

The chromosomal protein SMCHD1 regulates DNA methylation patterns by antagonizing 5-methylcytosine oxidation.
Abstract
5-Methylcytosine (5mC) oxidases, the ten-eleven translocation (TET) proteins, initiate DNA demethylation, but it is unclear how 5mC oxidation is regulated. We show that the protein SMCHD1 (structural maintenance of chromosomes flexible hinge domain containing 1) is found in complexes with TET proteins and negatively regulates TET activities. Removal of SMCHD1 from mouse embryonic stem (ES) cells induces DNA hypomethylation, preferentially at SMCHD1 target sites and accumulation of 5-hydroxymethylcytosine (5hmC), along with promoter demethylation and activation of the Dux double-homeobox gene. In the absence of SMCHD1, ES cells acquire a two-cell (2c) embryo–like state characterized by activation of an early embryonic transcriptome that is substantially imposed by Dux. Using Smchd1/Tet1/Tet2/Tet3 quadruple-knockout cells, we show that DNA demethylation, activation of Dux, and other genes upon SMCHD1 loss depend on TET proteins. These data identify SMCHD1 as an antagonist of the 2c-like state of ES cells and of TET-mediated DNA demethylation.
INTRODUCTION
Methylation at the 5-position of DNA cytosines within CpG sequences is a critical event in development and cell lineage differentiation (1–5). The methylation patterns are established by DNA methyltransferases (6) and can be modulated or erased by 5-methylcytosine (5mC) oxidases, the ten-eleven translocation (TET) proteins (7–9). In mammals, a small family of three TET proteins (TET1/2/3) catalyzes the oxidation of 5mC by generating 5-hydroxymethylcytosine (5hmC), 5-formylcytosine, and 5-carboxylcytosine (5caC) in three sequential oxidation steps (8, 10, 11). The latter two modified bases are excised from DNA via base excision repair leading to DNA demethylation, which can occur even in the absence of DNA replication (10). Certain cell types, such as neurons and embryonic stem (ES) cells contain relatively high levels of 5hmC (12). Although the involvement of 5mC oxidation has been demonstrated during genome-wide reprogramming of DNA methylation patterns (9, 13–16) and during enhancer activation (17–21), it is mostly unknown how TET activities are regulated globally and site specifically.
Structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) was initially identified as a modifier of murine metastable epialleles (22) and is involved in X chromosome inactivation in female cells by promoting the hypermethylation of a set of CpG islands on the inactive X (22, 23) and by affecting the structural organization of the inactive X chromosome (24–26). However, it is unclear how SMCHD1 is involved in the establishment of DNA methylation patterns.
Here, we have found that SMCHD1 is present in complexes with all three mammalian TET proteins. We show that SMCHD1 is a negative regulator of TET-mediated 5mC oxidation. Inactivation of SMCHD1 in ES cells leads to DNA hypomethylation and activation of the Dux transcription factor gene and elicits an early [two-cell (2c)–like] embryonic transcriptional program, which is, to a large extent, dependent on TET activity. The data reveal a previously unknown mechanism of how DNA methylation patterns can be regulated in mammals.
RESULTS
Identification of SMCHD1 as a protein associated with TET proteins
Using mass spectrometry (MS), we identified SMCHD1 as a protein interacting with FLAG-tagged TET3 in 293T cells (Fig. 1, A and B, and table S1), where it scored among the eight most significantly enriched proteins, which included TET3 itself and the known TET3 binding partner O-linked β-N-acetylglucosamine transferase (OGT) (27–29). To verify the SMCHD1-TET interaction further, we created ES cell lines by homologous recombination that carried a FLAG tag at the C terminus of the endogenous Smchd1 coding sequence (fig. S1A). We carried out anti-FLAG pulldown with one of the clones and performed proteomics analysis. Among the identified proteins were SMCHD1 itself as the highest-scoring protein (54% coverage) (fig. S1B and table S2). We detected the known SMCHD1-interacting protein, LRIF1 (12.3% coverage) (30). There were also several components of the PRC2 complex (EZH2, SUZ12, and MTF2; 4.5 to 15% coverage) but no PRC1 components. SMCHD1 has been shown to be a protecting factor against formation of histone H3 lysine 27 trimethylation (H3K27me3) by the Polycomb complex (25). In this proteomics experiment, we specifically recovered the TET2 protein (4.92% coverage), as associated with SMCHD1, but not TET1. TET3 is expressed at very low levels in ES cells. The known TET-interacting protein OGT (27–29) was also identified (9.37%). We then transfected FLAG-tagged TET2 into 293T cells. Although TET2 itself was identified at only 4.0% coverage (fig. S1C and table S2), we still found SMCHD1 (11.4% coverage) and OGT (46%) as TET2-interacting proteins.
Fig. 1. Interaction of SMCHD1 and TET proteins.

(A) Flag purification of TET3FL and TET3S from 293T cells. The purified samples were subjected to Coomassie blue staining (left) and Western blotting (right). The gel segments indicated were analyzed by MS. M, molecular weight markers; IB, immunoblot. (B) Identification of SMCHD1 as a binding partner of TET3 by MS. TET3S was expressed in 293T cells and immunoprecipitated with anti-FLAG beads. Gel segment 2 (A) was subjected to LC-MS/MS (liquid chromatography–tandem MS) analysis (see Materials and Methods and table S1). The top eight highest-scoring proteins are shown. M.W., molecular weight. (C) Endogenous coimmunoprecipitation (co-IP) of SMCHD1 with TET1, TET2, and TET3FL. (D) Interaction between TET proteins and SMCHD1 by co-IP using expression of tagged proteins in 293T cells. (E) Different domains of TET3 were cotransfected with full-length SMCHD1 into 293T cells. After IP, the interacting proteins were identified by Western blotting. Stars indicate IgG (immunoglobulin G) bands. aa, amino acids. (F) Different domains of SMCHD1 were cotransfected with TET3FL into 293T cells. After IP, the interacting proteins were identified by Western blotting. Stars indicate IgG bands. HATPase, histidine kinase-like ATPase domain.
Using coimmunoprecipitation (co-IP), we found that all mouse TET proteins, including the long and short isoforms of TET3 [TET3FL (full-length TET3) and TET3S, respectively (31)], and TET1 and TET2 interact with SMCHD1 as endogenous proteins in cell types in which the proteins are expressed at substantial levels (TET1 and TET2 in ES cells and TET3 in Neuro2a cells) (Fig. 1C). These interactions were confirmed by cotransfection of the respective expression constructs and IP experiments (Fig. 1D).
To further substantiate the SMCHD1-TET interactions in cells, we performed bimolecular fluorescence complementation (BiFC) experiments (32, 33) with SMCHD1 and TET3 (fig. S2). The data suggest an efficient interaction of TET3 and SMCHD1 at a level similar to a positive control (P.C.) experiment with TET3 and OGT. The latter two proteins are interaction partners, as reported previously (27, 28). Although this assay does not determine that the two proteins interact directly, it is an independent confirmation of the in vivo TET-SMCHD1 interaction, for example, within a protein complex.
We then determined the interacting domains of TET3 and SMCHD1 by cotransfection experiments. We found that the C-terminal double-stranded β-helix domain of TET3, which represents the core catalytic region conserved between all three TET proteins (8, 20, 34), interacts strongly with SMCHD1 (Fig. 1E). Analyzing SMCHD1 domains, we found that the N-terminal region including the GHKL adenosine triphosphatase (ATPase) domain interacted most efficiently with TET3FL (fragment F1; Fig. 1F), but the long central domain and the C-terminal hinge domain did not show any appreciable binding.
We then examined whether SMCHD1 and TET proteins can interact directly in vitro. We initially failed to observe a direct interaction between the recombinant N-terminal (ATPase) domain of SMCHD1 and the TET2 catalytic domain (TET2-CD). Next, we prepared recombinant full-length SMCHD1 and recombinant TET2-CD or TETFL proteins. These proteins were purified from either baculovirus-infected cells (SMCHD1 and TET2-CD) or from mammalian cells (TET2FL or TET1FL). We used the AlphaScreen system (fig. S3A) for assessing binding reactions in a quantitative biophysical assay. In these assays, SMCHD1-FLAG was biotinylated via an introduced C-terminal biotinylation sequence (AviTag) and was coexpressed along with biotin ligase (BirA) in baculovirus-infected insect cells. This SMCHD1 protein could be collected on streptavidin beads indicating that it was biotinylated successfully (fig. S3B). The mammalian TET proteins were expressed as C-terminal His-tagged proteins (fig. S3B). In the AlphaScreen assays, binding between a biotinylated protein captured on streptavidin AlphaScreen donor beads and a His-tagged protein captured on nickel chelate (Ni–nitrilotriacetic acid) AlphaScreen acceptor beads is measured. After performing these assays, we did not observe any direct binding between SMCHD1 and TET2 proteins (TET1 tested negatively as well) (fig. S3C). We also performed in vitro biotinylation of SMCHD1 using biotinylation kits, or we biotinylated the anti-SMCHD1 antibody followed by SMCHD1 binding. We concluded that our consistently observed interactions between SMCHD1 and TET proteins in cells are most likely based on an indirect interaction, perhaps involving a larger protein complex or a bridging protein. One potential candidate for such a protein is OGT, which we recovered in SMCHD1 and TET protein complexes and which is known to interact with the TET2/3-CDs (27, 29, 35, 36).
SMCHD1 inhibits TET activity
When SMCHD1 was coexpressed in 293T cells together with TET3FL, TET activity was inhibited in an SMCHD1 dose-dependent manner, leading to the formation of lower amounts of the TET reaction product 5hmC (Fig. 2A), while total levels of 5mC did not change appreciably. Another in vivo assay of TET activity is based on demethylation and reactivation of a luciferase vector that is methylated in vitro at all CpG sites before transfection. In this assay, TET3S is more active than TET3FL (31). The luciferase activity of the fully CpG-methylated luciferase reporter vector was increased by cotransfection of TET3S, as reported previously (Fig. 2B) (31). This TET-induced activity was inhibited by SMCHD1, which did not reduce the activity of an unmethylated control reporter (Fig. 2B).
Fig. 2. Inhibition of TET activity by SMCHD1.
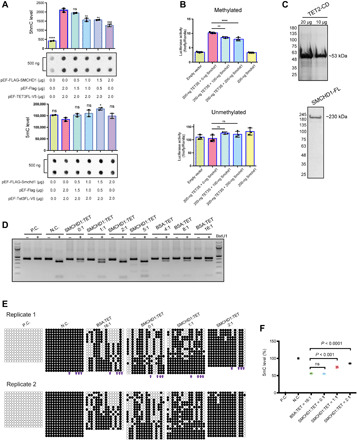
(A) Reduction in 5hmC levels by coexpression of SMCHD1 with TET3 in 293T cells. 5hmC and 5mC contents were assessed using antibody-based dot blots. One-way analysis of variance (ANOVA) was performed comparing the mean of each group with the mean of the second group (**P < 0.01 and ***P < 0.001; mean ± SEM). ns, not significant. (B) Inhibition of TET3S-induced reactivation of a methylation-silenced luciferase construct by SMCHD1 in 293T cells (top). One-way ANOVA was performed (**P < 0.01 and ****P < 0.0001). Data are for means ± SEM of three independent experiments. An unmethylated luciferase vector was used as a control (bottom). (C) FLAG purification of TET2-CD and SMCHD1 full length (SMCHD1-FL) from Sf9 insect cells. Coomassie blue staining. (D) Inhibition of TET2-CD activity on fully methylated DNA in the presence of SMCHD1 as shown by combined bisulfite restriction analysis (COBRA) assay (BstU I cleavage indicates methylation). P.C., positive control with excess TET protein (18 μg); N.C., negative control without TET treatment. Different molar ratios of SMCHD1 and TET protein (1.15 μg) are shown. The H19 imprinting control region was analyzed. (E) Bisulfite sequencing analysis of H19 methylation analyzed in duplicates. Solid black circles indicate modified CpGs; open circles indicate TET-oxidized mCpGs. The purple arrows indicate BstU I sites. (F) Percentages of modified cytosines (%Me) of the different samples. P values were determined by Fisher’s exact test (two sided).
We then proceeded to purify recombinant active TET proteins (TET2-CD and TET2FL) and full-length SMCHD1 from baculovirus-infected cells (Fig. 2C). TET2-CD was catalytically more active than TET2FL and was therefore used in our in vitro activity assays. The in vitro activity of TET2 was initially tested using combined bisulfite restriction analysis (COBRA) (37), an assay in which cleavage with BstU I (5′CGCG) indicates methylation at those sites. In this assay, addition of SMCHD1 inhibited TET activity in a dose-dependent manner (Fig. 2D). We further verified this effect by sodium bisulfite sequencing (Fig. 2, E and F). This assay monitors the end-product of TET activity, 5caC, which scores as unmodified cytosine in bisulfite sequencing due to decarboxylation and deamination of 5caC. SMCHD1 inhibited TET activity effectively at a 1:1 molar ratio and caused almost complete inhibition at a ratio of 2:1 (Fig. 2E). SMCHD1 is a DNA-binding protein with binding likely mediated through its hinge domain (38–40). We propose that binding of SMCHD1 to DNA leads to an occlusion of TET activity from its DNA target.
Effect of SMCHD1 inactivation on DNA methylation patterns
Next, we created and verified several Smchd1 knockout (KO) clones of male mouse ES cells (mESCs) using CRISPR-Cas9 technology (Fig. 3A and fig. S4A). Using immuno-dot blots, we determined that these KO clones have moderately increased levels of 5hmC (fig. S4B), suggesting that the lack of SMCHD1 leads to stimulation of the 5mC oxidation process, which is consistent with the in vitro data showing that SMCHD1 inhibits TET activity. Globally, TET and DNMT protein expression was not significantly altered in SMCHD1-deficient cells (fig. S4, C and D).
Fig. 3. Transcription activation and 2c-like gene signature in the absence of SMCHD1.
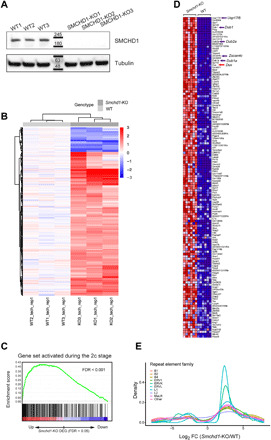
(A) Absence of SMCHD1 protein in three CRISPR-Cas9 KO ES cell clones. (B) Heatmap of RNA-seq data indicates differentially expressed genes between WT (n = 3 clones) and SMCHD1 KO (n = 3 clones) ES cells. (C) Gene set enrichment analysis (GSEA) of the 2c-like ES cell signature. The gene set represents genes activated during zygotic genome activation in 2c mouse embryos and enriched in 2C::tomato+ cells (42). The x axis shows the log2 fold change of the KO/WT-ranked transcriptome. GSEA analysis was performed as previously described (49). (D) The heatmap indicates the differentially expressed 2c-like genes between WT (n = 6) and SMCHD1 KO (n = 6) ES cells including two technical replicates for each clone. Typical 2c-like genes, such as Dux (indicated by red arrow), Zscan4c, Dub1, and Usp17l family members (indicated by purple arrows) are indicated. (E) The density plot indicates activation of repeat elements in SMCHD1 KO cells. The x axis shows the log2 (fold change of KO/WT) of repeat element expression. The y axis shows the density.
Next, we performed whole-genome bisulfite sequencing (WGBS) on three wild-type (WT) and three Smchd1 KO ES cell clones. We observed lower levels of modified cytosines in the knockouts on all chromosomes except for the Y chromosome, which became hypermethylated (fig. S5A). This moderate global reduction of modified cytosines affected all genomic compartments except for CpG islands, which have constitutively very low levels of methylation (fig. S5B). Using DMRseq analysis (41), we identified 283 hypomethylated and 223 hypermethylated differentially methylated regions (DMRs) in the clones lacking SMCHD1 (fig. S5C and table S3). One extensively hypomethylated genomic region was the Pcdha gene cluster, which is a known binding region for SMCHD1 (40).
SMCHD1 controls a 2c-like gene expression program in ES cells
To look for functionally relevant epigenetic changes upon loss of SMCHD1, we performed RNA sequencing (RNA-seq) and identified 1236 up-regulated and 256 down-regulated genes [fold change > 2, false discovery rate (FDR) < 0.05] in the Smchd1 KO cells compared to WT cells (Fig. 3B and table S3). This is consistent with a role of SMCHD1 as a transcriptional repressor. Up-regulated genes, but not down-regulated or unchanged genes, had slightly reduced levels of DNA methylation near the transcription start sites (TSSs) (fig. S5, D to F). There was an overall negative correlation between the direction of methylation change in the SMCHD1 knockouts and the expression change of the DMR-associated genes (fig. S3G). Gene ontology analysis for DMR-associated differentially expressed genes pointed to an enrichment of pattern-specific and organ development–specific processes (fig. S3H).
Using gene set enrichment analysis (GSEA), one notably up-regulated set of genes was identified as “2c embryo–like genes” (Fig. 3, C and D), which reflected the up-regulation of 136 genes normally expressed in 2c mouse embryos as a feature of the initial wave of zygotic genome activation (ZGA) (42). A similar set of genes found in ZSCAN4+ mESCs (43) or in CAF1 knockdown ES cells (44) was also enriched in the group of genes up-regulated in SMCHD1 KO cells. A small percentage of WT ES cells sporadically express 2c embryo stage–specific (2C) transcripts, such as Zscan4, and cycle in and out of this specialized state (42). Among the genes up-regulated in the absence of SMCHD1 was the Zscan4 gene cluster (Fig. 4A). The up-regulation of Zscan4, which encodes a protein involved in telomere maintenance (45), was confirmed at the protein level using a pan-ZSCAN4 antibody (Fig. 4B). The fraction of ZSCAN4-positive cells in the ES cell population increased from ~1.5% in WT cells to about 12% in the SMCHD1 KO cells (Fig. 4, C and D). Various repetitive element families, such as murine endogenous retroviruses (MERVK and MERVL), which are activated by ZSCAN4 (46) and also derepressed during ZGA (42), showed increased expression upon loss of SMCHD1 (Fig. 3E).
Fig. 4. Loss of SMCHD1 causes up-regulation of Dux and the Zscan4 gene cluster, leading to the appearance of 2c-like cells.
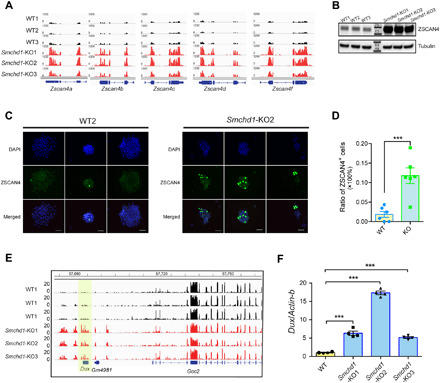
(A) Integrative Genomics Viewer screenshots of RNA-seq track peaks across all Zscan4 family members in WT and SMCHD1 KO ES cells. (B) Up-regulation of ZSCAN4 protein in SMCHD1 KO cell lines. (C) The fraction of ZSCAN4-positive cells in the ES cell population is increased in the absence of SMCHD1. ES cells were immunostained for ZSCAN4 (green). DNA was counterstained with DAPI (4′,6-diamidino-2-phenylindole) (blue). Scale bars, 50 μm. (D) Fractions of ZSCAN4+ cells in WT ES cells and Smchd1-KO ES cells. t test was performed for statistical analysis (P < 0.001). Error bars indicate SEM (six independent experiments). (E) Browser view of RNA-seq tracks across the Dux locus in WT and SMCHD1 KO ES cells. The Dux gene itself is shaded in yellow. (F) Quantitative real-time polymerase chain reaction (qRT-PCR) data confirm Dux activation upon SMCHD1 loss. β-Actin was used as a control. One-way ANOVA was performed for statistical analysis, comparing the mean of each group with the mean of the WT group (***P < 0.001). Data are for means ± SEM of three independent KO clones.
Activation of Dux in absence of SMCHD1 involves demethylation and 5hmC formation
We then focused our attention on the Dux locus, which encodes a double-homeobox transcription factor (DUX) implicated in ZGA and in 2c-like transcriptomes (47–49). We found that Dux is strongly (>10-fold) activated in SMCHD1-deficient ES cells (Fig. 4, E and F). Within the same locus, several other transcripts including the 5′UTR (5′ untranslated region) of a Dux pseudogene (Gm4981) were also up-regulated upon loss of SMCHD1 (Fig. 4E). WGBS (Fig. 5A) and manual bisulfite sequencing (Fig. 5B) revealed substantial demethylation of the Dux promoter in the SMCHD1 KO clones (WGBS, P < −1 × 105; manual bisulfite sequencing, P < 0.01; t test). We then incorporated a MERVL-promoter-dTomato reporter construct into the SMCHD1 KO and WT ES cells and purified dTomato-expressing (2c-like) cells by fluorescence-activated cell sorting (FACS) (Fig. 5C). In this cell population, methylation of the Dux promoter was even further reduced compared to unsorted cell populations, and methylation levels were lowest in the sorted SMCHD1 KO cells (Fig. 5, D and E). Using chromatin IP (ChIP) and quantitative polymerase chain reaction (qPCR), we observed that the SMCHD1 protein is present at the two different locations examined in the mouse Dux promoter in WT, but not in SMCHD1 KO ES cells (fig. S6A).
Fig. 5. Loss of SMCHD1 leads to changes of modified cytosine levels at the Dux locus.
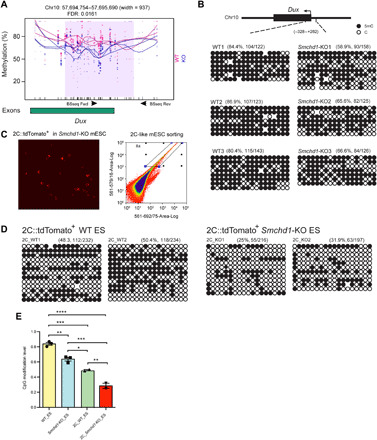
(A) Single CpG modification levels of WT and SMCHD1 KO samples at the Dux locus, as determined by WGBS. The differential methylation region is shaded in purple. CpGs are denoted with tick marks. Red circles, WT; blue circles, SMCHD1 KO. Circle size is proportional to coverage. A smoothed line is shown for each sample. (B) Manual bisulfite sequencing of the Dux promoter in WT and SMCHD1 KO cells. Solid black circles indicate modified CpG sites; open circles indicate unmodified CpG sites. Total percentages of modified cytosines (%Me) are shown. The primers are also indicated in (A). (C) Representative fluorescence image and FACS plot of 2C::tdTomato+ cells in the Smchd1 KO ES cell populations. (D) Methylation of the Dux promoter in the dTomato-expressing (FACS-sorted) Smchd1 KO cell population is further reduced. The data show bisulfite sequencing analysis of the Dux promoter in reporter-expressing WT and SMCHD1 KO ES cells. Total percentages of modified cytosines (%Me) are shown. (E) Percentages of modified cytosines at the Dux promoter determined from (B) and (D). One-way ANOVA was performed (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). Error bars indicate SEM from triplicate clones (WT and Smchd1-KO) or duplicate samples (FACS-sorted 2c-Smchd1-KO cells).
To examine whether TET-mediated 5mC oxidation is involved in demethylation of the Dux promoter when SMCHD1 is not functional, we analyzed 5hmC by a pulldown method following derivatization of the hydroxymethyl group with biotin (fig. S6B) (50) or by single-base resolution TET-assisted bisulfite sequencing [TAB sequencing; (51)] (fig. S6C; see fig. S7 for complete results). Both methods indicated a significant increase in 5hmC at the Dux promoter in SMCHD1-deficient cells. Using ChIP, we observed enhanced binding of TET1 to the R2 region of the Dux promoter in SMCHD1 KO cells (fig. S6D). This is the same location where SMCHD1 is normally bound (fig. S6A) in WT ES cells, suggesting a shielding effect of SMCHD1 toward the 5mC oxidase.
The contribution of Dux to the 2c-like program
To delineate the contribution of Dux to the entire set of genes up-regulated after loss of SMCHD1, we biallelically inactivated Dux in Smchd1 KO ES cells and also in WT ES cells (confirmed by extensive DNA sequencing because no reliable anti-DUX antibody was available; Fig. 6A). The subsequent RNA-seq analysis showed that 47 of the 136 up-regulated “2c-like” genes in SMCHD1 KO cells were no longer up-regulated in the absence of a functional Dux gene (Fig. 6, B and C). One example of such Dux-dependent genes is the Zscan4 gene cluster, the expression of which was strictly dependent on WT Dux (Fig. 6D). In a published study (48), 5738 genes were linked to HA-DUX peaks. On the basis of this gene set, we found that still, 49 of the 2c-like genes that are up-regulated in our Smchd1 single KO were not bound by DUX. So, we can confirm that a fraction of up-regulated 2c-like genes (49 of 136) in Smchd1-KO cells may not be directly regulated by DUX. The data indicate that DUX is a substantial but not exclusive contributor to the 2c-like transcriptome induced in the absence of SMCHD1 (Fig. 6).
Fig. 6. The 2c-like transcriptome in the absence of SMCHD1 is partially dependent on Dux.
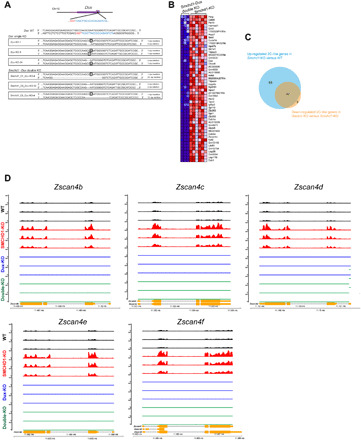
(A) The small guide RNA (gRNA) targeting region is immediately downstream of the start codon (ATG) of the Dux gene. Sanger sequencing confirmed frameshift mutations. Sequences targeted by the gRNA are in blue, and the PAM (protospacer adjacent motif) sequence is shown in red. Biallelic frameshift mutation was shown for each clone. The gRNA was applied in WT and in SMCHD1 KO ES cells to obtain the Dux single-knockout and Smchd1/Dux double-knockout ES cell clones, respectively. (B) A heatmap indicates the differentially expressed 2c-like genes between Dux/Smchd1 double-knockout (n = 3 clones) and Smchd1 single-knockout (n = 3 clones, two replicates each) ES cells. The blue color indicates genes no longer up-regulated in the double knockouts. (C) The pie chart shows that 47 of the 136 single-KO up-regulated 2c-like genes were no longer up-regulated in the absence of Dux in the Smchd1/Dux double KO. (D) RNA-seq tracks generated by the GVIZ package across the Zscan4 gene family members in WT ES cells (black), Smchd1 KO ES cells (red), Dux KO ES cells (blue), and Smchd1 plus Dux (double-KO) ES cells (green).
DNA hypomethylation and the 2c-like program depend on TET activity in the absence of SMCHD1
Our data showed an interaction of SMCHD1 with TET proteins within cells (Fig. 1 and figs. S1 and S2) and an inhibition of TET-induced 5mC oxidation by SMCHD1 (Fig. 2). In the absence of SMCHD1, 5hmC levels are increased (fig. S4; fig. S6, B and C; and fig. S7), suggesting that SMCHD1 is a negative regulator of TET activities. To obtain genetic support for this interaction, we used Tet1/2/3 triple-knockout ES cells (52) and deleted SMCHD1 from these cells, as confirmed by Western blotting and DNA sequencing (Fig. 7A and fig. S8A). The Dux gene could no longer be activated upon loss of SMCHD1 in TET-TKO cells (Fig. 7, B and C; compare to Fig. 4, E and F). Consequently, DUX target genes such as Usp17lc, Zscan4f, and Zfp352 (47–49), as well as other SMCHD1-regulated genes such as 1700013H16Rik and Gm12690, could also no longer be activated (Fig. 7D). Of the 136 2c-like genes up-regulated in the absence of SMCHD1, only 69 were still up-regulated in the absence of TET activities (Fig. 7E). In TET-TKO cells, the Dux promoter was >90% methylated at CpG sites. However, unlike in WT ES cells (Fig. 5B), loss of SMCHD1 in these cells did not elicit significant DNA demethylation (Fig. 7F). These experiments demonstrate that TET proteins are required to cause demethylation of the Dux promoter and activation of Dux when SMCHD1 is dysfunctional, genetically confirming that SMCHD1 operates as a negative regulator of TET activity.
Fig. 7. The aberrant transcriptome and DNA hypomethylation in the absence of SMCHD1 depend on the presence of TET proteins.
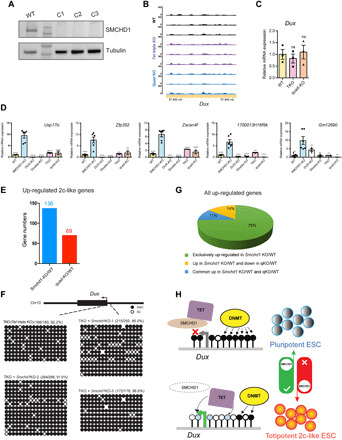
(A) Absence of SMCHD1 protein in three CRISPR-Cas9–targeted Tet triple-knockout ES cell clones. (B) RNA-seq tracks across Dux in WT, Tet triple-knockout ES cells, and Tet-Smchd1 quadruple-KO cells. (C) Quantitative RT-PCR analysis of Dux expression in WT, Tet triple-knockout and Tet-Smchd1 quadruple-knockout cells. One-way ANOVA was performed. Data are for means ± SEM of three independent clones. (D) RNA-seq analysis across different genes in WT, Smchd1 KO, Dux KO, Smchd1 and Dux (double) KO, Tet triple-knockout, and Tet-Smchd1 quadruple-knockout ES cells. One-way ANOVA was performed, comparing the mean of each group (n = 3 clones each) with the mean of the Smchd1-KO group (**P < 0.01 and ****P < 0.0001). Error bars indicate SEM. (E) The number of up-regulated 2c-like genes is decreased in the absence of TET proteins. (F) Bisulfite sequencing analysis of the Dux promoter in Tet-TKO cells and quadruple-knockout cells. Percentages of modified cytosines (%Me) are shown. (G) Eighty-nine percent (75 and 14%) of significantly up-regulated genes in the SMCHD1 single KO are no longer up-regulated in the absence of TET proteins in the Tet-Smchd1 quadruple-KO (qKO) cells. (H) Model of SMCHD1 as a negative regulator of TET proteins at the Dux promoter. Black circles, 5mC; light blue circles, 5hmC; white circles, unmethylated CpGs.
In the Smchd1, Tet1, Tet2, and Tet3 quadruple-knockout cells, a total of 921 of 1236 genes (75%), which were up-regulated in Smchd1 single-knockout cells, could no longer be activated (Fig. 7G and fig. S8B). Examples are shown in Fig. 7D. An additional 179 genes (14%), normally activated upon SMCHD1 loss, were even down-regulated in these quadruple knockouts (Fig. 7G). These data indicate that the aberrant transcriptome in the absence of SMCHD1 depends to a substantial extent on the presence of TET proteins (fig. S8, C and D).
DISCUSSION
In this study, we identified SMCHD1 as a TET-interacting protein initially by MS. There have been several previous studies in which TET- or SMCHD1-interacting proteins were analyzed by proteomics, but this interaction was not found (27, 28, 30, 53). One recent publication did identify SMCHD1 as a TET2-interacting protein in their proteomics data (54). Some studies used higher salt concentrations (300 mM) for cell lysis or washing steps (30, 53), but others used conditions similar to ours (27, 54). It is possible that the relatively milder extraction conditions we used may explain that we did find the TET-SMCHD1 association. The SMCHD1-TET complexes may be disrupted by 300 mM NaCl. Extraction and washing steps are certainly a determining factor for identification of interacting proteins. Using higher salt concentrations (>150 mM) will increase the risk of disassembling protein complexes. Other data in our manuscript, including endogenous co-IP, BiFC (which does not use cell extraction or salt washes), and the genetic studies, further support the TET-SMCHD1 interaction.
From our data, we propose a model in which SMCHD1 acts as a negative regulator of TET proteins by inhibiting their activity at target sequences (Fig. 7H). This regulation likely involves a “shielding” mechanism because DNA hypomethylation is most pronounced at known SMCHD1-bound genomic regions (e.g., Dux and Pcdha gene cluster). SMCHD1 may inhibit TET either by direct DNA binding or via its presence in heterochromatin. Localized TET inhibition or “trapping” of TET by SMCHD1 may also lead to a slight reduction in global 5mC oxidation, which is reversed upon SMCHD1 depletion leading to a moderate global DNA hypomethylation. Our data are conceptually consistent with other models that have posited that SMCHD1 functions in chromatin as an antagonistic protein against CCCTC binding factor (CTCF) binding (40), either by a shielding mechanism or by promoting DNA methylation that interferes with binding of CTCF. SMCHD1 also has been shown to be a protecting factor against formation of H3K27me3 by the Polycomb complex (25).
SMCHD1 loss-of-function mutation, often affecting its ATPase domain, is a hallmark of the human muscular dystrophy disease facioscapulohumeral dystrophy (FSHD2), which is characterized by inappropriate activation of the DUX4 gene, which is the human homolog of mouse Dux (55–57). It is of interest to speculate that loss of TET restriction by dysfunction of SMCHD1 may lead to TET-induced hypomethylation of DUX4 control regions and unscheduled expression of DUX4, perhaps starting already during human embryonic development and later manifesting itself in muscle disease.
Our data suggest that SMCHD1 is critical for Dux suppression in mESCs, thus controlling the 2c-like state. Furthermore, SMCHD1 plays a key role in de novo methylation of CpG islands on the inactive X chromosome during mouse development (23). Recent data have shown, however, that loss of SMCHD1 in somatic cells does not lead to X chromosome reactivation (24, 25). Together, the existing data suggest that SMCHD1 functions in promoting de novo DNA methylation during development rather than in mediating methylation maintenance, and we propose the following mechanism: SMCHD1 operates in these critical DNA methylation events by inhibiting TET-mediated 5mC oxidation and demethylation at its target regions, as we show in this study, thereby shifting the balance of methylation versus demethylation toward the methylated state (Fig. 7H). When SMCHD1 is lost, but DNA methylation remains high in the absence of TETs (reduced DNA methylation dynamics), Dux expression may not be occurring because of inhibition of transcription by DNA methylation. On the other hand, in absence of SMCHD1 and presence of TET activity, and thus higher methylation-demethylation dynamics, Dux will be activated. This pathway is important in inhibiting the totipotent (2c-like state) of ES cells. Although a role of SMCHD1 in inactivation of Dux in late 2c mouse embryos has recently been proposed (58), this event seems initially independent of DNA methylation inasmuch as the Dux promoter region is almost completely unmethylated in 2c and 4c mouse embryos (59); therefore, the de novo methylation events must occur later during development. Further studies are needed to confirm whether SMCHD1 is responsible for the remethylation of the Dux locus during early embryo development and whether inhibition of TET proteins plays an important role in this process.
METHODS
Mass spectrometry
FLAG-tagged TET3FL or FLAG-tagged TET3S plasmids (31) were transfected into 293T cells. After 48 hours, cells were lysed in 10 mM tris-HCl (pH 7.4), 150 mM NaCl, 0.125% NP-40, and 2.5 mM EDTA. The lysate was added to M2 anti-FLAG affinity beads (Sigma-Aldrich), which were agitated overnight at 4°C. After extensive washing with lysis buffer containing 200 mM NaCl followed by washing with 20 mM tris-HCl (pH 7.6) and 200 mM NaCl, the IP beads were mixed with 5× SDS loading buffer and heated for 10 min at 80°C. Each protein sample was loaded onto 12% SDS–polyacrylamide gel electrophoresis (PAGE) gels. After visualization using Coomassie blue, the gel lanes were cut into eight segments and sliced into small pieces for in-gel digestion. Gel pieces were washed three times with distilled water to remove SDS and dehydrated using 100% acetonitrile. Proteins were treated with 10 mM dithiothreitol (DTT) in 50 mM NH4HCO3 for 45 min at 56°C. After washing with 100% acetonitrile, alkylation of cysteines was performed with 55 mM iodoacetamide in 50 mM NH4HCO3 for 30 min in the dark. Last, each dehydrated gel piece was treated with sequencing-grade modified trypsin (12.5 ng/μl; Promega, Madison, WI) in 50 mM NH4HCO3 buffer (pH 7.8) at 37°C overnight. Following digestion, tryptic peptides were extracted with 5% formic acid in 50% acetonitrile solution at room temperature for 20 min. The supernatants were collected and dried in a SpeedVac. Samples were resuspended in 0.1% formic acid and were purified and concentrated using C18 ZipTips (Millipore, MA) before MS analysis.
Peptide separation was performed using a Dionex UltiMate 3000 RSLCnano system (Thermo Fisher Scientific). Tryptic peptides from bead columns were reconstituted using 0.1% formic acid and separated on a 50-cm EASY-Spray column with a 75-μm inner diameter packed with 2-μm C18 resin (Thermo Scientific, USA) over 120 min (300 nl/min) using a 0 to 45% acetonitrile gradient in 0.1% formic acid at 50°C. The liquid chromatography (LC) was coupled to a Q Exactive Plus mass spectrometer with a nano-ESI source (Thermo Fisher Scientific). Mass spectra were acquired in a data-dependent mode with an automatic switch between a full scan with 10 data-dependent tandem MS (MS/MS) scans. Target value for the full-scan MS spectra was 3,000,000 with a maximum injection time of 120 ms and a resolution of 70,000 at mass/charge ratio (m/z) of 400. The ion target value for MS/MS was set to 1,000,000 with a maximum injection time of 120 ms and a resolution of 17,500 at m/z 400. Dynamic exclusion of repeated peptides was applied for 20 s.
The acquired MS/MS spectra were searched using SequestHT on Proteome Discoverer (version 2.2, Thermo Fisher Scientific) against the Swiss-Prot database. Briefly, precursor mass tolerance was set to ±10 ppm (parts per million) and MS/MS tolerance was set at 0.02 Da. FDRs were set at 1% for each analysis using “Percolator.” From the Sequest search output, peptide data were default values of Proteome Discoverer. Label-free quantitation was performed using peak intensity for unique and razor peptides of each protein. Normalization was done using total peptide amount.
To identify TET2 interaction partners, we transfected FLAG-tagged TET2FL with N-terminal FLAG and HA tags (Addgene plasmid no. 41710; a gift from A. Rao) into 293T cells, harvested the cells after 48 hours, and processed them similar as described below for ES cells. ES cells in which the SMCHD1 protein was tagged endogenously with a C-terminal FLAG tag were prepared as follows: We followed the protocol of CETCh-seq (60). Briefly, we designed guide RNA (gRNA; 5′GTCTTCAGAAATGCTCAGTT) and cloned it into pSpCas9-2A-puromycin (PX459, Addgene; a gift from F. Zhang) to target and cut near SMCHD1’s stop codon. We cloned 700– to 800–base pair–(bp)–long homology arms of Smchd1 into the pFETCh-donor backbone vector (Addgene plasmid no. 63934; gift from E. Mendenhall and R. M. Myers) by Gibson assembly reaction. Then, we cotransfected the donor plasmid and gRNA plasmid at a ratio of 2:1. The single FLAG-tagged cell clones were selected in puromycin (1.5 μg/ml) and G418 (200 μg/ml). The DNA of selected clones was extracted and sequenced to detect the presence of the FLAG tag. Tagged SMCHD1 protein was detected by Western blot with anti-FLAG antibody and anti-SMCHD1 antibody.
The cells were harvested and lysed in ice-cold lysis buffer consisting of 10 mM tris-HCl (pH 7.4), 150 mM NaCl, 0.125% (v/v) NP-40, cOmplete protease inhibitor tablets (Sigma-Aldrich; 1 tablet/10 ml), and 2.5 mM EDTA at 4°C for 1 hour. We centrifuged the cell lysate at 40,000g for 60 min at 4°C, then transferred the supernatant onto equilibrated anti-FLAG M2 affinity beads (Sigma-Aldrich), and incubated the slurry on a rotation wheel overnight at 4°C. We washed the beads with ice-cold wash buffer containing 10 mM tris-HCl (pH 7.4), 250 mM NaCl, 0.125% (v/v) NP-40, cOmplete (1 tablet/10 ml), and 2.5 mM EDTA on a rotation wheel at 4°C for 5 min and repeated the washing five times. Following the final wash, the beads were then eluted twice with elution buffer containing 20 mM tris-HCl (pH 7.5), 150 mM NaCl, 0.02% (v/v) Tween 20, and 3× FLAG peptide (150 μg/ml) for 5 min. The eluted samples were then mixed with 5× SDS loading buffer and denatured for 10 min at 99°C. The protein samples were loaded and separated on a mini gel (Bio-Rad Mini-PROTEAN TGX 4 to 20%). The gel was stained using Coomassie blue and destained water. The cut gel samples were digested with trypsin and injected into a Thermo Orbitrap Fusion Lumos mass spectrometer at the University of Massachusetts Proteomics Core Facility. The data were searched against the Swiss-Prot human/mouse database using the Mascot search engine through Proteome Discoverer software.
Purification of proteins and in vitro TET activity assays
Full-length SMCHD1 was cloned into the pFastBac vector (Thermo Fisher Scientific) with a FLAG tag. We confirmed all expression vectors by Sanger sequencing. For FLAG-tagged SMCHD1 and TET2 protein expression, the bacmid DNA was transfected into Sf9 cells (Bac-to-Bac baculovirus expression system; Thermo Fisher Scientific) to obtain the passage 0 (P0) baculovirus at 96 hours after transfection. Then, we continued to generate P1 baculovirus by infecting the cells with P0 baculovirus. Proteins were expressed for 72 hours using 1000 ml of insect cells (2 million cells/ml) after transfecting P1 virus, and the cell pellet was resuspended in lysis buffer [50 mM Hepes (pH 7.5), 300 mM NaCl, 0.2% (v/v) NP-40, cOmplete, EDTA-free Protease Inhibitor Cocktail (Roche, 1 tablet/10 ml), and Benzonase Nuclease (10 U/ml) (Millipore) to destroy nucleic acids]. The lysate was cleared by centrifugation at 20,000g for 60 min. Anti-FLAG M2 affinity gel (Sigma-Aldrich) was equilibrated in lysis buffer following the manufacturer’s instructions. We incubated the cleared lysate with equilibrated FLAG M2 affinity gel at 4°C for 2 hours. Bound protein was then washed five times with wash buffer [50 mM Hepes (pH 7.5), 150 mM NaCl, and 15% (v/v) glycerol]. We eluted the protein with the wash buffer containing 3× FLAG peptide (100 μg/ml) (Sigma-Aldrich). Purified FLAG-tagged proteins were concentrated by Amicon Ultra Centrifugal Filters and DTT was added to 1 mM, then aliquots were flash-frozen in liquid nitrogen, and stored at −80°C. The purification of TET proteins from mammalian cells was performed as described using anti-FLAG purification, as described above.
We performed TET protein in vitro assays on the basis of the established TET oxidation reaction (see TAB sequencing reactions for details). For a P.C., 18 μg of TET2-CD protein was used to treat 500 ng of Sss I–methylated genomic DNA to get fully oxidized DNA containing 5caC. For a negative control (N.C.), no TET protein was used. For the testing samples, 1.15 μg of TET protein was used to treat Sss I–methylated genomic DNA, and different amounts of recombinant full-length SMCHD1 were added into the TET oxidation reaction. Bovine serum albumin (BSA) was used as a control. For a blank control, only the elution buffer for protein purification was used to keep the volume of the reactions identical. After the TET oxidation reaction, we performed bisulfite conversion treatment on the purified DNA with the EZ DNA Methylation-Gold Kit (Zymo Research). This treatment converts the TET reaction product 5caC to uracil. For COBRA, BstU I was used to digest the PCR products obtained after bisulfite conversion. For sequence analysis, the PCR products obtained after bisulfite conversion were cloned into the Topo TA cloning vector, and clones were sequenced.
Cell culture and generation of KO mESC lines using CRISPR-Cas9
J1 mESCs (from American Type Culture Collection) were cultured under feeder-free conditions on 0.1% gelatin–coated tissue culture plates in KO DMEM (Dulbecco’s modified Eagle’s medium; Gibco, 10829-018) supplemented with 15% fetal bovine serum, LIF (1000 U/ml) (Millipore, ESG1106), 1× nonessential amino acids (Gibco, 11140-050), 100 μM β-mercaptoethanol (Invitrogen, 21985-203), and 2 mM l-glutamine (Gibco, 25030-081).
mESCs were transfected with pSpCas9-2A-puromycin (PX459) plasmids (Addgene plasmid no. 62988; a gift from F. Zhang) carrying the appropriate Smchd1 sgRNAs, by using the BioT transfection reagent (Bioland, B01-01) according to the manufacturer’s instructions. Single-cell clones were selected in puromycin (1.5 μg/ml). To inactivate the Dux gene in WT and Smchd1 KO ES cell clones, we transfected WT cells or Smchd1 KO ES cells with a pSpCas9-2A-blasticidin plasmid carrying the appropriate Dux sgRNA. Single-cell clones were selected with blasticidin (8 μg/ml). We used the same pSpCas9-2A-puromycin-gRNA vector to knock out Smchd1 in Tet1/Tet2/Tet3 triple-knockout ES cells. The DNA was extracted and sequenced to detect the presence of WT and/or mutant alleles. Three independently derived WT, three homozygous mutant Smchd1-KO clones, three homozygous Dux-knockout clones, three homozygous Smchd1/Dux double-knockout clones, and three homozygous mutant Tet1/Tet2/Tet3/Smchd1 quadruple-knockout clones were selected and used in this study.
Plasmid constructs and interaction studies
For mammalian expression vectors, the Tet3FL, Tet3S, and Tet1 expression vectors were constructed as previously described (31). The pEF-Smchd1-FLAG expression vector was a gift from M. Blewitt. The pFastBac1-hTET2-CS construct was provided by R. Kohli (61). Fragments of TET3 and SMCHD1 were cloned into pEF-DEST51 expression vectors (Invitrogen, 430106). For co-IP of exogenously expressed full-length proteins, 293T cells were transfected by using a BioT transfection regent with plasmids expressing the appropriate FLAG- or V5-tagged proteins (5 μg of each plasmid on a 10-cm dish). 293T cells were harvested at 48 hours after transfection, and nuclear lysates were purified by NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, 78835) according to the manufacturer’s instructions. The nuclear lysates were incubated with 2 μg of the appropriate antibody for 2 hours and then incubated with 20 μl of Dynabeads Protein G (Invitrogen, 00671375) overnight to collect the immune complexes. We washed the immune complexes with ice-cold wash buffer containing 10 mM tris-HCl (pH 7.4), 150 mM NaCl, 0.125% (v/v) NP-40, cOmplete (1 tablet/10 ml), and 2.5 mM EDTA. The samples were boiled in SDS-PAGE loading buffer, followed by SDS-PAGE, and Western blotting. For co-IP of protein domains, 293T cells were transfected with plasmids expressing the appropriate FLAG- or V5-tagged proteins (5 μg of each plasmid on a 10-cm dish). 293T cells were harvested 48 hours after transfection and lysed in IP buffer [10 mM tris-HCl (pH 7.4), 150 mM NaCl, 0.125% NP-40, and 2.5 mM EDTA], supplemented with protease inhibitor cocktail (Roche).The cell lysate was centrifuged at 12,000g for 15 to 30 min at 4°C, incubated with 2 μg of the appropriate antibody for 2 hours, and then incubated with 20 μl of Dynabeads Protein G (Invitrogen, 00671375) overnight. The beads were then washed six times with IP buffer [10 mM tris-HCl (pH 7.4), 150 mM NaCl, 0.125% NP-40, and 2.5 mM EDTA]. Last, the samples were boiled in SDS-PAGE sample loading buffer, followed by SDS-PAGE, and Western blotting with the indicated antibodies. For endogenous co-IP, preparation of nuclear extract, buffer preparation, and co-IP, we used the Nuclear Complex Co-IP Kit (Active Motif, 54001) according to the manufacturer’s instructions. We used 5 μg of antibody for endogenous co-IP: SMCHD1 (Bethyl, A302-871A), TET1 antibody (GeneTex, GTX124207), TET2 antibody (Cell Signaling Technology, 92529), and TET3 antibody (31). After the IP reactions, we performed Western blotting with the indicated antibodies.
Immunofluorescence assays
ES cells were washed twice with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde in PBS for 15 min at room temperature. The fixed cells were permeabilized in 0.4% Triton X-100 in PBS at room temperature for 30 min, washed twice with PBS, and blocked for 30 min with 1% BSA in PBS. Cells were then incubated for 1 hour with anti-ZSCAN4 antibody (1:1000; Millipore, ab4340). After washing several times in 0.05% Tween 20 in PBS, the cells were incubated with Alexa Fluor 488 goat anti-rabbit (1:1000; Invitrogen, A27034) secondary antibody at room temperature for 1 hour and washed again three times. Then, we treated the cells with ProLong Gold anti-fade reagent with DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen, P36935) and acquired fluorescence images using a Nikon TE300 microscope with NIS-Elements AR 4.20.01.
Fluorescence-activated cell sorting
FACS analysis was performed with a Beckman cell sorting system. Smchd1-KO mESCs or WT cells containing the 2C::tdTomato reporter (Addgene plasmid no. 40281; a gift from S. Pfaff) were subjected to FACS sorting with a MoFlo Astrios instrument (Beckman Coulter).
Luciferase reporter assays
We seeded 1 × 105 293T cells into 24-well plates before transfection. Cells were transfected with the TET3S and SMCHD1 expression vectors, 47.5 ng of pGL3 luciferase reporter vector (methylated or unmethylated), and 2.5 ng of internal control Renilla luciferase reporter vector (pRL-CMV, Promega, Madison, WI). We harvested the cells 48 hours after transfection. All transfections were carried out at least in three independent experiments and in triplicate. Firefly and Renilla luciferase activities were assayed with the Dual-Luciferase assay kit (Promega) according to the manufacturer’s instructions. The firefly luciferase activities were normalized relative to Renilla activity.
BiFC assays
BiFC assays were performed for determining the in vivo interaction between SMCHD1 and TET3. The assay is based on interactions between bait and prey proteins that bring together two nonfluorescent fragments of a fluorescent protein (GFP) and then form a functional chromophore. In this study, all recombinant expression vectors were constructed on empty backbones of HA-GFP1-10-pDEST-C and FLAG-GFP11-pDEST-C (Addgene plasmid nos. 118369 and 118367; gifts from M. Vartiainen). We used human embryonic kidney 293T cells, which were cotransfected with pEF-SMCHD1-HA-GFP1-10 and pEF-TET3-FLAG-GFP11 expression vectors using the BioT transfection regent. At 48 hours after transfection, the cells were analyzed by confocal microscopy (Zeiss LSM 880 microscope) and by Cell Cytometer Counter (Celigo) to identify the interactions. Cotransfection of pEF-OGT-HA-GFP1-10 and pEF-TET3-FLAG-GFP11 was used as a P.C. Cotransfection of pEF-SMCHD1-HA-GFP1-10 and pEF-ccdB-FLAG-GFP11 and cotransfection of pEF-ccdB-HA-GFP1-10 and pEF-TET3-FLAG-GFP11 were the N.C.s.
AlphaScreen assays
AlphaScreen (PerkinElmer) assays were performed for determining the in vitro interaction between biotin-SMCHD1 and His-tagged TET proteins following the manufacturer’s protocol. Briefly, 200 nM SMCHD1-biotin and 200 nM TET2FL-His or TET2-CD-His were incubated at room temperature for 1 hour. The protein sample was then incubated with streptavidin-coated donor beads (final concentration of 10 μg/ml) and nickel-chelate acceptor beads (final concentration of 10 μg/ml) in a total volume of 100 μl of AlphaScreen buffer containing 50 mM MOPS (pH 7.4), 50 mM NaF, 50 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, and BSA (0.1 mg/ml) for 1 hour in the dark at room temperature. The photon counts were detected in 384-well plates by the EnVision Alpha reader (PerkinElmer).
Western blot and dot blot analysis
Western blots and dots blot were performed as previously described (31), with minor modifications. For Western blots, we lysed the cells in buffer containing 50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and proteinase inhibitor cocktail (Roche, 11873580001) on ice for 60 min followed by centrifugation at 12,000g for 15 min at 4°C. The lysates were separated on 4 to 15% SDS-polyacrylamide gels and transferred onto PVDF (polyvinylidene difluoride) membranes (Bio-Rad) by wet transfer at 4°C. We incubated the membranes with blocking buffer (5% nonfat milk and 0.1% Tween 20 in PBS) for 1 hour at room temperature and then with the indicated primary antibody at 4°C overnight. After washing with PBS-Tween (0.1%), the membranes were incubated with peroxidase-conjugated secondary antibodies for 1 hour at room temperature. Signals were detected using an ECL Prime detection reagent (GE Healthcare). Antibodies used for Western blots were as follows: anti-SMCHD1 (1:2500; Abcam, ab31865), anti-SMCHD1 (1:2500; Bethyl, A302-871A), anti-TET1 (1:1000; GeneTex, GTX124207), anti-TET2 (1:2000; Cell Signaling Technology, 92529) or anti-TET2 (1:1000; ProteinTech, 21207-1-AP), anti-DNMT1 (1:1000; Novus Biologicals, NB100-56519), anti-DNMT3A (1:1000; Novus Biologicals, NB120-13888), anti-DNMT3B (1:1000; Novus Biologicals, NB300-516), anti–α-tubulin (1:10,000; Abcam, ab7291), anti-ZSCAN4 (1:2500; Millipore, AB4340), HRP (horseradish peroxidase) goat anti-rabbit immunoglobulin G (IgG) (1:10,000; Active Motif, 15015), and HRP goat anti-mouse IgG (1:10,000; Active Motif, 15014). For dot blots, genomic DNA was purified with Quick-DNA Miniprep Plus kits (Zymo Research, D4070) followed by ribonuclease A treatment. The DNAs were then sonicated and purified using a QIAquick PCR purification kit (Qiagen, 28104). The purified DNAs were serially diluted and denatured in TE buffer at 98°C for 10 min and then immediately chilled on ice for 10 min. The DNAs were then spotted onto a wetted GeneScreen Plus hybridization nylon membrane (PerkinElmer, NEF988001PK) with a Bio-Dot apparatus (96-well; Bio-Rad). The blotted membranes were ultraviolet cross-linked. After incubation with the blocking buffer (5% nonfat milk and 0.15% Tween 20 in PBS) for 2 hours at room temperature, the membranes were then incubated with anti-5hmC antibody (1:8000; Active Motif, 39769) or anti-5mC antibody (1:1000; Active Motif, 39649) for 1 hour at room temperature. After washing with PBS-Tween (0.15%), the membranes were incubated with peroxidase-conjugated secondary antibodies for 1 hour at room temperature. Signals were detected using an ECL Prime detection reagent (GE Healthcare).
Bisulfite sequencing and TAB sequencing
DNA was purified with a Quick-DNA Miniprep Plus kit (Zymo Research, D4070). The bisulfite conversion was performed with the EZ DNA Methylation-Gold Kit (Zymo Research, D5005) according to the manufacturer’s instructions. PCR primer sequences for amplification of specific targets in bisulfite-treated DNA were 5′TTTGTTAGGGATGAGGAGTT (forward) and 5′AAACCTCTAATAAACCTCTTTA (reverse) for the Dux promoter. For sequence analysis, the PCR products obtained after bisulfite conversion were cloned into the Topo TA cloning vector (Thermo Fisher Scientific, 450030), and clones were sequenced. For TAB sequencing, 500 ng of genomic DNA was incubated with T4–β-glucosyltransferase (10 U/μl) [New England Biolabs (NEB)], 2 mM uridine diphosphate–glucose (NEB), and 10× CutSmart Buffer (NEB) at 37°C overnight, and then DNA was purified using standard phenol chloroform extraction followed by ethanol precipitation. Next, we performed the TET oxidation reaction as follows: The purified DNA was incubated with 12.5 μg of in-house purified TET2-CD protein, gelatin (1600 μg/ml), TET oxidation buffer 1 [1.5 mM Fe(NH4)2(SO4)2], and TET oxidation buffer 2 [83 mM NaCl, 167 mM Hepes (pH 7.5), 4 mM ATP, 8.3 mM DTT, 3.3 mM α-ketoglutarate, and 6.7 mM sodium ascorbate] at 37°C for 2 hours. Then, we added 1 μl of proteinase K (20 mg/ml) to the reaction, mixed well, and incubated at 50°C for 10 min. We performed phenol/chloroform purification and ethanol precipitation and dissolved the purified DNA in TE buffer [10 mM tris-HCl (pH 8.0) and 0.1 mM EDTA]. Last, we performed the bisulfite conversion treatment of the purified DNA with the EZ DNA Methylation-Gold Kit (Zymo Research). For sequence analysis, the PCR products obtained after bisulfite conversion were cloned into the Topo TA cloning vector and clones were sequenced.
RNA sequencing
Total RNA was extracted from whole cells with a PureLink RNA mini kit (Ambion, 12183020), according to the manufacturer’s instructions. Total RNA integrity was verified with an Agilent 2100 Bioanalyzer (Agilent Technologies) and quantified with a NanoDrop 8000 instrument (Thermo Fisher Scientific). RNA-seq libraries were prepared from total RNA with the KAPA RNA HyperPrep kit with RiboErase (KAPA Biosystems). Library size distributions were validated on the Bioanalyzer (Agilent Technologies). Sequencing was performed with an Illumina NextSeq500 machine and 75-bp single-end reads were obtained. Library demultiplexing was performed following Illumina standards.
RNA-seq data analysis
Trim Galore (version 0.4.0) was used to trim the 75-bp single-end reads. Reads were aligned to the mouse genome mm9 with STAR (version 2.5.1), and gene count was performed with STAR. Gene counts matrix was imported into R (version 3.5.1). Differential gene expression was determined with the Limma (version 3.38.2) statistical package19. Differential expression P values were adjusted for multiple testing correction using the Benjamini-Hochberg method in the stats package. Statistical significance for differentially expressed genes was fold change > 2 with q < 0.05. Heatmaps were generated with Pheatmap package. GSEA was performed with the GSEA preranked module of the Broad Institute’s GenePattern algorithm (62). For the GSEA analysis, all data were compared with the 2c-like gene set of Macfarlan et al. (42). One thousand gene-list permutations were used to determine the FDR value and the classic scoring scheme, according to methods previously described (49). Repeat element analysis was done by calculating read counts falling completely within RepeatMasker-annotated repeat elements, and the density plot was generated with R.
Whole-genome bisulfite library preparation and sequencing
For the WGBS library preparations, we used Swift, Accel-NGS Methyl-Seq DNA Library Kit (Swift Biosciences, 30024), and Zymo’s EZ DNA Methylation-Lightning kit (Zymo Research, D5030), according to the manufacturers’ instructions. Sequencing was performed with an Illumina HiSeq X with 150-bp paired-end read runs.
WGBS data analysis
Paired-end whole-genome bisulfite reads were trimmed using TrimGalore!, version 0.5.0 (https://github.com/FelixKrueger/TrimGalore) with the following parameters to remove library preparation artifacts and low quality bases: --length 50, --clip_R1 10, --clip_R2 18, --three_prime_clip_R1 10, and --three_prime_clip_R2 10. Trimmed reads were aligned to the mm9 primary chromosomes using Bismark version 0.19.0 (63) and Bowtie2 version 2.3.3.1 (64) with the following parameters: -X 1000, --nucleotide_coverage, and --bowtie2. Duplicates were marked and removed using the deduplicate_bismark script provided with Bismark. CpG methylation values were extracted using the bismark_methylation_extractor script provided with Bismark and the following parameters: --no_overlap, --comprehensive, --merge_non_CpG, and --cytosine_report. We used DMRseq version 0.99.0 (41) to identify DMRs. Briefly, CpG loci with fewer than five reads were not considered for DMR calling, and a single CpG coefficient cutoff of 0.05 was used for candidate regions. Significant DMRs were identified using a q value < 0.05. Each CpG methylation value was averaged based on groups. t test showed a significant methylation difference between WT and KO (P < 2.2 × 10−16). DMR-related genes were determined by defining a DMR within a gene’s proximity. DMRs were identified by bedtools (TSS ± 2K) and the Genomic Regions Enrichment Annotations Tool (GREAT) (65) and by long-range interaction between the DMRs and differentially expressed genes. We identified long-range interactions between the DMRs and differentially expressed genes by analyzing the Hi-C data in J1 ESC downloaded from GEO (Gene Expression Omnibus) dataset GSM862720 (SRR443885). Trim Galore (version 0.4.3) was used for adapter trimming for Hi-C data; HICCUP (version 0.5.9) was used for mapping and performing quality control. Significant interactions (default: P < 0.001 and z score > 1.0) were identified with HOMER, with a 40-kb resolution. Hi-C gene annotation involved identifying interactions with gene promoters, defined as ±2 kb of a gene TSS (fig. S5G).
5hmC pulldown qPCR and reverse transcription qPCR (RT-qPCR)
5hmC containing DNA was enriched by the EpiJET 5hmC Enrichment Kit (Thermo Fisher Scientific, K1491BID), according to the manufacturer’s instructions. The enriched DNA was then used for qPCR analysis of the Dux locus. qPCR reactions with target-specific primers included the forward (5′GCTTTGCTACCAGGGAGGAG) and reverse (5′GATCTTGAGCTGTGGGCCTG) primers for Dux region 1 and the forward (5′CTAGCGACTTGCCCTCCTTC) and reverse (5′GCTGATCAAGGAGGGGTTCC) primers for Dux region 2. PCR reactions were performed at 95°C for 10 min followed by 50 cycles at 95°C for 15 s, 57°C for 30 s, and 72°C for 30 s, using Power SYBR Green master mix (Applied Biosystems, 1809579) on a CFX96 real-time PCR cycler (Bio-Rad).
Total RNAs were isolated from cultured cells by using the PureLink RNA Mini Kit (Ambion). The SuperScriptIII reverse transcriptase (Invitrogen, 18080051) was used for reverse transcription of RNA, according to the manufacturer’s instructions. Real-time qPCR reactions with target-specific primers (available upon request) were performed at 50°C for 2 min and 95°C for 10 min followed by 50 cycles at 95°C for 15 s and 60°C for 1 min using TaqMan Gene Expression master mix (Applied Biosystems, 4369016) on a CFX96 real-time PCR cycler (Bio-Rad). The cDNA levels of target genes were analyzed using comparative Ct methods and normalized to internal standard, β-actin.
ChIP qPCR
ChIP was performed as previously described (31), with minor modifications. Briefly, cells were cross-linked with 1% formaldehyde (Thermo Fisher Scientific, 28908) in fixing buffer [50 mM Hepes (pH 7.5), 100 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA] for 10 min at room temperature, and chromatin from lysed nuclei was sheared to 300- to 500-bp fragments using a Covaris E220 sonicator (Covaris; Woburn, MA). Chromatin fragments were incubated with 5 μg of the appropriate antibody [SMCHD1 (Abcam, ab31865), TET1 (GeneTex, GTX125888), or IgG control (Santa Cruz Biotechnology; SC-2027)] overnight at 4°C with rotation. For ChIP-qPCR, real-time qPCR was carried out with a CFX96 real-time PCR cycler (Bio-Rad). Each sample was analyzed in quadruplicate. Data were analyzed according to the 2−(Ct of IP sample − Ct of IgG sample) method and are presented as −fold change of a percentage of input. PCR primer sequences are available upon request.
Supplementary Material
Acknowledgments
We thank G. Xu for providing Tet1/2/3 triple-knockout ES cells and H. Liu, P. Li, Z. Yuan, M. Du, K. Melcher, and S.-G. Jin for the advice and discussions. We thank the genomics, flow cytometry, high-throughput computing, and bioinformatics core facilities at the Van Andel Institute for the support. Funding: This work was supported by an Innovation Award from the Van Andel Institute. Author contributions: Z.H., J.Y., and G.P.P. designed and initiated the study and planned experiments; Z.H. performed interaction studies, genetic knockouts, and epigenomics and transcriptomics experiments; J.Y. and K.K. performed MS and analyzed the data; Z.H. analyzed the DNA methylation and gene expression data, with support from B.K.J.; W.C. provided experimental support; Z.H. and G.P.P. prepared the manuscript. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Genome-wide datasets generated in this study were deposited at the GEO database under the accession numbers GSE126468.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabb9149/DC1
REFERENCES AND NOTES
- 1.Iurlaro M., von Meyenn F., Reik W., DNA methylation homeostasis in human and mouse development. Curr. Opin. Genet. Dev. 43, 101–109 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Schubeler D., Function and information content of DNA methylation. Nature 517, 321–326 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Smith Z. D., Meissner A., DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 14, 204–220 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Suzuki M. M., Bird A., DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Greenberg M. V. C., Bourc’his D., The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Edwards J. R., Yarychkivska O., Boulard M., Bestor T. H., DNA methylation and DNA methyltransferases. Epig. Chrom. 10, 23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito S., D’Alessio A. C., Taranova O. V., Hong K., Sowers L. C., Zhang Y., Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tahiliani M., Koh K. P., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., Agarwal S., Iyer L. M., Liu D. R., Aravind L., Rao A., Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu T. P., Guo F., Yang H., Wu H. P., Xu G. F., Liu W., Xie Z. G., Shi L., He X., Jin S. G., Iqbal K., Shi Y. G., Deng Z., Szabó P. E., Pfeifer G. P., Li J., Xu G. L., The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610 (2011). [DOI] [PubMed] [Google Scholar]
- 10.He Y.-F., Li B.-Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., Sun Y., Li X., Dai Q., Song C.-X., Zhang K., He C., Xu G.-L., Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., He C., Zhang Y., Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruzov A., Tsenkina Y., Serio A., Dudnakova T., Fletcher J., Bai Y., Chebotareva T., Pells S., Hannoun Z., Sullivan G., Chandran S., Hay D. C., Bradley M., Wilmut I., de Sousa P., Lineage-specific distribution of high levels of genomic 5-hydroxymethylcytosine in mammalian development. Cell Res. 21, 1332–1342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iqbal K., Jin S. G., Pfeifer G. P., Szabo P. E., Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. U.S.A. 108, 3642–3647 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wossidlo M., Nakamura T., Lepikhov K., Marques C. J., Zakhartchenko V., Boiani M., Arand J., Nakano T., Reik W., Walter J., 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2, 241 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Hackett J. A., Sengupta R., Zylicz J. J., Murakami K., Lee C., Down T. A., Surani M. A., Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339, 448–452 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill P. W. S., Leitch H. G., Requena C. E., Sun Z., Amouroux R., Roman-Trufero M., Borkowska M., Terragni J., Vaisvila R., Linnett S., Bagci H., Dharmalingham G., Haberle V., Lenhard B., Zheng Y., Pradhan S., Hajkova P., Epigenetic reprogramming enables the transition from primordial germ cell to gonocyte. Nature 555, 392–396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hahn M. A., Jin S.-G., Li A. X., Liu J., Huang Z., Wu X., Kim B.-W., Johnson J., Bilbao A.-D. V., Tao S., Yim J. A., Fong Y., Goebbels S., Schwab M. H., Lu Q., Pfeifer G. P., Reprogramming of DNA methylation at NEUROD2-bound sequences during cortical neuron differentiation. Sci. Adv. 5, eaax0080 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L., Ozark P. A., Smith E. R., Zhao Z., Marshall S. A., Rendleman E. J., Piunti A., Ryan C., Whelan A. L., Helmin K. A., Morgan M. A., Zou L., Singer B. D., Shilatifard A., TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 4, eaau6986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bogdanovic O., Smits A. H., de la Calle Mustienes E., Tena J. J., Ford E., Williams R., Senanayake U., Schultz M. D., Hontelez S., van Kruijsbergen I., Rayon T., Gnerlich F., Carell T., Veenstra G. J. C., Manzanares M., Sauka-Spengler T., Ecker J. R., Vermeulen M., Gómez-Skarmeta J. L., Lister R., Active DNA demethylation at enhancers during the vertebrate phylotypic period. Nat. Genet. 48, 417–426 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X., Zhang Y., TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 18, 517–534 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Hon G. C., Song C.-X., du T., Jin F., Selvaraj S., Lee A. Y., Yen C. A., Ye Z., Mao S.-Q., Wang B. A., Kuan S., Edsall L. E., Zhao B. S., Xu G.-L., He C., Ren B., 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol. Cell 56, 286–297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blewitt M. E., Gendrel A. V., Pang Z., Sparrow D. B., Whitelaw N., Craig J. M., Apedaile A., Hilton D. J., Dunwoodie S. L., Brockdorff N., Kay G. F., Whitelaw E., SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 40, 663–669 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Gendrel A. V., Apedaile A., Coker H., Termanis A., Zvetkova I., Godwin J., Tang Y. A., Huntley D., Montana G., Taylor S., Giannoulatou E., Heard E., Stancheva I., Brockdorff N., Smchd1-dependent and -independent pathways determine developmental dynamics of CpG island methylation on the inactive X chromosome. Dev. Cell 23, 265–279 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gdula M. R., Nesterova T. B., Pintacuda G., Godwin J., Zhan Y., Ozadam H., McClellan M., Moralli D., Krueger F., Green C. M., Reik W., Kriaucionis S., Heard E., Dekker J., Brockdorff N., The non-canonical SMC protein SmcHD1 antagonises TAD formation and compartmentalisation on the inactive X chromosome. Nat. Commun. 10, 30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jansz N., Keniry A., Trussart M., Bildsoe H., Beck T., Tonks I. D., Mould A. W., Hickey P., Breslin K., Iminitoff M., Ritchie M. E., McGlinn E., Kay G. F., Murphy J. M., Blewitt M. E., Smchd1 regulates long-range chromatin interactions on the inactive X chromosome and at Hox clusters. Nat. Struct. Mol. Biol. 25, 766–777 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Wang C. Y., Jegu T., Chu H. P., Oh H. J., Lee J. T., SMCHD1 merges chromosome compartments and assists formation of super-structures on the inactive X. Cell 174, 406–421.e25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deplus R., Delatte B., Schwinn M. K., Defrance M., Méndez J., Murphy N., Dawson M. A., Volkmar M., Putmans P., Calonne E., Shih A. H., Levine R. L., Bernard O., Mercher T., Solary E., Urh M., Daniels D. L., Fuks F., TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 32, 645–655 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Q., Chen Y., Bian C., Fujiki R., Yu X., TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vella P., Scelfo A., Jammula S. G., Chiacchiera F., Williams K., Cuomo A., Roberto A., Christensen J., Bonaldi T., Helin K., Pasini D., Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell 49, 645–656 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Brideau N. J., Coker H., Gendrel A. V., Siebert C. A., Bezstarosti K., Demmers J., Poot R. A., Nesterova T. B., Brockdorff N., Independent mechanisms target SMCHD1 to trimethylated histone H3 lysine 9-modified chromatin and the inactive X chromosome. Mol. Cell. Biol. 35, 4053–4068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin S. G., Zhang Z.-M., Dunwell T. L., Harter M. R., Wu X., Johnson J., Li Z., Liu J., Szabó P. E., Lu Q., Xu G. L., Song J., Pfeifer G. P., Tet3 reads 5-carboxylcytosine through its CXXC domain and is a potential guardian against neurodegeneration. Cell Rep. 14, 493–505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu C. D., Chinenov Y., Kerppola T. K., Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Cabantous S., Terwilliger T. C., Waldo G. S., Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 23, 102–107 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Rasmussen K. D., Helin K., Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 30, 733–750 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q., Liu X., Gao W., Li P., Hou J., Li J., Wong J., Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-Linked β-N-Acetylglucosamine transferase (OGT). J. Biol. Chem. 289, 5986–5996 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ito R., Katsura S., Shimada H., Tsuchiya H., Hada M., Okumura T., Sugawara A., Yokoyama A., TET3-OGT interaction increases the stability and the presence of OGT in chromatin. Genes Cells 19, 52–65 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Xiong Z., Laird P. W., COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 25, 2532–2534 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen K., Birkinshaw R. W., Gurzau A. D., Wanigasuriya I., Wang R., Iminitoff M., Sandow J. J., Young S. N., Hennessy P. J., Willson T. A., Heckmann D. A., Webb A. I., Blewitt M. E., Czabotar P. E., Murphy J. M., Crystal structure of the hinge domain of Smchd1 reveals its dimerization mode and nucleic acid-binding residues. Sci. Signal. 13, eaaz5599 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Gurzau A. D., Blewitt M. E., Czabotar P. E., Murphy J. M., Birkinshaw R. W., Relating SMCHD1 structure to its function in epigenetic silencing. Biochem. Soc. Trans. 48, 1751–1763 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen K., Hu J., Moore D. L., Liu R., Kessans S. A., Breslin K., Lucet I. S., Keniry A., Leong H. S., Parish C. L., Hilton D. J., Lemmers R. J. L. F., van der Maarel S. M., Czabotar P. E., Dobson R. C. J., Ritchie M. E., Kay G. F., Murphy J. M., Blewitt M. E., Genome-wide binding and mechanistic analyses of Smchd1-mediated epigenetic regulation. Proc. Natl. Acad. Sci. U.S.A. 112, E3535–E3544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korthauer K., Chakraborty S., Benjamini Y., Irizarry R. A., Detection and accurate false discovery rate control of differentially methylated regions from whole genome bisulfite sequencing. Biostatistics 20, 367–383 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macfarlan T. S., Gifford W. D., Driscoll S., Lettieri K., Rowe H. M., Bonanomi D., Firth A., Singer O., Trono D., Pfaff S. L., Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akiyama T., Xin L., Oda M., Sharov A. A., Amano M., Piao Y., Cadet J. S., Dudekula D. B., Qian Y., Wang W., Ko S. B. H., Ko M. S. H., Transient bursts of Zscan4 expression are accompanied by the rapid derepression of heterochromatin in mouse embryonic stem cells. DNA Res. 22, 307–318 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishiuchi T., Enriquez-Gasca R., Mizutani E., Bošković A., Ziegler-Birling C., Rodriguez-Terrones D., Wakayama T., Vaquerizas J. M., Torres-Padilla M. E., Early embryonic-like cells are induced by downregulating replication-dependent chromatin assembly. Nat. Struct. Mol. Biol. 22, 662–671 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Zalzman M., Falco G., Sharova L. V., Nishiyama A., Thomas M., Lee S. L., Stagg C. A., Hoang H. G., Yang H. T., Indig F. E., Wersto R. P., Ko M. S. H., Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature 464, 858–863 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W., Chen F., Chen R., Xie D., Yang J., Zhao X., Guo R., Zhang Y., Shen Y., Göke J., Liu L., Lu X., Zscan4c activates endogenous retrovirus MERVL and cleavage embryo genes. Nucleic Acids Res. 47, 8485–8501 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Iaco A., Planet E., Coluccio A., Verp S., Duc J., Trono D., DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 49, 941–945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hendrickson P. G., Doráis J. A., Grow E. J., Whiddon J. L., Lim J. W., Wike C. L., Weaver B. D., Pflueger C., Emery B. R., Wilcox A. L., Nix D. A., Peterson C. M., Tapscott S. J., Carrell D. T., Cairns B. R., Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 49, 925–934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whiddon J. L., Langford A. T., Wong C. J., Zhong J. W., Tapscott S. J., Conservation and innovation in the DUX4-family gene network. Nat. Genet. 49, 935–940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song C. X., Szulwach K. E., Fu Y., Dai Q., Yi C., Li X., Li Y., Chen C. H., Zhang W., Jian X., Wang J., Zhang L., Looney T. J., Zhang B., Godley L. A., Hicks L. M., Lahn B. T., Jin P., He C., Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29, 68–72 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu M., Hon G. C., Szulwach K. E., Song C. X., Zhang L., Kim A., Li X., Dai Q., Shen Y., Park B., Min J. H., Jin P., Ren B., He C., Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 149, 1368–1380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu X., Zhang L., Mao S. Q., Li Z., Chen J., Zhang R. R., Wu H. P., Gao J., Guo F., Liu W., Xu G. F., Dai H.-Q., Shi Y. G., Li X., Hu B., Tang F., Pei D., Xu G.-L., Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell 14, 512–522 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Williams K., Christensen J., Pedersen M. T., Johansen J. V., Cloos P. A. C., Rappsilber J., Helin K., TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 473, 343–348 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen L. L., Smith M. D., Lv L., Nakagawa T., Li Z., Sun S. C., Brown N. G., Xiong Y., Xu Y. P., USP15 suppresses tumor immunity via deubiquitylation and inactivation of TET2. Sci. Adv. 6, eabc9730 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daxinger L., Tapscott S. J., van der Maarel S. M., Genetic and epigenetic contributors to FSHD. Curr. Opin. Genet. Dev. 33, 56–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tawil R., Facioscapulohumeral muscular dystrophy. Handb. Clin. Neurol. 148, 541–548 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Jansz N., Chen K., Murphy J. M., Blewitt M. E., The epigenetic regulator SMCHD1 in development and disease. Trends Genet. 33, 233–243 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Ruebel M. L., Vincent K. A., Schall P. Z., Wang K., Latham K. E., SMCHD1 terminates the first embryonic genome activation event in mouse two-cell embryos and contributes to a transcriptionally repressive state. Am. J. Physiol. Cell Physiol. 317, C655–C664 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang L., Zhang J., Duan J., Gao X., Zhu W., Lu X., Yang L., Zhang J., Li G., Ci W., Li W., Zhou Q., Aluru N., Tang F., He C., Huang X., Liu J., Programming and inheritance of parental DNA methylomes in mammals. Cell 157, 979–991 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Savic D., Partridge E. C., Newberry K. M., Smith S. B., Meadows S. K., Roberts B. S., Mackiewicz M., Mendenhall E. M., Myers R. M., CETCh-seq: CRISPR epitope tagging ChIP-seq of DNA-binding proteins. Genome Res. 25, 1581–1589 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu M. Y., Torabifard H., Crawford D. J., DeNizio J. E., Cao X. J., Garcia B. A., Cisneros G. A., Kohli R. M., Mutations along a TET2 active site scaffold stall oxidation at 5-hydroxymethylcytosine. Nat. Chem. Biol. 13, 181–187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reich M., Liefeld T., Gould J., Lerner J., Tamayo P., Mesirov J. P., GenePattern 2.0. Nat. Genet. 38, 500–501 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Krueger F., Andrews S. R., Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M., Bejerano G., GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabb9149/DC1