

We provide an update of the specific plant secondary metabolites that shape the microbiome, emphasizing newly discovered links between root chemistry and microbiome composition.
Keywords: Benzoxazinoids, camalexin, coumarins, glucosinolates, microbial community assembly, plant–microbe interactions, secondary metabolites, triterpenes
Abstract
One of the major questions in contemporary plant science involves determining the functional mechanisms that plants use to shape their microbiome. Plants produce a plethora of chemically diverse secondary metabolites, many of which exert bioactive effects on microorganisms. Several recent publications have unequivocally shown that plant secondary metabolites affect microbiome composition and function. These studies have pinpointed that the microbiome can be influenced by a diverse set of molecules, including: coumarins, glucosinolates, benzoxazinoids, camalexin, and triterpenes. In this review, we summarize the role of secondary metabolites in shaping the plant microbiome, highlighting recent literature. A body of knowledge is now emerging that links specific plant metabolites with distinct microbial responses, mediated via defined biochemical mechanisms. There is significant potential to boost agricultural sustainability via the targeted enhancement of beneficial microbial traits, and here we argue that the newly discovered links between root chemistry and microbiome composition could provide a new set of tools for rationally manipulating the plant microbiome.
Introduction
Historically, the vast majority of the literature on plant–microbe interactions studied a small set of nutrient-mobilizing symbionts and agricultural pathogens (Glazebrook et al., 1997; Udvardi and Poole, 2013). However, there has been a conceptual shift during the last 20 years, with new sequencing technologies revealing that all non-sterile plants are colonized by a diverse microbiome, which exerts a strong influence on plant productivity in both natural and agricultural settings (Bulgarelli et al., 2013; Müller et al., 2016). Akin to medical science, plant microbiome research has rapidly emerged as a major scientific frontier, because it is increasingly appreciated that microbial science will play a growing role in future agricultural systems (Finkel et al., 2017).
Functional significance of the microbiome for plant and crop performance
Metagenomics surveys show that the number of enzymatic functions encoded in the microbiome vastly outweighs the enzymatic capability of the plant itself (Busby et al., 2017). Some of the functional activities undertaken by the microbiome can directly benefit plant performance, with most attention being paid to three distinct mechanisms: (i) the improvement of plant nutrition (Jacoby et al., 2017); (ii) the suppression of pathogen outbreaks (Pieterse et al., 2014); or (iii) the modulation of abiotic stress tolerance (Cheng et al., 2019). In agriculture, the targeted enhancement of these desirable microbial activities offers a potential avenue towards maintaining crop yields while reducing the application of synthetic fertilizers and pesticides (Bender et al., 2016).
Although there is significant potential to boost agricultural sustainability by incorporating microbiome science into farm practice, there are still major knowledge gaps that restrict our capacity to rationally manipulate the plant microbiome (Tosi et al., 2020). For instance, one option to promote desirable plant–microbe interactions involves selecting crop varieties according to their ability to recruit beneficial microbes into the rhizosphere. However, this strategy is not yet commercially feasible due to an incomplete knowledge of how plant genetics affects the microbiome, as well as a lack of high-throughput screening tools for phenotyping microbial-mediated traits in large breeding populations (Cordovez et al., 2019; Wille et al., 2019). Another option involves inoculating seeds or crops with beneficial microbes. Although a wide range of microbial crop inoculants are commercially available, their uptake by farmers is relatively limited, with the exception of N2-fixing Rhizobium inocula that are routinely applied onto legumes (O’Callaghan, 2016). One of the criticisms of crop inoculants is that they often deliver unpredictable results in field settings, frequently because the inoculated strains are poorly adapted to local soil conditions, or because they fail to colonize the host plant in competition against environmental strains (Kaminsky et al., 2019). Moreover, there is still significant uncertainty surrounding what constitutes a ‘healthy’ or ‘beneficial’ microbiome (Trivedi et al., 2020). In order to improve the efficacy of microbiome-based amendments in agriculture, a future research priority involves unravelling how final crop yield is influenced by the complex plant genotype×microbiome×environment×management interaction (Busby et al., 2017).
Dissecting the factors shaping microbiome assembly
Over the last 20 years, a plethora of studies have quantified the factors that influence plant microbiome composition. Integrating these results together, it is evident that soil is the main source of inoculum and therefore exerts the strongest effect upon microbiome composition, whereas the host genotype fine-tunes what the soil provides the plant with (Bulgarelli et al., 2012; Lundberg et al., 2012). Although this plant genotype effect is relatively weak, it is often linked to specific microbial taxa which can dramatically modulate host fitness. Mechanistically, microbiome assembly can be modulated by multiple plant traits, such as immune responses, root morphology, and metabolite composition (Rodriguez et al., 2019). Each of these traits offers a potential target for crop breeding strategies aiming to recruit desirable microbial strains, and this review will focus on the role played by secondary metabolites.
Metabolic interdependence between plants and microbes
When considering plant–microbe interactions from a metabolic perspective, it is well understood that plants fuel the proliferation of microbial life in the rhizosphere by depositing carbon substrates below-ground (Sasse et al., 2018). These rhizodeposits account for ~10% of the plant’s carbon budget (Pausch and Kuzyakov, 2018), and the microbial utilization of plant-derived growth substrates explains why the rhizosphere contains a dramatically higher abundance of microbes compared with bulk soil (Rovira, 1965). The microbial strains recruited to the rhizosphere can exert a spectrum of effects upon plant performance, ranging from pathogenic to mutualistic. Several adaptive plant phenotypes are actually mediated by microbial associations, such as nutrient uptake, pathogen suppression, and stress tolerance (Mendes et al., 2011; Haney et al., 2015; Castrillo et al., 2017). Over evolutionary time, it is postulated that the ecological success of plants was dependent upon their ability to recruit cooperative strains to the rhizosphere (Vandenkoornhuyse et al., 2015). Therefore, scientific research that defines the metabolic mechanisms used by plants to recruit beneficial microbes could provide a new set of breeding targets for crop improvement (Wei and Jousset, 2017).
A substantial field of literature has investigated the specific molecules that are exchanged between plants and microbes (Sasse et al., 2018; Stringlis et al., 2018; Cotton et al., 2019; Huang et al., 2019; Jacoby et al., 2020). Historically, the overwhelming focus of these studies involved documenting the microbial consumption of the primary metabolites contained in root exudates, particularly sugars, amino acids, and organic acids (Canarini et al., 2019). This focus is supported by a solid physiological basis, because these abundant primary metabolites are loaded into the phloem, transported to the root, and then exuded at the root apical meristem, which is the major site of below-ground sugar deposition (Farrar et al., 2003). Once released into the soil, primary metabolites serve as labile growth substrates that are rapidly consumed by fast-growing generalist microbial strains (Goldfarb et al., 2011). However, the microbial consumption of primary metabolites does not provide a comprehensive explanation accounting for microbial community assembly, because recent investigations of rhizosphere microbiome composition have revealed that a huge diversity of taxa assemble on and around plant roots, including a relatively high proportion of slow-growing strains adapted to specialized metabolic niches (Zhalnina et al., 2018). This suggests that the metabolic interplay between plants and microbes involves a wider spectrum of metabolites, comprising both primary and secondary metabolites.
Physiologically, there are two major reasons why rhizosphere microbes would have access to a wide diversity of plant-derived secondary metabolites. First, many rhizosphere microbial strains are actually endophytes that colonize internal spaces within the root (Gaiero et al., 2013), where they would have access to the full chemical diversity of living plant cells. Secondly, a significant amount of rhizodeposition occurs via the lysis of sloughed-off root cells (Dennis et al., 2010), and these lysates would presumably contain a diverse mixture of metabolic classes. Both of these mechanisms imply that plant secondary metabolites would play a prominent role in shaping microbial community assembly.
Evolution and ecology of plant secondary metabolites
Secondary metabolites are broadly defined as molecules that are not essential to organismal growth and development, which differentiates them from primary metabolites (Wink, 2003; Kliebenstein and Osbourn, 2012). Compared with other organisms, plants produce a rich and diverse array of secondary metabolites. Many of these molecules exert pharmacological activity, which explains why drug discovery pipelines have a long history of researching bioactive plant natural products (Schmidt et al., 2008). Biochemically, secondary metabolites derive from precursors in primary metabolism, whereby the molecular structure of the precursor is usually modified via the successive action of specialized enzymes. Many of these enzymes appear to have resulted from gene duplication events, whereby an essential gene from primary metabolism was duplicated, providing a redundant copy that could subsequently evolve a new function under relaxed selection pressure (Ober, 2005). Gene duplication is extensive amongst the angiosperms, and this massive expansion of the genetic repertoire occurring in higher plant evolution seems to be a central factor contributing to the tremendous diversification of secondary metabolite profiles across plant species adapted to various ecological niches (De Bodt et al., 2005).
Plant secondary metabolites were widely considered as metabolic waste products until roughly the 1970s, when the emergence of chemical ecology as a scientific discipline enabled researchers to study how secondary metabolites were a central mechanism mediating the interactions between plants and other organisms (Hartmann, 2007). This includes cooperative interactions, such as plants recruiting insect pollinators via volatile emissions, and also antagonistic interactions, such as plants deterring herbivores via production of unpalatable or toxic metabolites. Generally, the majority of the early chemical ecology literature focused on plant–animal interactions rather than plant–microbe interactions. Amongst those studies that did focus on microbes, most of the literature investigated how plant secondary metabolites can modulate the interactions with individual microbial strains, particularly pathogens and symbionts (Table 1). However, this situation is now changing, because new methodological tools developed for microbiome-scale studies have recently been applied to study how secondary metabolites affect the composition and function of entire microbial communities.
Table 1.
Overview of chemical structures, metabolic precursors, and microbial effects for secondary metabolites that shape the root microbiome
| Secondary metabolite | Example chemical structure | Precursor primary metabolite | Mechanistic action on microbes |
|---|---|---|---|
| Benzoxazinoids |
|
• Chorismate | • Chemoattractant (Neal et al., 2012a) • Modification of -SH and -NH2 groups in proteins, leading to enzyme inactivation (Wouters et al., 2016) |
| Camalexin |
|
• Tryptophan | • Disruption of membrane integrity (Rogers et al., 1996) |
| Coumarins |
|
• Phenylalanine | • Disruption of transcription (Yang et al., 2016) • Disruption of quorum sensing and biofilm formation (Yang et al., 2016) • Damage to membranes (Yang et al., 2016) |
| Flavonoids |
|
• Phenylalanine | • Induction of nod gene expression in Rhizobium (Redmond et al., 1986) • Damage to membranes (Tsuchiya and Iinuma, 2000) • Inhibition of enzymes (Zhang and Rock, 2004) • Disruption of nucleic acid synthesis (Mori et al., 1987) • Disruption of biofilm formation (Vikram et al., 2010) |
| Glucosinolates |
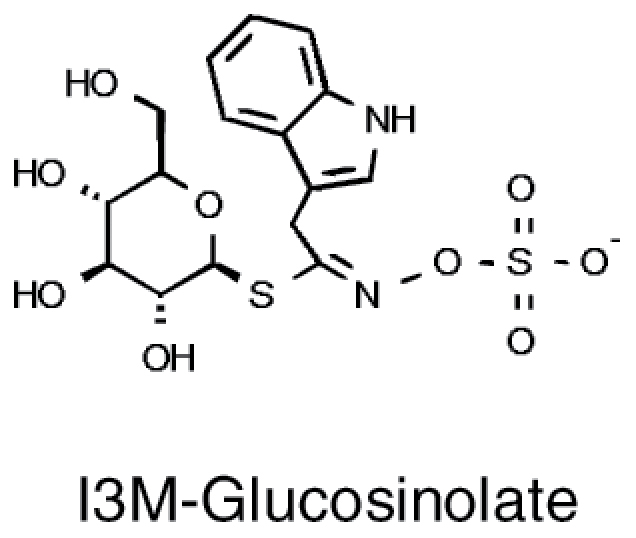
|
• Tryptophan • Phenylalanine • Methionine |
• Isothiocyanate-mediated enzyme inactivation (Aires et al., 2009) • Isothiocyanate-mediated disruption of membrane integrity (Borges et al., 2015) |
| Strigolactones |
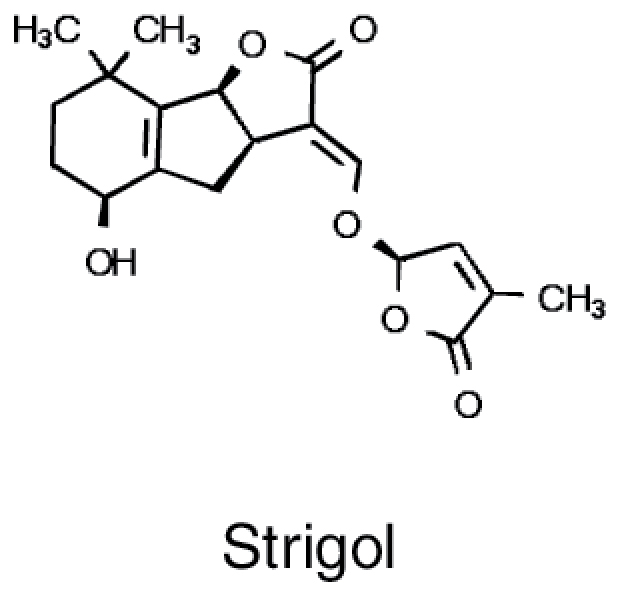
|
• Isopentenyl pyrophosphate • β-Carotene |
• Induction of hyphal branching in mycorrhizal fungi (Akiyama et al., 2005) |
| Triterpenes |
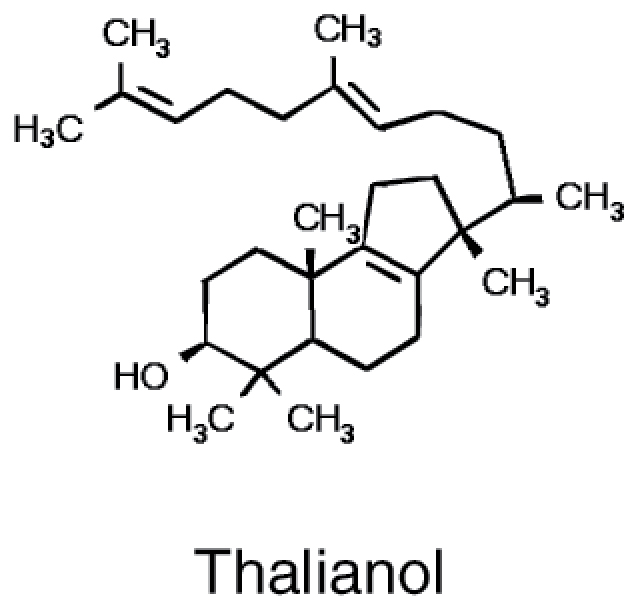
|
• Isopentenyl pyrophosphate • Squalene |
• Disruption of membrane integrity (de León et al., 2010) |
New tools and approaches to characterize specific metabolites that shape the microbiome
For several decades, it has been known that legumes secrete flavonoid secondary metabolites as a mechanism to recruit nitrogen-fixing Rhizobia symbionts (Redmond et al., 1986). In recent years, a suite of new publications has characterized how specific secondary metabolites can shape the root microbiome of Arabidopsis and maize, by modulating the abundance or function of distinct microbial strains (Fig. 1) (Lebeis et al., 2015; Stringlis et al., 2018; Cotton et al., 2019; Huang et al., 2019; Koprivova et al., 2019; Voges et al., 2019). Methodologically, these new studies have utilized a set of emerging tools and approaches for investigating plant–microbe interactions. Specifically, this includes screening microbial responses across diverse panels of plant genotypes using panels of natural accessions or mutants in specific metabolic pathways (Stringlis et al., 2018; Koprivova et al., 2019). On the microbial side, natural soil or defined synthetic communities (SynComs) have been successfully used. In particular, the availability of microbial collections offers great opportunities to generate microbial SynComs with a defined and controlled diversity to address various research questions (Bai et al., 2015; Vorholt et al., 2017).
Fig. 1.

Secondary metabolites are a mechanistic link between plant genetics and microbiome functions. The top panel shows genetic tools for generating plants with altered secondary metabolite profiles. The middle panel illustrates four secondary metabolites recently shown to affect microbiome composition and function. The bottom panel shows microbial responses elicited by variations in secondary metabolite abundance.
Synthetic communities as simplification of complex microbial assemblies
Most of the reports use SynComs that represent the taxonomic assembly of the microbiota (Bodenhausen et al., 2014; Lebeis et al., 2015; Niu et al., 2017). This enables a simplified analysis of the effects of plant genotypes on the microbial assembly. Using a community of the seven most abundant taxa, Bodenhausen et al. (2014) revealed that changes in cuticular properties have a large impact on the associated bacteria in the leaves. Interestingly, the effects were different for different taxa, showing the ability of plants to shape not just the overall quantity of associated microbes, but also the quality (i.e. the composition of the communities). This has been confirmed by analyses of several accessions, which also showed a different impact on the growth of the SynComs (Bodenhausen et al., 2014). A more complex SynCom of 38 strains isolated from Arabidopsis roots was used to dissect the contribution of plant hormones to root microbiome assembly (Lebeis et al., 2015). Analysis of a panel of phytohormone mutants showed the importance of salicylic acid for exclusion of certain bacterial strains from colonizing the root. This was confirmed by treatment with exogenous salicylate, showing that, indeed, metabolites in the rhizosphere affect the ability of microbes to interact with the plant host (Lebeis et al., 2015). A similar SynCom with 35 members was instrumental in finding a coordination between phosphate starvation response, immunity, and microbiome assembly, and the identification of PHOSPHATE STARVATION RESPONSE 1 (PHR1) as the key regulator of this integration (Castrillo et al., 2017). A Syncom based on a similar principle but reduced to 22 members helped to dissect a role for coumarins in shaping the root microbiome (Voges et al., 2019).
The SynComs used in these and other studies were chosen to represent a simplified assembly of strains based on the taxonomical composition of the full microbiome. Across the literature, the SynComs used differ in their size, from small communities of ~7 members (Bodenhausen et al., 2014) to multi-kingdom communities of >200 strains (Durán et al., 2018). So what is the optimal SynCom size? While greater size of the SynComs is a better proxy for the functional diversity of the endogenous plant microbiome, it may also affect the stability of the community. This has been rarely explicitly tested, and to date probably only one SynCom can reliably be called stable, based on rigorous testing (Niu et al., 2017). The maize root SynCom is composed of seven strains representing all the major phyla, which were obtained through host-driven selection from more complex inocula (Niu et al., 2017). The community is stable and develops consistently in independent replicates, but only when complete. Removals of single strains lead to changes in the composition, and removal of one particular strain, Enterobacter cloacae, leads to a complete domination of another strain, Curtobacterium pusillum. One limitation of SynComs is that they only grasp a small fraction of the functional traits that would be contained in a natural microbiome, and are therefore poorly suited to unbiased discovery of microbiome functions. However, the work of Niu et al. (2017) shows the power of SynComs for identifying putative microbial hubs that govern community assembly, thus demonstrating that SynComs are an excellent resource to study the mechanisms that underlie stability of bacterial communities.
Exometabolomics
Such studies will be feasible due to recent developments in exometabolomics (Erbilgin et al., 2017; Jacoby and Kopriva, 2019). Exometabolomics, also called metabolic footprinting, uses the methods of metabolic analysis—LC-MS, GS-MS, or NMR—to analyse extracellular metabolites (Allen et al., 2003). It is a simplified way to provide information about metabolic effects of gene mutations in microorganisms or their communication mechanisms, and has been successfully used to support metabolic engineering and industrial biotechnological processes (Allen et al., 2003; Mapelli et al., 2008; Zha et al., 2014). In the context of microbiome research, exometabolomics can be used to identify metabolites used for cross-feeding among strains in a SynCom in a similar way to how it was used to identify strains capable of complementation of auxotroph mutants in a biotechnological setting (Kosina et al., 2016). Exometabolomics in combination with growth assays on spent media is a powerful approach to identify metabolites enabling or inhibiting growth and so dissect the dynamics of the microbial communities. To move this approach towards better fit for plant microbe research, root extracts or exudates have been successfully used as the carbon source for the microbes (Jacoby et al., 2018; Zhalnina et al., 2018). In experiments using Arabidopsis root extracts, Jacoby et al. (2018) showed that root-associated bacteria are capable of using a much more diverse spectrum of metabolites than Escherichia coli, and that the individual strains also widely differ in this capability. Zhalnina et al. (2018) used root exudates from Avena barbata to reveal that changes in exudate composition during plant development together with bacterial preferences for uptake of certain metabolites, such as organic acids, determine root microbiome assembly. These reports show the power of this approach and the directions in which it could be developed to dissect the metabolic interdependencies in the plant microbiome, both between individual strains and between the host and the microbiota. In particular, the analyses using microbes growing on exudates or extracts from plant mutants, which display differences in microbiome assembly and/or function, have the potential to discover the key metabolites shaping the communities.
Natural variation and genome-wide association studies
The mutants are just one part of genetic resources that can be used to disentangle the metabolic dependencies in plant–microbe interactions. Their power was clearly demonstrated in the identification of several metabolites important for microbiome assembly, such as salicylic acid, coumarins, and other secondary metabolites (Lebeis et al., 2015; Stringlis et al., 2018; and see below). However, to use mutant collections requires previous knowledge and is biased towards investigations of rather obvious candidate signals. In contrast, using natural variation for genome-wide association studies (GWAS) allows identification of the mechanisms in an unbiased manner and possibly can uncover unexpected links (Weigel, 2012). Microbiome composition differs not only between species but also between accessions/varieties of the same species, such as Arabidopsis or maize (Micallef et al., 2009; Bulgarelli et al., 2012; Lundberg et al., 2012; Peiffer et al., 2013). In addition, clear heritable differences in root exudate composition have been determined among Arabidopsis accessions (Mönchgesang et al., 2016). Thus, as plant microbiomes are shaped by metabolites in the rhizosphere, it is feasible to use microbiota traits as phenotypes to identify plant genes through GWAS.
A number of GWAS have addressed various immunity-related phenotypes after inoculation of plants with pathogens (summarized in Bartoli and Roux, 2017 and Xiao et al., 2017). In the context of the microbiome, and in line with most of the research focusing on taxonomic composition, Horton et al. (2014) used operational taxonomic unit (OTU) abundance in leaves of 196 Arabidopsis accessions grown in a common garden to identify genes responsible for controlling leaf microbiome composition. Indeed, several significant single nucleotide polymorphism (SNP) associations were found (Horton et al., 2014). They mapped into regions with genes responsible for cell wall synthesis, defence response, and kinase activity; however, no specific genes have been functionally characterized. The function of the candidate genes, however, agrees well with confirmed effects of these processes on the microbiome composition (Bulgarelli et al., 2012; Lebeis et al., 2015) and also with human microbiome studies that also often find association of microbiome composition with immunity (Hall et al., 2017). In a similar approach with 300 accessions from the extended Goodman Maize Association Panel (Flint-Garcia et al., 2005), Wallace et al. (2018) showed that only two OTUs and three higher order taxa from leaf microbiomes demonstrated significant heritability. Using the OTUs to infer metagenome content yielded a further 222 heritable traits, represented by predicted functional gene annotations (Wallace et al., 2018). Less than 25% of the features produced significant associations, and the corresponding quantitative trait loci (QTLs) were mostly of a small effect and in the vicinity of genes with unknown function (Wallace et al., 2018).
The experiment using a panel of 196 Arabidopsis accessions has been sampled for composition of root microbiome and analysed in the same way as in Horton et al. (2014). Interestingly, this study revealed that the host exerts a larger effect on fungal communities than on bacterial communities (Bergelson et al., 2019). Candidate genes potentially affecting the composition of root microbiome identified in a GWAS were annotated as related to cell wall integrity and immunity, similar to leaves, but also to root and vasculature development (Bergelson et al., 2019).
Only a few studies so far have addressed the effect of plant genetic variation on the function of the microbiome beyond its taxonomic composition. In a pioneering study, using metatranscriptomics, Turner et al. (2013) revealed vast differences in abundance and function of microorganisms in the rhizosphere of three different crop species. Most importantly, the rhizospheres of some species were specifically enriched for different metabolic capabilities, such as cellulose degradation in cereals or hydrogen oxidation in pea (Turner et al., 2013). Similarly, rhizoplane bacterial communities of wheat and cucumber grown in the same soil were enriched in expression of genes for nitrate and sulfate reduction, respectively (Ofek-Lalzar et al., 2014). However, the wide phylogenetic distances between the studied species probably prevents any further attempt at identifying the causative genes underpinning the observed microbial diversification. This has been possible in a study by Koprivova et al. (2019), who measured microbial aryl-sulfatase activity in soils after cultivation of Arabidopsis accessions. While several studies showed only a small impact of the accessions on microbiome structure (Bulgarelli et al., 2012), this analysis revealed up to a 10-fold difference of sulfatase activity in soil from different genotypes (Koprivova et al., 2019), showing the importance of focusing on functional rather than taxonomic traits. Furthermore, the microbial sulfatase activities were used for GWAS to identify Arabidopsis genes affecting the microbial community of the rhizosphere. The candidate genes included genes involved in sulfur metabolism and, as expected, in secondary metabolism. Among them one candidate has been analysed in detail to show that the phytoalexin camalexin, which is an important component of plant immunity against leaf fungal pathogens, has a role in plant–microbe interactions in the rhizosphere (Koprivova et al., 2019). In the functionality context, the approach of Wallace et al. (2018) to predict microbiome function from host genetics has strong potential to formulate working hypotheses, and it would be interesting to see it applied more widely and to compare derived predictions against the measured metabolic functions of the microbial community.
One agriculturally relevant function of soil microbiota is the plant growth promotion (PGP) effect, which can be caused by different mechanisms. Haney et al. (2015) found that Arabidopsis accessions varied in their ability to host the root-associated bacterium Pseudomonas fluorescens WCS365, and also in the ability to gain from the PGP properties of other Pseudomonas strains. The variation in growth promotion was also associated with variation in protection against the root pathogen Fusarium oxysporum (Haney et al., 2015). In a more detailed study, Wintermans et al. (2016) compared the extent to which Arabidopsis accessions profited from incubations with the PGP rhizobacterium Pseudomonas simiae WCS417r. The accessions showed large variation in fresh weight gain, proliferation of lateral roots, and elongation of the primary root upon exposure to the bacterium (Wintermans et al., 2016). GWAS analysis yielded several highly significant associations, and consequently candidate genes potentially influencing the susceptibility of Arabidopsis to the PGP effects of this strain; unfortunately, these were not further tested (Wintermans et al., 2016). However, these analyses together with the multiple GWAS on plant–pathogen interaction and on human microbiome show very clearly that the exploitation of natural variation is a powerful approach to identify the mechanisms by which plants shape their microbiomes.
Plant secondary metabolites shown to affect the microbiome
Several recent studies have shown that secondary metabolites play a distinct role in fine-tuning the composition and function of the rhizosphere microbiome (Table 2) (Lebeis et al., 2015; Stringlis et al., 2018; Cotton et al., 2019; Koprivova et al., 2019; Voges et al., 2019). Using sensitive analytical techniques, untargeted metabolomic profiling has revealed that root tissues and exudates contain hundreds of secondary metabolites from diverse molecular classes (Neal et al., 2012a; Mönchgesang et al., 2016; Hu et al., 2018; Yuan et al., 2018; Zhalnina et al., 2018). Mechanistically, plant secondary metabolites can exert a wide spectrum of effects upon individual microbial strains, by functioning as signalling molecules, nutrient sources, or as toxins. This provides a wide scope for secondary metabolites to act as causative agents shaping the biochemical ecology of the rhizosphere (Jacoby et al., 2020; Pascale et al., 2020). Indeed, a number of secondary metabolites have recently been unequivocally shown to affect microbiome composition and/or function. They belong to different chemical and functional classes and, therefore, they presumably have different mechanisms of action on the communities. Compounds involved in plant immunity and response to pathogens seem, however, to be the most common group of the metabolites active in the rhizosphere.
Table 2.
Summary of recent literature giving new insights into the specific secondary metabolites shaping the root microbiome
| Secondary metabolite | Effects on microbiome | Growth system | Functional mechanism | Target microorganism | Reference |
|---|---|---|---|---|---|
| Coumarins | • Altered microbial community assembly | • Arabidopsis; pots | • Differential microbial toxicity | • Verticillium dahliae JR2 • Fusarium oxysporum f. sp. raphani |
• Stringlis et al. (2018) |
| • Altered SynCom assembly | • Arabidopsis; hydroponics | • ROS production | • Pseudomonas sp. Root329 | • Voges et al. (2019) | |
| Benzoxazinoids | • Altered microbial community assembly • Soil legacy of pathogen suppression |
• Maize; field and pots | • Plant–soil feedback | • OTUs belonging to Proteobacteria and Chloroflexi | • Hu et al. (2018) |
| • Altered microbial community assembly | • Maize; pots | • Metabolic regulation | • OTUs belonging to Methylophilaceae and Xanthomonadaceae | • Cotton et al. (2019) | |
| • Altered microbial community assembly | • Maize; pots | • Gatekeeper effects | • OTUs belonging to Proteobacteria and Chloroflexi | • Kudjordjie et al. (2019) | |
| Camalexin | • Altered associations with plant growth-promoting bacteria | • Arabidopsis; plates | • Differential microbial toxicity | • Pseudomonas sp. CH267 | • Koprivova et al. (2019) |
| Glucosinolates | • Altered association with a plant growth-promoting fungus | • Arabidopsis; plates | • Integration of immune and nutrition status | • Colletotrichum tofieldiae | • Hiruma et al. (2016) |
| • Restriction of excessive fungal proliferation | • Arabidopsis; plates | • Toxicity of glucosinolate breakdown products | • Serendipita indica • Sebacina vermifera |
• Lahrmann et al. (2015) | |
| Triterpenes | • Altered microbial community assembly | • Arabidopsis; pots | • Differential impact on microbial growth rates | • Arthrobacter sp. strain A224 • Agromyces sp. strain A475-1 |
• Huang et al. (2019) |
Glucosinolates
The glucosinolates are one of the best studied classes of defence compounds found in the Brassicaceae (Halkier and Gershenzon, 2006). These sulfur-containing metabolites were originally described as defence against herbivores, as after tissue damage they are metabolized by myrosinase into toxic and deterrent isothiocyanates, nitriles, or other products. However, they are also part of antifungal and antibacterial machinery and are found in root exudates, all prerequisites for compounds shaping the microbiome (Bednarek et al., 2009; Mönchgesang et al., 2016). It has been long known that brassica plants affect soil microbiota and it was exploited for disease control (Papavizas, 1966). These effect were attributed to the degradation products of glucosinolates, for example showing a correlation between the concentration of phenylethylisothiocyanate in the rhizosphere and bacterial community structures (Rumberger and Marschner, 2003). Similarly, engineering Arabidopsis to produce p-hydroxybenzylglucosinolate has resulted in significant changes in the microbial community (Bressan et al., 2009). Glucosinolates also have an impact on association of Arabidopsis with endophytic fungi. Indolic glucosinolates, derived from tryptophan, accumulate upon inoculation with Serendipita indica or Sebacina vermifera, which naturally colonize Arabidopsis roots (Lahrmann et al., 2015). Mutants in indolic glucosinolate synthesis show a highly increased colonization by the fungi, pointing to their important role in maintaining the mutualistic interaction with these fungi (Lahrmann et al., 2015). Also Colletotrichum tofieldiae, another naturally occurring colonizer of Arabidopsis, requires indolic glucosinolates for exerting its PGP effect, and at least some indolic phytoalexins to prevent it from turning into a pathogen (Hiruma et al., 2016). Glucosinolate patterns of Arabidopsis accessions are highly diverse, also affecting the root exudates (Mönchgesang et al., 2016); therefore, glucosinolates are good candidates for metabolic signals to drive the composition of the microbiomes. This has, however, not been apparent in the available GWAS data so far.
Camalexin
On the other hand, the importance of camalexin, another sulfur-containing indolic phytoalexin, for shaping the root microbiome was revealed first by GWAS. Camalexin, 3-thiazol-2'-yl-indole, derived from tryptophan, is one of the major phytoalexins of Arabidopsis (Glawischnig, 2007). Camalexin plays an important role in the response to the necrotrophic pathogens Alternaria brassicicola and Botrytis cinerea, and the oomycete Phytophthora brassicae (Thomma et al., 1999; Rowe and Kliebenstein, 2008; Schlaeppi et al., 2010). Most of the research on camalexin focused on its role in pathogen defence in the leaves. However, camalexin was also shown to be exuded from roots upon elicitation with flagellin (Millet et al., 2010) and to affect several root-specific plant–microbe interaction-related traits (Koprivova et al., 2019). A gene for a new isoform of cytochrome P450 was found in a GWAS screen for variation in microbial sulfatase activity in the rhizosphere soil. Loss of function of this gene, and other genes for camalexin synthesis, was associated with lower sulfatase activity and lower accumulation of camalexin in roots of soil-grown plants. Both phenotypes could be complemented by exogenous camalexin, providing evidence for its function in the soil (Koprivova et al., 2019). In addition, these camalexin-deficient Arabidopsis mutants were unable to benefit from PGP effects, which several mutualistic bacterial strains confer on the wild-type plants, shown by increased biomass. While the direct effect of camalexin on the microbial community structure remains to be shown, the loss of all indolic secondary metabolites in the mutant with disrupted function of both CYP79B2 and CYP79B3 affected the abundance of individual strains in a SynCom (Voges et al., 2019).
Benzoxazinoids
Glucosinolates and camalexin are prominent examples of metabolites affecting microbiome assembly, because they are synthesized by the model plant Arabidopsis; however, they are specific to Brassicaceae. Another group of secondary metabolites that has been shown to shape the soil microbiota are the benzoxazinoids (de Bruijn et al., 2018). Benzoxazinoids are indole-derived compounds found mainly in the grasses and crops, such as maize, where they are important for insect resistance (Niemeyer, 2009). They are also exuded from the roots and serve as allelochemicals or protectants against pathogens, similar to camalexin or glucosinolates (de Bruijn et al., 2018). However, at the same time, they can also act as chemoattractants for PGP bacteria in the rhizosphere (Neal et al., 2012b). It is therefore not surprising that benzoxazinoids affect the composition of the root microbiome (Hu et al., 2018; Cotton et al., 2019; Kudjordjie et al., 2019). These three reports employed maize mutants in benzoxazinoid synthesis to reveal alteration in both bacterial and fungal communities. Interestingly, Hu et al. (2018) was able to link the shifts in microbiota with pathogen resistance of the next plant generation, revealing benzoxazinoids to be part of the plant–soil feedback mechanism, at least for cereals.
Coumarins
Another group of secondary metabolites involved in shaping the root microbiome are ubiquitous in the plant kingdom. Coumarins were first studied in the context of the human microbiome, because many health-promoting natural products as well as toxins belong to this class of metabolites. For example, feeding rats ochratoxin A, one of the major mycotoxins derived from a coumarin backbone, resulted in increased abundance of a Lactobacillus strain that is able to absorb this metabolite (Guo et al., 2014). In plant roots, secreted coumarins have been recognized as important for increasing bioavailability of iron, through reduction and chelation (Tsai and Schmidt, 2017), and also for their antimicrobial functions (Beyer et al., 2019; Stringlis et al., 2019). Interestingly, a transcription factor, MYB72, which controls expression of several genes in coumarin synthesis, is also important for induced systemic resistance against pathogens triggered by the root bacterium Pseudomonas simiae WCS417 (Zamioudis et al., 2014). However, exudation of coumarins also has an impact on the microbial communities, as shown by analyses of microbiota assemblies of Arabidopsis myb72 and f6′h1 mutants that do not secrete scopoletin (Stringlis et al., 2018; Voges et al., 2019). As predicted from the function of coumarins in iron mobilization, the effects on the communities were apparent in iron-deficient soils, and were shown to inhibit a selection of pathogenic fungi, whereas a number of beneficial PGP rhizobacteria were tolerant (Stringlis et al., 2018). There is some evidence that a scopoletin-tolerant Pseudomonas strain can stimulate iron uptake in Arabidopsis (Verbon et al., 2019), so a promising target for future research could involve boosting plant iron nutrition via coumarin-mediated shaping of the root microbial community.
Triterpenes
Triterpenes are an abundant and ubiquitous class of secondary metabolites with known antimicrobial effects (Papadopoulou et al., 1999). A large variety of triterpenes are produced in Arabidopsis roots and some of their biosynthetic genes are induced by jasmonate, pointing to a possible role in plant–microbe interactions (Huang et al., 2019). Indeed, mutants in key genes of triterpene synthesis, affecting thalianin, arabidin, and several triterpene fatty acid esters, showed distinct alterations in microbiome assembly when grown in natural soil (Huang et al., 2019). Interestingly, the OTUs affected by disruption of triterpene synthesis were enriched in bacterial OTUs specific for Arabidopsis. It is thus possible that plant species-specific triterpenes are instrumental in selection of species-specific strains for root microbiome assembly. To test this hypothesis more plant species need to be analysed.
Conclusion and perspectives
One of the major questions in the field of plant–microbe interactions involves defining the functional mechanisms that plants use to shape their microbiome. This is a strategic priority, because these traits could be targeted in crop breeding programmes to develop more sustainable agriculture. Secondary metabolites have often been framed as a plausible mechanism for fine-tuning the plant microbiome, because these chemically diverse molecules frequently exhibit bioactive properties against microbes, and the extensive variation in metabolite profiles between plant species could explain some of the interspecies differences in microbial community assembly (Fitzpatrick et al., 2018). However, until recently, there was a lack of empirical evidence defining the specific plant metabolites that modulate the root microbiome.
There is now a rapidly expanding body of literature unequivocally showing that plant secondary metabolites affect microbiome composition and function (Table 2). Methodologically, these recent studies have synthesized plant genetics with microbiological techniques, often by analysing the altered microbial communities that assemble on the roots of mutant plants impaired in the biosynthesis of a specific secondary metabolite. Increasingly, synthetic community approaches are being used to define the individual microbial strains that are enriched or depleted according to specific plant metabolites. Although synthetic communities will always under-represent the true diversity of a natural microbiome, their key advantage is the ability to address specific questions. For instance, if one individual strain exhibits a particularly strong enrichment or depletion according to plant genotypes differing in secondary metabolite profiles, then this strain can be studied in follow-up experiments, to define the biochemical mode of action exerted by the metabolite on the microorganism, such as reactive oxygen species-mediated toxicity (Voges et al., 2019).
Over the next few years, we anticipate that further studies will continue to advance the mechanistic understanding of how secondary metabolites affect the microbiome. The existing literature has only scratched the surface of plant metabolic diversity, particularly because the majority of them are focused on Arabidopsis. Therefore, genetic manipulation of other metabolite biosynthesis pathways across diverse plant species is almost certain to generate plants with altered microbial associations. Furthermore, untargeted metabolomics measurements are becoming more widespread and accessible, with new approaches now enabling the integration of root metabolomic profiles with rhizosphere microbiome composition (Cotton et al., 2019).
In the longer term, it is tempting to speculate that secondary metabolites could be used as a tool for tailoring the plant microbiome. For example, breeding programmes could aim to rationally manipulate root chemistry by incorporating the entire biosynthetic pathway required to produce a targeted secondary metabolite, in order to recruit a beneficial strain or to deter a pathogen. To achieve this aim, one option could involve large-scale genome editing to introduce the necessary alleles into cultivated plants. Of course, efforts to move from discovery science to translation will probably encounter obstacles, particularly if the secondary metabolites which have the strongest influence on the microbiome were counter-selected during domestication and breeding due to their negative impact on palatability. Despite this, we still feel that secondary metabolites represent promising targets for rationally manipulating the plant microbiome, particularly because one of the selection pressures which favoured the evolution of these bioactive molecules was probably their capacity to influence the composition and function of the root microbial community.
Acknowledgements
Research in SK’s lab is funded by the Deutsche Forschungsgemeinschaft (DFG) under Germany´s Excellence Strategy—EXC 2048/1—project 390686111 and within the SPP 2125 DECRyPT. RPJ is funded by a Humboldt Research Fellowship from the Alexander von Humboldt Foundation.
Author contributions
Conceptualization: RPJ and SK; writing—original draft: RPJ, AK, and SK; writing—review and editing: RPJ, AK, and SK; funding acquisition: RPJ and SK
References
- Aires A, Mota VR, Saavedra MJ, Monteiro AA, Simoes M, Rosa EAS, Bennett RN. 2009. Initial in vitro evaluations of the antibacterial activities of glucosinolate enzymatic hydrolysis products against plant pathogenic bacteria. Journal of Applied Microbiology 106, 2096–2105. [DOI] [PubMed] [Google Scholar]
- Akiyama K, Matsuzaki K, Hayashi H. 2005. Plant sesquiterpenes induce hyphal branching in arbuscular mycorrhizal fungi. Nature 435, 824–827. [DOI] [PubMed] [Google Scholar]
- Allen J, Davey HM, Broadhurst D, Heald JK, Rowland JJ, Oliver SG, Kell DB. 2003. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nature Biotechnology 21, 692–696. [DOI] [PubMed] [Google Scholar]
- Bai Y, Müller DB, Srinivas G, et al. 2015. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528, 364–369. [DOI] [PubMed] [Google Scholar]
- Bartoli C, Roux F. 2017. Genome-wide association studies in plant pathosystems: toward an ecological genomics approach. Frontiers in Plant Science 8, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarek P, Pislewska-Bednarek M, Svatos A, et al. 2009. A glucosinolate metabolism pathway in living plant cells mediates broad-spectrum antifungal defense. Science 323, 101–106. [DOI] [PubMed] [Google Scholar]
- Bender SF, Wagg C, van der Heijden MGA. 2016. An underground revolution: biodiversity and soil ecological engineering for agricultural sustainability. Trends in Ecology & Evolution 31, 440–452. [DOI] [PubMed] [Google Scholar]
- Bergelson J, Mittelstrass J, Horton MW. 2019. Characterizing both bacteria and fungi improves understanding of the Arabidopsis root microbiome. Scientific Reports 9, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer SF, Beesley A, Rohmann PFW, Schultheiss H, Conrath U, Langenbach CJG. 2019. The Arabidopsis non-host defence-associated coumarin scopoletin protects soybean from Asian soybean rust. The Plant Journal 99, 397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenhausen N, Bortfeld-Miller M, Ackermann M, Vorholt JA. 2014. A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genetics 10, e1004283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges A, Abreu AC, Ferreira C, Saavedra MJ, Simões LC, Simões M. 2015. Antibacterial activity and mode of action of selected glucosinolate hydrolysis products against bacterial pathogens. Journal of Food Science and Technology 52, 4737–4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan M, Roncato MA, Bellvert F, Comte G, Haichar FZ, Achouak W, Berge O. 2009. Exogenous glucosinolate produced by Arabidopsis thaliana has an impact on microbes in the rhizosphere and plant roots. The ISME Journal 3, 1243–1257. [DOI] [PubMed] [Google Scholar]
- Bulgarelli D, Rott M, Schlaeppi K, et al. 2012. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488, 91–95. [DOI] [PubMed] [Google Scholar]
- Bulgarelli D, Schlaeppi K, Spaepen S, Ver Loren van Themaat E, Schulze-Lefert P. 2013. Structure and functions of the bacterial microbiota of plants. Annual Review of Plant Biology 64, 807–838. [DOI] [PubMed] [Google Scholar]
- Busby PE, Soman C, Wagner MR, Friesen ML, Kremer J, Bennett A, Morsy M, Eisen JA, Leach JE, Dangl JL. 2017. Research priorities for harnessing plant microbiomes in sustainable agriculture. PLoS Biology 15, e2001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canarini A, Kaiser C, Merchant A, Richter A, Wanek W. 2019. Corrigendum: root exudation of primary metabolites: mechanisms and their roles in plant responses to environmental stimuli. Frontiers in Plant Science 10, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillo G, Teixeira PJ, Paredes SH, et al. 2017. Root microbiota drive direct integration of phosphate stress and immunity. Nature 543, 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YT, Zhang L, He SY. 2019. Plant–microbe interactions facing environmental challenge. Cell Host & Microbe 26, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordovez V, Dini-Andreote F, Carrión VJ, Raaijmakers JM. 2019. Ecology and evolution of plant microbiomes. Annual Review of Microbiology 73, 69–88. [DOI] [PubMed] [Google Scholar]
- Cotton TEA, Pétriacq P, Cameron DD, Meselmani MA, Schwarzenbacher R, Rolfe SA, Ton J. 2019. Metabolic regulation of the maize rhizobiome by benzoxazinoids. The ISME Journal 13, 1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bodt S, Maere S, Van de Peer Y. 2005. Genome duplication and the origin of angiosperms. Trends in Ecology & Evolution 20, 591–597. [DOI] [PubMed] [Google Scholar]
- de Bruijn WJC, Gruppen H, Vincken JP. 2018. Structure and biosynthesis of benzoxazinoids: plant defence metabolites with potential as antimicrobial scaffolds. Phytochemistry 155, 233–243. [DOI] [PubMed] [Google Scholar]
- de León L, López MR, Moujir L. 2010. Antibacterial properties of zeylasterone, a triterpenoid isolated from Maytenus blepharodes, against Staphylococcus aureus. Microbiological Research 165, 617–626. [DOI] [PubMed] [Google Scholar]
- Dennis PG, Miller AJ, Hirsch PR. 2010. Are root exudates more important than other sources of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiology Ecology 72, 313–327. [DOI] [PubMed] [Google Scholar]
- Durán P, Thiergart T, Garrido-Oter R, Agler M, Kemen E, Schulze-Lefert P, Hacquard S. 2018. Microbial interkingdom interactions in roots promote arabidopsis survival. Cell 175, 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbilgin O, Bowen BP, Kosina SM, Jenkins S, Lau RK, Northen TR. 2017. Dynamic substrate preferences predict metabolic properties of a simple microbial consortium. BMC Bioinformatics 18, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar J, Hawes M, Jones D, Lindow S. 2003. How roots control the flux of carbon to the rhizosphere. Ecology 84, 827–837. [Google Scholar]
- Finkel OM, Castrillo G, Herrera Paredes S, Salas González I, Dangl JL. 2017. Understanding and exploiting plant beneficial microbes. Current Opinion in Plant Biology 38, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick CR, Copeland J, Wang PW, Guttman DS, Kotanen PM, Johnson MTJ. 2018. Assembly and ecological function of the root microbiome across angiosperm plant species. Proceedings of the National Academy of Sciences, USA 115, E1157–E1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint-Garcia SA, Thuillet AC, Yu J, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich S, Goodman MM, Buckler ES. 2005. Maize association population: a high-resolution platform for quantitative trait locus dissection. The Plant Journal 44, 1054–1064. [DOI] [PubMed] [Google Scholar]
- Gaiero JR, McCall CA, Thompson KA, Day NJ, Best AS, Dunfield KE. 2013. Inside the root microbiome: bacterial root endophytes and plant growth promotion. American Journal of Botany 100, 1738–1750. [DOI] [PubMed] [Google Scholar]
- Glawischnig E 2007. Camalexin. Phytochemistry 68, 401–406. [DOI] [PubMed] [Google Scholar]
- Glazebrook J, Rogers EE, Ausubel FM. 1997. Use of Arabidopsis for genetic dissection of plant defense responses. Annual Review of Genetics 31, 547–569. [DOI] [PubMed] [Google Scholar]
- Goldfarb KC, Karaoz U, Hanson CA, Santee CA, Bradford MA, Treseder KK, Wallenstein MD, Brodie EL. 2011. Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Frontiers in Microbiology 2, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo MZ, Huang KL, Chen SY, Qi XZ, He XY, Cheng WH, Luo YB, Xia K, Xu WT. 2014. Combination of metagenomics and culture-based methods to study the interaction between ochratoxin A and gut microbiota. Toxicological Sciences 141, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halkier BA, Gershenzon J. 2006. Biology and biochemistry of glucosinolates. Annual Review of Plant Biology 57, 303–333. [DOI] [PubMed] [Google Scholar]
- Hall AB, Tolonen AC, Xavier RJ. 2017. Human genetic variation and the gut microbiome in disease. Nature Reviews. Genetics 18, 690–699. [DOI] [PubMed] [Google Scholar]
- Haney CH, Samuel BS, Bush J, Ausubel FM. 2015. Associations with rhizosphere bacteria can confer an adaptive advantage to plants. Nature Plants 1, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann T 2007. From waste products to ecochemicals: fifty years research of plant secondary metabolism. Phytochemistry 68, 2831–2846. [DOI] [PubMed] [Google Scholar]
- Hiruma K, Gerlach N, Sacristán S, et al. 2016. Root endophyte Colletotrichum tofieldiae confers plant fitness benefits that are phosphate status dependent. Cell 165, 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton MW, Bodenhausen N, Beilsmith K, et al. 2014. Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nature Communications 5, 5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Robert CAM, Cadot S, et al. 2018. Root exudate metabolites drive plant–soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nature Communications 9, 2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ACC, Jiang T, Liu YX, Bai YC, Reed J, Qu BY, Goossens A, Nutzmann HW, Bai Y, Osbourn A. 2019. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 364, eaau6389. [DOI] [PubMed] [Google Scholar]
- Jacoby R, Peukert M, Succurro A, Koprivova A, Kopriva S. 2017. The role of soil microorganisms in plant mineral nutrition—current knowledge and future directions. Frontiers in Plant Science 8, 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby RP, Chen L, Schwier M, Koprivova A, Kopriva S. 2020. Recent advances in the role of plant metabolites in shaping the root microbiome. F1000Res 9, F1000 Faculty Rev-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby RP, Kopriva S. 2019. Metabolic niches in the rhizosphere microbiome: new tools and approaches to analyse metabolic mechanisms of plant–microbe nutrient exchange. Journal of Experimental Botany 70, 1087–1094. [DOI] [PubMed] [Google Scholar]
- Jacoby RP, Martyn A, Kopriva S. 2018. Exometabolomic profiling of bacterial strains as cultivated using arabidopsis root extract as the sole carbon source. Molecular Plant-Microbe Interactions 31, 803–813. [DOI] [PubMed] [Google Scholar]
- Kaminsky LM, Trexler RV, Malik RJ, Hockett KL, Bell TH. 2019. The inherent conflicts in developing soil microbial inoculants. Trends in Biotechnology 37, 140–151. [DOI] [PubMed] [Google Scholar]
- Kliebenstein DJ, Osbourn A. 2012. Making new molecules—evolution of pathways for novel metabolites in plants. Current Opinion in Plant Biology 15, 415–423. [DOI] [PubMed] [Google Scholar]
- Koprivova A, Schuck S, Jacoby RP, et al. 2019. Root-specific camalexin biosynthesis controls the plant growth-promoting effects of multiple bacterial strains. Proceedings of the National Academy of Sciences, USA 116, 15735–15744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosina SM, Danielewicz MA, Mohammed M, Ray J, Suh Y, Yilmaz S, Singh AK, Arkin AP, Deutschbauer AM, Northen TR. 2016. Exometabolomics assisted design and validation of synthetic obligate mutualism. ACS Synthetic Biology 5, 569–576. [DOI] [PubMed] [Google Scholar]
- Kudjordjie EN, Sapkota R, Steffensen SK, Fomsgaard IS, Nicolaisen M. 2019. Maize synthesized benzoxazinoids affect the host associated microbiome. Microbiome 7, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahrmann U, Strehmel N, Langen G, Frerigmann H, Leson L, Ding Y, Scheel D, Herklotz S, Hilbert M, Zuccaro A. 2015. Mutualistic root endophytism is not associated with the reduction of saprotrophic traits and requires a noncompromised plant innate immunity. New Phytologist 207, 841–857. [DOI] [PubMed] [Google Scholar]
- Lebeis SL, Paredes SH, Lundberg DS, et al. 2015. Plant microbiome. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349, 860–864. [DOI] [PubMed] [Google Scholar]
- Lundberg DS, Lebeis SL, Paredes SH, et al. 2012. Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli V, Olsson L, Nielsen J. 2008. Metabolic footprinting in microbiology: methods and applications in functional genomics and biotechnology. Trends in Biotechnology 26, 490–497. [DOI] [PubMed] [Google Scholar]
- Mendes R, Kruijt M, de Bruijn I, et al. 2011. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332, 1097–1100. [DOI] [PubMed] [Google Scholar]
- Micallef SA, Shiaris MP, Colón-Carmona A. 2009. Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. Journal of Experimental Botany 60, 1729–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet YA, Danna CH, Clay NK, Songnuan W, Simon MD, Werck-Reichhart D, Ausubel FM. 2010. Innate immune responses activated in Arabidopsis roots by microbe-associated molecular patterns. The Plant cell 22, 973–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mönchgesang S, Strehmel N, Schmidt S, Westphal L, Taruttis F, Müller E, Herklotz S, Neumann S, Scheel D. 2016. Natural variation of root exudates in Arabidopsis thaliana—linking metabolomic and genomic data. Scientific Reports 6, 29033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori A, Nishino C, Enoki N, Tawata S. 1987. Antibacterial activity and mode of action of plant flavonoids against Proteus vulgaris and Staphylococcus aureus. Phytochemistry 26, 2231–2234. [Google Scholar]
- Müller DB, Vogel C, Bai Y, Vorholt JA. 2016. The plant microbiota: systems-level insights and perspectives. Annual Review of Genetics 50, 211–234. [DOI] [PubMed] [Google Scholar]
- Neal AL, Ahmad S, Gordon-Weeks R, Ton J. 2012a Benzoxazinoids in root exudates of maize attract Pseudomonas putida to the rhizosphere. PLoS One 7, e35498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal AL, Ahmad S, Gordon-Weeks R, Ton J. 2012b Benzoxazinoids in root exudates of maize attract Pseudomonas putida to the rhizosphere. PLoS One 7, e35498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer HM 2009. Hydroxamic acids derived from 2-hydroxy-2H-1,4-benzoxazin-3(4H)-one: key defense chemicals of cereals. Journal of Agricultural and Food Chemistry 57, 1677–1696. [DOI] [PubMed] [Google Scholar]
- Niu B, Paulson JN, Zheng X, Kolter R. 2017. Simplified and representative bacterial community of maize roots. Proceedings of the National Academy of Sciences, USA 114, E2450–E2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan M 2016. Microbial inoculation of seed for improved crop performance: issues and opportunities. Applied Microbiology and Biotechnology 100, 5729–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober D 2005. Seeing double: gene duplication and diversification in plant secondary metabolism. Trends in Plant Science 10, 444–449. [DOI] [PubMed] [Google Scholar]
- Ofek-Lalzar M, Sela N, Goldman-Voronov M, Green SJ, Hadar Y, Minz D. 2014. Niche and host-associated functional signatures of the root surface microbiome. Nature Communications 5, 4950. [DOI] [PubMed] [Google Scholar]
- Papadopoulou K, Melton RE, Leggett M, Daniels MJ, Osbourn AE. 1999. Compromised disease resistance in saponin-deficient plants. Proceedings of the National Academy of Sciences, USA 96, 12923–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papavizas GC 1966. Suppression of Aphanomyces root rot of peas by cruciferous soil amendments. Phytopathology 56, 1071–1075. [Google Scholar]
- Pascale A, Proietti S, Pantelides IS, Stringlis IA. 2020. Modulation of the root microbiome by plant molecules: the basis for targeted disease suppression and plant growth promotion. Frontiers in Plant Science 10, 1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pausch J, Kuzyakov Y. 2018. Carbon input by roots into the soil: quantification of rhizodeposition from root to ecosystem scale. Global Change Biology 24, 1–12. [DOI] [PubMed] [Google Scholar]
- Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE. 2013. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proceedings of the National Academy of Sciences, USA 110, 6548–6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieterse CM, Zamioudis C, Berendsen RL, Weller DM, Van Wees SC, Bakker PA. 2014. Induced systemic resistance by beneficial microbes. Annual Review of Phytopathology 52, 347–375. [DOI] [PubMed] [Google Scholar]
- Redmond JW, Batley M, Djordjevic MA, Innes RW, Kuempel PL, Rolfe BG. 1986. Flavones induce expression of nodulation genes in Rhizobium. Nature 323, 632–635. [Google Scholar]
- Rodriguez PA, Rothballer M, Chowdhury SP, Nussbaumer T, Gutjahr C, Falter-Braun P. 2019. Systems biology of plant–microbiome interactions. Molecular Plant 12, 804–821. [DOI] [PubMed] [Google Scholar]
- Rogers EE, Glazebrook J, Ausubel FM. 1996. Mode of action of the Arabidopsis thaliana phytoalexin camalexin and its role in Arabidopsis–pathogen interactions. Molecular Plant-Microbe Interactions 9, 748–757. [DOI] [PubMed] [Google Scholar]
- Rovira AD 1965. Interactions between plant roots and soil microorganisms. Annual Review of Microbiology 19, 241–266. [DOI] [PubMed] [Google Scholar]
- Rowe HC, Kliebenstein DJ. 2008. Complex genetics control natural variation in Arabidopsis thaliana resistance to Botrytis cinerea. Genetics 180, 2237–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumberger A, Marschner P. 2003. 2-Phenylethylisothiocyanate concentration and microbial community composition in the rhizosphere of canola. Soil Biology and Biochemistry 35, 445–452. [DOI] [PubMed] [Google Scholar]
- Sasse J, Martinoia E, Northen T. 2018. Feed your friends: do plant exudates shape the root microbiome? Trends in Plant Science 23, 25–41. [DOI] [PubMed] [Google Scholar]
- Schlaeppi K, Abou-Mansour E, Buchala A, Mauch F. 2010. Disease resistance of Arabidopsis to Phytophthora brassicae is established by the sequential action of indole glucosinolates and camalexin. The Plant Journal 62, 840–851. [DOI] [PubMed] [Google Scholar]
- Schmidt B, Ribnicky DM, Poulev A, Logendra S, Cefalu WT, Raskin I. 2008. A natural history of botanical therapeutics. Metabolism 57, S3–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringlis IA, de Jonge R, Pieterse CMJ. 2019. The age of coumarins in plant–microbe interactions. Plant & Cell Physiology 60, 1405–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringlis IA, Yu K, Feussner K, de Jonge R, Van Bentum S, Van Verk MC, Berendsen RL, Bakker PAHM, Feussner I, Pieterse CMJ. 2018. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proceedings of the National Academy of Sciences, USA 115, E5213–E5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomma BP, Nelissen I, Eggermont K, Broekaert WF. 1999. Deficiency in phytoalexin production causes enhanced susceptibility of Arabidopsis thaliana to the fungus Alternaria brassicicola. The Plant Journal 19, 163–171. [DOI] [PubMed] [Google Scholar]
- Tosi M, Mitter EK, Gaiero J, Dunfield K. 2020. It takes three to tango: the importance of microbes, host plant, and soil management to elucidate manipulation strategies for the plant microbiome. Canadian Journal of Microbiology 66, 413–433. [DOI] [PubMed] [Google Scholar]
- Trivedi P, Leach JE, Tringe SG, Sa TM, Singh BK. 2020. Plant–microbiome interactions: from community assembly to plant health. Nature Reviews. Microbiology doi: 10.1038/s41579-020-0412-1. [DOI] [PubMed] [Google Scholar]
- Tsai HH, Schmidt W. 2017. Mobilization of iron by plant-borne coumarins. Trends in Plant Science 22, 538–548. [DOI] [PubMed] [Google Scholar]
- Tsuchiya H, Iinuma M. 2000. Reduction of membrane fluidity by antibacterial sophoraflavanone G isolated from Sophora exigua. Phytomedicine 7, 161–165. [DOI] [PubMed] [Google Scholar]
- Turner TR, Ramakrishnan K, Walshaw J, Heavens D, Alston M, Swarbreck D, Osbourn A, Grant A, Poole PS. 2013. Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. The ISME Journal 7, 2248–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udvardi M, Poole PS. 2013. Transport and metabolism in legume–rhizobia symbioses. Annual Review of Plant Biology 64, 781–805. [DOI] [PubMed] [Google Scholar]
- Vandenkoornhuyse P, Quaiser A, Duhamel M, Le Van A, Dufresne A. 2015. The importance of the microbiome of the plant holobiont. New Phytologist 206, 1196–1206. [DOI] [PubMed] [Google Scholar]
- Verbon EH, Trapet PL, Kruijs S, Temple-Boyer-Dury C, Rouwenhorst TG, Pieterse CMJ. 2019. Rhizobacteria-mediated activation of the Fe deficiency response in arabidopsis roots: impact on Fe status and signaling. Frontiers in Plant Science 10, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikram A, Jayaprakasha GK, Jesudhasan PR, Pillai SD, Patil BS. 2010. Suppression of bacterial cell–cell signalling, biofilm formation and type III secretion system by citrus flavonoids. Journal of Applied Microbiology 109, 515–527. [DOI] [PubMed] [Google Scholar]
- Voges MJEEE, Bai Y, Schulze-Lefert P, Sattely ES. 2019. Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proceedings of the National Academy of Sciences, USA 116, 12558–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorholt JA, Vogel C, Carlström CI, Müller DB. 2017. Establishing causality: opportunities of synthetic communities for plant microbiome research. Cell Host & Microbe 22, 142–155. [DOI] [PubMed] [Google Scholar]
- Wallace JG, Kremling KA, Kovar LL, Buckler ES. 2018. Quantitative genetics of the maize leaf microbiome. Phytobiomes Journal 2, 208–224. [Google Scholar]
- Wei Z, Jousset A. 2017. Plant breeding goes microbial. Trends in Plant Science 22, 555–558. [DOI] [PubMed] [Google Scholar]
- Weigel D 2012. Natural variation in Arabidopsis: from molecular genetics to ecological genomics. Plant Physiology 158, 2–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille L, Messmer MM, Studer B, Hohmann P. 2019. Insights to plant–microbe interactions provide opportunities to improve resistance breeding against root diseases in grain legumes. Plant, Cell & Environment 42, 20–40. [DOI] [PubMed] [Google Scholar]
- Wink M 2003. Evolution of secondary metabolites from an ecological and molecular phylogenetic perspective. Phytochemistry 64, 3–19. [DOI] [PubMed] [Google Scholar]
- Wintermans PC, Bakker PA, Pieterse CM. 2016. Natural genetic variation in Arabidopsis for responsiveness to plant growth-promoting rhizobacteria. Plant Molecular Biology 90, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters FC, Gershenzon J, Vassao DG. 2016. Benzoxazinoids: reactivity and modes of action of a versatile class of plant chemical defenses. Journal of the Brazilian Chemical Society 27, 1379–1397. [Google Scholar]
- Xiao Y, Liu H, Wu L, Warburton M, Yan J. 2017. Genome-wide association studies in maize: praise and stargaze. Molecular Plant 10, 359–374. [DOI] [PubMed] [Google Scholar]
- Yang L, Ding W, Xu YQ, Wu DS, Li SL, Chen JN, Guo B. 2016. New insights into the antibacterial activity of hydroxycoumarins against Ralstonia solanacearum. Molecules 21, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Zhao J, Wen T, et al. 2018. Root exudates drive the soil-borne legacy of aboveground pathogen infection. Microbiome 6, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamioudis C, Hanson J, Pieterse CM. 2014. β-Glucosidase BGLU42 is a MYB72-dependent key regulator of rhizobacteria-induced systemic resistance and modulates iron deficiency responses in Arabidopsis roots. New Phytologist 204, 368–379. [DOI] [PubMed] [Google Scholar]
- Zha Y, Westerhuis JA, Muilwijk B, Overkamp KM, Nijmeijer BM, Coulier L, Smilde AK, Punt PJ. 2014. Identifying inhibitory compounds in lignocellulosic biomass hydrolysates using an exometabolomics approach. BMC Biotechnology 14, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhalnina K, Louie KB, Hao Z, et al. 2018. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nature Microbiology 3, 470–480. [DOI] [PubMed] [Google Scholar]
- Zhang YM, Rock CO. 2004. Evaluation of epigallocatechin gallate and related plant polyphenols as inhibitors of the FabG and FabI reductases of bacterial type II fatty-acid synthase. Journal of Biological Chemistry 279, 30994–31001. [DOI] [PubMed] [Google Scholar]