

MNA contributes to the immune suppression of T cells and represents a potential immunotherapy target to treat human cancer.
Abstract
Immune regulatory metabolites are key features of the tumor microenvironment (TME), yet with a few exceptions, their identities remain largely unknown. Here, we profiled tumor and T cells from tumor and ascites of patients with high-grade serous carcinoma (HGSC) to uncover the metabolomes of these distinct TME compartments. Cells within the ascites and tumor had pervasive metabolite differences, with a notable enrichment in 1-methylnicotinamide (MNA) in T cells infiltrating the tumor compared with ascites. Despite the elevated levels of MNA in T cells, the expression of nicotinamide N-methyltransferase, the enzyme that catalyzes the transfer of a methyl group from S-adenosylmethionine to nicotinamide, was restricted to fibroblasts and tumor cells. Functionally, MNA induces T cells to secrete the tumor-promoting cytokine tumor necrosis factor alpha. Thus, TME-derived MNA contributes to the immune modulation of T cells and represents a potential immunotherapy target to treat human cancer.
INTRODUCTION
Tumor-derived metabolites can have profound suppressive effects on antitumor immunity, with increasing evidence that they can also function as key drivers of disease progression (1). Beyond the Warburg effect, recent work has begun to characterize the metabolic states of tumor cells and their relationship to the immunological state of the tumor microenvironment (TME). Studies in murine models and human T cells have revealed that glutamine metabolism (2), oxidative metabolism (3), and glucose metabolism (4) are able to independently act on various immune cell subsets. Several metabolites in these pathways inhibit the antitumor function of T cells. Blockade of the enzyme cofactor tetrahydrobiopterin (BH4) was shown to impair T cell proliferation, and augmenting BH4 in vivo enhanced CD4- and CD8-mediated antitumor immune responses. Furthermore, the immune-suppressive effects of kynurenine could be rescued by administration of BH4 (5). In isocitrate dehydrogenase (IDH) mutant glioblastoma, secretion of the oncometabolite (R)-2-hydroxyglutarate (R-2-HG) suppresses T cell activation, proliferation, and cytolytic activity (6). More recently, methylglyoxal, a by-product of glycolysis, was shown to be produced by myeloid-derived suppressor cells, and T cell transfer of methylglyoxal can inhibit effector T cell function. Therapeutically, neutralization of methylglyoxal could overcome myeloid-derived suppressor cell (MDSC) activity and synergistically enhance checkpoint blockade therapy in mouse models (7). Collectively, these studies highlight the critical role for TME-derived metabolites in modulating the function and activity of T cells.
T cell dysfunction has been widely reported in ovarian cancer (8). This is due in part to the metabolic features inherently associated with hypoxia and aberrant tumor vasculature (9), leading to the conversion of glucose and tryptophan into by-products such as lactate and kynurenine. Excess extracellular lactate decreases the production of interferon-γ (IFN-γ) and drives the differentiation of myeloid suppressor subsets (10, 11). Depletion of tryptophan directly inhibits T cell proliferation and suppresses T cell receptor signaling (12–14). Despite these observations, much of the work surrounding immunometabolism has been conducted on in vitro T cell cultures using optimized media or limited to in vivo syngeneic murine models, both of which do not fully reflect the heterogeneity and physiological macro- and microenvironments in human cancer.
A common feature of ovarian cancer is peritoneal dissemination and the presentation of ascites, a buildup of cellular fluid in the peritoneal cavity that is associated with advanced disease and poor prognosis (15). This unique compartment has been reported to lack oxygen, have high levels of vascular endothelial growth factor (VEGF) and indolamine 2,3-dioxygenase (IDO), and be infiltrated by T regulatory and myeloid suppressor cells (15–18). How the metabolic environment of the ascites might differ from that of the tumor itself and, consequently, reprogramming of T cells in the peritoneal space is unknown. Furthermore, critical differences and heterogeneity between the metabolites that are present within the ascites and tumor environments that might hamper the infiltration of immune cells and their function to the tumor require further investigation.
To begin addressing these questions, we devised a sensitive cell isolation and liquid chromatography tandem mass spectrometry (LC-MS/MS) method to interrogate the repertoire of metabolites within and between distinct cell types, including CD4+ and CD8+ T cells, as well as tumor cells across the ascites and tumor environments in the same patients. We coupled this approach with high-dimensional flow cytometry and single-cell RNA sequencing (scRNA-seq) to provide a highly resolved portrait of the metabolic status of these key populations. This approach uncovered profound increases in levels of 1-methylnicotinamide (MNA) in T cells in the tumor, while in vitro assays revealed a previously unknown immunomodulatory effect of MNA on T cell function. Collectively, this approach revealed reciprocal metabolic interactions between tumor and immune cells and provided a unique insight into immune regulatory metabolites that may yield therapeutic opportunities to manipulate T cell–based immunotherapies for ovarian cancer.
RESULTS
Metabolism of T cells and tumor cells in the ascites and TMEs
We evaluated the phenotypic and metabolic states of cells in the matched ascites and tumor environments from six patients with high-grade serous carcinoma (HGSC) (table S1) using high-dimensional flow cytometry to synchronously quantify glucose uptake [2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG)] and mitochondrial activity [MitoTracker Deep Red (MT DR)] (7, 19, 20) alongside canonical markers to distinguish immune and tumor cell populations (table S2 and fig. S1A). This analysis revealed high levels of glucose uptake in tumor cells relative to T cells in both the ascites and tumor, but more modest differences in mitochondrial activity. Tumor cells [CD45−Epithelial cell adhesion molecule (EpCAM)+] had, on average, three to four times the glucose uptake of T cells, whereas CD4+ T cells had, on average, 1.2 times the glucose uptake of CD8+ T cells, suggesting that tumor-infiltrating lymphocytes (TILs) have different metabolic requirements even within the same TME (Fig. 1A). In contrast, the mitochondrial activity in tumor cells was similar to CD4+ T cells, and both cell types had greater mitochondrial activity than CD8+ T cells (Fig. 1B). Collectively, these results reveal a metabolic hierarchy, with tumor cells more metabolically active than CD4+ T cells, and CD4+ T cells more metabolically active than CD8+ T cells. Despite these effects across cell types, there were no consistent differences in the metabolic states of CD4+ and CD8+ T cells, or their relative proportions, in the ascites compared with the tumor (Fig. 1C). Conversely, within the CD45− cell fraction, there was an increase in the proportion of EpCAM+ cells in the tumor compared with the ascites (Fig. 1D). We also observed clear metabolic differences among EpCAM+ and EpCAM− cell fractions. EpCAM+ (tumor) cells had substantially greater glucose uptake and mitochondrial activity than EpCAM− cells, consistent with much higher metabolic activity in tumor cells than fibroblasts in the TME (Fig. 1, E and F).
Fig. 1. Tumor cells have greater glucose uptake but similar mitochondrial activity to T cells.
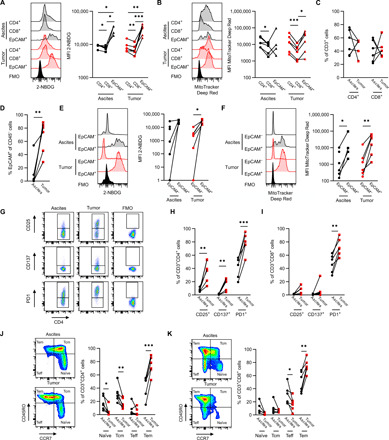
(A and B) Representative plot (left) and tabulated data (right) for median fluorescence intensity (MFI) of glucose uptake (2-NBDG) (A) and mitochondrial activity (MitoTracker Deep Red) (B) of CD4+ T cells, CD8+ T cells, and EpCAM+CD45− tumor cells from ascites and tumor. (C) Proportion of CD4+ and CD8+ cells (of CD3+ T cells) within ascites and tumor. (D) Proportion of EpCAM+ (of CD45−) tumor cells within ascites and tumor. (E and F) Representative plot (left) and tabulated data (right) for glucose uptake (2-NBDG) (E) and mitochondrial activity (MitoTracker Deep Red) (F) of EpCAM+CD45− tumor and EpCAM−CD45− stromal cells from ascites and tumor. (G) Representative plots for CD25, CD137, and PD1 expression by flow cytometry. (H and I) CD25, CD137, and PD1 expression on CD4+ T cells (H) and CD8+ T cells (I). (J and K) Naïve, central memory (Tcm), effector (Teff), and effector memory (Tem) phenotype based on CCR7 and CD45RO expression. Representative plot (left) and tabulated data (right) for CD4+ T cells (J) and CD8+ T cells (K) from ascites and tumor. P values determined by paired t test (*P < 0.05, **P < 0.01, and ***P < 0.001). Lines indicate matched patients (n = 6). FMO, fluorescence minus one; MFI, median fluorescence intensity.
Further analysis revealed other clear differences across highly resolved phenotypic states of T cells. Activated (Fig. 1, G to I) and effector memory (Fig. 1, J and K) T cells were much more frequent (as a proportion of CD3+ T cells) in the tumor than ascites. Similarly, resolving phenotypes by the expression of activation markers (CD25 and CD137) and exhaustion markers [Programmed cell death protein 1 (PD1)] revealed that while these populations showed some differences in metabolic features (fig. S1, B to E), no consistently significant metabolic differences were observed between naïve, effector, or memory subsets (fig. S1, F to I). These results were confirmed through automated assignment of cell phenotypes using a machine learning method (21), which further revealed an abundant myeloid cell population (CD45+/CD3−/CD4+/CD45RO+) predominately in patient ascites (fig. S2A). This myeloid population displayed the highest glucose uptake and mitochondrial activity of any identified cell type (fig. S2, B to G). These results underscore strong metabolic differences across a broad range of cell types found in the ascites and tumors of patients with HGSC.
Cellular enrichment and metabolite profiling by mass spectrometry
A major challenge in understanding the metabolomic profiles of TIL has been the need to isolate samples of T cells of sufficient purity, quality, and quantity from tumors. Recent studies have shown that flow cytometry–based sorting and bead enrichment methods can cause alterations in cellular metabolite profiles (22–24). To overcome this, we optimized a bead enrichment approach to isolate and separate TIL from surgically resected human ovarian cancers before analysis by LC-MS/MS (see Materials and Methods; Fig. 2A). To assess the overall impact of this protocol on metabolite changes, we compared the metabolite profiles of healthy donor activated T cells following the above bead isolation steps with cells that did not undergo bead isolation but remained on ice. This quality control analysis found high correlation among these two conditions (r = 0.77), as well as high reproducibility among technical replicates for this panel of 86 metabolites (Fig. 2B). Thus, these methods enabled accurate metabolite profiling in cells undergoing cell type enrichment to provide a first high-resolution platform for the identification of specific metabolites in HGSC, thereby allowing deeper insight into cell-specific metabolic programs.
Fig. 2. Metabolite profiling of matched ascites and tumor reveals key differences between tumor cells and T cells.
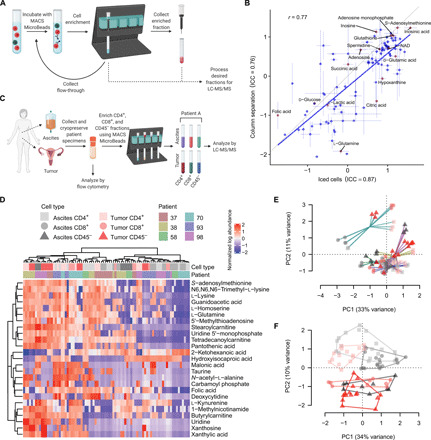
(A) Schematic of magnetic bead enrichment. Cells underwent three consecutive rounds of magnetic bead enrichment or remained on ice before analysis by LC-MS/MS. (B) Impact of enrichment type on metabolite abundance. Means of triplicate measurements for each enrichment type ±SE shown. Gray line represents 1:1 relationship. Intraclass correlation (ICC) for replicate measurements shown in axis labels. NAD, nicotinamide adenine dinucleotide. (C) Schematic of patient metabolite profiling workflow. Ascites or tumor was collected from patients and cryopreserved. A fraction of each sample was analyzed by flow cytometry, while the remaining sample underwent three rounds of enrichment for CD4+, CD8+, and CD45− cells. These cell fractions were analyzed using LC-MS/MS. (D) Heatmap of normalized metabolite abundance, with dendrograms representing Ward’s clustering of Euclidean distances among samples. (E) Principal components analysis (PCA) of sample metabolite profiles, showing triplicate replicates of each sample, with samples from the same patients joined by lines. (F) PCA of sample metabolite profiles conditioned on patient (i.e., using partial redundancy); sample types are circumscribed by convex hulls. PC1, principal component 1; PC2, principal component 2.
Metabolite profiling revealed differences in MNA
Next, we applied this enrichment method to profile 99 metabolites in CD4+, CD8+, and CD45− cell fractions from the primary ascites and tumor of six patients with HGSC (Fig. 2C, fig. S3A, and tables S3 and S4). The populations of interest ranged from 2 to 70% of live cells of the original bulk sample, with high variability in cellular proportions between patients. After bead isolations, the enriched fractions of interest (CD4+, CD8+, or CD45−) consisted, on average, of greater than 85% of the total live cells within the sample. This enrichment approach allowed us to metabolically profile cell populations from human tumor tissues that would not otherwise be possible from bulk samples. Using this protocol, we identified l-kynurenine and adenosine, two well-characterized immunosuppressive metabolites that were elevated in T cells from the tumor or in tumor cells (fig. S3, B and C). Thus, these results demonstrate the fidelity and capacity of our cell isolation and mass spectrometry technique to uncover biologically important metabolites in patient tissues.
Our profiling also revealed strong metabolic separation of cell types within and across patients (Fig. 2D and fig. S4A). In particular, patient 70 exhibited distinct metabolic profiles compared with the other patients (Fig. 2E and fig. S4B), indicating the potential for substantial metabolic heterogeneity among patients. Notably, patient 70 had a smaller total volume of ascites collected (80 ml) compared with the other patients (1.2 to 2 liters; table S1). Controlling for interpatient heterogeneity during principal components analysis (e.g., using partial redundancy analysis) revealed consistent changes among cell types, with clear clustering of cell types and/or microenvironments based on metabolite profile (Fig. 2F). Analyses of single metabolites underscored these effects and revealed marked differences among cell types and microenvironments. Notably, the most extreme difference observed was for MNA, which was enriched in CD45− cells in general, and in tumor-infiltrating CD4+ and CD8+ cells (Fig. 3A). This effect was most pronounced for CD4+ cells, while MNA in CD8+ cells also appeared to be strongly affected by the environment. However, this was not significant as tumor CD8+ fractions were only evaluable for three of the six patients. In addition to MNA, other metabolites less well-characterized in TIL were found to be differentially abundant across cell types in the ascites and tumor (figs. S3 and S4). Thus, these data reveal a promising resource set of immunoregulatory metabolites for further study.
Fig. 3. MNA is more abundant in T cells from the tumor compared with ascites.

(A) Normalized abundance of MNA in CD4+, CD8+, and CD45− cells from ascites and tumor. Boxplots show medians (lines), interquartile range (box hinges), and range of data up to 1.5× interquartile range (box whiskers). P values are determined using limma with patient as a random effect, as described in Materials and Methods (*P < 0.05 and **P < 0.01). (B) Schematic of MNA metabolism (60). Metabolites: S-adenosyl-l-methionine; SAH, S-adenosyl-l-homocysteine; NA, nicotinamide; MNA, 1-methylnicotinamide; 2-PY, 1-methyl-2-pyridone-5-carboxamide; 4-PY, 1-methyl-4-pyridone-5-carboxamide; NR, nicotinamide riboside; NMN, nicotinamide mononucleotide. Enzymes (green): NNMT, nicotinamide N-methyl transferase; SIRT, sirtuins; NAMPT, nicotinamide phosphoribosyltransferase; AOX1, aldehyde oxidase 1; NRK, nicotinamide riboside kinase; NMNAT, nicotinamide mononucleotide adenylyltransferase; Pnp1, purine nucleoside phosphorylase. (C) t-SNE of scRNA-seq of ascites (gray) and tumor (red; n = 3 patients). (D) NNMT expression in different cellular populations identified using scRNA-seq. (E) Expression of NNMT and AOX1 in SK-OV-3, human embryonic kidney (HEK) 293T, T cells, and T cells treated with MNA. Fold expression is shown relative to SK-OV-3. Means of expression (n = 6 healthy donors) with SEM shown. A Ct values greater than 35 is considered undetectable (U.D.). (F) Expression of SLC22A1 and SLC22A2 in SK-OV-3, HEK293T, T cells, and T cells treated with 8 mM MNA. Fold expression is shown relative to SK-OV-3. Means of expression (n = 6 healthy donors) with SEM shown. A Ct value greater than 35 is considered undetectable (U.D.). (G) Cellular MNA content in activated healthy donor T cells after 72-hour incubation with MNA. Means of expression (n = 4 healthy donors) with SEM shown.
Nicotinamide N-methyltransferase is primarily expressed in tumor cells and fibroblasts
MNA is produced by the transfer of a methyl group from S-adenosyl-l-methionine (SAM) to nicotinamide (NA) by nicotinamide N-methyltransferase (NNMT; Fig. 3B). NNMT is overexpressed in multiple human cancers and has been linked to proliferation, invasion, and metastasis (25–27). To better understand the source of MNA in T cells in the TME, we used scRNA-seq to characterize NNMT expression across cell types in the ascites and tumor of three patients with HGSC (table S5). Profiling ~6500 cells revealed that NNMT expression was confined to presumptive fibroblast and tumor cell populations in both the ascites and tumor environments (Fig. 3, C and D). Notably, there was no appreciable NNMT expression in any PTPRC-expressing (CD45+) populations (Fig. 3D and fig. S5A), suggesting the MNA detected in the metabolite profiling is imported into T cells. The expression of aldehyde oxidase 1 (AOX1), which converts MNA to 1-methyl-2-pyridone-5-carboxamide (2-PYR) or 1-methyl-4-pyridone-5-carboxamide (4-PYR; Fig. 3B), was likewise restricted to COL1A1-expressing fibroblast populations (fig. S5A), collectively suggesting that T cells lack the capacity for conventional MNA metabolism. The expression pattern of these MNA-related genes was validated using a second independent dataset of cells from the ascites of patients with HGSC (fig. S5B; n = 6) (16). Furthermore, quantitative polymerase chain reaction (qPCR) analysis of healthy donor T cells treated with MNA showed little to no expression of either NNMT or AOX1 relative to control SK-OV-3 ovarian tumor cells (Fig. 3E). These unexpected results suggest that MNA may be secreted from fibroblasts or tumors to neighboring T cells in the TME.
T cells uptake exogenous MNA
Potential transporters of MNA remain poorly defined, although candidates include the family of organic cation transporters 1 to 3 (OCT1, OCT2, and OCT3) encoded by the soluble carrier 22 (SLC22) family (SLC22A1, SLC22A2, and SLC22A3) (28). qPCR of mRNA from healthy donor T cells revealed low levels of SLC22A1 expression and undetectable levels of SLC22A2, confirming what has previously been reported in the literature (Fig. 3F) (29). In contrast, the SK-OV-3 ovarian tumor cell line expressed high levels of both transporters (Fig. 3F).
To test the possibility that T cells have the capacity to take up exogenous MNA, healthy donor T cells were cultured in the presence of varying concentrations of MNA for 72 hours. In the absence of exogenous MNA, the cellular content of MNA was undetectable (Fig. 3G). However, activated T cells treated with exogenous MNA exhibited a dose-dependent increase in cellular MNA content up to 6 mM MNA (Fig. 3G). This result suggests that TIL could take up MNA despite a low level of transporter expression and lack of the major enzymes responsible for intracellular MNA metabolism.
MNA induces up-regulation of tumor necrosis factor–α in T cells and suppresses T cell activity
The metabolite profiles in T cells from patients and the in vitro MNA uptake experiments raised the possibility that secretion of MNA by cancer-associated fibroblasts (CAFs) and tumor cells could modulate TIL phenotype and function. To determine the impact of MNA on T cells, healthy donor T cells were activated in vitro in the presence or absence of MNA and assessed for proliferation and cytokine production. Addition of MNA for 7 days led to a modest decrease in the number of population doublings at the highest dose, while viability was maintained across all doses (Fig. 4A). Furthermore, treatment with exogenous MNA led to an increased proportion of CD4+ and CD8+ T cells that expressed tumor necrosis factor–α (TNFα; Fig. 4B). In contrast, there was a significant decrease in intracellular production of IFN-γ in CD4+ T cells, but not CD8+ T cells, with no significant change in interleukin-2 (IL-2; Fig. 4, C and D). Accordingly, enzyme-linked immunosorbent assay (ELISA) of supernatants from these MNA-treated T cell cultures showed a significant increase in TNFα, a decrease in IFN-γ, and no change in IL-2 (Fig. 4, E to G). The reduction in IFN-γ suggested that MNA may function to suppress T cell antitumor activity. To model the impact of MNA on T cell–mediated cytotoxicity, folate-receptor alpha targeting chimeric antigen receptor T (FRα-CAR-T) cells and green fluorescent protein (GFP)–control CAR-T (GFP-CAR-T) cells were generated from healthy donor peripheral blood mononuclear cells (PBMCs). CAR-T cells were cultured in the presence of MNA for 24 hours and subsequently cocultured with folate receptor alpha–expressing human SK-OV-3 ovarian tumor cells at a 10:1 effector-to-target ratio. MNA treatment resulted in a significant reduction in FRα-CAR-T cell killing activity that was similar to adenosine-treated FRα-CAR-T cells (Fig. 4H).
Fig. 4. Exogenous MNA enhances TNFα expression and inhibits IFN-γ production in T cells.
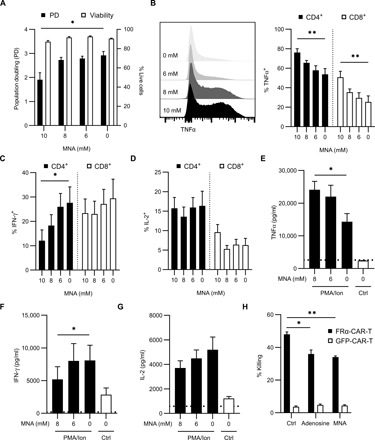
(A) Total live cell count and population doubling (PD) directly from culture on day 7. Bar graphs represent means + SEM of six healthy donors. Data representative of at least n = 3 independent experiments. (B to D) T cells were activated using CD3/CD28 with IL-2 in respective concentrations of MNA for 7 days. Cells were stimulated with PMA/ionomycin with GolgiStop for 4 hours before analysis. TNFα (B) expression in T cells. Example plot of TNFα expression in live cells (left) and tabulated data (right). IFN-γ (C) and IL-2 (D) expression in T cells. Cytokine expression was measured by flow cytometry. Bar graphs represent means (n = 6 healthy donors) + SEM. P value determined using a one-way ANOVA with repeated measures (*P < 0.05 and **P < 0.01). Data representative of at least n = 3 independent experiments. (E to G) T cells were activated using CD3/CD28 with IL-2 in respective concentrations of MNA for 7 days. Media was collected before (Ctrl) and after PMA/ionomycin stimulation for 4 hours. Concentration of TNFα (E), IFN-γ (F), and IL-2 (G) was measured by ELISA. Bar graphs represent means (n = 5 healthy donors) + SEM. P value determined using a one-way ANOVA with repeated measures (*P < 0.05). Dotted line indicates the limit of detection for the assay. (H) Cytolytic assay. FRα-CAR-T or GFP-CAR-T cells were conditioned with adenosine (250 μM) or MNA (10 mM) for 24 hours, or left untreated (Ctrl). Percentage of killing was measured against SK-OV-3 cells. P value determined by Welch’s t test (*P < 0.5 and **P < 0.01).
MNA increases TNFα mRNA expression through promoter occupancy
To gain mechanistic insight into MNA-dependent regulation of TNFα expression, MNA-treated T cells were assessed for changes in TNFα mRNA (Fig. 5A). Healthy donor T cells treated with MNA showed a twofold increase in TNFα transcripts, suggesting MNA-dependent transcriptional regulation of TNFα. To investigate this possible mechanism of regulation, two transcription factors that are known to regulate TNFα, nuclear factor of activated T cells (NFAT) and specificity protein 1 (Sp1), were assessed for binding to the proximal TNFα promoter in response to MNA (30). The TNFα promoter contains six identified NFAT-binding sites and two Sp1-binding sites, with overlap at one site [−55 base pairs (bp) from the 5′ cap] (30). Chromatin immunoprecipitation (ChIP) revealed a threefold increase in Sp1 binding to the TNFα promoter when treated with MNA. Binding of NFAT also increased and approached significance (Fig. 5B). These data suggest that MNA transcriptionally regulates the expression of TNFα through Sp1 and, to a lesser extent, NFAT.
Fig. 5. MNA increases binding of Sp1 to the promoter of TNFα, increasing TNFα transcription and cytokine production.

(A) Fold change in expression of TNFα in T cells treated with MNA over T cells cultured without MNA. Means of expression (n = 5 healthy donors) with SEM shown. Data representative of at least n = 3 independent experiments. (B) NFAT and Sp1 binding to the TNFα promoter of T cells treated with or without 8 mM MNA before (Ctrl) and after 4-hour stimulation by PMA/ionomycin. Immunoglobulin G (IgG) and H3 were used as negative and positive controls, respectively, for the immunoprecipitation. Quantification of ChIP shows the fold increase in Sp1 and NFAT binding to the TNFα promoter in MNA-treated cells compared with control. Data representative of at least n = 3 independent experiments. P value determined by multiple t tests (***P < 0.01). (C) T cells (noncytotoxic) show increased expression of TNF in the tumor relative to the ascites of HGSC. Colors represent different patients. Displayed cells have been randomly subsampled to 300 and jittered to limit overplotting (**Padj = 0.0076). (D) Proposed model of MNA in ovarian cancer. MNA is produced in tumor cells and fibroblasts in the TME and taken up by the T cells. MNA increases binding of Sp1 to the promoter of TNFα, leading to increased transcription of TNFα and cytokine production of TNFα. MNA also leads to a decrease in IFN-γ. The resulting inhibition of T cell function leads to decreased killing capacity and increased tumor growth.
While TNFα has been reported to have context-dependent pro- and antitumor effects, it has a well-described role in promoting ovarian cancer growth and metastasis (31–33). Patients with ovarian cancer have been reported to have higher concentrations of TNFα within their ascites and tumor tissue than benign tissues (34–36). Mechanistically, TNFα can modulate activation, function, and proliferation of leukocytes and change the phenotype of cancer cells (37, 38). Consistent with these findings, differential gene expression analysis revealed a significant up-regulation of TNF in T cells from tumor tissue compared with ascites (Fig. 5C). Increased TNF expression was only apparent in T cell populations with a noncytotoxic phenotype (fig. S5A). Together, these data support the notion of a dual immune-suppressive and tumor-promoting role for MNA in HGSC.
DISCUSSION
Flow cytometry–based fluorescent markers have been the primary method used to study the metabolism of TIL. These studies have shown that murine and human TILs have a higher propensity for glucose uptake (4, 39) and a progressive loss of mitochondria function (19, 40) when compared with peripheral blood lymphocytes or T cells from secondary lymphoid organs. Although we observed similar results in this study, a key advance was the comparison of the metabolism of tumor cells and TIL from the same resected tumor tissue. Consistent with some of these previous reports, tumor (CD45−EpCAM+) cells from both the ascites and tumor had greater glucose uptake than both CD8+ and CD4+ T cells, supporting the concept that high glucose uptake by tumor cells can compete with T cells in the TME. However, tumor cells had greater mitochondrial activity than CD8+ T cells but similar mitochondrial activity to CD4+ T cells. These results reinforce the emerging theme that oxidative metabolism is important to tumor cells (41, 42). They also imply that CD8+ T cells may be more susceptible to dysfunctions in oxidative capacity than CD4+ T cells or that CD4+ T cells may use carbon sources other than glucose to maintain mitochondrial activity (43, 44). It should be noted that we observed no differences in glucose uptake or mitochondrial activity between CD4+ T effector, T effector memory, and T central memory cells from the ascites. Similarly, the differentiation state of CD8+ T cells in the tumor was not associated with changes in glucose uptake, highlighting the prominent differences between in vitro cultured T cells compared with in vivo human TIL (22). These observations were also confirmed using an unbiased automated cell population assignment that further uncovered a prevalent yet metabolically active population of CD45+/CD3−/CD4+/CD45RO+ cells with greater glucose uptake and mitochondrial activity than tumor cells. This population may represent a putative myeloid suppressor cell subset or plasmacytoid dendritic cells that were identified in the scRNA-seq analysis, both of which have been reported in human ovarian tumors (45), although further work will be needed to delineate this myeloid subset.
While flow cytometry–based methods permit the elucidation of general differences in glucose and oxidative metabolism between cell types, the precise metabolites that are produced from glucose or other carbon sources used for mitochondrial metabolism in the TME are largely undetermined. Assigning the presence or absence of a metabolite to a given TIL subset requires purified populations of cells from a resected tissue. Thus, our cell enrichment method combined with mass spectrometry provides insight into metabolites that are differentially enriched in T cell and tumor cell populations from matched patient specimens. Although this approach has advantages over fluorescence-activated cell sorting, some metabolite pools are likely affected, because of inherent stability and/or rapid turnover rates (22). Despite this, our approach was able to identify two well-established immunosuppressive metabolites, adenosine and kynurenine, as being differentially abundant across sample types.
Our metabolome profiling of tumor and TIL subsets provides additional insight into the role of metabolites within the ovarian TME. First, using flow cytometry, we determined no differences in mitochondrial activity between tumor and CD4+ T cells. However, LC-MS/MS profiling uncovered significant changes in metabolite abundance between these populations, suggesting that conclusions regarding the metabolism of TIL and their overall metabolic activities require careful interpretation. Second, MNA was the most differentially abundant metabolite between the CD45− cells and T cells in the ascites, but not the tumor. Compartmentalization and tumor location may therefore have differential impacts on TIL metabolism, which highlights the heterogeneity that likely exists within a given microenvironment. Third, expression of NNMT, the enzyme that generates MNA, was predominately restricted to CAFs and, to a lesser extent, tumor cells, yet detectable levels of MNA were observed in T cells from the tumor. The overexpression of NNMT in ovarian CAFs has a known protumorigenic role in part by promoting CAF metabolism, tumor invasion, and metastasis (27). NNMT expression in CAFs is strongly associated with The Cancer Genome Atlas (TCGA) mesenchymal subtype, which is associated with poor prognosis, despite moderate overall levels of TIL (27, 46, 47). Last, the expression of AOX1, the enzyme responsible for the degradation of MNA, was also restricted to CAF populations, suggesting that T cells lack the capacity to metabolize MNA. These results support the notion that high levels of MNA in T cells may be indicative of an immune-suppressive CAF microenvironment, although further work will be required to verify this finding.
The presence of MNA in T cells was unexpected given the low level of MNA transporter expression and undetectable levels of key proteins that are involved in MNA metabolism. Both NNMT and AOX1 were undetectable by scRNA-seq analysis of two independent cohorts as well as by targeted qPCR. These results indicate that MNA is not synthesized by T cells but rather taken up from the surrounding TME. In vitro assays showed that T cells readily accumulate exogenous MNA.
Our in vitro studies demonstrate that exogenous MNA induces expression of TNFα and increased binding of Sp1 to the TNFα promoter in T cells. Although TNFα has both pro- and antitumor functions, in ovarian cancer, TNFα is known to promote ovarian cancer growth (31–33). Neutralization of TNFα in ovarian tumor cell culture or abolishing TNFα signaling in murine models ameliorates TNFα-mediated production of inflammatory cytokines and suppresses tumor growth (32, 35). Thus, in this context, TME-derived MNA may be acting as a proinflammatory metabolite through an autocrine loop via a TNFα-dependent mechanism to drive ovarian cancer tumorigenesis and dissemination (31). In accordance with this possibility, TNFα blockade is under investigation as a potential therapeutic for ovarian cancer (37, 48, 49). In addition, MNA impaired the cytotoxicity of CAR-T cells against ovarian tumor cells, providing further evidence of an MNA-mediated immune-suppressive role. Overall, these results imply a model whereby tumor and CAF cells secrete MNA into the extracellular TME. This may have dual protumor effects through (i) TNF-induced stimulation of ovarian cancer growth and (ii) MNA-induced suppression of T cell cytotoxic activity (Fig. 5D).
In conclusion, by applying a combined approach of rapid cellular enrichment, single-cell sequencing, and metabolic profiling, this study revealed large immune metabolome differences between cells within the tumor and ascites of patients with HGSC. This integrated analysis demonstrated differences in glucose uptake and mitochondrial activity between T cells and identified MNA as a non–cell autonomous immune regulatory metabolite. These data have implications for how the TME influences T cell metabolism in human cancer. While direct competition for nutrients between T cells and cancer cells has been reported, metabolites may also serve as indirect modulators to promote tumor progression and likely to suppress endogenous immune responses. Further delineation of the functional effects of these regulatory metabolites may open alternative strategies to enhance antitumor immune responses.
MATERIALS AND METHODS
Patient sample collection and processing
Patient specimens and clinical data were obtained through the BC Cancer Tumour Tissue Repository, certified by the Canadian Tissue Repository Network. All specimens and clinical data were obtained with either informed written consent or a formal waiver of consent under protocols approved by the Research Ethics Board of BC Cancer and the University of British Columbia (H07-00463). Samples are stored in a certified BioBank (BRC-00290). Detailed patient characteristics are shown in tables S1 and S5. For cryopreservation, patient tumor samples were mechanically disaggregated using a scalpel and pushed through a 100-μm filter to obtain a single-cell suspension. Patient ascites were centrifuged at 1500 rpm for 10 min at 4°C to pellet cells and remove supernatant. Cells obtained from tumor and ascites were cryopreserved in 50% heat-inactivated human AB serum (Sigma-Aldrich), 40% RPMI-1640 (Thermo Fisher Scientific), and 10% dimethyl sulfoxide. These preserved single-cell suspensions were thawed for metabolomic and metabolite assays described below.
Cell culture reagents
Complete media consisted of a 0.22-μm filtered 50:50 supplemented RPMI 1640:AimV. RPMI 1640 + 2.05 mM l-glutamine (Thermo Fisher Scientific) was supplemented with 10% heat-inactivated human AB serum (Sigma-Aldrich), 12.5 mM Hepes (Thermo Fisher Scientific), 2 mM l-glutamine (Thermo Fisher Scientific), 1× penicillin-streptomycin (PenStrep) solution (Thermo Fisher Scientific), and 50 μM B-mercaptoethanol. AimV (Invitrogen) was supplemented with 20 mM Hepes (Thermo Fisher Scientific) and 2 mM l-glutamine (Thermo Fisher Scientific). Flow cytometry staining buffer consisted of 0.22-μm filtered phosphate-buffered saline (PBS; Invitrogen) supplemented with 3% heat-inactivated AB human serum (Sigma). Cell enrichment buffer consisted of 0.22-μm filtered PBS supplemented with 0.5% heat-inactivated human AB serum (Sigma-Aldrich).
Flow cytometry for metabolic profiling
Cells were stained with 10 nM MT DR and 100 μM 2-NBDG for 30 min in complete media at 37°C. Next, cells were stained with viability dye eF506 for 15 min at 4°C. Cells were resuspended in FC block (eBioscience) and Brilliant Stain Buffer (BD Biosciences), diluted in flow cytometry staining buffer (according to the manufacturer’s instructions), and incubated for 10 min at room temperature. Cells were stained with a panel of antibodies (table S2) in flow cytometry staining buffer for 20 min at 4°C. Cells were resuspended in flow cytometry staining buffer before analysis (Cytek Aurora; 3L-16V-14B-8R configuration). Cytometry data were analyzed using SpectroFlo and FlowJo V10, and figures were created using GraphPad Prism 8. Median fluorescence intensity (MFI) of 2-NBDG and MT DR were log normalized before statistical analysis using paired t test to account for matched patients. Any population with less than 40 events was removed from the analysis; an MFI value of 1 was inputted for any negative values before statistical analysis and data visualization.
Unbiased discovery of cell populations in flow cytometry data
To supplement our manual gating strategy for the above flow panel, we used full annotation using shape-constrained trees (FAUST) (21) to automatically assign cells to populations after dead cell exclusion in FlowJo. We manually curated outputs to merge populations that appeared to be misassigned (merged PD1+ with PD1− tumor cells) and retained populations comprising, on average, more than 2% of cells in each sample, for a total of 11 populations.
Cell activation and enrichment for metabolite profiling optimization
PBMCs were isolated from a leukapheresis product (STEMCELL Technologies) using Ficoll gradient density centrifugation. CD8+ T cells were isolated from the PBMCs using CD8 MicroBeads (Miltenyi) and expanded using TransAct (Miltenyi) for 2 weeks in complete media according to the manufacturer’s instructions. Cells were rested for 5 days in complete media with IL-7 (10 ng/ml; PeproTech) and then restimulated with TransAct. On day 7, cells were enriched using Human CD45 MicroBeads (Miltenyi) in three rounds of sequential enrichment according to the manufacturer’s instructions. Cells were aliquoted for analysis by flow cytometry (described above), and 1 million cells were aliquoted in triplicate for analysis by LC-MS/MS. Samples were processed for LC-MS/MS as described below. We imputed missing metabolite values with an ion count of 1000. Each sample was sum normalized by total ion count (TIC), log transformed, and auto-normalized in MetaboAnalystR before analysis.
Cell type enrichment of patient samples
Single-cell suspensions were thawed for each patient and filtered through a 40-μm filter into complete media (described above). Samples were enriched for CD8+, CD4+, and CD45− cells (on ice) using three sequential rounds of positive selection by magnetic bead separation using MicroBeads (Miltenyi) according to the manufacturer’s protocol. Briefly, cells were resuspended in cell enrichment buffer (described above) and counted. Cells were incubated at 4°C for 15 min with Human CD8 MicroBeads, Human CD4 MicroBeads, or Human CD45 MicroBeads (Miltenyi) and then washed with cell enrichment buffer. Samples were passed through the LS column (Miltenyi), and both the positive and negative fractions were collected. To reduce the duration and maximize the cell recovery steps, CD8− fraction was subsequently used for a second round CD4+ enrichment, and the CD4− fraction was used for the subsequent CD45− enrichment. Solutions were kept on ice throughout the duration of the separation.
Metabolite extraction for mass spectrometry
To prepare samples for metabolite profiling, cells were washed once with ice-cold saline solution and 1 ml of 80% methanol added to each sample before vortexing and snap freezing in liquid nitrogen. Samples were subjected to three freeze-thaw cycles and centrifuged at 14,000 rpm for 15 min at 4°C. The metabolite-containing supernatant was evaporated until dry. Metabolites were reconstituted in 50 μl of 0.03% formic acid, vortex-mixed, and centrifuged to remove debris.
LC-MS/MS metabolite profiling for patient biospecimens
Metabolite extraction was carried out as described above. The supernatant was transferred to a high-performance liquid chromatography vial for the metabolomics study. Each sample was processed with similar numbers of cells using a randomized processing scheme to prevent batch effects. We performed qualitative assessment of global metabolites as previously published on the AB SCIEX QTRAP 5500 triple-quadrupole mass spectrometer (50). Chromatogram review and peak area integration were performed using MultiQuant software version 2.1 (Applied Biosystems SCIEX).
Characterizing metabolic differences across cell types and microenvironments
Missing metabolite values were imputed with an ion count of 1000 and normalized peak area calculated for each detected metabolite using the TIC from each sample to correct variations introduced from sample handling through instrument analysis. TIC normalization was followed by log transformation and autoNorm row-scaling using MetaboAnalystR (51) (default parameters). We conducted exploratory analysis of metabolome differences across sample types using PCA with the vegan R package and conditioned the analysis on patient using partial redundancy analysis. Heatmap dendrograms were constructed using Ward’s method to cluster Euclidean distances among samples. We identified differentially abundant metabolites across cell types and microenvironments using limma (52) on the normalized metabolite abundances. To simplify interpretation, we specified the model using the group means parameterization, treating cell types within microenvironments as each group (n = 6 groups); for significance testing, we took the average of triplicate measurements for each metabolite to avoid pseudoreplication and included patient as a block in the limma design. To examine metabolites that differed across patients, we refit models in limma including patient as a fixed effect. We reported significance at Padj < 0.05 (Benjamini-Hochberg correction) for prespecified contrasts among cell types and microenvironments.
scRNA-seq
Single-cell transcriptome sequencing was performed on total viably frozen ascites and tumor samples using the 10× 5′ Gene Expression protocol, following viability enrichment with the Miltenyi Dead Cell Removal Kit (>80% viability). Five cases with matched tumor and ascites available were profiled, although low viability from one tumor sample prevented its inclusion. To enable multiplexing of patients, we combined samples from each patient in lanes of the 10× chromium controller, with separate runs for ascites and tumor fractions. Following sequencing [Illumina HiSeq 4000 28 × 98 bp paired end (PE), Genome Quebec; mean of 73,488 and 41,378 reads per cell for tumor and ascites, respectively], we assigned donor identities using CellSNP and Vireo (53) (based on the common human SNP VCF provided by CellSNP for GRCh38). We excluded unassigned cells and those identified as doublets and matched donors between ascites and tumor samples based on the nearest identity by state (IBS) of inferred patient genotypes using SNPRelate (54). On the basis of this assignment, we retained three cases with abundant cellular representation in both tumor and ascite fractions for downstream analysis. Following quality-filtering steps in the scater (55) and scran (56) BioConductor packages, this yielded 6975 cells (2792 and 4183 from tumor and ascites, respectively) for analysis. We clustered cells by expression using igraph’s (57) Louvain clustering implementation of the shared nearest neighbor network (SNN) based on Jaccard distance. Clusters were manually annotated into presumptive cell types based on marker gene expression and visualized with t-SNE. The cytotoxic T cells are defined by the expression of CD8A and GZMA, excluding a subcluster with low ribosomal protein expression. We accessed the published data of Izar et al. (16), including their t-SNE embedding, to rule overlap in expression between immune cell markers and NNMT expression.
T cell functional assay
PBMCs were isolated from a leukapheresis product (STEMCELL Technologies) by Ficoll gradient density centrifugation. CD3+ cells were isolated from the PBMCs using CD3 Microbeads (Miltenyi). The CD3+ cells were activated with plate-bound CD3 (5 μg/ml), soluble CD28 (3 μg/ml), and IL-2 (300 U/ml; Proleukin) in the presence or absence of MNA. On the final day of expansion, viability (Fixable Viability Dye eFluor450, eBioscience) and proliferation (123count eBeads, Thermo Fisher Scientific) were assessed by flow cytometry. Effector function was assessed by stimulating cells for 4 hours with PMA (20 ng/ml) and ionomycin (1 μg/ml) with GolgiStop and monitored for CD8-PerCP (RPA-T8, BioLegend), CD4-AF700 (RPA-T4, BioLegend), and TNFα–fluorescein isothiocyanate (FITC) (MAb11, BD). Cells for qPCR and ChIP were stimulated for 4 hours with PMA (20 ng/ml) and ionomycin (1 μg/ml). Supernatant for ELISA was collected before and after 4 hours of stimulation with PMA (20 ng/ml) and ionomycin (1 μg/ml).
Quantitative polymerase chain reaction
RNA was isolated using the RNeasy Plus Mini Kit (QIAGEN) following the manufacturer’s protocol. Samples were homogenized using the QIAshredder (QIAGEN). Complementary DNA (cDNA) was synthesized using the High-Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific). TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific) was used to quantify gene expression (following the manufacturer’s protocol) with the following probes: Hs00196287_m1 (NNMT), Hs00154079_m1 (AOX1), Hs00427552_m1 (SLC22A1), Hs02786624_g1 [glyceraldehyde-3-phosphate dehydrogenase (GAPDH)], and Hs01010726_m1 (SLC22A2). Samples were run on the StepOnePlus Real-Time PCR System (Applied Biosystems) in the MicroAmp Fast Optical 96-Well Reaction Plate (Applied Biosystems) with MicroAmp Optical Adhesive Film (Applied Biosystems). Any Ct value over 35 is considered above the detection threshold and identified as undetectable.
Chromatin immunoprecipitation
ChIP was performed as described previously (58). Briefly, cells were treated with formaldehyde (1.42% final concentration) and incubated at room temperature for 10 min. A supplemental swelling buffer (25 mM Hepes, 1.5 mM MgCl2, 10 mM KCl, and 0.1% NP-40) was used for 10 min on ice before resuspension in immunoprecipitation buffer as described (58). Samples were then sonicated with the following cycle: 10 cycles of 20 1-s pulses with 40-s rest periods. ChIP grade immunoglobulin G (Cell Signaling Technology; 1 μl), histone H3 (Cell Signaling Technology; 3 μl), NFAT (Invitrogen; 3 μl), and SP1 (Cell Signaling Technology; 3 μl) antibodies were incubated with samples at 4°C overnight while rocking. Protein A beads (Thermo Fisher Scientific) were incubated with the samples for 1 hour at 4°C with gentle rocking followed by DNA enrichment with chelex beads (Bio-Rad) and protein digestion with proteinase K (Thermo Fisher). TNFα promoter was detected by PCR: forward, GGG TAT CCT TGA TGC TTG TGT; reverse, GTG CCA ACA ACT GCC TTT ATA TG (207-bp product). Images were produced by Image Lab (Bio-Rad), and quantification was performed with ImageJ software.
Enzyme-linked immunosorbent assay
Cell culture supernatants were collected as described above. Assays were carried out according to the manufacturer’s protocol for the Human TNFα ELISA Kit (Invitrogen), the Human IL-2 ELISA Kit (Invitrogen), and the Human IFN-γ ELISA Kit (Abcam). Supernatants were diluted 1:100 for detection of TNFα and IL-2, and 1:3 for IFN-γ according to the manufacturer’s protocol. Absorbance was measured at 450 nm using the EnVision 2104 Multilabel Reader (PerkinElmer).
Cellular MNA content
PBMCs were isolated from a leukapheresis product (STEMCELL Technologies) by Ficoll gradient density centrifugation. CD3+ cells were isolated from the PBMCs using CD3 Microbeads (Miltenyi). The CD3+ cells were activated with plate-bound CD3 (5 μg/ml), soluble CD28 (3 μg/ml), and IL-2 (300 U/ml; Proleukin) in the presence or absence of MNA for 3 days. After 3 days, cells were collected and washed with 0.9% saline, and the pellet was snap frozen. Cell counts were carried out using 123count eBeads by flow cytometry (Cytek Aurora; 3L-16V-14B-8R configuration).
Metabolite extraction was performed as described above. Dried extracts were reconstituted at a concentration of 4000 cell equivalents/μl. Samples were analyzed via reversed-phase chromatography (1290 Infinity II, Agilent Technologies, Santa Clara, CA) with a CORTECS T3 column (2.1 × 150 mm, 1.6-μm particle size, 120-Å pore size; #186008500, Waters) coupled to a triple quadrupole mass spectrometer (6470, Agilent) with electrospray ionization operated in positive mode. Mobile phase A was 0.1% formic acid (in H2O), and mobile phase B was 90% acetonitrile, 0.1% formic acid. The LC gradient was 0 to 2 min 100% A, 2 to 7.1 min ramp to 99% B, and 7.1 to 8 min hold at 99% B. The column was then reequilibrated with mobile phase A for 3 min at 0.6 ml/min. Flow rate was 0.4 ml/min, and the column compartment was heated to 50°C. A neat chemical standard (M320995, Toronto Research Chemicals, North York, ON, CAN) of MNA was used to establish retention time (RT) and transitions (RT = 0.882 min, transition 1 = 137 → 94.1, transition 2 = 137 → 92, transition 3 = 137 → 78). Transition 1 was used for quantitation when all three transitions were present at the correct retention time to ensure specificity. A standard curve for MNA (Toronto Research Chemicals) was generated from six subsequent serial dilutions of a stock solution (1 mg/ml) to yield 0.1, 1.0, 10, and 100 ng/ml, and 1.0 and 10 μg/ml standards. The lower limit of detection was 1 ng/ml, with a linear response between 10 ng/ml and 10 μg/ml. Two microliters of each sample and standard was injected for LC/MS analysis, and a pooled quality control sample was run every eighth injection to ensure stability of the analytical platform. The MNA response of all MNA-treated cell samples was within the linear range of the assay. Data analysis was completed with MassHunter Quantitative Analysis Software (v9.0, Agilent).
CAR design and vector cloning
The second-generation αFR-CAR construct was taken from Song et al. (59). Briefly, the construct contained the following: CD8a leader sequence, single-chain variable fragment specific to human αFR, CD8a hinge and transmembrane region, CD27 intracellular domain, and CD3z intracellular domain. The full CAR sequence was synthesized by GenScript and then cloned into a second-generation lentiviral expression vector upstream of a GFP expression cassette used to assess transduction efficiency.
Lentivirus production
Lentivirus was generated by transfecting HEK293T cells [American Type Culture Collection (ATCC); grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and 1% PenStrep] with CAR-GFP vector and packaging plasmids (psPAX2 and pMD2.G, Addgene) at a 1:1:1 ratio using Lipofectamine (Sigma-Aldrich). Virus-containing supernatant was collected at 48 and 72 hours posttransfection, filtered, and concentrated by ultracentrifugation. Concentrated virus supernatants were stored at −80°C until transduction.
CAR-T cell generation
PBMCs were isolated from a healthy donor leukapheresis product (STEMCELL Technologies) by Ficoll gradient density centrifugation. CD8+ cells were isolated from PBMCs using positive selection CD8 microbeads (Miltenyi). T cells were stimulated with TransAct (Miltenyi) and expanded in TexMACS medium [Miltenyi; supplemented with 3% heat-inactivated human serum, 1% PenStrep, and IL-2 (300 U/ml)] according to the manufacturer’s instructions. Twenty-four hours poststimulation, T cells were transduced with lentivirus (10 μl of concentrated virus supernatant per 106 cells). Cells were assessed for GFP expression 1 to 3 days following transduction on a Cytek Aurora (gated on FSC (forward scatter)/SSC (side scatter), singlets, GFP+) to demonstrate at least 30% transduction efficiency.
Cytolytic assay
CAR-T cells were cultured in Immunocult (STEMCELL Technologies; supplemented with 1% PenStrep) under the following conditions for 24 hours: without treatment, with 250 μM adenosine, or with 10 mM MNA. After pretreatment, CAR-T cells were washed with PBS and cocultured with 20,000 SK-OV-3 cells [ATCC; expanded in McCoy 5A medium (Sigma-Aldrich) supplemented with 10% FBS and 1% PenStrep] in supplemented Immunocult media in triplicate at an effector-to-target ratio of 10:1. SK-OV-3 cells and SK-OV-3 cells lysed with digitonin (0.5 mg/ml; Sigma-Aldrich) were used as negative and positive controls, respectively. Following 24 hours of coculture, supernatants were collected, and lactate dehydrogenase (LDH) was measured according to the manufacturer’s instructions (LDH Glo Cytotoxicity Assay Kit, Promega). LDH supernatant was diluted 1:50 in LDH buffer. Percent killing was measured using the following formula: % killing = corrected killing/maximum killing × 100%, where corrected killing = coculture − T cells alone, and maximum killing = positive control − negative control.
Quantification and statistical analysis
Statistical analysis was carried out as described in the text or Materials and Methods using GraphPad Prism 8, Microsoft Excel, or R v3.6.0. Where multiple samples were taken from the same patient (e.g., ascites and tumor), we used paired t tests or included patient as a random effect in linear or generalized models, as appropriate. For metabolomic analysis, significance testing was done on means of triplicate measurements.
Supplementary Material
Acknowledgments
We thank the patients who donated specimens to make this study possible. We also thank M. Lesperance and F. Nathoo for advice on statistical analysis. Funding: This work was supported by the research grants to J.J.L. from the U.S. Department of Defense Ovarian Cancer Research Program Pilot Award (W81XWH-18-1-0264) and Canadian Institutes of Health Research (MOP-142351 and PJT-162279). P.T.H. is supported by research grants from the Carraresi Family Foundation Award. B.H.N. is supported by research grants from the BC Cancer Foundation and the Canadian Foundation for Innovation. R.G.J. is supported by grants from the Canadian Institutes of Health Research (MOP-142259) and funds from the Van Andel Institute. M.K.K. is supported by a University of Victoria Graduate Award. P.T.H. is supported by a Canadian Institutes for Health Research Postdoctoral Fellowship. S.K. is supported by BioCanRx and BC Cancer scholarships. Biospecimens were obtained with assistance from the IROC-TTR Biobank. Single-cell library preparations and RNA sequencing were provided by Genome Quebec. P.T.H. is supported by research grants from the Carraresi Family Foundation Award. Carraresi Foundation OVCARE Research Grants are supported by the VGH & UBC Hospital Foundation. B.H.N. is supported by research grants from the BC Cancer Foundation and the Canadian Foundation for Innovation. Author contributions: M.K.K. and P.T.H. designed and performed the experiments, analyzed the data, and wrote the manuscript. S.M. preformed the ChIP. E.Y.L. and G.C. generated the CAR-T cells. E.Y.L. performed the cytolytic assay. S.M., S.K., and B.P. helped perform the metabolite profiling experiments. P.T.H., J.Sm., B.A., J.St., and B.H.N. generated the scRNA-seq data. L.G.Z. and R.J.D. generated and analyzed the metabolite profiling data. A.E.E., R.D.S., K.S.W., and R.G.J. generated and analyzed the cellular MNA content data. P.H.W. oversaw biospecimen collection through the IROC-TTR. J.J.L. conceived the project and wrote the manuscript. Competing interests: J.St. is a permanent member and owns stocks of Surface Oncology. R.J.D. is a member of the Scientific Advisory Board at Agios Pharmaceuticals. R.G.J. is a consultant for Agios Pharmaceuticals and is a member of the scientific advisory board and owns equity in ImmunoMet Therapeutics. Data and materials availability: Raw sequencing data are deposited at the European Genome Phenome Archive (EGAS00001004829). Processed data files and scripts to reproduce metabolomics and scRNA-seq analyses are available at https://github.com/vicDRC/BCCJJL01_ovarian. Flow cytometry data is deposited at Flow Repository (FR-FCM-Z2NH). Schematics were created with BioRender.com. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabe1174/DC1
REFERENCES AND NOTES
- 1.Pavlova N. N., Thompson C. B., The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leone R. D., Zhao L., Englert J. M., Sun I.-M., Oh M.-H., Sun I.-H., Arwood M. L., Bettencourt I. A., Patel C. H., Wen J., Tam A., Blosser R. L., Prchalova E., Alt J., Rais R., Slusher B. S., Powell J. D., Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Najjar Y. G., Menk A. V., Sander C., Rao U., Karunamurthy A., Bhatia R., Zhai S., Kirkwood J. M., Delgoffe G. M., Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight. 4, e124989 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang C.-H., Qiu J., O’Sullivan D., Buck M. D., Noguchi T., Curtis J. D., Chen Q., Gindin M., Gubin M. M., van der Windt G. J. W., Tonc E., Schreiber R. D., Pearce E. J., Pearce E. L., Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cronin S. J. F., Seehus C., Weidinger A., Talbot S., Reissig S., Seifert M., Pierson Y., McNeill E., Longhi M. S., Turnes B. L., Kreslavsky T., Kogler M., Hoffmann D., Ticevic M., da Luz Scheffer D., Tortola L., Cikes D., Jais A., Rangachari M., Rao S., Paolino M., Novatchkova M., Aichinger M., Barrett L., Latremoliere A., Wirnsberger G., Lametschwandtner G., Busslinger M., Zicha S., Latini A., Robson S. C., Waisman A., Andrews N., Costigan M., Channon K. M., Weiss G., Kozlov A. V., Tebbe M., Johnsson K., Woolf C. J., Penninger J. M., The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature 563, 564–568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bunse L., Pusch S., Bunse T., Sahm F., Sanghvi K., Friedrich M., Alansary D., Sonner J. K., Green E., Deumelandt K., Kilian M., Neftel C., Uhlig S., Kessler T., von Landenberg A., Berghoff A. S., Marsh K., Steadman M., Zhu D., Nicolay B., Wiestler B., Breckwoldt M. O., Al-Ali R., Karcher-Bausch S., Bozza M., Oezen I., Kramer M., Meyer J., Habel A., Eisel J., Poschet G., Weller M., Preusser M., Nadji-Ohl M., Thon N., Burger M. C., Harter P. N., Ratliff M., Harbottle R., Benner A., Schrimpf D., Okun J., Herold-Mende C., Turcan S., Kaulfuss S., Hess-Stumpp H., Bieback K., Cahill D. P., Plate K. H., Hänggi D., Dorsch M., Suvà M. L., Niemeyer B. A., von Deimling A., Wick W., Platten M., Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 24, 1192–1203 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Baumann T., Dunkel A., Schmid C., Schmitt S., Hiltensperger M., Lohr K., Laketa V., Donakonda S., Ahting U., Lorenz-Depiereux B., Heil J. E., Schredelseker J., Simeoni L., Fecher C., Körber N., Bauer T., Hüser N., Hartmann D., Laschinger M., Eyerich K., Eyerich S., Anton M., Streeter M., Wang T., Schraven B., Spiegel D., Assaad F., Misgeld T., Zischka H., Murray P. J., Heine A., Heikenwälder M., Korn T., Dawid C., Hofmann T., Knolle P. A., Höchst B., Regulatory myeloid cells paralyze T cells through cell–cell transfer of the metabolite methylglyoxal. Nat. Immunol. 21, 555–566 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Anderson K. G., Stromnes I. M., Greenberg P. D., Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell 31, 311–325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Townsend K. N., Spowart J. E., Huwait H., Eshragh S., West N. R., Elrick M. A., Kalloger S. E., Anglesio M., Watson P. H., Huntsman D. G., Lum J. J., Markers of T cell infiltration and function associate with favorable outcome in vascularized high-grade serous ovarian carcinoma. PLOS ONE 8, e82406 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colegio O. R., Chu N.-Q., Szabo A. L., Chu T., Rhebergen A. M., Jairam V., Cyrus N., Brokowski C. E., Eisenbarth S. C., Phillips G. M., Cline G. W., Phillips A. J., Medzhitov R., Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Husain Z., Huang Y., Seth P., Sukhatme V. P., Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 191, 1486–1495 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Munn D. H., Bronte V., Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 39, 1–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Platten M., von Knebel Doeberitz N., Oezen I., Wick W., Ochs K., Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front. Immunol. 5, 673 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mellor A. L., Munn D. H., IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 4, 762–774 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Kipps E., Tan D. S. P., Kaye S. B., Meeting the challenge of ascites in ovarian cancer: New avenues for therapy and research. Nat. Rev. Cancer 13, 273–282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Izar B., Tirosh I., Stover E. H., Wakiro I., Cuoco M. S., Alter I., Rodman C., Leeson R., Su M.-J., Shah P., Iwanicki M., Walker S. R., Kanodia A., Melms J. C., Mei S., Lin J.-R., Porter C. B. M., Slyper M., Waldman J., Jerby-Arnon L., Ashenberg O., Brinker T. J., Mills C., Rogava M., Vigneau S., Sorger P. K., Garraway L. A., Konstantinopoulos P. A., Liu J. F., Matulonis U., Johnson B. E., Rozenblatt-Rosen O., Rotem A., Regev A., A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 26, 1271–1279 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rådestad E., Klynning C., Stikvoort A., Mogensen O., Nava S., Magalhaes I., Uhlin M., Immune profiling and identification of prognostic immune-related risk factors in human ovarian cancer. Oncoimmunology 8, e1535730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanizaki Y., Kobayashi A., Toujima S., Shiro M., Mizoguchi M., Mabuchi Y., Yagi S., Minami S., Takikawa O., Ino K., Indoleamine 2,3-dioxygenase promotes peritoneal metastasis of ovarian cancer by inducing an immunosuppressive environment. Cancer Sci. 105, 966–973 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scharping N. E., Menk A. V., Moreci R. S., Whetstone R. D., Dadey R. E., Watkins S. C., Ferris R. L., Delgoffe G. M., The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 374–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho P.-C., Bihuniak J. D., Macintyre A. N., Staron M., Liu X., Amezquita R., Tsui Y.-C., Cui G., Micevic G., Perales J. C., Kleinstein S. H., Abel E. D., Insogna K. L., Feske S., Locasale J. W., Bosenberg M. W., Rathmell J. C., Kaech S. M., Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greene E., Finak G., D’Amico L. A., Bhardwaj N., Church C. D., Morishima C., Ramchurren N., Taube J. M., Nghiem P. T., Cheever M. A., Fling S. P., Gottardo R., New interpretable machine learning method for single-cell data reveals correlates of clinical response to cancer immunotherapy. bioRxiv, 702118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma E. H., Verway M. J., Johnson R. M., Roy D. G., Steadman M., Hayes S., Williams K. S., Sheldon R. D., Samborska B., Kosinski P. A., Kim H., Griss T., Faubert B., Condotta S. A., Krawczyk C. M., DeBerardinis R. J., Stewart K. M., Richer M. J., Chubukov V., Roddy T. P., Jones R. G., Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity 51, 856–870.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Binek A., Rojo D., Godzien J., Rupérez F. J., Nuñez V., Jorge I., Ricote M., Vázquez J., Barbas C., Flow cytometry has a significant impact on the cellular metabolome. J. Proteome Res. 18, 169–181 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Llufrio E. M., Wang L., Naser F. J., Patti G. J., Sorting cells alters their redox state and cellular metabolome. Redox Biol. 16, 381–387 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ulanovskaya O. A., Zuhl A. M., Cravatt B. F., NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 9, 300–306 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanakkanthara A., Kurmi K., Ekstrom T. L., Hou X., Purfeerst E. R., Heinzen E. P., Correia C., Huntoon C. J., O’Brien D., Wahner Hendrickson A. E., Dowdy S. C., Li H., Oberg A. L., Hitosugi T., Kaufmann S. H., Weroha S. J., Karnitz L. M., BRCA1 deficiency upregulates NNMT, which reprograms metabolism and sensitizes ovarian cancer cells to mitochondrial metabolic targeting agents. Cancer Res. 79, 5920–5929 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eckert M. A., Coscia F., Chryplewicz A., Chang J. W., Hernandez K. M., Pan S., Tienda S. M., Nahotko D. A., Li G., Blaženović I., Lastra R. R., Curtis M., Yamada S. D., Perets R., McGregor S. M., Andrade J., Fiehn O., Moellering R. E., Mann M., Lengyel E., Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 569, 723–728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.A. T. Nies, H. Koepsell, K. Damme, M. Schwab, Handbook of experimental pharmacology, in Drug Transporters, M. F. Fromm, R. B. Kim, Eds. (Springer, 2011), pp. 105–167. [DOI] [PubMed] [Google Scholar]
- 29.Minuesa G., Purcet S., Erkizia I., Molina-Arcas M., Bofill M., Izquierdo-Useros N., Casado F. J., Clotet B., Pastor-Anglada M., Martinez-Picado J., Expression and functionality of anti-human immunodeficiency virus and anticancer drug uptake transporters in immune cells. J. Pharmacol. Exp. Ther. 324, 558–567 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Falvo J. V., Tsytsykova A. V., Goldfeld A. E., Transcriptional control of the TNF gene. Curr. Dir. Autoimmun. 11, 27–60 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kulbe H., Thompson R., Wilson J. L., Robinson S., Hagemann T., Fatah R., Gould D., Ayhan A., Balkwill F., The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 67, 585–592 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charles K. A., Hoare S. A., Hagemann T., Scott D., Aird R., Smyth J., Balkwill F. R., Infliximab, a humanised anti-TNF-a monoclonal antibody, exhibits biological activity in the ovarian tumor microenvironment in patients. Cancer Res. 66, 1055–1056 (2006). [Google Scholar]
- 33.Salako M. A., Kulbe H., Ingemarsdotter C. K., Pirlo K. J., Williams S. L., Lockley M., Balkwill F. R., McNeish I. A., Inhibition of the inflammatory cytokine TNF-α increases adenovirus activity in ovarian cancer via modulation of cIAP1/2 expression. Mol. Ther. 19, 490–499 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sipak-Szmigiel O., Włodarski P., Ronin-Walknowska E., Niedzielski A., Karakiewicz B., Słuczanowska-Głąbowska S., Laszczyńska M., Malinowski W., Serum and peritoneal fluid concentrations of soluble human leukocyte antigen, tumor necrosis factor alpha and interleukin 10 in patients with selected ovarian pathologies. J. Ovarian Res. 10, 25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szlosarek P. W., Grimshaw M. J., Kulbe H., Wilson J. L., Wilbanks G. D., Burke F., Balkwill F. R., Expression and regulation of tumor necrosis factor α in normal and malignant ovarian epithelium. Mol. Cancer Ther. 5, 382–390 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Piura B., Medina L., Rabinovich A., Dyomin V., Levy R. S., Huleihel M., Distinct expression and localization of TNF system in ovarian carcinoma tissues: Possible involvement of TNF-α in morphological changes of ovarian cancerous cells. Anticancer Res 34, 745–752 (2014). [PubMed] [Google Scholar]
- 37.Montfort A., Colacios C., Levade T., Andrieu-Abadie N., Meyer N., Ségui B., Corrigendum: The TNF paradox in cancer progression and immunotherapy. Front. Immunol. 10, 2515 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charles K., Kulbe H., Soper R., Escorcio Correia M., Lawrence T., Schultheis A., Chakravarty P., Thompson R., Kollias G., Smyth J., Balkwill F., Hagemann T., The tumor-promoting actions of TNF-α involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J. Clin. Invest. 119, 3011–3023 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sukumar M., Liu J., Ji Y., Subramanian M., Crompton J. G., Yu Z., Roychoudhuri R., Palmer D. C., Muranski P., Karoly E. D., Mohney R. P., Klebanoff C. A., Lal A., Finkel T., Restifo N. P., Gattinoni L., Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 123, 4479–4488 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siska P. J., Beckermann K. E., Mason F. M., Andrejeva G., Greenplate A. R., Sendor A. B., Chiang Y.-C. J., Corona A. L., Gemta L. F., Vincent B. G., Wang R. C., Kim B., Hong J., Chen C.-L., Bullock T. N., Irish J. M., Rathmell W. K., Rathmell J. C., Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight. 2, e93411 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martínez-Reyes I., Chandel N. S., Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hensley C. T., Faubert B., Yuan Q., Lev-Cohain N., Jin E., Kim J., Jiang L., Ko B., Skelton R., Loudat L., Wodzak M., Klimko C., McMillan E., Butt Y., Ni M., Oliver D., Torrealba J., Malloy C. R., Kernstine K., Lenkinski R. E., DeBerardinis R. J., Metabolic heterogeneity in human lung tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ron-Harel N., Santos D., Ghergurovich J. M., Sage P. T., Reddy A., Lovitch S. B., Dephoure N., Satterstrom F. K., Sheffer M., Spinelli J. B., Gygi S., Rabinowitz J. D., Sharpe A. H., Haigis M. C., Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 24, 104–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma E. H., Bantug G., Griss T., Condotta S., Johnson R. M., Samborska B., Mainolfi N., Suri V., Guak H., Balmer M. L., Verway M. J., Raissi T. C., Tsui H., Boukhaled G., Henriques da Costa S., Frezza C., Krawczyk C. M., Friedman A., Manfredi M., Richer M. J., Hess C., Jones R. G., Serine is an essential metabolite for effector T cell expansion. Cell Metab. 25, 345–357 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Zou W., Machelon V., Coulomb-L’Hermin A., Borvak J., Nome F., Isaeva T., Wei S., Krzysiek R., Durand-Gasselin I., Gordon A., Pustilnik T., Curiel D. T., Galanaud P., Capron F., Emilie D., Curiel T. J., Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat. Med. 7, 1339–1346 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Bowtell D. D., Böhm S., Ahmed A. A., Aspuria P.-J., Bast R. C. Jr., Beral V., Berek J. S., Birrer M. J., Blagden S., Bookman M. A., Brenton J. D., Chiappinelli K. B., Martins F. C., Coukos G., Drapkin R., Edmondson R., Fotopoulou C., Gabra H., Galon J., Gourley C., Heong V., Huntsman D. G., Iwanicki M., Karlan B. Y., Kaye A., Lengyel E., Levine D. A., Lu K. H., McNeish I. A., Menon U., Narod S. A., Nelson B. H., Nephew K. P., Pharoah P., Powell D. J. Jr., Ramos P., Romero I. L., Scott C. L., Sood A. K., Stronach E. A., Balkwill F. R., Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 15, 668–679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tothill R. W., Tinker A. V., George J., Brown R., Fox S. B., Lade S., Johnson D. S., Trivett M. K., Etemadmoghadam D., Locandro B., Traficante N., Fereday S., Hung J. A., Chiew Y.-E., Haviv I.; Australian Ovarian Cancer Study Group, Gertig D., DeFazio A., Bowtell D. D. L., Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 14, 5198–5208 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Bertrand F., Montfort A., Marcheteau E., Imbert C., Gilhodes J., Filleron T., Rochaix P., Andrieu-Abadie N., Levade T., Meyer N., Colacios C., Ségui B., TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 8, 2256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monk J. P., Phillips G., Waite R., Kuhn J., Schaaf L. J., Otterson G. A., Guttridge D., Rhoades C., Shah M., Criswell T., Caligiuri M. A., Villalona-Calero M. A., Assessment of tumor necrosis factor alpha blockade as an intervention to improve tolerability of dose-intensive chemotherapy in cancer patients. J. Clin. Oncol. 24, 1852–1859 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Mullen A. R., Hu Z., Shi X., Jiang L., Boroughs L. K., Kovacs Z., Boriack R., Rakheja D., Sullivan L. B., Linehan W. M., Chandel N. S., DeBerardinis R. J., Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 7, 1679–1690 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chong J., Xia J., MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 34, 4313–4314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K., limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang Y., McCarthy D. J., Stegle O., Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol. 20, 273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng X., Levine D., Shen J., Gogarten S. M., Laurie C., Weir B. S., A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCarthy D. J., Campbell K. R., Lun A. T. L., Wills Q. F., Scater: Pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lun A. T. L., Bach K., Marioni J. C., Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 17, 75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Csardi G., Nepusz T., The Igraph software package for complex network research. InterJournal. Com. Syst. 1695, 1695 (2006). [Google Scholar]
- 58.Nelson J. D., Denisenko O., Bomsztyk K., Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 1, 179–185 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Song D.-G., Ye Q., Poussin M., Harms G. M., Figini M., Powell D. J. Jr., CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 119, 696–706 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Neelakantan H., Brightwell C. R., Graber T. G., Maroto R., Wang H.-Y. L., McHardy S. F., Papaconstantinou J., Fry C. S., Watowich S. J., Small molecule nicotinamide N-methyltransferase inhibitor activates senescent muscle stem cells and improves regenerative capacity of aged skeletal muscle. Biochem. Pharmacol. 163, 481–492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabe1174/DC1