindness | < 5 | 10* | CVP, S, CSA, rituximab | Rem-Rel-Rem | 48 |
| 13 | 21 | WF | Idiopathic | Initial | 9 | 0.129 | 1489 | No | 132.6 | No | AMS | < 5 | 5* | P | Res-Rem | 18 |
| 14 | 26 | WF | Idiopathic | Multi | 18 | 0.25 | 680 | No | 79.56 | Yes | Headache | < 5 | 1* | P | Rem-Rel-Rem | 16 |
| 15 | 42 | WF | Idiopathic | Initial | 42 | 0.249 | 1584 | No | 159.12 | No | None | 25 | Neg | P | Res-Rem | 32 |
| 16 | 42 | AAF | Idiopathic | Initial | 16 | 0.238 | 1262 | No | 150.28 | No | None | 14 | Neg | None | Res-Rem | 32 |
| 17 | 73 | AAF | Idiopathic | Multi | 35 | 0.28 | 939 | No | 61.88 | No | None | <5 | 5* | P, S | Rem-Rel-Died | 0.03 |
| 18 | 46 | AAF | Idiopathic | Multi | 40 | 0.32 | 409 | No | 238.68 | No | Aphasia | < 5 | Neg | P, V, S | Rem-Rel-Died | 0.5 |
| 19 | 37 | WF | Idiopathic | Multi | 80 | 0.37 | 428 | Yes | 380.12 | No | AMS | 46 | Neg | None | Rem-Rel-Rem | 22 |
| 20 | 59 | WF | Idiopathic | Initial | 47 | 0.247 | 638 | No | 265.2 | No | None | < 5 | 1* | None | Res-Rem | 7 |
| 21 | 54 | WF | HSCT | Initial | 25 | 0.326 | 1150 | No | 44.2 | No | AMS | 41 | Neg | None | NR-Died | 0.6 |
| 22 | 54 | WF | HSCT | Initial | 11 | 0.225 | 463 | No | 132.6 | No | AMS | 16 | Neg | None | Res-Rem | 25 |
| 23 | 19 | WM | HSCT | Initial | 34 | 0.283 | 328 | No | 70.72 | Yes | None | 56 | Neg | P | Res-Rem | 30 |
| 24 | 45 | AsiF | HSCT | Initial | 15 | 0.296 | 1483 | No | 141.44 | No | None | 66 | Neg | None | NR | 25 |
| 25 | 44 | WM | HSCT | Initial | 8 | 0.31 | 1902 | Yes | 565.76 | Yes | Slurred speech | 64 | Neg | P | NR-Died | 1.3 |
| 26 | 25 | WM | HSCT | Initial | 13 | 0.236 | 1678 | No | 424.32 | No | AMS | 84 | Neg | None | NR-Died | ? |
| 27 | 30 | WM | HSCT | Multi | 16 | 0.263 | 1362 | No | 291.72 | Yes | AMS | 32 | Neg | P | Rem-Rel- Died |
3 |
| 28 | 47 | WF | HSCT | Initial | 4 | 0.261 | 648 | Yes | 194.48 | No | AMS | 32 | Neg | P | Res-Rem | 35 |
| 29 | 69 | WF | FK506 | Initial | 23 | 0.252 | 832 | No | 150.28 | Yes | None | 34 | Neg | P | Res-Died | 1.3 |
| 30 | 44 | WM | Cancer | Initial | 147 | 0.25 | 839 | No | 344.76 | No | None | 66 | Neg | P | NR-Died | 2 |
| 31 | 52 | AAF | Cancer | Multi | 18 | 0.24 | 1477 | Yes | 238.68 | Yes | Syncope | 48 | Neg | P | Res-Died | 5 |
| 32 | 59 | WF | Mitomycin | Initial | 2 | 0.19 | 2383 | Yes | 97.24 | No | AMS | 29 | Neg | None | NR-Died | 36 |
| 33 | 74 | WM | Clopidogrel | Multi | 26 | 0.25 | 263 | Yes | 247.52 | Yes | None | 96 | Neg | P, V | NR-Died | 1.6 |
| 34 | 16 | AAM | Lupus | Initial | 41 | 0.238 | ND | No | 176.8 | No | None | 52 | Neg | P, S, V | NR-Died | 2 |
| 35 | 17 | AAF | SCD | Initial | 39 | 0.221 | 28000 | Yes | 309.4 | No | AMS | 74 | Neg | None | Res-Rem | 19 |
| 36 | 29 | WF | Postpartum | Initial | 36 | 0.28 | 2401 | No | 256.36 | No | None | 48 | Neg | None | Res-Rem | 32 |
| 37 | 36 | WF | Pregnancy | Initial | 46 | 0.266 | 1118 | No | 53.04 | Yes | None | 116 | Neg | None | Res-Rem | 28 |
Entries correspond to values on the day of admission. All patients received plasma exchange therapy. For ADAMTS13 inhibitor, Neg indicates a negative inhibitor screen with 1:1 mixing study as described in “Patients, materials, and methods”; low titer (< 5 units/mL) of inhibitor was detected in patients 14 and 20. High titer (> 5 units/mL) was detected in patients 10, 11, 12, 13, and 17 on admission.
Plt indicates platelet count; Hct, hematocrit; Cr, creatinine; CNS, central nervous system; F/U, the length of follow-up in months since patients were enrolled into the study; AAF, African American female; Res, response; Rem, remission; P, prednisone; V, vincristine; S, splenectomy; WF, white female; Multi, history of multiple episodes of TTP at presentation; AMS, altered mental status; IVIg, intravenous immunoglobulin; CVP, a combination of cyclophosphamide, vincristine, and prednisone; Rel, relapse; CSA, cyclosporine; HSCT, hematopoietic stem cell transplantation; NR, no response; WM, white male; AsiF, Asian female; Lupus, positive antinuclear antibody; ND, not done; ?, serum LDH not done for this patient; and SCD, sickle cell disease.
Positive inhibitor identified.
The time in months between enrollment into the study and patient death.
Table 2.
Clinical and laboratory characteristics of patients
| Characteristic | |
|---|---|
| Sex, F/M | 4/1 |
| Race, no. of AA/W/other | 16/20/1 |
| Median age, y (range) | 47 (16–79) |
| Mean lactatedehydrogenase level, IU/dL (range) | 939 (328–28000) |
| Median hematocrit (range) | 0.28 (0.129–0.37) |
| Median platelet count, × 109/L (range) | 18(2–147) |
| No. of patients with renal dysfunction, creatinine level more than 132.6 μM | 17 |
| Median creatinine level, μM (range) | 176.8 (44.2–302.56) |
| No. of patients with initial acute event | 26 |
| No. of patients with multiple acute events | 11 |
| No. of patients with fever | 13 |
| No. of patients with neurologic symptoms | 21 |
| No. of patients who required dialysis during the hospitalization | 7 |
| No. of patients who received corticosteroid treatment | 23 |
| No. of patients who received vincristine treatment | 10 |
| No. of patients who received cyclosporine treatment | 1 |
| No. of patients who received cyclophosphamide treatment | 4 |
| No. of patients who received rituximab treatment | 1 |
| No. of patients who had splenectomy | 8 |
Race is indicated as African American (AA), white (W), or other. Signs and symptoms were recorded on the date of admission. Fever was defined as temperature of at least 37.5°C.
Severe deficiency of plasma ADAMTS13 (< 5%) was found in 16 (80%) of 20 patients with idiopathic TTP. Of these, 7 (≈ 44%) had an ADAMTS13 inhibitor detected (Tables 1–3). Plasma ADAMTS13 activity was essentially normal (> 50% of normal human plasma) or slightly decreased (20%−50%) in patients with either hematopoietic stem cell transplantation or cancer-, drug-, or pregnancy-associated TTP, and no inhibitor was detected in any of these patients (Tables 1–3). The serum creatinine level on admission correlated inversely with idiopathic TTP (P < .05) and with severe ADAMTS13 deficiency (P < .005). In this series, all patients with idiopathic TTP were female. The sex distribution of patients with nonidiopathic TTP was approximately equal, comprising 7 males and 10 females, and significantly different from that of idiopathic TTP (P < .01). As observed in previous studies3,34 African American race also correlated with severe ADAMTS13 deficiency (P < .05) and idiopathic TTP (P < .01).
Table 3.
Association between etiology of TTP and ADAMTS13 activity
| Etiology | ADAMTS13activity lowerthan 5% | ADAMTS13 activity from 5%−50% | ADAMTS13activity higher than 50% | +ADAMTS13 inhibitor | Total |
|---|---|---|---|---|---|
| Idiopathic | 16 | 4 | 0 | 7 | 20 |
| HSCT and cyclosporine | 0 | 4 | 4 | 0 | 8 |
| Cancer and chemotherapy | 0 | 2 | 1 | 0 | 3 |
| Clopidogrel | 0 | 0 | 1 | 0 | 1 |
| Pregnancy | 0 | 1 | 1 | 0 | 2 |
| Other | 0 | 1 | 2 | 0 | 3 |
| Total | 16 | 12 | 9 | 7 | 37 |
Values in the table indicate the number of patients in each category. ADAMTS13 activity and inhibitors were measured on plasma collected immediately before the first plasma exchange treatment as described in “Patients, materials, and methods.” HSCT indicates hematopoietic stem cell transplantation.
Serial data were obtained for 8 of 9 patients with idiopathic TTP who had severe deficiency of ADAMTS13 but no detectable inhibitor (Figure 1). The exception (patient 18; Table 1) had multiple previous episodes of TTP and received prednisone, vincristine, and splenectomy; she died 12 days after admission. Plasma exchange in these 8 patients increased the ADAMTS13 level with a corresponding clinical improvement, including resolution of neurologic symptoms and normalization of the low platelet count (Figure 1) and elevated serum LDH level (data not shown); all 8 achieved a complete response (Table 1). The mean duration of daily plasma exchange required to achieve sustained normal platelet count (> 150 × 109/L [> 150 × 103/μL]) for 3 days was approximately 14 days (range, 4–35 days) and was correlated with the time required for a sustained increase in ADAMTS13 levels (Figure 1). Patients with a delayed clinical response often received additional immune therapies such as corticosteroids, splenectomy, vincristine, and cyclophosphamide at attending physicians’ discretion (Figure 1).
Figure 1. Clinical response in patients with idiopathic TTP and severe ADAMTS13 deficiency but without detectable inhibitor.
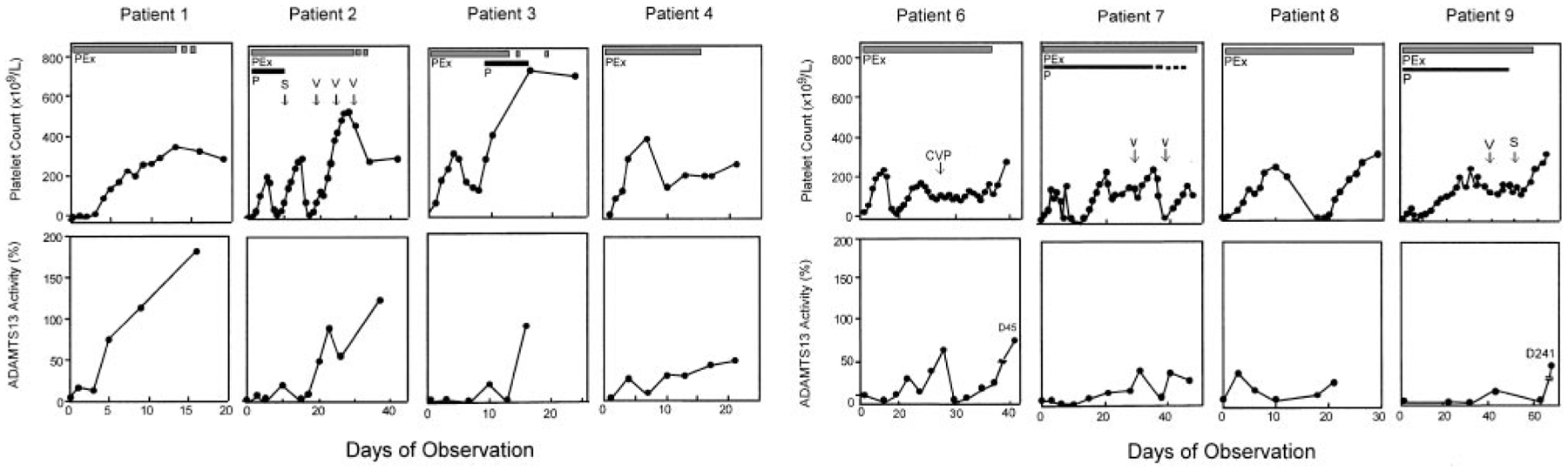
Platelet count is shown in the top row and ADAMTS13 activity in bottom row as labeled. All data points of ADAMTS13 in each patient represented the means of 2 separate assays. The thick solid bar indicates daily plasma exchange (PEx); thick dashed bar, taper of plasma exchange; thin solid line, prednisone (P) administration; and thin dashed line, taper of prednisone. Other adjunctive therapies such as vincristine (V), splenectomy (S), and a combination of cyclophosphamide, vincristine and prednisone (CVP) are indicated with arrows in the top panel of the figure of each patient.
Four patients with severe deficiency of ADAMTS13 had a high-titer inhibitor (> 5 units/mL), and plasma exchange therapy did not result in any detectable level of plasma ADAMTS13 or consistently reduce the inhibitor titer (Figure 2). In one patient, the inhibitor titer was transiently reduced but increased rapidly to more than 16 units/mL upon continuing plasma exchange (Figure 2 patient 13). However, even though ADAMTS13 remained undetectable in this patient, plasma exchange led to clinical remission indicated by a normalization of the platelet count (Figure 2) and reduction of serum LDH (data not shown). Review and follow-up of these 4 patients indicated that 3 had a prolonged and recurrent course of TTP. Patient 10 had a 10-year history of chronic TTP with frequent exacerbations and was treated with plasma exchange, spleen irradiation, splenectomy, prednisone, cyclophosphamide, and vincristine. Patient 11 died shortly after discharge home following 43 plasma exchanges, along with prednisone, vincristine, and cyclophosphamide therapy (Figure 2). Patient 12, who was reported previously,33 experienced multiple cerebral infarctions resulting in blindness and deafness. She had received more than 200 procedures of plasma exchange in addition to cyclosporine, cyclophosphamide, vincristine, splenectomy, and rituximab therapy. She remains in complete remission more than 2 years later. Patient 13 remains in remission. Serial samples from patients 14 through 20 were not available for ADAMTS13 analysis because they were studied before the protocol was modified to include multiple blood sampling. However, one of them (patient 17) who had an inhibitor titer of more than 5 units/mL in her initial plasma sample died within 24 hours of admission. She had chronic relapsing TTP for 3 years and had been treated with prednisone and splenectomy prior to this admission (Table 1).
Figure 2. Clinical response in patients with idiopathic TTP, severe ADAMTS13 deficiency, and a high-titer inhibitor.
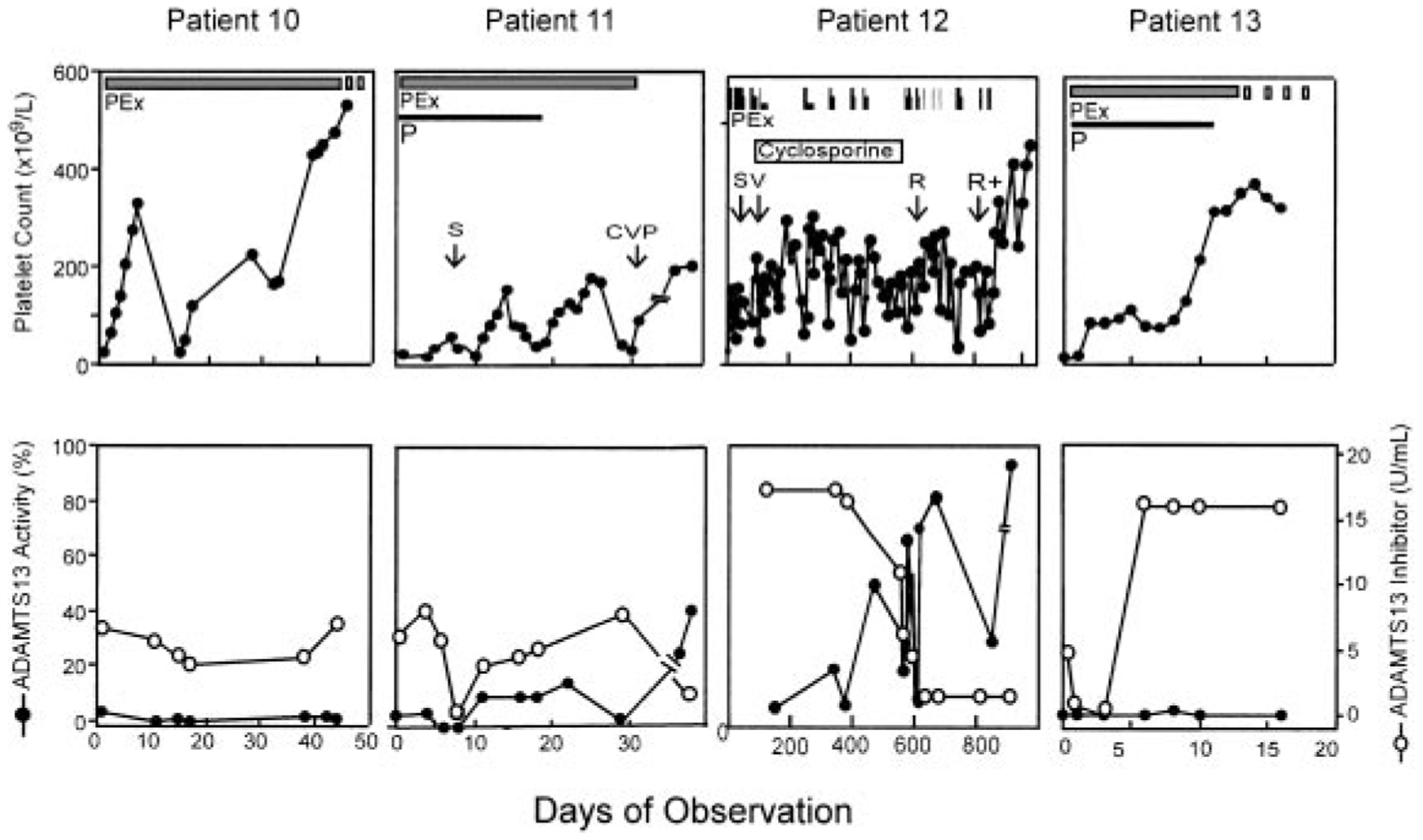
ADAMTS13 inhibitor screen was positive on the initial plasma samples prior to plasma exchange therapy and inhibitor titer was determined. All patients represented in this figure had inhibitor titer of more than 5 units/mL on presentation. The thick solid bar indicates daily plasma exchange; and thick dashed bar, tapering of plasma exchange. Other adjunctive therapies were given during the period of observation as indicated. However, patient 10 had a more than 10-year history of TTP and received plasma exchange (PEx) in addition to prednisone (P), splenectomy (S), and a combination of prednisone, vincristine, and cyclophosphamide (CVP) prior to this admission. Patient 12 was reported previously33 and whose therapies with splenectomy (S), vincristine (V), rituximab (R), and rituximab plus cyclophosphamide (R+) are shown as indicated. Patient 13 received only plasma exchange and prednisone and is in remission.
In 17 patients whose TTP was not idiopathic but instead was associated with hematopoietic stem cell transplantation (patients 21 through 28), drugs (patients 29, 32, and 33), cancer (patients 30 and 31), and pregnancy (patients 36 and 37), etc, (Table 1), plasma exchange had a variable effect on platelet count, and the plasma ADAMTS13 activity was always higher than 25% of normal (Table 1; Figure 3). The mortality for patients with idiopathic TTP was 15% (3 of 20), which was significantly lower than the mortality of 59% (10 of 17) for patients with nonidiopathic TTP (P < .02). Similarly, the prevalence of undetectable ADAMTS13 activity was significantly higher in idiopathic TTP (80%) than in nonidiopathic TTP (0%; P < .00001).
Figure 3. Clinical response in patients with nonidiopathic and normal or mildly reduced ADAMTS13 activity.
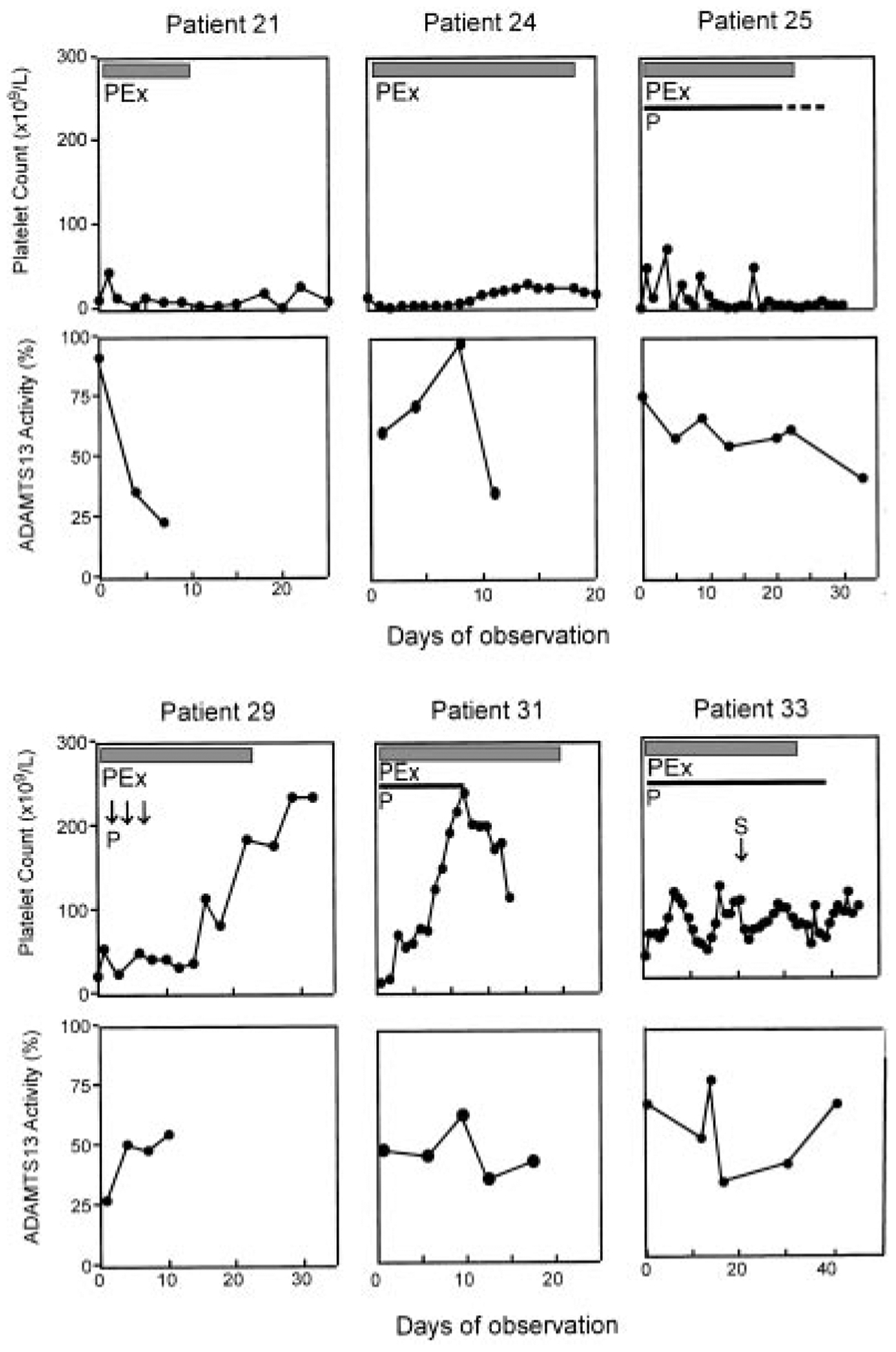
Six patients in this category were serially studied. All patients had plasma ADAMTS13 activity higher than 25% and no ADAMTS13 inhibitor on admission. Patients 21, 24, and 25 had hematopoietic stem cell transplantation–associated TTP. Patient 29 had idiopathic pulmonary sclerosis on FK506. Patients 31 and 33 had cancer- and clopidogrel-associated TTP, respectively. Patient 33 received 3 doses of vincristine (V) treatment prior to the study. All patients (except patient 24) died despite temporary recovery of platelet count in some patients (patients 29 and 31). The data shown are from the initial hospitalization. The ADAMTS13 activity shown in the figure was the mean of 2 independent analyses. PEx indicates plasma exchange; P, prednisone; and S, splenectomy.
Discussion
The efficacy of plasma therapy for TTP has been established for adult patients presenting with thrombocytopenia and microangiopathic anemia but without other potential causes such as disseminated intravascular coagulation, cancer, or eclampsia.4,37 Patients in these studies were not required to have fever, renal failure, or neurologic abnormalities (Table 1).2,4 Plasma exchange has reduced the mortality of patients with idiopathic TTP from more than 90% to approximately 25%.4,37–41 Although the prognosis for patients with TTP has improved markedly, the mortality for some subgroups has not changed appreciably. For example, TTP associated with bone marrow transplantation, cancer chemotherapy, or the use of certain drugs has an extremely poor outcome with a mortality of 86%42 to 100%,34 despite plasma exchange therapy. These large differences in prognosis suggest that TTP is caused by several different mechanisms that respond differently to standard treatments.
Severe ADAMTS13 deficiency reportedly is present in almost all patients with idiopathic TTP who lack both pre-existing medical conditions and acute renal failure,13,30 but the correspondence between ADAMTS13 deficiency and TTP may be less perfect in less highly selected patient groups.34,43,44 For example, a recent prospective study found severe ADAMTS13 deficiency in only 16 of 48 patients with idiopathic thrombotic microangiopathy, even when those with acute renal insufficiency were excluded.34 In our prospective series, however, 80% of patients with idiopathic TTP had severe ADAMTS13 deficiency (Tables 1 and 3), similar to the higher prevalence observed in several earlier reports.13,30 Some of the differences among studies probably reflect distinct referral patterns and patient populations. Nevertheless, severe ADAMTS13 deficiency is associated with a favorable prognosis, with death occurring in only 3 of 18 patients34 and 3 of 16 patients in our series (Table 1), respectively, in the 2 prospective studies that describe outcome, for an average mortality of approximately 18%. In contrast, the mortality for patients with detectable ADAMTS13 activity is much higher, with deaths reported by the study in 68 of 124 patients34 and 10 of 21 patients (Table 1), for an average mortality of approximately 54%. Thus, assays of ADAMTS13 activity appear to provide significant prognostic information for patients with thrombotic microangiopathies, as does the clinical category such as idiopathic or nonidiopathic TTP.
Although severe ADAMTS13 deficiency correlates with idiopathic TTP and a relatively good prognosis with plasma exchange, the therapeutic implications of detectable ADAMTS13 activity at diagnosis are less certain. The high mortality for TTP with detectable ADAMTS13 activity might have several causes. Many patients have associated medical conditions that might limit their survival, such as cancer, sepsis, or complications of hematopoietic stem cell transplantation. In these cases, thrombotic microangiopathy without ADAMTS13 deficiency usually would be a secondary consequence of the underlying disorder and need not respond to plasma exchange therapy. For example, TTP after hematopoietic stem cell transplantation has a high mortality of approximately 80%, and any potential effect of plasma exchange has not been distinguishable from that of discontinuing cyclosporine or spontaneous improvement.45,46 The rationale for plasma exchange in patients with ADAMTS13 deficiency is clear, but at present there is no compelling experimental or clinical support for the use of plasma exchange in thrombotic microangiopathy caused by other mechanisms.
Although acquired ADAMTS13 deficiency is thought to be an autoimmune disease, many patients do not have easily detected autoantibody inhibitors. The prevalence of ADAMTS13 inhibitors in patients with severe ADAMTS13 deficiency was 44% in our study (Table 2), which may be compared with values of 65% to 95% in other reports using different assay methods.34,43,44,47 It is possible that the current assays for inhibitor detection are not sufficiently sensitive. Alternatively, some patients may have antibodies that bind ADAMTS13 and facilitate clearance without inhibiting ADAMTS13 activity, and such nonneutralizing antibodies have been reported.48
Unexpectedly, patients with ADAMTS13 inhibitors often respond to plasma exchange with resolution of their thrombocytopenia and hemolysis, even though the inhibitor titer stays elevated and the ADAMTS13 activity remains undetectable (Figure 2). Such findings were also obtained by Kremer-Hovinga et al.49 These results suggest that ADAMTS13 deficiency is not always sufficient to cause thrombotic microangiopathy, which is consistent with the variable clinical course of patients with congenital ADAMTS13 deficiency. As summarized by Furlan and Lämmle50 in a recent review, many patients with congenital ADAMTS13 deficiency have long intervals without disease but develop thrombotic microangiopathy acutely in association with infection, surgery, pregnancy, or another condition that may induce increased von Willebrand factor secretion from endothelial cells. After the stress resolves, they may return to a state of good health without a need for prophylactic plasma infusions. Patients with TTP and persistent autoimmune ADAMTS13 deficiency also can have prolonged remissions punctuated by acute exacerbations, and plasma exchange may sustain the patient until the inciting stress subsides. If so, then TTP caused by autoimmune ADAMTS13 deficiency might benefit from anti-inflammatory therapy, providing an additional theoretical rationale for the use of corticosteroids.
We conclude that severe ADAMTS13 deficiency is associated with idiopathic TTP and low mortality. Conversely, thrombotic microangiopathies with detectable ADAMTS13 activity are associated with a very high mortality. ADAMTS13 deficiency caused by inhibitors is associated with a relapsing course of TTP. So far no single prospective study has been of sufficient size, but data pooled from Veyradier et al43 and Vesely et al34 and this study (Table 1) indicate that patients without inhibitors rarely relapse (1 of 19, ≈ 5%), but patients with inhibitors often relapse (15 of 35, ≈ 43%; P < .01). Although patients with relatively high-titer inhibitors frequently do respond to therapy (Figure 2), these data and other reports34,43 suggest that high-titer inhibitors are associated with delayed response to plasma exchange or refractory disease. Therefore, ADAMTS13 inhibitor titers may prove to be useful for identifying patients with a high likelihood of relapse who may be candidates for more intensive immunosuppressive therapy in addition to therapeutic plasma exchange.
Acknowledgments
We thank Claudine Mazurier (CRTS, Lille, France) for providing purified human plasma VWF; the residents of the Division of Laboratory Medicine; the nurses of the Donor-Apheresis Center, Barnes-Jewish Hospital; and Drs Samuel Santoro, Douglas Lublin, George Despotis, Morey Blinder, and Charles Eby at the Department of Pathology and Immunology, Washington University School of Medicine for advice and assistance in identifying patients and collecting plasma samples.
This work is supported in part by the Faculty Development Fund from the Children’s Hospital of Philadelphia, Philadelphia, PA
References
- 1.Moschcowitz E Hyaline thrombosis of the terminal arterioles and capillaries: a hitherto undescribed disease. Proc N Y Pathol Soc. 1924;24:21–24. [Google Scholar]
- 2.Amorosi EL, Ultmann JE. Thrombocytopic purpura: report of 16 cases and review of the literature. Medicine (Baltimore). 1966;45:139–159. [Google Scholar]
- 3.Torok TJ, Holman RC, Chorba TL. Increasing mortality from thrombotic thrombocytopenic purpura in the United States—analysis of national mortality data, 1968–1991. Am J Hematol. 1995;50:84–90. [DOI] [PubMed] [Google Scholar]
- 4.Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura: Canadian Apheresis Study Group. N Engl J Med. 1991;325:393–397. [DOI] [PubMed] [Google Scholar]
- 5.May HV Jr, Harbert GM Jr, Thornton WN Jr. Thrombotic thrombocytopenic purpura associated with pregnancy. Am J Obstet Gynecol. 1976;126:452–458. [DOI] [PubMed] [Google Scholar]
- 6.Porta C, Caporali R, Montecucco C. Thrombotic thrombocytopenic purpura and autoimmunity: a tale of shadows and suspects. Haematologica. 1999;84:260–269. [PubMed] [Google Scholar]
- 7.Leaf AN, Laubenstein LJ, Raphael B, Hochster H, Baez L, Karpatkin S. Thrombotic thrombocytopenic purpura associated with human immunodeficiency virus type 1 (HIV-1) infection. Ann Intern Med. 1988;109:194–197. [DOI] [PubMed] [Google Scholar]
- 8.Medina PJ, Sipols JM, George JN. Drug-associated thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Curr Opin Hematol. 2001;8:286–293. [DOI] [PubMed] [Google Scholar]
- 9.Elliott MA, Nichols WL Jr, Plumhoff EA, et al. Posttransplantation thrombotic thrombocytopenic purpura: a single-center experience and a contemporary review. Mayo Clin Proc. 2003;78:421–430. [DOI] [PubMed] [Google Scholar]
- 10.Furlan M, Robles R, Solenthaler M, Wassmer M, Sandoz P, Lämmle B. Deficient activity of von Willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1997;89:3097–3103. [PubMed] [Google Scholar]
- 11.Furlan M, Lämmle B. Deficiency of von Willebrand factor-cleaving protease in familial and acquired thrombotic thrombocytopenic purpura. Baillieres Clin Haematol. 1998;11:509–514. [DOI] [PubMed] [Google Scholar]
- 12.Furlan M, Robles R, Solenthaler M, Lämmle B. Acquired deficiency of von Willebrand factor-cleaving protease in a patient with thrombotic thrombocytopenic purpura. Blood. 1998;91:2839–2846. [PubMed] [Google Scholar]
- 13.Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339:1585–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujikawa K, Suzuki H, McMullen B, Chung D. Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 2001;98:1662–1666. [DOI] [PubMed] [Google Scholar]
- 15.Gerritsen HE, Robles R, Lämmle B, Furlan M. Partial amino acid sequence of purified von Willebrand factor-cleaving protease. Blood. 2001;98:1654–1661. [DOI] [PubMed] [Google Scholar]
- 16.Soejima K, Mimura N, Hirashima M, et al. A novel human metalloprotease synthesized in the liver and secreted into the blood: possibly, the von Willebrand factor-cleaving protease? J Biochem (Tokyo). 2001;130:475–480. [DOI] [PubMed] [Google Scholar]
- 17.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276:41059–41063. [DOI] [PubMed] [Google Scholar]
- 18.Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–494. [DOI] [PubMed] [Google Scholar]
- 19.Furlan M, Robles R, Lämmle B. Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood. 1996;87:4223–4234. [PubMed] [Google Scholar]
- 20.Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. 1996;87:4235–4244. [PubMed] [Google Scholar]
- 21.Moake JL, Rudy CK, Troll JH, et al. Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med. 1982;307:1432–1435. [DOI] [PubMed] [Google Scholar]
- 22.Brass L VWF meets the ADAMTS family. Nat Med. 2001;7:1177–1178. [DOI] [PubMed] [Google Scholar]
- 23.Sadler JE. A new name in thrombosis, ADAMTS13. Proc Natl Acad Sci U S A. 2002;99:11552–11554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kokame K, Matsumoto M, Soejima K, et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc Natl Acad Sci U S A. 2002;99:11902–11907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Assink K, Schiphorst R, Allford S, et al. Mutation analysis and clinical implications of von Willebrand factor-cleaving protease deficiency. Kidney Int. 2003;63:1995–1999. [DOI] [PubMed] [Google Scholar]
- 26.Antoine G, Zimmermann K, Plaimauer B, et al. ADAMTS13 gene defects in two brothers with constitutional thrombotic thrombocytopenic purpura and normalization of von Willebrand factor-cleaving protease activity by recombinant human ADAMTS13. Br J Haematol. 2003;120:821–824. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto M, Kokame K, Soejima K, et al. Molecular characterization of ADAMTS13 gene mutations in Japanese patients with Upshaw-Schulman syndrome. Blood. 2004;103:1305–1310. [DOI] [PubMed] [Google Scholar]
- 28.Pimanda JE, Maekawa A, Wind T, Paxton J, Chesterman CN, Hogg PJ. Congenital thrombotic thrombocytopenic purpura in association with a mutation in the second CUB domain of ADAMTS13. Blood. 2004;103:627–629. [DOI] [PubMed] [Google Scholar]
- 29.Schneppenheim R, Budde U, Oyen F, et al. von Willebrand factor cleaving protease and ADAMTS13 mutations in childhood TTP. Blood. 2003;101:1845–1850. [DOI] [PubMed] [Google Scholar]
- 30.Furlan M, Robles R, Galbusera M, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolyticuremic syndrome. N Engl J Med. 1998;339:1578–1584. [DOI] [PubMed] [Google Scholar]
- 31.Tsai HM. High titers of inhibitors of von Willebrand factor-cleaving metalloproteinase in a fatal case of acute thrombotic thrombocytopenic purpura. Am J Hematol. 2000;65:251–255. [DOI] [PubMed] [Google Scholar]
- 32.Knobl P, Haas M, Laczika K, Varadi K, Turecek PL. Immunoadsorption for the treatment of a patient with severe thrombotic thrombocytopenic purpura resistant to plasma exchange: kinetics of an inhibitor of ADAMTS13. J Thromb Haemost. 2003;1:187–189. [DOI] [PubMed] [Google Scholar]
- 33.Zheng X, Pallera AM, Goodnough LT, Sadler JE, Blinder MA. Remission of chronic thrombotic thrombocytopenic purpura after treatment with cyclophosphamide and rituximab. Ann Intern Med. 2003;138:105–108. [DOI] [PubMed] [Google Scholar]
- 34.Vesely SK, George JN, Lämmle B, et al. ADAMTS13 activity in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients. Blood. 2003;102:60–68. [DOI] [PubMed] [Google Scholar]
- 35.Gerritsen HE, Turecek PL, Schwarz HP, Lämmle B, Furlan M. Assay of von Willebrand factor (vWF)-cleaving protease based on decreased collagen binding affinity of degraded vWF: a tool for the diagnosis of thrombotic thrombocytopenic purpura (TTP). Thromb Haemost. 1999;82:1386–1389. [PubMed] [Google Scholar]
- 36.Zheng X, Nishio K, Majerus EM, Sadler JE. Cleavage of von Willebrand factor requires the spacer domain of the metalloprotease ADAMTS13. J Biol Chem. 2003;278:30136–30141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: clinical experience in 108 patients. N Engl J Med. 1991;325:398–403. [DOI] [PubMed] [Google Scholar]
- 38.Goodnough LT, David S, Dorothea V, Charles F. Morbidity and mortality in adults with “idiopathic thrombotic thrombocytopenic purpura/hemolytic uremic syndrome.” J Inten Care Med. 1994;9:167–171. [Google Scholar]
- 39.Hayward CP, Sutton DM, Carter WH Jr, et al. Treatment outcomes in patients with adult thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Arch Intern Med. 1994;154:982–987. [PubMed] [Google Scholar]
- 40.Elkins SL, Wilson PP Jr, Files JC, Morrison FS. Thrombotic thrombocytopenic purpura: evolution across 15 years. J Clin Apheresis. 1996;11:173–175. [DOI] [PubMed] [Google Scholar]
- 41.Lara PN Jr, Coe TL, Zhou H, Fernando L, Holland PV, Wun T. Improved survival with plasma exchange in patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Am J Med. 1999;107:573–579. [DOI] [PubMed] [Google Scholar]
- 42.Fuge R, Bird JM, Fraser A, et al. The clinical features, risk factors and outcome of thrombotic thrombocytopenic purpura occurring after bone marrow transplantation. Br J Haematol. 2001;113:58–64. [DOI] [PubMed] [Google Scholar]
- 43.Veyradier A, Obert B, Houllier A, Meyer D, Girma JP. Specific von Willebrand factor-cleaving protease in thrombotic microangiopathies: a study of 111 cases. Blood. 2001;98:1765–1772. [DOI] [PubMed] [Google Scholar]
- 44.Studt JD, Kremer Hovinga JA, Alberio L, Bianchi V, Lämmle B. Von Willebrand factor-cleaving protease (ADAMTS-13) activity in thrombotic microangiopathies: diagnostic experience 2001/2002 of a single research laboratory. Swiss Med Wkly. 2003;133:325–332. [DOI] [PubMed] [Google Scholar]
- 45.Allford SL, Bird JM, Marks DI. Thrombotic thrombocytopenic purpura following stem cell transplantation. Leuk Lymphoma. 2002;43:1921–1926. [DOI] [PubMed] [Google Scholar]
- 46.Daly AS, Xenocostas A, Lipton JH. Transplantation-associated thrombotic microangiopathy: twenty-two years later. Bone Marrow Transplant. 2002;30:709–715. [DOI] [PubMed] [Google Scholar]
- 47.Mori Y, Wada H, Gabazza EC, et al. Predicting response to plasma exchange in patients with thrombotic thrombocytopenic purpura with measurement of vWF-cleaving protease activity. Transfusion. 2002;42:572–580. [DOI] [PubMed] [Google Scholar]
- 48.Scheiflinger F, Knobl P, Trattner B, et al. Nonneutralizing IgM and IgG antibodies to von Willebrand factor-cleaving protease (ADAMTS-13) in a patient with thrombotic thrombocytopenic purpura. Blood. 2003;102:3241–3243. [DOI] [PubMed] [Google Scholar]
- 49.Kremer-Hovinga JA, Studt JD, Alberio L, Lämmle B. Von Willebrand factor-cleaving protease (ADAMTS-13γ activity determination in diagnosis of thrombotic microangiopathies: the Swiss experience. Semin Hematol. 2004;41:75–82. [DOI] [PubMed] [Google Scholar]
- 50.Furlan M, Lämmle B. Aetiology and pathogenesis of thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome: the role of von Willebrand factor-cleaving protease. Best Pract Res Clin Haematol. 2001;14:437–454. [DOI] [PubMed] [Google Scholar]