

Abstract
Acute pancreatitis (AP), an acute inflammatory disorder of the exocrine pancreas, is one of the most common gastrointestinal diseases encountered in emergency departments with no specific treatments. Laboratory-based research has formed the cornerstone of endeavours to decipher the pathophysiology of AP, because of the limitations of such study in human beings. While this has provided us with substantial understanding, we cannot answer several pressing questions. These are: (a) Why is it that only a minority of individuals with gallstones, or who drink alcohol excessively, or are exposed to other causative factors develop AP? (b) Why do only some develop more severe manifestations of AP with necrosis and/or organ failure? (c) Why have we been unable to find an effective therapeutic for AP? This manuscript provides a state-of-the-art review of our current understanding of the pathophysiology of AP providing insights into the unanswered clinical questions. We describe multiple protective factors operating in most people, and multiple stressors that in a minority induce AP, independently or together, via amplification loops. We present testable hypotheses aimed at halting progression of severity for the development of effective treatments for this common unpredictable disease.
INTRODUCTION
Acute pancreatitis (AP) is a leading cause for emergency hospital admission globally. While often self-limiting, 20% of patients with AP progress to a more severe form with organ failure1 and mortality between 15% and 35%.2 In the absence of specific treatment, current management centres around early fluid resuscitation, pain control and nutrition.3
There is general consensus that human AP can be defined by at least two of three criteria: (1) characteristic abdominal pain; (2) serum amylase and/or lipase levels three or more times the upper limit of normal and (3) characteristic abdominal imaging findings.4 Much research has been undertaken to decipher the mechanisms underlying the pathogenesis of AP. Most of this has used animal models,5 largely because of the limitations of pathophysiological studies of AP in human beings. Moreover, our lack of understanding of the natural history of AP5 and the relative inaccessibility of pancreatic tissue in patients with AP have resulted in an incomplete understanding of AP pathogenesis. We do not know if the many variations5 between human and experimental animals account for the failure of clinical trials testing agents effective in animal models of AP. Unlike in experimental AP, clinical trials have initiated treatments up to 72 hours after admission,6 but efficacy is likely to require earlier administration.
We aim to provide a state-of-the-art, up-to-date review of current understanding of the pathophysiology of AP, proposing ways to address unanswered clinical questions. We describe how the balance of protective factors is disrupted by factors that induce AP in a minority of individuals, stressors that act independently or together, including via amplification loops. Two critical thresholds in the development of AP are proposed: threshold 1 is exceeded at the onset of AP and threshold 2 is exceeded at the onset of severe AP with persistent organ failure (hereon ‘severe’ AP describes AP with organ failure for ≥48 hours, whether using the Revised Atlanta7 or Determinant-Based8 Classifications, see figure 1). We present testable hypotheses and a framework for future research to develop effective therapeutics to ameliorate and prevent this unpredictable disorder.
Figure 1.
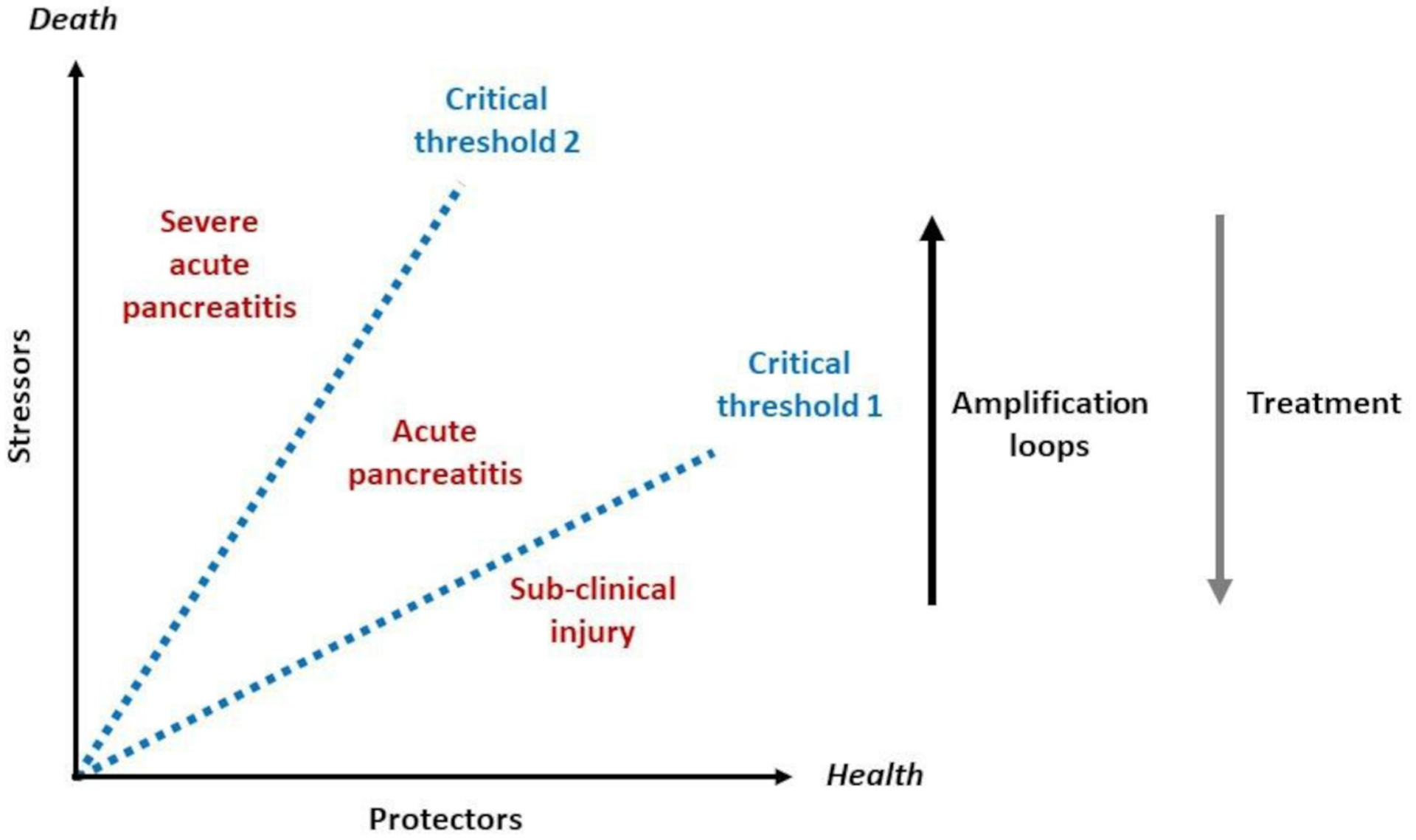
Critical thresholds in the balance between protective factors and stressors that determine the onset of clinically apparent AP and progression to severe AP, increasing the risk of death. Amplification loops, for example, diminished ATP supply leading to necrosis and necrosis leading to inflammation, increase the likelihood of an individual exceeding critical threshold 1 (AP) and critical threshold 2 (severe AP). Effective interventions (treatment) will reduce the impact of stressors on the onset and progression of AP and reduce the likelihood to exceeding either threshold. AP, acute pancreatitis.
THE PATHOBIOLOGY OF EXPERIMENTAL AP
We posit that exposure of the pancreas to factors associated with AP does not usually lead to AP because of adaptive and protective mechanisms in place or deployed in response to stressors. When adaptive and protective mechanisms are overwhelmed, AP ensues, and severity is accelerated by feed forward and amplification pathways that interact stochastically (see figure 2). Many factors may contribute, including toxic fatty acids locally,9 and mesenteric lymph components systemically.10
Figure 2.
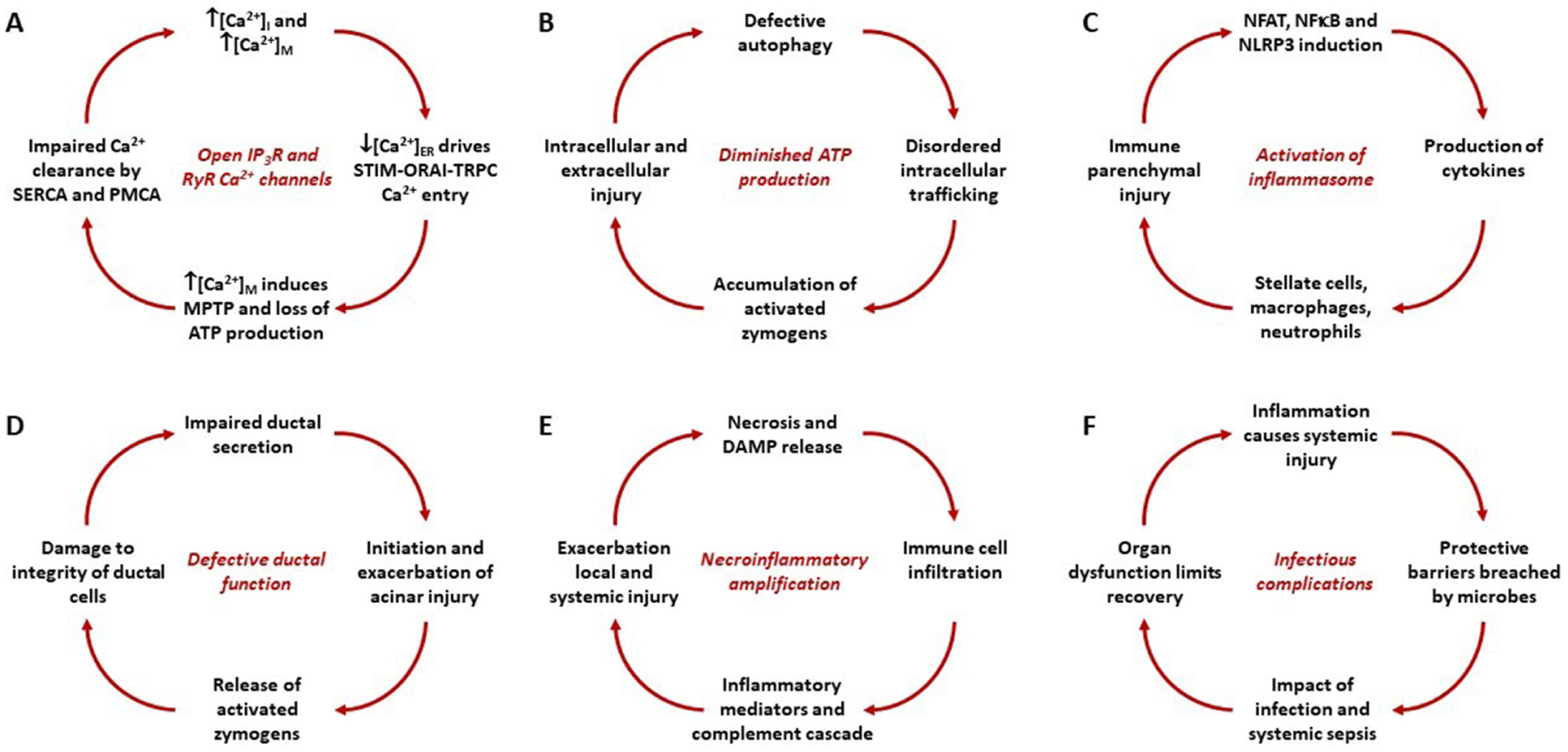
Amplification loops that contribute to an individual exceeding critical threshold 1 (Acute pancreatitis (AP)) and critical threshold 2 (severe AP) in the pathogenesis and progression of AP. While distinct, the loops interact and drive each other. (A) Toxins that increase the open probability of inositol trisphosphate receptor (IP3R) and ryanodine receptor (RyR) Ca2+ channels induce release of Ca2+ from the endoplasmic reticulum (ER), raising the concentration of Ca2+ in the cytosol ([Ca2+]i) and mitochondria ([Ca2+]m) while lowering that in the ER ([Ca2+]ER), which drives formation of stromal interaction molecule-calcium release-activated calcium channel-transient receptor potential canonical cation channel (STIM-ORAI-TRPC) puncta for Ca2+ entry. Continued elevation of [Ca2+]m induces the mitochondrial permeability transition pore (MPTP) with loss of mitochondrial membrane potential and ATP production, which impairs Ca2+ clearance by the sarcoendoplasmic reticulum and plasma membrane Ca2+ ATPases (SERCA and PMCA) pumps, further increasing [Ca2+]i and [Ca2+]m. (B) Diminished ATP production induces defective autophagy, disordered endolysosomal trafficking, building up activated digestive enzymes and further driving intracellular and extracellular injury. (C) Sustained elevations of [Ca2+]i and [Ca2+]m induce nuclear factor of activated T cells (NFAT), nuclear factor kappa B (NF-κB) and the Nucleotide-binding oligomerisation domain, Leucine rich Repeat and Pyrin domain containing (NLRP) inflammasome system resulting in cytokine release, stimulating resident and infiltrating leucocytes, causing further parenchymal injury. (D) Acinar-ductal tango: ductal secretion may be diminished by inherited or acquired defects of cystic fibrosis transmembrane conductance regulator (CFTR) function or ductal obstruction that in turn injures acinar cells that release activated zymogens which further diminish ductal function. (E) Necrosis releases damage-associated molecular patterns (DAMPs, for example, histones, DNA) that attract and activate leucocytes which release cytokines, inflammatory mediators and neuropeptides. Parenchymal and circulating inflammatory mediators exacerbate local pancreatic and distant systemic organ injury resulting in further necrosis. (F) Inflammatory injury of the gut, lung and other organs damage mucosal and epithelial protective barriers, which bacteria breach to infect local and distant sites. Systemic sepsis impacts on organ function, prompting further inflammation and tissue injury.
Disruption of acinar cell homeostasis
Premature digestive enzyme activation
The pancreatic acinar cell is the functional unit of the exocrine pancreas that undertakes highly regulated synthesis, storage and secretion of digestive enzymes.11 These zymogens are packaged as proenzymes in secretory granules, separated from lysosomal hydrolases, which would otherwise induce intracellular zymogen activation. Numerous further mechanisms within the acinar cell and interstitium, including α1-antitrypsin, α1-macroglobulin, α1-intertrypsin, protease-activated receptor 2,12 and pancreatic secretory trypsin inhibitor13 protect the cell from zymogen activation and AP. Experimental evidence has confirmed that the first changes following application of noxious stimuli include a block in zymogen secretion from progressive disassembly of microtubules and actin filaments,14 and premature activation of trypsinogen to trypsin15 from colocalisation of trypsinogen with lysosomal enzymes such as cathepsin B within fragile vacuoles,16 damaging the acinar cell.
Intracellular calcium overload
Excessive intracellular Ca2+ concentrations within the cytosolic ([Ca2+]i) and mitochondrial ([Ca2+]m) compartments induced by toxins are critical in premature digestive enzyme activation during human and murine acinar cell injury and experimental AP.17 The central role of physiological Ca2+ signalling in the synthesis and secretion of pancreatic zymogens, reviewed elsewhere,18 leaves the pancreas vulnerable to disordered Ca2+ signalling induced by toxins that cause AP. One breakthrough has been unravelling disordered acinar cell Ca2+ signalling in AP pathogenesis, offering new avenues for therapy.19–21 Ca2+ stored in the endoplasmic reticulum (ER) is released into the cytosol and mitochondria via inositol trisphosphate receptor and ryanodine receptor Ca2+ release channels, normally elicited by cholecystokinin and/or acetylcholine secretagogue stimulation. Ca2+ release is excessive following exposure to pancreatitis toxins, including bile acids, fatty acid ethyl esters, fatty acids and hormonal hyperstimulants.21 Unsaturated fatty acids (eg, linoleic, linolenic and oleic) result in higher Ca2+ release than saturated fatty acids (eg, palmitic and stearic), increase cytoplasmic leakage of lactate dehydrogenase and cytochrome C, upregulate inflammatory mediators and inhibit mitochondrial complexes I and V, causing a drop in ATP levels to induce necrosis.9,22 In response to decreased ER Ca2+ concentrations, punctate ER-plasma membrane connections form from stromal interaction molecule, Ca2+ release-activated Ca2+ channel protein 1 (Orai1) and transient receptor potential cation channel complexes, to replenish stores from the cell exterior.23 Continued toxic stimulation sustains ER Ca2+ release and cellular Ca2+ entry, overloading mitochondria via the Ca2+ uniporter and triggering high conductance opening of the mitochondrial permeability transition pore (MPTP).17 Hydrogen ions enter the mitochondrial matrix through the MPTP with loss of the electrochemical gradient required for ATP production.17 Decreased ATP impairs Ca2+ clearance by plasma membrane Ca2+ ATPase (PMCA) and sarco/ER Ca2+-ATPase pumps, exacerbating [Ca2+]i and [Ca2+]m elevations.17
Sustained overload of [Ca2+]i and [Ca2+]m is also induced by high pressure and Piezo1-mediated Ca2+ entry of sufficient magnitude and/or duration to prompt transient receptor potential vanilloid type 4 (TRPV4) channel Ca2+ entry.20 ATP production is again impaired and autophagy, secretion and endolysosomal trafficking disrupted; activated zymogens are dispersed in the cytosol; calcineurin, nuclear factor of activated T cells (NFAT) and the Nucleotide-binding oligomerisation domain, Leucine rich Repeat and Pyrin domain containing (NLRP)3 inflammasome are activated; inflammatory cytokines are released; reactive oxygen species accumulate, initiating apoptosis; with deficient ATP, necrosis supervenes; and multiple damage-associated molecular patterns (DAMPs) enter the interstitium, lymphatic, portal and systemic circulations, driving immune responses that exacerbate pancreatic and systemic injury.10,17,20
Variable threshold of injury
There is mounting evidence to suggest that repeated exposure to stressors results in adaptive, protective responses but that AP occurs when these are inhibited or overwhelmed.24 In animal models, cigarette smoking inhibits the protective unfolded protein response (UPR) and the transcription factor X-box binding protein 1 (XBP-1) generated by ethanol exposure, resulting in ER stress and necrosis not observed with either alcohol or smoking alone.24 Moreover, pharmacologic or genetic inhibition of XBP-1s leads to acinar cell damage, disordered autophagy and AP.24 Thus, when the normally protective UPR, upregulated by cellular stresses, is inhibited or overwhelmed, pathologic responses ensue.24
The inflammatory cascade
Parenchymal and leucocyte pathways
Nuclear factor kappa B (NF-κB) is activated by acinar cell injury25 independently of trypsinogen,26 triggering transcription of key pronflammatory mediators that recruit leucocytes into the pancreas and distant organs. NF-κB is comprised of homodimers and heterodimers of the Rel family that exert multiple, often conflicting, effects varying between acinar and myeloid cells. The NF-kB homodimer RelA/p50, rapidly induced on pancreatic injury, contributes to resolution of inflammation.27 Acinar RelA/p65 also protects against chronic inflammation,28 whereas myeloid RelA/p65 promotes fibrogenesis characteristic of chronic pancreatitis (CP). Several mechanisms are common to parenchymal and immune cells: cytosolic Ca2+ elevations activate calcineurin and NFAT,29 driving cytokine production and release; the NLRP inflammasome system is activated, coordinating immune responses, notably interleukin release.30 Tumour necrosis factor alpha (TNFα) is central, produced by injured parenchymal cells, resident macrophages and infiltrating leucocytes. Through chemotactic cytokine leucocyte (CCL) and chemokine receptor interactions, for example, CCL2/chemotactic cytokine receptor-2, monocytes are recruited and differentiate into TNFα-activated macrophages. Neutrophils are among the first immune cells recruited into the pancreas and lung, demonstrated by cellular, chemokine and adhesion molecule blockade studies.31 Neutrophils activate trypsinogen in acinar cells32 as do trypsinogen in endocytosing macrophages, contributing to AP severity.33 These cells amplify the inflammatory cascade, generating many chemokines and cytokines including interleukins 1 and 6 (IL-1 and IL-6), and intercellular adhesion molecule 1 (ICAM-1) to promote pancreatic and extrapancreatic multiorgan injury. Recent results, together with findings made from other forms of organ injury, suggest significant interactions between innate and adaptive immune responses.30 NF-κB activation in myeloid cells and expression of IL6 by macrophages mediatethe development of murine AP34 by binding to its membrane receptor (IL6R) or complexing with soluble IL6R, both of which activate IL6 signal transducer (glycoprotein 130 (GP130)). GP130 activates the Janus kinase 2 to phosphorylate signal transducer and activator of transcription 3 (STAT3) at Y705. In infiltrating myeloid cells, NF-κB regulates production of IL6 at sites of inflammation; the ensuing persistent activation of STAT3 results in high levels of CXCL1, which together with ICAM1 in lung endothelial cells mediates granulocyte infiltration into the lung, promoting lethal acute lung injury.34,35
Pattern recognition and pattern recognition receptors (PRRs)
DAMPs, for example, histones, high-mobility group box protein 1 (HMGB1), DNA, ATP, hyaluronic released from acinar cell injury and death, are recognised by innate immune cells of the monocyte macrophage system to induce further local and systemic inflammation.36 Widespread organ injury in severe AP notably of the gastrointestinal permeability barrier is associated with infectious complications and bacterial products, for example, lipopolysaccharide (LPS), other endotoxins and flagellin termed pathogen-associated molecular patterns (PAMPs), which also activate the innate immune system.37 DAMPs and PAMPs ligate a growing array of PRRs, including toll-like receptors (TLRs), nucleotide-binding oligomerisation domain-like receptors (NLRs and NLRP receptors), C-type lectin receptors and sensor stimulator of interferon genes.38 Crosstalk between these receptors determines the overall shape of the immune response. It is of direct interest that circulating histones, HMGB1 and DNA correlate with clinical AP severity.39
Autophagy
Autophagy is the major lysosome-mediated catabolic process by which cells eliminate damaged, defective, or unwanted cytoplasmic organelles, proteins and lipids, and recycle constituents for energy and biogenesis needs.40 Deregulation of autophagy is associated with AP pathogenesis.41 Both human and experimental pancreatitis display severe defects in acinar cell lysosomal functions with reduced enzymatic activities of lysosomal proteases, cathepsins and degradation of integral lysosome-associated membrane proteins (LAMPs) critical to the structure and function of lysosomes. The inhibition of autophagy manifests by the accumulation in acinar cells of autophagic vacuoles, filled with poorly degraded material, regarded as an early marker of experimental and human AP.
Recent studies have used genetic models to target autophagy/lysosomal pathways in the maintenance of acinar cell homeostasis and initiation of AP. Mice with pancreas-specific knockouts of key autophagy mediators autophagy related protein (Atg) 542 or Atg743 or the transcription factors TFEB and TFE3 that mediate lysosomal biogenesis and autophagy,44 and LAMP2 knockout mice,45 all develop spontaneous AP and subsequent features of CP, with trypsinogen activation, fibrosis and acinar-to-ductal metaplasia. Collectively, these findings confirm an essential role of autophagy/lysosomal pathways in maintaining pancreatic acinar cell homeostasis, and strongly implicate their disorders in initiation and development of AP.
Ductal bicarbonate (HCO3−) secretion protects the pancreas
Pancreatic ductal epithelial cells have a prominent role in maintaining the integrity of the pancreas. They secrete HCO3−-rich fluid into the ductal lumen that prevents premature activation of trypsinogen inside the lumen and washes out digestive enzymes from the pancreas. Deficient ductal function changes the composition and volume of the fluid, leading to acinar cell damage. In recent years, it has been demonstrated that low concentrations of ethanol and bile acids stimulate HCO3− secretion while high concentrations are inhibitory46,47 (see table 1). Human studies suggest moderate alcohol intake (less than 40 g/day) might be protective and reduce the risk of AP, more so in women.48 When the concentration of ethanol or bile acids reaches a toxic level, HCO3− secretion is reduced and irreversibly elevated intracellular Ca2+ results in mitochondrial damage and cell death.49 Ductal fluid secretion is also decreased due to inhibition of aquaporin-1 by bile acids.50 Taken together, the impaired ductal function contributes to the development of AP (see figure 2).
Table 1.
Protective factors that prevent acute pancreatitis throughout life except when one or more stressors build up, increasing the likelihood of acute pancreatitis, which will occur if the critical threshold is exceeded
| Protective factors | Stressors |
|---|---|
| Systemic toxin protection and clearance mechanisms | Genetic variants imposing limits on cell function |
| Facilitatory metabolic effects of islet hormones | High ductal pressure or parenchymal trauma |
| Separated, discrete acini and parenchymal lobules | Toxin exposure to which acinar cells are vulnerable |
| High output secretory units with robust acinar cells | Reduced O2 supply for oxidative phosphorylation |
| Ca2+ supply ducted within the endoplasmic reticulum | Untimely and/or excessive stimulation of secretion |
| Prompt unfolded protein response on cellular stresses | Impaired ductal function and zymogen clearance |
| Transcription/release of danger signals suppressed | Prolonged opening of inositol trisphosphate receptor and ryanodine receptor Ca2+ channels |
| Highly active acinar cells with abundant functional mitochondria | Sustained Ca2+ entry via calcium release-activated calcium channel, transient receptor potential canonical cation channel, transient receptor potential vanilloid |
| Sufficient ATP for energy-dependent functions | High conductance opening of mitochondrial permeability transition pore with loss of ΔΨM |
| Stimulus-secretion and stimulus-metabolism coupling | Diminished oxidative phosphorylation and ATP supply |
| Proper zymogen synthesis that prevents intracellular activation | Zymogen and lysosomal-protease colocalisation |
| Zymogens packaged in exclusive subcellular compartments condensed with enzyme inhibitors | Defective autophagy and endolysosomal cycling |
| Flow away with ductal secretion at high pH | Build-up of activated digestive enzymes within acinar cells |
| Enhanced ductal flow with low doses of toxins | Activation of nuclear factor of activated T cells, nuclear factor kappa B, Nucleotide-binding oligomerisation domain, Leucine rich Repeat and Pyrin domain containing signalling |
| Zymogens activated distantly in duodenum with enterokinase | Stellate and resident macrophage danger responses |
| Infiltration of activated innate immune cells | |
| Necroinflammatory amplification loops |
The acinar-ductal tango
The altered HCO3− secretion is primarily due to inhibition or mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel, which transports HCO3− into the lumen, in close association with solute carrier family 26 (SLC26) anion exchanger (Cl−/HCO3− exchanger). In patients with cystic fibrosis, the type of mutation plays a significant role in determining the risk of AP. Two cohort studies demonstrated that with mild mutations the incidence of AP is much higher.51,52 Ductal obstruction can also lead to AP. Pancreatic cancer or bile duct stones can cause obstruction and prevent the outflow of pancreatic fluid causing AP.53 On the other hand, impairment of acinar cell function induces ductal damage, which aggravates the course of AP (figure 3). Intra-acinar or luminal trypsinogen activation markedly reduces ductal HCO3− secretion through inhibition of the SLC26 anion exchanger and CFTR Cl− channels expressed on the duct cell apical membrane, lowering luminal pH. This low pH enhances trypsinogen autoactivation that, in a vicious cycle, further reduces HCO3− secretion54 (see figure 2).
Figure 3.
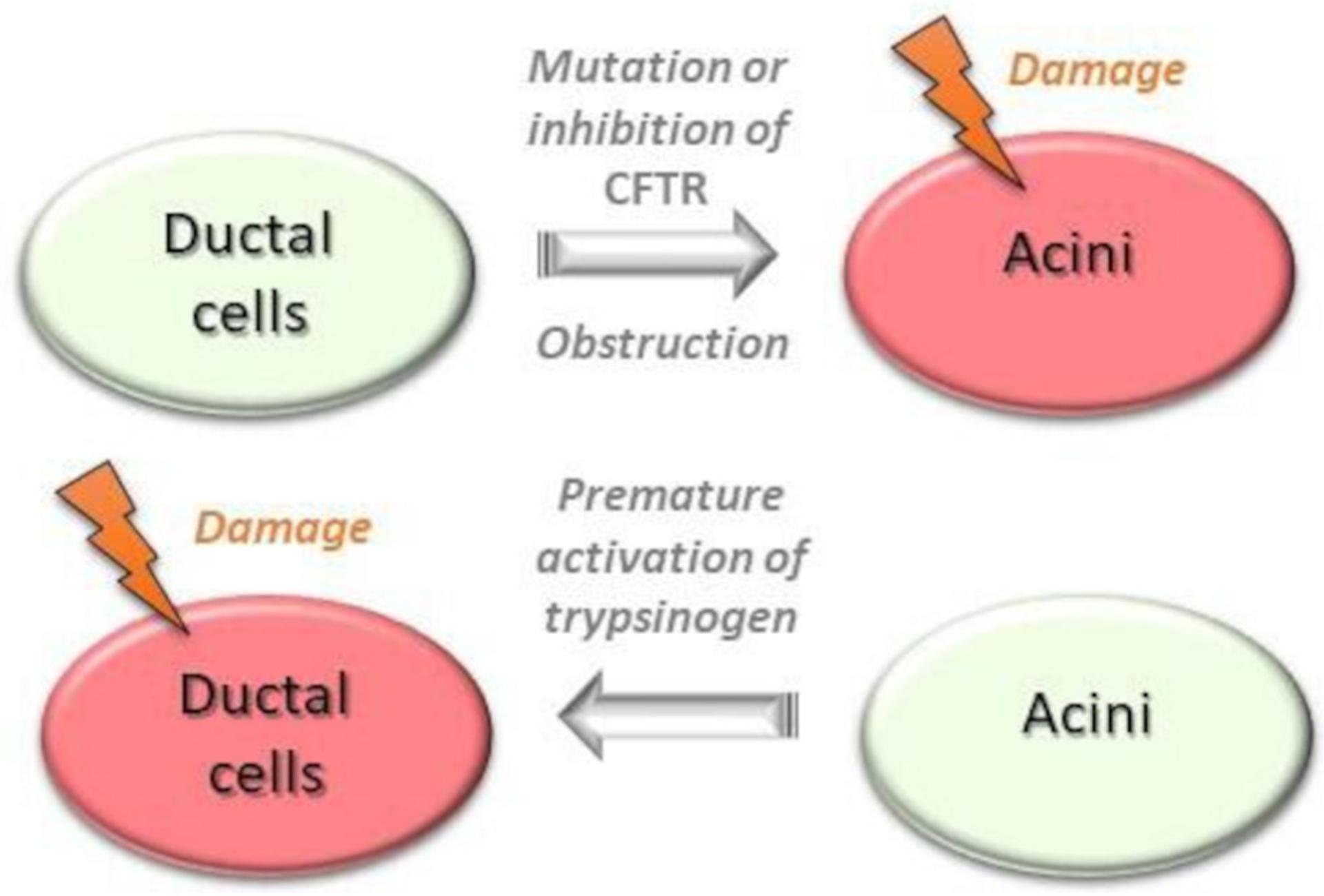
The acinar-ductal tango depicting the ductal changes that precipitate acinar injury and the effect of resultant acinar injury on ductal cell dysfunction. CFTR, cystic fibrosis transmembrane conductance regulator.
Islet hormones and neuropeptides
Beneficial effects
As there is complex interplay between the endocrine and exocrine pancreas,55 notably arterial flow from islets to acini, investigators have studied effects of islet hormones on AP. Somatostatin and its analogues inhibit basal and stimulated exocrine pancreatic secretion,56 reduce lipid peroxidation, leucocyte infiltration and necrosis in experimental AP, and promote tissue organisation and healing. It is disappointing that meta-analysis of randomised clinical trials have shown that neither somatostatin nor its analogues have efficacy in the treatment of clinical AP.57 Insulin, however, protects pancreatic acinar cells by facilitating PMCA pump Ca2+ clearance, limiting Ca2+ overload and consequent cellular injury, likely increasing nutrient supply for ATP production.58 While these findings require confirmation in vivo, early phase trials assessing the hyperinsulinaemic euglycemic clamp technique can be envisaged, if justified by accumulating preclinical data.
Deleterious effects
Neuropeptides and neuroinflammation have been a focus in AP research for many years,59,60 also pursued avidly in brain research (there are now several thousand neuropeptides identified); earlier AP work investigated the kinin-kallikrein system,59 later substance P60 and galanin.61 Kinins are derived from kininogens that modulate blood pressure, vascular permeability and pain transmission through the bradykinin receptor. Seminal work 60 years ago identified a key role for bradykinin in hypotension, pancreatic oedema, fluid shifts and haemoconcentration in AP,59 leading to early trials of trasylol. Unfortunately, preclinical efficacy was not confirmed to confer clinical benefit.57 Substance P, a prototypical tachykinin, contributes to AP through neurogenic inflammation,62 mediated via neurokinin-1 (NK1) receptors60 present in acinar cells upregulated in AP.60 Substance P increases vascular permeability and interstitial oedema,60 potentiates effects of other secretagogues,63 upregulates production of proinflammatory mediators, and increases NF-κB activation,64 pancreatic myeloperoxidase activity and necrosis in experimental AP.65 Galanin, another neuropeptide ubiquitously present in the nervous system, contributes to AP,61 potentiating effects of other secretagogues,66 promoting neuroinflammation,65 reducing pancreatic microcirculation, enhancing neutrophil function67 and inhibiting ductal HCO3− secretion.68 Neuropeptides and neuroinflammation contribute to pain in AP, implicating bradykinin and TRPV1.62
Genetic mutations increasing the susceptibility to AP
Variations in several genes in many populations have been linked to increased susceptibility to CP, which may be heralded by a sentinel attack of AP. Genetic mutations affect different processes and pathways, including trypsin activation (PRSS1, serine protease inhibitor Kazal type 1 (SPINK1) and chymotrypsin C (CTRC)),69 chloride and HCO3− transport (CFTR),70 protein misfolding response (carboxypeptidase A1, CPA1),71 and Ca2+ signalling (TRPV6)72 and suggest a shared pathobiology between AP and CP. Variations in the claudin 2 gene (CLDN2) alter the inflammatory response to acinar cell destruction and fibrosis.73 The inheritance varies from autosomal dominant for PRSS1, autosomal recessive for CFTR, to complex and sporadic inheritance patterns for other genes. Apte et al74 suggested that investigation into protective mechanisms associated with CFTR mutations may uncover pancreatic susceptibility to alcohol toxicity. Maléth et al47 provided clinical evidence of lower levels of CFTR messenger RNA and protein expression in pancreatic tissue in patients with AP compared with healthy volunteers and evidence that alcohol and fatty acids inhibit CFTR activity and fluid and bicarbonate secretion in pancreatic ductal epithelial cells. In patient populations, variations in CFTR and SPINK1 are the most frequently detected. Other than an earlier age at presentations, clinical features in patients with genetic mutations are similar to those without.75 Koziel et al76 found mutations in SPINK1 (especially p.N34S) predispose individuals to AP, especially with alcohol abuse, and promote more severe disease. Polonikov et al77 confirmed a robust association of polymorphism rs10273639 at PRSS1-PRSS2 with AP modified by alcohol consumption. Since AP, recurrent AP and CP represent a disease continuum,78 it is reasonable to conclude that variations in the described genes play a role in AP pathogenesis. A recent meta-analysis observed significant risk of AP with SPINK1 variants; the lack of association with PRSS1 was attributed to exclusion of paediatric populations, and few data were available for CFTR and CTRC variants.79 Interestingly, the meta-analysis also identified an increased risk of AP related to variation in genes linked to IL-1B and IL-6 cytokine production.79 Genome-wide association studies have identified CLDN2 and PRSS1-PRSS2 variants altering the risk of alcohol-related and sporadic pancreatitis.73 It is, thus, plausible that AP due to different risk factors might have a distinct genetic background making the interpretation of genetic results difficult (as cohorts in the subgroups are small and phenotypes not well characterised). These associations need replication and must be reported currently with caution, including data on IL6 and other immune genes.
Pancreatic stellate cells in necroinflammatory amplification
Stellate cells are resident pancreatic cells next to acinar cells. The teams of Apte and Bachem80,81 first identified stellate cells and their role in pancreatic pathophysiology. Evidence has emerged to support the existence of a ‘necrotic amplification loop’ in AP between acinar and stellate cells.82 It has been demonstrated in experimental AP that initiation of an inflammatory signal potentiates acinar cell injury, further activating inflammatory signals and acinar cytokine/adipokine secretion that amplify the inflammatory response. The resultant necrosis in a proportion of acinar cells leads to the release of activated proteases and bradykinin that act on neighbouring stellate cells by evoking Ca2+ signals. Trypsin-induced and bradykinin-induced Ca2+ signals within stellate cells, in turn, activate nitric oxide (NO) synthase to produce NO. NO diffuses into neighbouring acinar and endothelial cells exacerbating parenchymal cellular damage, decreasing the pancreatic microcirculation and increasing adherent leucocytes in the pancreas.83 This results in a vicious circle termed a ‘necrotic amplification loop’82 that intensifies local and systemic inflammation in AP. Stellate cells may be activated directly by ethanol and its metabolites to secrete proinflammatory84 and fibroinflammatory19 signals. Stellate cells express many membrane receptors including TLRs85 that can be stimulated by growth factors, cytokines and DAMPs released by damaged acinar or immune cells. LPS binds to TLRs on stellate cells, activating fibroinflammatory signals mediated by extracellular Ca2+ entry.19 There are high levels of circulating LPS in alcohol-fed animals, in alcoholics and patients with AP,86 likely due to alcohol-induced increases in gut permeability; furthermore, LPS injection induces AP in alcohol-fed animals.74
Pancreatic stellate cells contribute to reorganisation of the pancreatic environment by producing or degrading extracellular matrix (ECM) molecules. ECM components (eg, fibronectin, collagen I, etc) are critical to multiple physiological and pathological signalling pathways including inflammation, tissue damage regulation and might provide a scaffold for tissue repair through interaction with acinar cell integrin receptors.87,88 In this context, they may influence the balance between protectors and stressors in determining the severity of AP, and the risk of recurrent episodes.
What critical threshold of injury triggers AP?
Some or all above events may unfold when the pancreas is exposed to aetiological triggers. Mechanisms deciphered from preclinical experiments have clinical parallels, notably disordered Ca2+ signalling within human pancreatic acinar cells and their consequent injury, innate immune responses, mediation by DAMPs, PAMPs and PRRs, and necroinflammatory amplification.20 Substantial gaps remain, as translation is necessarily incomplete. Unlike clinical AP, experimental AP is predictable and consistently replicated, although with significant differences between species, aetiologies and assessment methods.5 Preclinical models differ from human disease, but preclinical investigations remain essential to advance our understanding of and therapies for AP, through a translational pipeline. While preclinical AP displays a continuum of injury, clinical AP is defined by two of the three criteria outlined earlier, after an emergency presentation to hospital or complicating an inpatient stay for other reasons. This indicates a critical threshold has been exceeded in patients developing clinical AP. While this threshold will typically have been reached prior to diagnosis, the time of diagnosis is preferred to represent when this has occurred, to enable and facilitate areas of research. One important area would be more nearly complete and rapid diagnosis of all AP globally, for example, from the onset of symptoms, essential to improved outcomes.
While Gryshchenko et al suggested AP is characterised pathobiologically by injury of ‘a portion of acinar cells’,82 we proposed previously that AP occurs on injury of a ‘critical mass’ of acinar cells,89 accurate measurement of which, for example, using activation peptides requires further development. Mounting evidence supports a balance between protective systems and multiple stressors (see table 1); when stressors overwhelm protectors, parenchymal injury occurs. If inflammatory signals from injured acinar cells are amplified by stellate cells, resident macrophages and infiltrating leucocytes, with release of neuropeptides and other inflammatory mediators, classical clinical features develop, namely, pain, swelling and loss of function. In AP, feed forward necroinflammatory amplification can be so profound that extensive necrosis and organ failure may supervene. Therefore, we propose that an attack of clinically evident AP is caused by a critical level of acinar cell injury. This first critical threshold is reached when local protective responses are exceeded, a nidus of inflammation propagates, and multiple inflammatory mediators are released. Feed forward necroinflammatory amplification loops increase local pancreatic damage and spill into a systemic inflammatory response, which above a second critical threshold induces distant organ injury that can be profound (figure 4).
Figure 4.
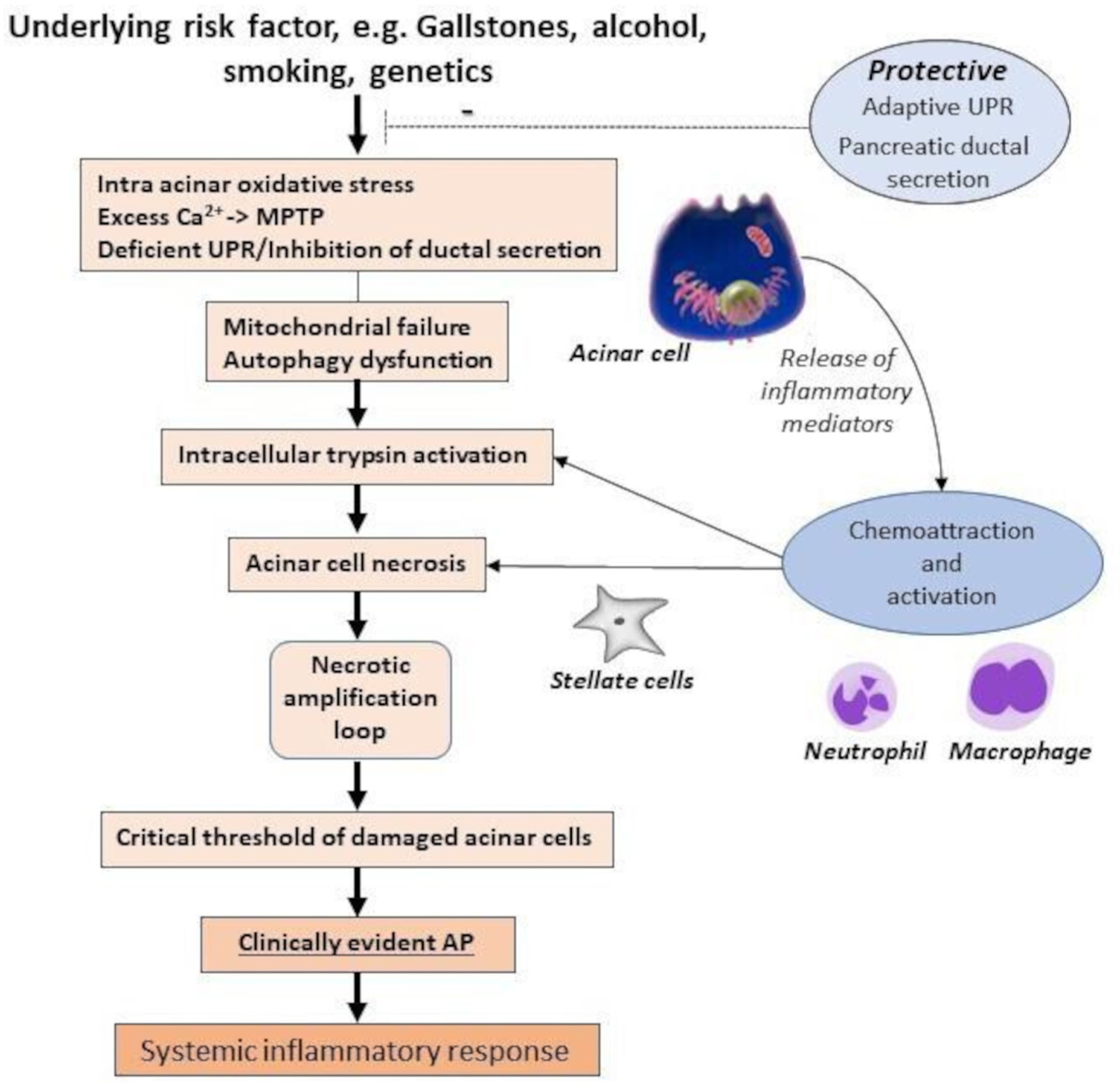
The multifactorial ‘critical threshold’ hypothesis in the causation of AP. AP, acute pancreatitis; Ca2+, free cytosolic calcium; MPTP, mitochondrial permeability transition pore; UPR, unfolded protein response.
IMPLICATIONS FOR CLINICAL AP
Epidemiology and health services research
Much population-based research can address how individuals exceed the critical thresholds proposed, which may require analysis of large data sets. Basic to this are figures for the incidence of AP, estimated at ~35 individuals per 100 000 people/year, with marked variation between countries, and mortality of 2%–5%.90 Detailed data on the epidemiology of AP outside Europe and North America are required to assess global burden, including age, gender, lifestyle, aetiology, disease course and outcome. Hospital admission underpins the diagnosis of AP, that is, when the first critical threshold can be identified to have been exceeded, but the determinants and timing of hospital admission are largely undefined, including individual biological and psychosocial factors, and the nature, availability and accessibility of health service provision.90 Factors affecting access to and quality of hospital care are important in addressing health inequalities, health service policy and financing globally. AP is the most common disease of the pancreas, yet there have been relatively few large-scale genomic studies including many patients with typical etiologies,77,79 unlike for CP. DNA collection and storage from patients with well-characterised AP should be a priority to feed into well designed and funded consortia studies. The most common cause of AP is gallstones followed closely by heavy alcohol consumption, although reversed in some countries with notable differences to explore, for example, hypertriglyceridaemia in China,91 related to life style and increasing in incidence.92 Addressing the aetiology of AP prevents future attacks, but much investigation may be left for varying periods after onset, when it may be too late to detect for example, high triglyceride levels or other aetiologies. Comparative trials of alternative diagnostic and preventative algorithms would strengthen focus on appropriate workup and preventative strategies.
Alcohol consumption is the most common cause of recurrent AP, the risk of which can be reduced substantially by abstinence. Fewer than 5% of heavy drinkers, however, develop pancreatitis,93 which remains largely unexplained. Short-term heavy drinking increases the risk of AP. A 52% (RR 1.52, 95% CI: 1.12 to 2.06) increased risk of AP has been estimated for every five standard spirit drink increment consumed on a single occasion.94 More recently, the effects of smoking have been noted; smoking ≥1 pack/day is associated with recurrent AP (and CP), independent of alcohol.95 Other health-related behaviours such as high-fat diets may push individuals over the first critical threshold. Animal studies demonstrate that while alcohol increases the vulnerability to AP, additional triggers, such as cholecystokinin (CCK) administration, are needed to induce AP.96 CCK is an endogenous hormone stimulated by diets high in fat and protein.97 Indeed, high intakes of saturated fat and cholesterol are positively associated with the incidence of gallstone AP.98 Studies are needed to investigate the short-term and long-term impact of diet on the risk of AP, including intake shortly before the onset of AP, when two or more factors may cause an individual to exceed the first or second critical threshold outlined.
Precision patient management
Despite our seemingly detailed understanding of the pathogenesis of AP derived from animal models, we have many unanswered questions in human AP, which is significantly more heterogeneous and may require substantial omics data to unravel. Figure 5 presents a multicausal model of AP with interventions to target gaps in understanding risk factors and their threshold for disease causation. Why does AP develop in some, but not other people exposed to known risk factors for AP? How precisely might the risk of AP from gallstone passage through the ampulla of Vater be determined, for example, number, size, shape, bile constituents, morphology and function of the biliary tree, Oddi’s sphincter and exocrine pancreas? If accurate determination of risk were possible, might prophylactic cholecystectomy be appropriate? We know cholecystectomy for gallstones99 and abstinence from smoking and/or alcohol reduce the risk of AP. Nevertheless, patients may develop recurrent AP following cholecystectomy for presumed gallstone AP.100 What impact has an episode of AP on the likelihood of a subsequent episode? How does this vary between individuals? Multiple factors including diet, quantity and type of alcohol, and pre-existing genetic variants may contribute to an episode of AP, each of which alone would not necessarily have triggered an episode. This is not uncommon in children, especially in those with recurrent AP.101 Obesity contributes to an amplified SIRS in AP and is associated with local and systemic complications, including mortality.102 How do we determine the primary cause to prevent future episodes? Postendoscopic retrograde cholangiopancreatography AP has presented a unique opportunity to institute and further develop preventative strategies. Fastidious patient selection and informed decision-making, prophylactic rectal non-steroidal anti-inflammatory drugs, careful guidewire-assisted cannulation, pancreatic stent placement and prompt intravenous fluid hydration have reduced but not removed the risk of AP,103 necessitating further trials.
Figure 5.
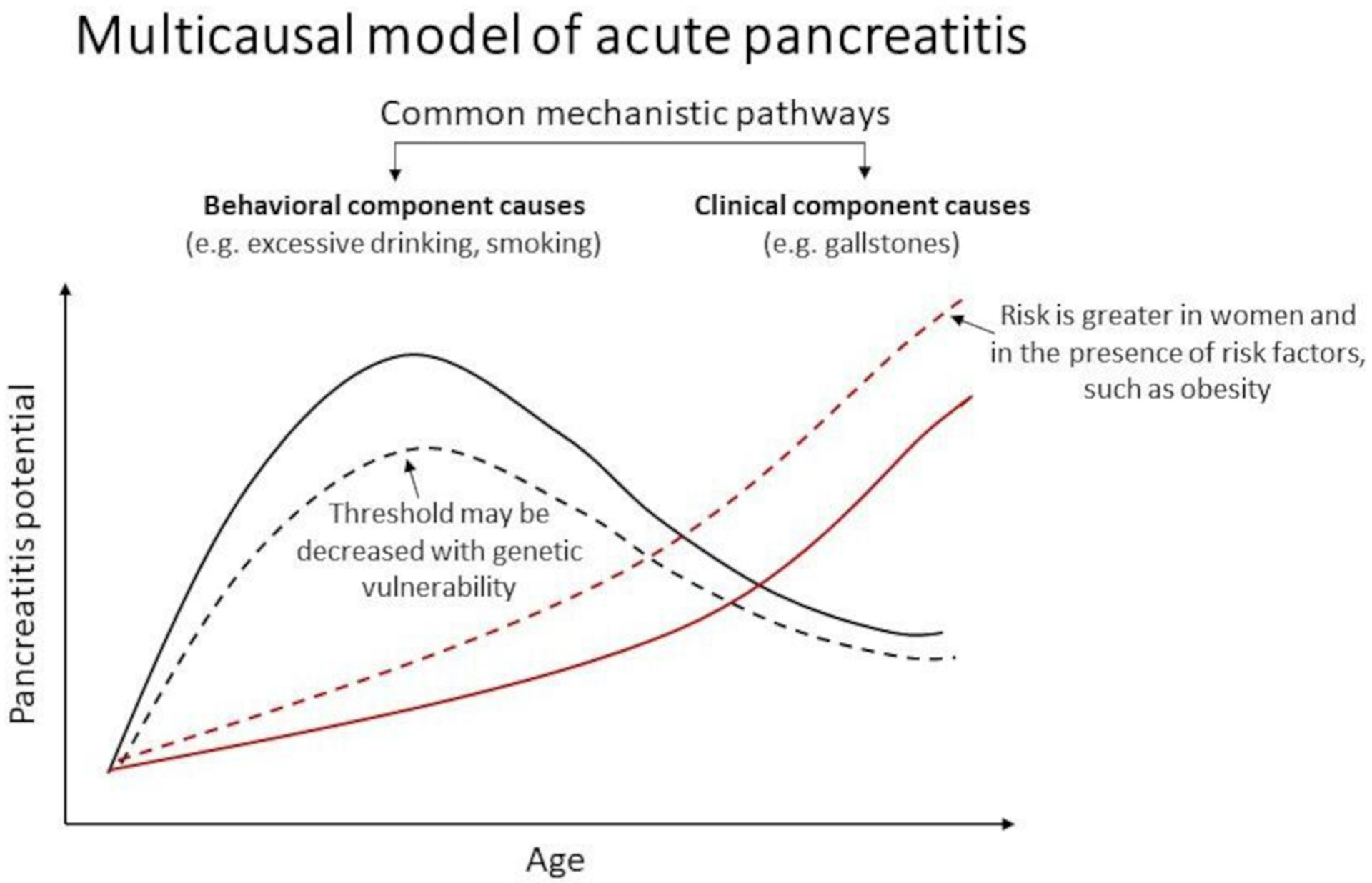
This hypothesised multicausal model of acute pancreatitis differentiates two main types of causes: behavioural and clinical. Behavioural component causes, such as excessive drinking, that are more common in young adults to middle aged people increase the potential for pancreatitis (solid black line). People with greater genetic vulnerability (eg, mutations in known genetic susceptibility factors, such as cystic fibrosis transmembrane conductance regulator and serine protease inhibitor Kazal) may experience pancreatitis at a lower threshold of drinking (dotted black line). Clinical component causes, such as gallstones, increase with age (solid red line). The risk is elevated in women, and in the presence of metabolic factors, such as obesity. Among other causes, metabolic factors, such as hypertriglyceridaemia-induced pancreatitis, are seen in individuals with an underlying lipid abnormality who have one of more secondary factors, such as diabetes.
Why do some but not other people with AP develop organ dysfunction and exceed the second threshold? Animal studies suggests the magnitude of noxious stimuli is key, but clinical quantification is elusive, as is accurate identification of severity on admission. Similar questions posed concerning the first critical threshold, including multiple cocontributory factors, apply to the second critical threshold. Despite our knowledge of the clinical course and underlying mechanisms of AP, we are unable to prevent its progression once the disease continuum has set in, treatment for which is primarily supportive and directed at complications. Early judicious intravenous fluid resuscitation corrects hypovolaemia from third-space fluid losses to maintain or restore pancreatic, splanchnic and systemic perfusion.99 The optimum fluid type and methods of monitoring to avoid overadministration are undetermined, however, and why only some patients develop pancreatic necrosis and distant organ dysfunction despite being adequately fluid resuscitated requires determination. Since a high haematocrit is associated with poorer outcome, earlier presentation and earlier fluid resuscitation is desirable; this being so, patients should be encouraged to present as soon as possible after symptom onset from AP. Lung injury, the most common distant organ dysfunction, has multiple potential contributors including circulating phospholipase A2, elastase, LPS and IL6-IL6R complexes. These pathways have been targeted effectively in animal models but clinical trials have failed to match expectations thus far, suggesting the need to test new treatments more rapidly after diagnosis for example, IL6 antagonists or target alternative pathways, for example, the kynurenine mono-oxygenase pathway.104 Hyperactive immune responses are probable instruments of injury, so unravelling any contribution of immune response gene variants is a priority, and immune targeting may be appropriate. A correlated persistent inflammation, immunosuppression and catabolism syndrome may contribute to late patient death.105 It is hoped that the results of the rapid treatment of acute pancreatitis with infliximab (RAPID-I) Trial (NCT03684278) testing early administration of the TNF-α inhibitor infliximab will be informative in this regard. Treatments are required to reduce the early and long-term impact of AP, including organ failure, necrotising pancreatitis, superadded infection, exocrine and endocrine failure,106,107 recurrent AP and CP. The risks of long-term effects are significantly higher in alcohol-related AP compared with gallstone-related disease.107 An encouraging strategy to prevent AP includes focusing on upregulating preventative pathways, such as reducing ER stress by targeting XBP-1 in the acinar cell thereby facilitating a robust UPR to stressors, namely alcohol.108
FUTURE DIRECTIONS
We have placed current understanding of the pathogenesis of AP into a framework that has two thresholds critical to the development and impact of AP on individuals. Our approach is designed to assist forward and reverse translation integrating animal and human studies to stimulate further clinical studies and trials in the prevention and treatment of AP. To date, animal studies have been used more to elucidate underlying mechanisms and monitor pathophysiologic events, less to evaluate clinical interventions and their effects. Greater focus is required on appropriate preclinical models to develop interventions after disease initiation, as in the clinical setting.
We propose the two critical thresholds and associated framework to refocus endeavour on improvement of the management and outcome of AP: to estimate and reduce risk, assess and modify contributing cofactors, capture more cases earlier, optimise and personalise resuscitation, analgesia, nutrition, aetiological and severity assessment, and develop effective prophylaxis and therapy. A pipeline of trials using ORAI inhibitors to reduce Ca2+ injury,109 simvastatin to reverse deregulated autophagy110 and agents to prevent MPTP opening or pathologic ER stress could confirm an agent that halts progression of early pancreatic injury and prevent systemic progression. Inhibition of inflammatory mediators including TNF-α or IL-6, particularly given early, could ameliorate AP even after exceeding the second threshold. Omics technologies and novel measurement systems offer fertile areas for the development of precision-based therapy. These approaches also apply to prevention of iatrogenic and recurrent AP. To complement this, population-based studies are needed to more accurately determine who develops AP, when and why, and to inform health service policy and delivery for those with AP.
Funding
DY support—UO1 DK108306; DoD PR182623. PH and VV support—National Research, Development and Innovation Office project grants (FK123982 to VV and K131996 to PH). PH support—GINOP 2.3.2-15-2016-00048 STAY ALIVE (PH). SJP support—US NIH: R01 AA024464, P01 DK098108, P50 AA0119991, U01 DK108314 US DoD: W81XWH1910888. RS support—funded by the U.K. Medical Research Council and National Institute for Health Research (EME 15/20/01), European Union Innovative Medicines Initiative, GlaxoSmithKline PLC and Hampson Trust, and is an NIHR Senior Investigator. AH support—NIH U01 DK108300, NIH DK092421 and DOD W81XWH-17-1-0339. CJ support—U01DK108314, DoD: W81XWH1910888.
Competing interests RS has received research support and/or funding from Calcimedica, Cypralis, GlaxoSmithKline, MSD/Merck and Novartis, has been a consultant for AbbVie, Calcimedica, Cypralis, Eagle Pharmaceuticals, Novartis and Takeda (all funds to the University of Liverpool), and is collaborating in the IMI2 TransBioLine project with multiple public and private institutions including Janssen, Lilly, MSD/Merck, Novartis, Pfizer, Roche and Sanofi-Aventis.
Footnotes
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice This article has been corrected since it published Online First. All author affiliations have been updated.
REFERENCES
- 1.Garg PK, Singh VP. Organ failure due to systemic injury in acute pancreatitis. Gastroenterology 2019;156:2008–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Santvoort HC, Bakker OJ, Bollen TL, et al. A conservative and minimally invasive approach to necrotizing pancreatitis improves outcome. Gastroenterology 2011;141:1254–63. [DOI] [PubMed] [Google Scholar]
- 3.Hines OJ, Pandol SJ. Management of severe acute pancreatitis. BMJ 2019;38:l6227. [DOI] [PubMed] [Google Scholar]
- 4.Working Group IAP/APA Acute Pancreatitis Guidelines. IAP/APA evidence-based guidelines for the management of acute pancreatitis. Pancreatology 2013;13:e1–15. [DOI] [PubMed] [Google Scholar]
- 5.Gorelick FS, Lerch MM. Do animal models of acute pancreatitis reproduce human disease? Cell Mol Gastroenterol Hepatol 2017;4:251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wen L, Voronina S, Javed MA, et al. Inhibitors of Orai1 prevent cytosolic Calcium-Associated injury of human pancreatic acinar cells and acute pancreatitis in 3 mouse models. Gastroenterology 2015;149:481–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banks PA, Bollen TL, Dervenis C, et al. Classification of acute pancreatitis--2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013;62:102–11. [DOI] [PubMed] [Google Scholar]
- 8.Dellinger EP, Forsmark CE, Layer P, et al. Determinant-based classification of acute pancreatitis severity: an international multidisciplinary consultation. Ann Surg 2012;256:875–80. [DOI] [PubMed] [Google Scholar]
- 9.Navina S, Acharya C, DeLany JP, et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci Transl Med 2011;3:107ra10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flint RS, Phillips ARJ, Power SE, et al. Acute pancreatitis severity is exacerbated by intestinal ischemia-reperfusion conditioned mesenteric lymph. Surgery 2008;143:404–13. [DOI] [PubMed] [Google Scholar]
- 11.Gukovskaya AS, Gorelick FS, Groblewski GE, et al. Recent insights into the pathogenic mechanism of pancreatitis: role of acinar cell organelle disorders. Pancreas 2019;48:459–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma A, Tao X, Gopal A, et al. Calcium dependence of proteinase-activated receptor 2 and cholecystokinin-mediated amylase secretion from pancreatic acini. Am J Physiol Gastrointest Liver Physiol 2005;289:G686–95. [DOI] [PubMed] [Google Scholar]
- 13.Naruse S Molecular pathophysiology of pancreatitis. Intern Med 2003;42:288–9. [PubMed] [Google Scholar]
- 14.O’Konski MS, Pandol SJ. Effects of caerulein on the apical cytoskeleton of the pancreatic acinar cell. J Clin Invest 1990;86:1649–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grady T, Saluja A, Kaiser A, et al. Edema and intrapancreatic trypsinogen activation precede glutathione depletion during caerulein pancreatitis. Am J Physiol 1996;271:G20–6. [DOI] [PubMed] [Google Scholar]
- 16.Hofbauer B, Saluja AK, Lerch MM, et al. Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am J Physiol 1998;275:G352–62. [DOI] [PubMed] [Google Scholar]
- 17.Mukherjee R, Mareninova OA, Odinokova IV, et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut 2016;65:1333–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petersen OH, Courjaret R, Machaca K. Ca2+ tunnelling through the ER lumen as a mechanism for delivering Ca2+ entering via store-operated Ca2+ channels to specific target sites. J Physiol 2017;595:2999–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waldron RT, Lugea A, Pandol SJ. Brake adjustment: Ca2+ entry pathway provides a novel target for acute pancreatitis therapy. Ann Transl Med 2019;7:S284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swain SM, Romac JM-J, Shahid RA, et al. TRPV4 channel opening mediates pressure-induced pancreatitis initiated by Piezo1 activation. J Clin Invest 2020;130:2527–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Criddle DN, Sutton R, Petersen OH. Role of Ca2+ in pancreatic cell death induced by alcohol metabolites. J Gastroenterol Hepatol 2006;21:S14–17. [DOI] [PubMed] [Google Scholar]
- 22.Noel P, Patel K, Durgampudi C, et al. Peripancreatic fat necrosis worsens acute pancreatitis independent of pancreatic necrosis via unsaturated fatty acids increased in human pancreatic necrosis collections. Gut 2016;65:100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutton R Parenchymal pressure injury Ca2+ entry mechanism in pancreatitis. Cell Calcium 2020;88:102208. [DOI] [PubMed] [Google Scholar]
- 24.Lugea A, Gerloff A, Su H-Y, et al. The combination of alcohol and cigarette smoke induces endoplasmic reticulum stress and cell death in pancreatic acinar cells. Gastroenterology 2017;153:1674–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gukovskaya AS, Gukovsky I, Zaninovic V, et al. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest 1997;100:1853–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hietaranta AJ, Saluja AK, Bhagat L, et al. Relationship between NF-kappaB and trypsinogen activation in rat pancreas after supramaximal caerulein stimulation. Biochem Biophys Res Commun 2001;280:388–95. [DOI] [PubMed] [Google Scholar]
- 27.Song L, Wörmann S, Ai J, et al. Bcl3 reduces the sterile inflammatory response in pancreatic and biliary tissues. Gastroenterology 2016;150:e20:499–512. [DOI] [PubMed] [Google Scholar]
- 28.Song J, Lee J, Kim J, et al. Pancreatic adenocarcinoma up-regulated factor (PAUF) enhances the accumulation and functional activity of myeloid-derived suppressor cells (MDSCs) in pancreatic cancer. Oncotarget 2016;7:51840–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen L, Javed TA, Yimlamai D, et al. Transient high pressure in pancreatic ducts promotes inflammation and alters tight junctions via calcineurin signaling in mice. Gastroenterology 2018;155:1250–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sendler M, van den Brandt C, Glaubitz J, et al. Nlrp3 inflammasome regulates development of systemic inflammatory response and compensatory anti-inflammatory response syndromes in mice with acute pancreatitis. Gastroenterology 2020;158:e14:253–69. [DOI] [PubMed] [Google Scholar]
- 31.Steele CW, Karim SA, Foth M, et al. CXCR2 inhibition suppresses acute and chronic pancreatic inflammation. J Pathol 2015;237:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gukovskaya AS, Vaquero E, Zaninovic V, et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology 2002;122:974–84. [DOI] [PubMed] [Google Scholar]
- 33.Sendler M, Weiss F-U, Golchert J, et al. Cathepsin B-mediated activation of trypsinogen in Endocytosing macrophages increases severity of pancreatitis in mice. Gastroenterology 2018;154:e10:704–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, Neuhöfer P, Song L, et al. Il-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J Clin Invest 2013;123:1019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gukovskaya AS, Gukovsky I, Algül H, et al. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology 2017;153:1212–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szatmary P, Huang W, Criddle D, et al. Biology, role and therapeutic potential of circulating histones in acute inflammatory disorders. J Cell Mol Med 2018;22:4617–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoque R, Farooq A, Ghani A, et al. Lactate reduces liver and pancreatic injury in Toll-like receptor-and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 2014;146:1763–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li G, Wu X, Yang L, et al. TLR4-mediated NF-κB signaling pathway mediates HMGB1-induced pancreatic injury in mice with severe acute pancreatitis. Int J Mol Med 2016;37:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kocsis ÁK, Szabolcs A, Hofner P, et al. Plasma concentrations of high-mobility group box protein 1, soluble receptor for advanced glycation end-products and circulating DNA in patients with acute pancreatitis. Pancreatology 2009;9:383–91. [DOI] [PubMed] [Google Scholar]
- 40.Gukovskaya AS, Gukovsky I. Autophagy and pancreatitis. Am J Physiol Gastrointest Liver Physiol 2012;303:G993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mareninova OA, Hermann K, French SW, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest 2009;119:3340–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diakopoulos KN, Lesina M, Wörmann S, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex-and nutrition-dependent processes. Gastroenterology 2015;148:e17:626–38. [DOI] [PubMed] [Google Scholar]
- 43.Antonucci L, Fagman JB, Kim JY, et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci U S A 2015;112:E6166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang S, Ni H-M, Chao X, et al. Impaired TFEB-mediated lysosomal biogenesis promotes the development of pancreatitis in mice and is associated with human pancreatitis. Autophagy 2019;15:1954–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mareninova OA, Sendler M, Malla SR, et al. Lysosome associated membrane proteins maintain pancreatic acinar cell homeostasis: lamp-2 deficient mice develop pancreatitis. Cell Mol Gastroenterol Hepatol 2015;1:678–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venglovecz V, Hegyi P, Rakonczay Z, et al. Pathophysiological relevance of apical large-conductance Ca²+-activated potassium channels in pancreatic duct epithelial cells. Gut 2011;60:361–9. [DOI] [PubMed] [Google Scholar]
- 47.Maléth J, Balázs A, Pallagi P, et al. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology 2015;148:e16:427–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Setiawan VW, Pandol SJ, Porcel J, et al. Prospective study of alcohol drinking, smoking, and pancreatitis: the Multiethnic cohort. Pancreas 2016;45:819–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maléth J, Rakonczay Z, Venglovecz V, et al. Central role of mitochondrial injury in the pathogenesis of acute pancreatitis. Acta Physiol 2013;207:226–35. [DOI] [PubMed] [Google Scholar]
- 50.Venglovecz V, Pallagi P, Kemény LV, et al. The importance of aquaporin 1 in pancreatitis and its relation to the CFTR Cl− channel. Front Physiol 2018;9:854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Durno C, Corey M, Zielenski J, et al. Genotype and phenotype correlations in patients with cystic fibrosis and pancreatitis. Gastroenterology 2002;123:1857–64. [DOI] [PubMed] [Google Scholar]
- 52.Ooi CY, Dorfman R, Cipolli M, et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology 2011;140:153–61. [DOI] [PubMed] [Google Scholar]
- 53.Li S, Tian B. Acute pancreatitis in patients with pancreatic cancer: timing of surgery and survival duration. Medicine 2017;96:e5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pallagi P, Venglovecz V, Rakonczay Z, et al. Trypsin reduces pancreatic ductal bicarbonate secretion by inhibiting CFTR Cl⁻ channels and luminal anion exchangers. Gastroenterology 2011;141:2228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barreto SG, Carati CJ, Toouli J, et al. The islet-acinar axis of the pancreas: more than just insulin. Am J Physiol Gastrointest Liver Physiol 2010;299:G10–22. [DOI] [PubMed] [Google Scholar]
- 56.Guan D, Maouyo D, Sarfati P, et al. Effects of SMS 201–995 on basal and stimulated pancreatic secretion in rats. Endocrinology 1990;127:298–304. [DOI] [PubMed] [Google Scholar]
- 57.Moggia E, Koti R, Belgaumkar AP, et al. Pharmacological interventions for acute pancreatitis. Cochrane Database Syst Rev 2017;4:CD011384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samad A, James A, Wong J, et al. Insulin protects pancreatic acinar cells from palmitoleic acid-induced cellular injury. J Biol Chem 2014;289:23582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ryan JW, Moffat JG, Thompson AG. Role of bradykinin in the development of acute pancreatitis. Nature 1964;204:1212–3. [DOI] [PubMed] [Google Scholar]
- 60.Bhatia M, Saluja AK, Hofbauer B, et al. Role of substance P and the neurokinin 1 receptor in acute pancreatitis and pancreatitis-associated lung injury. Proc Natl Acad Sci U S A 1998;95:4760–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barreto SG, Carati CJ, Bhandari M, et al. Galanin in the pathogenesis of acute pancreatitis. Pancreas 2011;40:156–7. [Google Scholar]
- 62.Liddle RA, Nathan JD. Neurogenic inflammation and pancreatitis. Pancreatology 2004;4:551–60. [DOI] [PubMed] [Google Scholar]
- 63.Katoh K, Murai K, Nonoyama T. Effects of substance P on fluid and amylase secretion in exocrine pancreas of rat and mouse. Res Vet Sci 1984;36:147–52. [PubMed] [Google Scholar]
- 64.Ramnath RD, Bhatia M. Substance P treatment stimulates chemokine synthesis in pancreatic acinar cells via the activation of NF-kappaB. Am J Physiol Gastrointest Liver Physiol 2006;291:G1113–9. [DOI] [PubMed] [Google Scholar]
- 65.Barreto SG, Carati CJ, Schloithe AC, et al. The combination of neurokinin-1 and galanin receptor antagonists ameliorates caerulein-induced acute pancreatitis in mice. Peptides 2010;31:315–21. [DOI] [PubMed] [Google Scholar]
- 66.Barreto SG, Carati CJ, Schloithe AC, et al. Galanin potentiates supramaximal caerulein-stimulated pancreatic amylase secretion via its action on somatostatin secretion. Am J Physiol Gastrointest Liver Physiol 2009;297:G1268–73. [DOI] [PubMed] [Google Scholar]
- 67.Barreto SG, Carati CJ, Schloithe AC, et al. The efficacy of combining feG and galantide in mild caerulein-induced acute pancreatitis in mice. Peptides 2010;31:1076–82. [DOI] [PubMed] [Google Scholar]
- 68.Hegyi P, Venglovecz V, Pallagi P, et al. Galanin, a potent inhibitor of pancreatic bicarbonate secretion, is involved in the induction and progression of cerulein-induced experimental acute pancreatitis. Pancreas 2011;40:155–6. [DOI] [PubMed] [Google Scholar]
- 69.Hegyi E, Sahin-Tóth M. Genetic risk in chronic pancreatitis: the trypsin-dependent pathway. Dig Dis Sci 2017;62:1692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.LaRusch J, Jung J, General IJ, et al. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet 2014;10:e1004376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sahin-Tóth M Genetic risk in chronic pancreatitis: the misfolding-dependent pathway. Curr Opin Gastroenterol 2017;33:390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Masamune A, Kotani H, Sörgel FL, et al. Variants that affect function of calcium channel TRPV6 are associated with early-onset chronic pancreatitis. Gastroenterology 2020;158:1626–41. [DOI] [PubMed] [Google Scholar]
- 73.Whitcomb DC, LaRusch J, Krasinskas AM, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet 2012;44:1349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Apte MV, Pirola RC, Wilson JS. Individual susceptibility to alcoholic pancreatitis. J Gastroenterol Hepatol 2008;23:S63–8. [DOI] [PubMed] [Google Scholar]
- 75.Schwarzenberg SJ, Uc A, Zimmerman B, et al. Chronic pancreatitis: pediatric and adult cohorts show similarities in disease progress despite different risk factors. J Pediatr Gastroenterol Nutr 2019;68:566–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koziel D, Gluszek S, Kowalik A, et al. Genetic mutations in SPINK1, CFTR, CTRC genes in acute pancreatitis. BMC Gastroenterol 2015;15:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Polonikov AV, Samgina TA, Nazarenko PM, et al. Alcohol consumption and cigarette smoking are important modifiers of the association between acute pancreatitis and the PRSS1-PRSS2 locus in men. Pancreas 2017;46:230–6. [DOI] [PubMed] [Google Scholar]
- 78.Whitcomb DC, Frulloni L, Garg P, et al. Chronic pancreatitis: an international draft consensus proposal for a new mechanistic definition. Pancreatology 2016;16:218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van den Berg FF, Kempeneers MA, van Santvoort HC, et al. Meta-analysis and field synopsis of genetic variants associated with the risk and severity of acute pancreatitis. BJS Open 2020;4:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Apte MV, Haber PS, Applegate TL, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 1998;43:128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bachem MG, Schneider E, Gross H, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998;115:421–32. [DOI] [PubMed] [Google Scholar]
- 82.Gryshchenko O, Gerasimenko JV, Peng S, et al. Calcium signalling in the acinar environment of the exocrine pancreas: physiology and pathophysiology. J Physiol 2018;596:2663–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hegyi P Necrotic amplification loop in acute pancreatitis: pancreatic stellate cells and nitric oxide are important players in the development of the disease. J Physiol 2018;596:2679–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sherman MH, Yu RT, Engle DD, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014;159:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Masamune A, Kikuta K, Watanabe T, et al. Pancreatic stellate cells express Toll-like receptors. J Gastroenterol 2008;43:352–62. [DOI] [PubMed] [Google Scholar]
- 86.Exley AR, Leese T, Holliday MP, et al. Endotoxaemia and serum tumour necrosis factor as prognostic markers in severe acute pancreatitis. Gut 1992;33:1126–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Apte M, Pirola RC, Wilson JS. Pancreatic stellate cell: physiologic role, role in fibrosis and cancer. Curr Opin Gastroenterol 2015;31:416–23. [DOI] [PubMed] [Google Scholar]
- 88.Vonlaufen A, Apte MV, Imhof BA, et al. The role of inflammatory and parenchymal cells in acute pancreatitis. J Pathol 2007;213:239–48. [DOI] [PubMed] [Google Scholar]
- 89.Barreto SG, Saccone GTP. Alcohol-induced acute pancreatitis: the ‘critical mass’ concept. Med Hypotheses 2010;75:73–6. [DOI] [PubMed] [Google Scholar]
- 90.Petrov MS, Yadav D. Global epidemiology and holistic prevention of pancreatitis. Nat Rev Gastroenterol Hepatol 2019;16:175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang R, Deng L, Jin T, et al. Hypertriglyceridaemia-associated acute pancreatitis: diagnosis and impact on severity. HPB 2019;21:1240–9. [DOI] [PubMed] [Google Scholar]
- 92.Song PK, Man QQ, Li H, et al. Trends in lipids level and dyslipidemia among Chinese adults, 2002–2015. Biomed Environ Sci 2019;32:559–70. [DOI] [PubMed] [Google Scholar]
- 93.Rebours V, Vullierme M-P, Hentic O, et al. Smoking and the course of recurrent acute and chronic alcoholic pancreatitis: a dose-dependent relationship. Pancreas 2012;41:1219–24. [DOI] [PubMed] [Google Scholar]
- 94.Sadr Azodi O, Orsini N, Andrén-Sandberg Å, et al. Effect of type of alcoholic beverage in causing acute pancreatitis. Br J Surg 2011;98:1609–16. [DOI] [PubMed] [Google Scholar]
- 95.Yadav D, Hawes RH, Brand RE, et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med 2009;169:1035–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pandol SJ, Periskic S, Gukovsky I, et al. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology 1999;117:706–16. [DOI] [PubMed] [Google Scholar]
- 97.Liddle RA, Green GM, Conrad CK, et al. Proteins but not amino acids, carbohydrates, or fats stimulate cholecystokinin secretion in the rat. Am J Physiol 1986;251:G243–8. [DOI] [PubMed] [Google Scholar]
- 98.Setiawan VW, Pandol SJ, Porcel J, et al. Dietary factors reduce risk of acute pancreatitis in a large multiethnic cohort. Clin Gastroenterol Hepatol 2017;15:257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Crockett SD, Wani S, Gardner TB, et al. American gastroenterological association institute guideline on initial management of acute pancreatitis. Gastroenterology 2018;154:1096–101. [DOI] [PubMed] [Google Scholar]
- 100.Yadav D, O’Connell M, Papachristou GI. Natural history following the first attack of acute pancreatitis. Am J Gastroenterol 2012;107:1096–103. [DOI] [PubMed] [Google Scholar]
- 101.Kumar S, Ooi CY, Werlin S, et al. Risk factors associated with pediatric acute recurrent and chronic pancreatitis: lessons from INSPPIRE. JAMA Pediatr 2016;170:562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Premkumar R, Phillips ARJ, Petrov MS, et al. The clinical relevance of obesity in acute pancreatitis: targeted systematic reviews. Pancreatology 2015;15:25–33. [DOI] [PubMed] [Google Scholar]
- 103.Zhang H, Cho J, Buxbaum J. Update on the prevention of post-ERCP pancreatitis. Curr Treat Options Gastroenterol 2018;16:428–40. [DOI] [PubMed] [Google Scholar]
- 104.Mole DJ, Webster SP, Uings I, et al. Kynurenine-3-monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis. Nat Med 2016;22:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mira JC, Brakenridge SC, Moldawer LL, et al. Persistent inflammation, immunosuppression and catabolism syndrome. Crit Care Clin 2017;33:245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhi M, Zhu X, Lugea A, et al. Incidence of new onset diabetes mellitus secondary to acute pancreatitis: a systematic review and meta-analysis. Front Physiol 2019;10:637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shimosegawa T, Ueda T, Kuroda Y, et al. Long-term outcome of acute pancreatitis. Pancreas 2006;33:87–8. [Google Scholar]
- 108.Pandol SJ, Gorelick FS, Gerloff A, et al. Alcohol abuse, endoplasmic reticulum stress and pancreatitis. Dig Dis 2010;28:776–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Waldron RT, Chen Y, Pham H, et al. The Orai Ca2+ channel inhibitor CM4620 targets both parenchymal and immune cells to reduce inflammation in experimental acute pancreatitis. J Physiol 2019;597:3085–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Piplani H, Marek-Iannucci S, Sin J, et al. Simvastatin induces autophagic flux to restore cerulein-impaired phagosome-lysosome fusion in acute pancreatitis. Biochim Biophys Acta Mol Basis Dis 2019;1865:165530. [DOI] [PubMed] [Google Scholar]