

Disease-causing mutations in a linkage-specific deubiquitylase provide insights into chromatin remodeling during embryogenesis.
Abstract
Reversible modification of proteins with linkage-specific ubiquitin chains is critical for intracellular signaling. Information on physiological roles and underlying mechanisms of particular ubiquitin linkages during human development are limited. Here, relying on genomic constraint scores, we identify 10 patients with multiple congenital anomalies caused by hemizygous variants in OTUD5, encoding a K48/K63 linkage–specific deubiquitylase. By studying these mutations, we find that OTUD5 controls neuroectodermal differentiation through cleaving K48-linked ubiquitin chains to counteract degradation of select chromatin regulators (e.g., ARID1A/B, histone deacetylase 2, and HCF1), mutations of which underlie diseases that exhibit phenotypic overlap with OTUD5 patients. Loss of OTUD5 during differentiation leads to less accessible chromatin at neuroectodermal enhancers and aberrant gene expression. Our study describes a previously unidentified disorder we name LINKED (LINKage-specific deubiquitylation deficiency–induced Embryonic Defects) syndrome and reveals linkage-specific ubiquitin cleavage from chromatin remodelers as an essential signaling mode that coordinates chromatin remodeling during embryogenesis.
INTRODUCTION
Determining genetic causes of diseases is a powerful approach to discover and study physiologic pathways critical for human health. Exome and genome sequencing have facilitated the rapid increase in genetic diagnoses, especially in pediatric populations with developmental disorders (1). Identifying and studying disease-causing variants in patients presenting with multiple congenital anomalies at birth not only allows for more accurate clinical management (2) but also enables discovery of important regulatory processes during embryonic development.
Cell fate decisions during human embryogenesis rely on signaling information encoded with ubiquitin, an essential posttranslational modifier that is typically attached to lysine residues of substrates (3–8). During ubiquitin signaling, cells use intricate enzymatic cascades to synthesize topologically unique ubiquitin signals that differentially affect substrate fates (3). Transfer of a single ubiquitin to one or multiple substrate sites often results in changes in the interaction landscape of the modified protein (9, 10). Ubiquitin itself is a target of ubiquitylation and can be modified at its N terminus or at one of its seven lysine residues, thus enabling the formation of uniquely linked ubiquitin chains that can adopt distinct structures and functions (11). Examples include K11- and K48-linked chains that mediate degradation via the 26S proteasome (12, 13), M1- and K63-linked chains that allow for formation of signaling complexes during nuclear factor κB activation and DNA repair (14, 15), K11/K48-branched chains that serve as priority signal for proteasomal degradation during the cell cycle and protein quality control (16, 17), and M1/K63- and K48/K63-branched chains that play important roles during immune signaling (18, 19).
Ubiquitylation is highly specific and reversible, two features that empower ubiquitin signaling to regulate cellular behavior and decision-making during human development (8). Specificity is ensured by more than 600 human E3 ligase enzymes that cooperate with 2 E1 enzymes and ~40 E2 enzymes to catalyze ubiquitin conjugation to particular substrates. Reversibility is provided by ~100 human deubiquitylases (DUBs), a family of enzymes that edits chain architecture or cleaves off ubiquitin signals from substrates to modulate and terminate signaling, respectively (8, 20, 21). There are six structurally distinct subclasses of DUBs, which have been well studied in terms of their mechanism of ubiquitin cleavage (22), regulatory principles governing their catalytic activity (23), and mechanisms of functional diversification through, e.g., differential isoform expression or localization (24). However, how the relatively small number of ~100 human DUBs can regulate thousands of cellular ubiquitin modifications to ensure proper signaling during human development is still an active area of investigation, and specific physiological examples of how such regulation occurs in space and time are limited.
The subfamily of ovarian tumor (OTU) DUBs have been implicated in many essential cellular processes and have emerged as crucial regulators of human physiology and development (25–27). OTU DUBs elicit their functions by hydrolyzing specific linkage types within polyubiquitin to modulate the stability, activity, or interaction landscapes of their substrates (28, 29). In particular, cleavage of K48-linked ubiquitin chains protects substrates from proteasomal degradation, and cleavage of M1- and K63-linked ubiquitin chains alters intracellular signaling. Five of the 16 OTU DUBs have been linked to human disease: Mutations in OTULIN and TNFAIP3 (encoding A20) cause autoinflammatory disorders (27, 30, 31), mutations in OTUD7A and ALG13 cause neurodevelopmental disorders (32–35), and mutations in OTUD6B cause multiple congenital anomalies (36). However, with the exception of OTULIN and A20, the underlying mechanisms and cognate substrates for these disease-associated enzymes are ill-defined, and the physiological functions for other OTU DUB members have remained largely unknown.
To help provide genetic diagnoses, recent bioinformatics efforts have leveraged the availability of large datasets from sequencing studies to determine the mutational constraint spectrum of human genes (37–40). By sequencing and comparing a large number of healthy individuals, genomic constraint quantifies the depletion of variation in every gene within control populations. These metrics allow for identification of genes intolerant to variation that, when mutated, have a high likelihood of causing embryonic lethality or disease. Such scores can thus help overcome limitations in current variant interpretation workflows and have the potential to uncover novel developmental pathways.
Here, by coupling human genetics and molecular studies, we identify a male-specific developmental disease caused by hypomorphic variants in OTUD5, an X-linked OTU DUB gene highly intolerant to genomic variation. This disorder is characterized by central nervous system (CNS), craniofacial, cardiac, skeletal, and genitourinary anomalies. We find that a subset of severe disease-causing mutations affect K48- but not K63-ubiquitin chain cleavage activity of OTUD5. By leveraging these variants as a mechanistic tool, we show that OTUD5 elicits its function through its K48 linkage–specific deubiquitylation activity and prevents the degradation of multiple chromatin remodelers to coordinate enhancer activation during neuroectodermal differentiation. Mutations in these same chromatin remodelers are the cause of distinct yet clinically overlapping developmental diseases. Together, our study describes a previously unidentified developmental disorder we name LINKED (LINKage-specific deubiquitylation deficiency–induced Embryonic Defects) syndrome, identifies a mechanistic connection between phenotypically overlapping diseases, and reveals K48-ubiquitin chain cleavage of functionally related substrates as an essential signaling mode coordinating chromatin remodeling during embryogenesis.
RESULTS
Identification of a developmental disease caused by mutations in OTUD5 by leveraging genomic constraint
We identified an index family (F1) with three affected sons (P1 to P3), all with severe multiple congenital anomalies, who had exome sequencing performed with no known pathogenic variant identified. By filtering coding variants based on inheritance and allele frequency, we found a novel, maternally inherited missense mutation in the gene OTUD5, which encodes a DUB (41), at p.Gly494Ser (Fig. 1A and Table 1). Although ubiquitin-dependent processes are known to control embryonic cell fate decisions (5, 42–46), predictive algorithms, such as SIFT (47) and REVEL (48), yielded contradicting results for OTUD5 p.Gly494Ser pathogenicity. Thus, we determined the likelihood of OTUD5 and other OTU DUB family members to be disease-causing by querying how often they are lost or subject to change in the exome of healthy individuals (Fig. 1B) (38). As expected, this genomic constraint approach captured genes already known to cause autosomal dominant developmental disorders [ALG13, TNFAIP3, and OTUD7A (31–35)]. Notably, we found that among all OTU DUBs, OTUD5 was the most restricted in heterozygous or hemizygous loss-of-function [pLI (probability of being loss-of-function intolerant) = 1, o/e (observed/expected) = 0] and missense mutations (Z = 3.99, o/e = 0.21) in healthy individuals, strongly supporting that mutations in OTUD5 could cause severe early-onset disease in our index family. Through combined resources at the National Institutes of Health and external collaborations, we identified 7 additional male patients, 10 total from seven families, all with novel variants in OTUD5 segregating with disease (Fig. 1, C to E; Table 1; and table S1). These mutations were not present in gnomAD databases (38, 40, 49). P4, who carried a novel variant, inherited de novo, at p.Leu352Pro, exhibited clinical features highly overlapping with our index patients P1 to P3, including brain malformations, congenital heart disease, distinctive facial features, postaxial polydactyly, and genitourinary anomalies (Fig. 1C). These patients have a unique identifiable phenotype, with partially overlapping features with chromatinopathies such as Coffin-Siris and Cornelia de Lange syndromes. The remaining patients (P5 to P10) exhibited attenuated phenotypes with conserved features (Fig. 1, A and B; Table 1; and table S1). All patients displayed global developmental delay with brain malformations, with a range of clinical characteristics from neonatal lethality and multiple congenital anomalies in severe cases to developmental delay, brain malformations, hirsutism, and genitourinary defects in mild cases (Fig. 1, A and C; fig. S1, A to D; Table 1; and table S1), strongly suggesting that OTUD5 variants were disease-causing. Corroborating this notion, we found that carrier mothers, although being heterozygous for the mutant allele, were unaffected and presented evidence of skewed X inactivation both by methylation-specific restriction enzyme testing (fig. S1E) and by RNA sequencing (RNA-seq) (see below). Further demonstrating that these OTUD5 missense variants are pathogenic and consistent with previous large-scale knockout screens (50), we found that CRISPR-mediated knockout of OTUD5 or knock-in of p.Gly494Ser or p.Leu352Pro patient alleles was lethal in mice (Fig. 1F). In these experiments, while we detected heterozygous knock-in females when isolating embryos at E12.5 (embryonic day 12.5), we were unable to recover them as live-born pups. Whether this might be due to preferential CRIPSR targeting of the X chromosome that escapes inactivation or differences in phenotypes associated with female carriers between mice and humans remains unclear and will require further investigation. Together, our genomic constraint–driven approach and mouse genetic data reveal that OTUD5 is required for embryonic development and, if mutated, leads to developmental disease.
Fig. 1. Hemizygous variants in OTUD5 cause multiple congenital anomaly disorder.
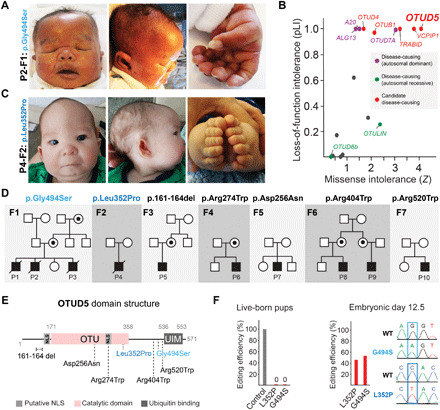
(A) Clinical photos showing craniofacial (retrognathia, midface hypoplasia, hypertelorism, low-set posteriorly rotated ears, and craniosynostosis) and digital anomalies (bilateral postaxial polydactyly of the hands and feet) of patient P2 carrying the p.Gly494Ser variant of OTUD5. Photo credit: With permission of the subjects’ legal guardian. (B) Among several OTU DUBs, OTUD5 is the strongest candidate for hypomorphic mutations leading to disease given high loss-of-function and missense intolerance scores. Loss-of-function intolerance (pLI) and missense intolerance (Z) were determined for all OTU DUBs using gnomAD. (C) Clinical photos showing craniofacial (retrognathia, midface hypoplasia, hypertelorism, low-set posteriorly rotated ears, and craniosynostosis) and digital anomalies (bilateral postaxial polydactyly of the hands and feet) of patient P4 carrying the p.Leu352Pro variant of OTUD5. (D) Genetic pedigrees of 10 patients from seven families with hemizygous variants in OTUD5, all with overlapping phenotypes. (E) Domain structure of OTUD5 indicating the location of the patient mutations. Variants associated with the most severe phenotypes, p.Gly494Ser and p.Leu352Pro, are highlighted in blue colors. NLS, nuclear localization sequence. (F) CRISPR-mediated knockout of OTUD5 or knock-in of the p.Gly494Ser or p.Leu352Pro patient variants results in embryonic lethality. Left: Mouse zygotes were injected with Cas9 complexed with gRNAs and respective repair oligos and transferred into pseudo-pregnant recipient mice. Percentage of live-born pups with edited alleles (knockout or knock-in) for a nonessential gene (control), OTUD5L352P, or OTUD5G494S is shown (n > 70 injected embryos per condition). Right: Mouse embryos were injected with gRNA-loaded Cas9 and respective repair oligos and implanted into mice. Pregnant mice were euthanized and embryos were isolated at E12.5. Percentage of pups with edited alleles (knockout or knock-in) for OTUD5L352P or OTUD5G494S injections are shown (n > 70 injected embryos per condition). Sanger sequencing depicting examples of E12.5 knock-in embryos is shown.
Table 1. Clinical table highlighting multiple congenital anomalies in patients with OTUD5 mutations.
Detailed manifestations for each category are listed in table S1. mo, months old; yo, years old.
| Variant | Inheritance | Gender | Status |
Global develop- mental delay |
Hirsutism | Anomalies | |||||
|
Central nervous system |
Cardiac | Genitourinary |
Postaxial polydactyly |
Cranio facial |
|||||||
| P1-F1 | No tissue available |
? | Male | Deceased in utero |
+ | + | + | + | + | + | + |
| P2-F1 | p.Gly494Ser, c.1480 G>A |
Maternal | Male | Deceased 1 mo |
+ | + | + | + | + | + | + |
| P3-F1 | p.Gly494Ser, c.1480 G>A |
Maternal | Male | Deceased 4 mo |
+ | + | + | + | + | + | + |
| P4-F2 | p.Leu352Pro, c.1055T>C |
De novo | Male | Deceased 1 yo |
+ | + | + | + | + | + | + |
| P5-F3 | p.161_164del, c. 482_490 del |
Maternal | Male | Alive 5 yo | + | − | + | + | + | − | − |
| P6-F4 | p.Arg274Trp, c.820C>T |
Maternal | Male | Alive 14 yo | + | + | + | − | + | − | + |
| P7-F5 | p.Asp256ASn, c.766G>A |
De novo | Male | Alive 4 yo | + | − | + | ? | + | − | − |
| P8-F6 | p.Arg404Trp, c.1210C>T |
Maternal | Male | Alive 2 yo | + | − | + | − | + | − | + |
| P9-F6 | p.Arg404Trp, c.1210C>T |
Maternal | Male | Alive 8 yo | + | − | + | + | + | − | + |
| P10-F7 | p.Arg520Trp, c. 1558C>T |
De novo | Male | Alive 13 yo | + | + | + | − | − | − | + |
Patient mutations are hypomorphic and alter OTUD5 levels, localization, and activity
Previous reports had demonstrated that OTUD5 is a nuclear (fig. S2, A and B), phospho-activated DUB that prefers cleavage of degradative K48- and non-degradative K63-ubiquitin chains over other linkage types (29, 41, 51, 52). To determine how the patient mutations affect OTUD5 function, we used cell-based and biochemical assays. These studies revealed three distinct loss-of-function mechanisms. First, the OTUD5 p.Gly494Ser (c.1480 G>A) found in one of the most severe cases is located at an exon-intron splice junction and caused a reduction in OTUD5 mRNA expression with intron retention in affected patient cells but not in those of the unaffected carrier mother or controls (Fig. 2, A and B). This reduction in mRNA resulted in a decrease in protein levels (Fig. 2C). Second, p.Arg274Trp, present in a conserved putative nuclear localization sequence (fig. S2C), caused a partial mis-localization of OTUD5 to the cytoplasm at steady-state expression in human embryonic stem cells (hESCs) (fig. S2, D and E). Third, most of the patient variants were located near or in the catalytic domain of OTUD5 and exhibited lower cleavage activity toward ubiquitin chains in vitro (Fig. 2, D and E, and fig. S3, A to E), without substantially affecting OTUD5’s activating phosphorylation (fig. S3F). In these experiments, we used the catalytically inactive C224S OTUD5 as control (41). Notably, p.Leu352Pro, associated with a severe phenotype, specifically impaired cleavage of K48- but not K63-ubiquitin chains (Fig. 2, D and E, and fig. S3, D and E), highlighting an important contribution of loss of degradative chain cleavage to the disease. Together, these results suggest an inverse correlation between residual biochemical activity and phenotype severity. We conclude that proper levels, nuclear localization, and specifically K48-ubiquitin chain cleavage activity of OTUD5 are critical for its role during development.
Fig. 2. OTUD5 patient mutations are hypomorphic and reduce OTUD5 levels or K48-ubiuqitin cleavage activity.
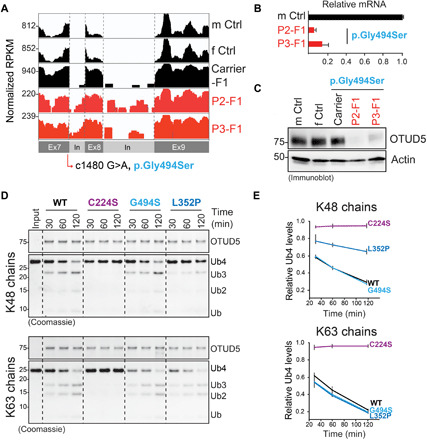
(A) The c.1480 G>A, p.Gly494Ser mutation is located in a 5′ splice site and leads to intron retention and reduction of OTUD5 mRNA levels as revealed by RNA-seq of patient-derived fibroblasts. RNA-seq reads were differentially scaled to visualize intron retention. m Ctrl, male control; f Ctrl, female control; mother carrier, mother F1. (B) The c.1480 G>A p.Gly494Ser mutation results in a decrease in OTUD5 mRNA levels in patient-derived fibroblasts as determined by qRT-PCR (n = 3 biological replicates, error bars denote SD). (C) The p.Gly494Ser mutation results in a decrease in OTUD5 protein levels as revealed by immunoblotting of lysates of patient-derived fibroblasts using indicated antibodies. (D) The Leu352Pro mutation specifically reduces OTUD5’s K48-ubiquitin chain cleavage activity. WT FLAGHAOTUD5 (WT), catalytically inactive FLAGHAOTUD5 (C224S), and patient variant FLAGHAOTUD5 (G494S and L352P) were purified from HEK 293T cells and incubated with K48- or K63-tetra ubiquitin chains for the indicated time periods and analyzed by colloidal Coomassie-stained SDS–polyacrylamide gel electrophoresis gels. (E) Quantification of three independent in vitro deubiquitylation experiments as shown in (D) (error bars denote SEM). Intensity of Ub4 band is relative to the sum of intensity of Ub3, Ub2, and Ub bands. RPKM, reads per kilobase per million mapped reads.
OTUD5 controls neuroectodermal differentiation through its K48-ubiquitin cleavage activity
OTUD5 had been predominantly investigated as a regulator of innate and adaptive immune signaling (41, 53). However, our discovery that hypomorphic mutations in OTUD5 cause a severe developmental disease, without any detectable immune manifestations, gave us the unique opportunity to study the role of OTUD5, in particular its K48-deubiquitylation activity, during early cell fate decisions of human embryogenesis. We focused on the splice site–altering p.Gly494Ser and K48 cleavage–deficient p.Leu352Ser variants, both associated with severe phenotypes. First, we established patient-derived induced pluripotent stem cells (iPSCs) and performed teratoma formation assays. Since affected patients had craniofacial and structural brain malformations, we concentrated on neuroectoderm differentiation and found defects in patient cells expressing the p.Gly494Ser allele (fig. S4A). On the basis of these observations, we subjected these iPSC lines to dual-SMAD inhibition (neural conversion), which directs differentiation toward CNS precursor and neural crest cells (54) (Fig. 3A). We observed a marked up-regulation of OTUD5 mRNA and protein levels during differentiation of iPSCs of the carrier mother, suggesting a functional role for OTUD5 during this process (Fig. 3B and fig. S4, B and C). While we observed no substantial differences in the expression of pluripotency markers OCT4 and NANOG, there was a notable defect in the neural differentiation capacity when comparing iPSCs of affected OTUD5-deficient patients to carrier mother (Fig. 3B). This was apparent by the loss of neural crest markers, including SOX10 and SNAIL2, and the aberrant expression of CNS markers, including increases in the forebrain marker FOXG1 and decreases in neural stem cell marker PAX6, as evidenced by immunoblotting and quantitative polymerase chain reaction (qPCR) (Fig. 3, B and C) or at single-cell resolution by immunofluorescence (Fig. 3D). To corroborate these results and exclude any off-target effects originating from clonal selection during iPSC reprogramming (55), we next generated control or OTUD5-depleted hES H1 cells and subjected them to neural conversion. These experiments revealed that short hairpin RNA (shRNA)– or small interfering RNA (siRNA)–mediated depletion of OTUD5 resulted in a similar reduction of neural crest cell progeny and aberrant formation of CNS precursor cells as observed in our patient-derived iPSC model (fig. S4, D to G). These effects could be rescued by shRNA-resistant wild-type (WT) OTUD5, but not by catalytically inactive C224S or K48 cleavage–deficient p.Leu352Ser OTUD5 (Fig. 3, E and F, and fig. S4H), demonstrating that it is OTUD5’s ability to cleave K48-ubiquitin chains that is required to support differentiation. We therefore conclude that during embryonic development, OTUD5 regulates cell fate decisions by cleaving degradative K48-ubiquitin linkages on substrates, and reduction of this activity through mutation leads to a multiple congenital anomaly disease we name LINKED syndrome.
Fig. 3. OTUD5 regulates CNS precursor and neural crest cell differentiation via its K48-ubiquitin chain–specific deubiquitylation activity.
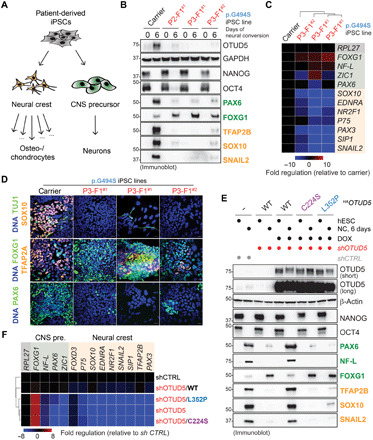
(A) Schematic overview of the neural conversion paradigm. (B) Reduction of OTUD5 levels causes aberrant neural conversion. iPSCs derived from OTUD5 p.Gly494Ser patients or the maternal carrier were subjected to neural conversion for 6 days. Differentiation was monitored by immunoblotting using indicated antibodies against hESC, CNS precursor, and neural crest markers. (C) Same experimental approach as described in (B), but cells were analyzed by qRT-PCR for expression of CNS precursor markers (green) and neural crest markers (orange). Marker expression was normalized to carrier control followed by hierarchical clustering. RPL27, endogenous control. (D) Same experimental approach as described in (B), but cells were subjected to neural conversion for 9 days and analyzed by immunofluorescence microscopy using antibodies against indicated CNS precursor and neuronal markers (green) or neural crest markers (orange). Scale bars, 20 μm. (E) K48-ubiquitin chain–specific deubiquitylation activity of OTUD5 is required for proper CNS precursor and neural crest differentiation. hES H1 cells stably expressing shRNA-resistant and doxycycline-inducible WT, catalytically inactive (C224S), or K48 chain cleavage–deficient (L352P) HAOTUD5 were generated. Cells were depleted of endogenous OTUD5 using shRNA as indicated, treated with or without doxycycline (DOX), and subjected to neural conversion (NC) for 6 days. This was followed by immunoblotting using the indicated antibodies against hESC, CNS precursor, and neural crest markers. (F) Same experimental approach as described in (E), but cells were analyzed by qRT-PCR analysis for expression of CNS precursor markers (green) and neural crest markers (orange). Marker expression was normalized to shcontrol followed by hierarchical cluster analysis. RPL27, endogenous control.
OTUD5 regulates neuroectodermal cell fate decisions by stabilizing a select group of chromatin remodelers during differentiation
To identify substrates through which OTUD5 controls embryonic development, we devised an experimental strategy that builds upon our observation that OTUD5 activity controls differentiation but is less important for self-renewal of hESCs (Fig. 3). We reasoned that any substrates through which OTUD5 regulates neural differentiation should be (i) more ubiquitylated in the absence of OTUD5 during neural conversion and (ii) found in a physical complex with OTUD5. To identify these essential substrates, we performed a series of proteomic experiments (Fig. 4A). First, we used Tandem Ubiquitin-Binding Entity (TUBE)–based mass spectrometry to capture high-confidence ubiquitylated proteins during neural conversion of control and OTUD5-depleted hESCs. Consistent with a role of OTUD5 during cell fate determination, these experiments revealed that OTUD5 regulates ubiquitylation networks preferentially during neural conversion and less in self-renewing hESCs (Fig. 4B and table S2). Second, we used mass spectrometry to identify proteins that bound to WT or catalytically inactive C224S OTUD5 during hESC differentiation (table S3). Combining these two datasets, we found ~40 candidate OTUD5 substrates that were more ubiquitylated in OTUD5-depleted differentiating hESCs and were OTUD5 interactors (Fig. 4B, fig. S5A, and table S4). While we detected some regulators of mRNA metabolism, transcription, and nucleocytoplasmic transport among these candidate substrates, Gene Ontology (GO) term analysis revealed that chromatin remodelers were the most enriched class of proteins (Fig. 4B and fig. S5B). These included the BAF complex components ARID1A/B, the histone deacetylase HDAC2, the transcriptional regulator HCF1, and the ubiquitin E3 ligase UBR5, the only previously identified OTUD5 substrate among our candidates (53). Intriguingly, most of these chromatin regulators had previously been shown to control neural cell fate decisions and had been implicated in neurodevelopmental diseases that exhibit phenotypic overlap with LINKED syndrome (fig. S5A) (45, 56–61). Notably, mutations in none of the other candidate substrates had been shown to cause similar developmental diseases. Thus, while we cannot exclude contributions of other candidate substrates, the GO enrichment of chromatin remodelers and their apparent genetic link to OTUD5 provided us with a strong rationale to focus on this group of proteins. We could confirm the interactions between OTUD5 and ARID1A/B, HDAC2, HCF1, and UBR5 by immunoblotting at the endogenous level in hES H1 cells (Fig. 4C). In addition, we expressed and immunoprecipitated flag-tagged OTUD5 truncation variants in human embryonic kidney (HEK) 293T cells and found that the C terminus of OTUD5 including the ubiquitin-interacting motif (UIM) is necessary and, with the exception of UBR5, sufficient for these binding events (Fig. 4D).
Fig. 4. Identification of chromatin remodelers as candidate substrates of OTUD5.
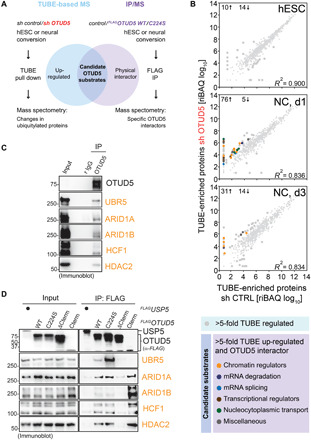
(A) Strategy used to isolate high-probability substrates of OTUD5. Two independent proteomic experiments were performed. First, control or OTUD5-depleted hESCs or hESCs undergoing neural conversion were lysed. Ubiquitylated proteins were isolated by TUBE pull down and identified by mass spectrometry. Second, self-renewing or differentiating control hESCs or hESCs expressing WT or catalytically inactive (C224S) FLAGOTUD5 were lysed, subjected to anti-FLAG immunoprecipitation, and interacting proteins were identified by mass spectrometry. Candidate OTUD5 substrates were defined as proteins that are more ubiquitylated upon OTUD5 depletion and specifically interact with OTUD5 WT or C224S. (B) OTUD5 preferentially controls ubiquitylation dynamics during neural conversion, and many OTUD5 candidate substrates are chromatin regulators. Relative iBAQ values of high-probability ubiquitylated proteins of control or OTUD5-depleted hESCs were plotted for each differentiation state (hESC, NC day 1, and NC day 3). More than fivefold regulated proteins are highlighted by larger circles. Total numbers of up-regulated (↑) or down-regulated (↓) proteins are indicated for each differentiation state. More than fivefold up-regulated proteins also found in FLAGOTUD5 IPs (i.e., candidate OTUD5 substrates) are highlighted in different colors according to their molecular function (bottom table). Note that candidate OTUD5 substrates are found specifically during differentiation. (C) OTUD5 endogenously interacts with chromatin regulators. hESCs were lysed and subjected to anti-OTUD5 immunoprecipitation followed by immunoblot analysis using indicated antibodies. Rabbit immunoglobulin G (r IgG), control. (D) OTUD5 interacts with chromatin regulators via its C terminus. HEK 293T cells expressing FLAGOTUD5 WT and indicated mutants were lysed and subjected to anti-FLAG immunoprecipitation followed by immunoblot analysis using indicated antibodies.
OTUD5 controls neuroectodermal differentiation via its K48-cleavage activity (Fig. 3, E and F), and K48-ubiquitin chains are a common targeting signal for proteasomal degradation (3, 12). Thus, if the interacting chromatin regulators were substrates of OTUD5, we expected to see changes in their stability in the absence of OTUD5. To test this hypothesis, we performed cycloheximide (CHX) chases in self-renewing and differentiating control or OTUD5-depleted hESCs. While reduction of OTUD5 had no substantial impact on the half-life of the chromatin regulators in self-renewing hESCs, it markedly decreased their stability during neural conversion (Fig. 5, A and B), a process that was dependent on the proteasome (Fig. 5C). Notably, OTUD5 depletion did not result in obvious changes in the protein levels of a subset of other tested candidate substrates that we had identified in our mass spectrometry approach (fig. S5C). To investigate whether OTUD5 regulates neuroectodermal differentiation through targeting chromatin regulator substrates, we next performed rescue experiments using a version of OTUD5 that lacked its C terminus (OTUD5ΔCterm) and thus was deficient in chromatin remodeler binding (Fig. 4D) yet retained K48-specific deubiquitylation activity (fig. S6A). Intriguingly, this separation-of-function mutant (OTUD5ΔCterm) failed to support neural crest differentiation and showed aberrant CNS precursor formation (Fig. 5D and fig. S6B), suggesting that OTUD5 needs to interact with and stabilize chromatin remodelers to exert its functions during cell fate determination. In line with this observation, individual depletion of interacting chromatin regulators resulted in a similar yet less pronounced dysregulation of the neural conversion program as the one observed upon loss of OTUD5 (Fig. 5E and fig. S6C). In contrast, we found that TRIM25 and TRAF3, two previously described K63-ubiquitin chain–modified substrates of OTUD5 (41, 62), are dispensable for neural conversion (Fig. 5E and fig. S6D). Further corroborating that the K48- but not K63-specific deubiquitylation activity is essential for the function of OTUD5 during embryonic development, we were able to purify K48- but not K63-ubiquitin chain–modified His-HDAC2 from HEK 293T cells (fig. S7A). These K48-linked chains on HDAC2 were reduced and shortened in length in the presence of WT but less by catalytically inactive OTUD5 (fig. S7A). In addition, we found that OTUD5 depletion in differentiating hESCs resulted in increases in endogenous UBR5 that was modified with K48 but not with K63 chains (fig. S7B). Together, these results suggest that during early differentiation, OTUD5 cleaves K48-ubiquitin chains deposited on a subset of chromatin regulators to prevent their degradation, thus coordinating their function in chromatin remodeling events required for neural cell fate commitment. Consistent with this notion, loss-of-function mutations in ARID1A/B, HDAC2, and HCF1 lead to different chromatinopathies (56, 57, 60, 61) that affect neuroectodermal differentiation and exhibit phenotypic overlap with patients with LINKED syndrome such as developmental delay and intellectual disability (Fig. 1 and fig. S8).
Fig. 5. OTUD5 controls early embryonic differentiation through regulating the stability of chromatin remodelers.
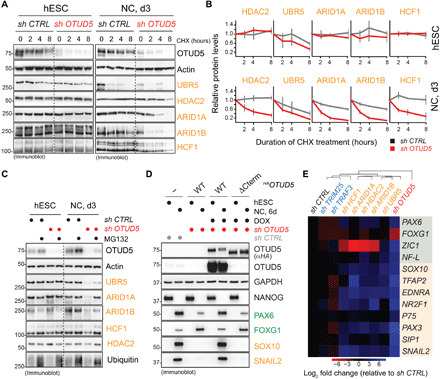
(A) OTUD5 stabilizes chromatin regulators in differentiating hESCs. Control or OTUD5-depleted hESCs or hESC subjected to neural conversion for 3 days were treated with CHX for indicated time periods. Protein stability of chromatin regulators was determined by immunoblotting using indicated antibodies. (B) Quantification of three biological replicates of the experiment shown in (A) (error bars denote SD, normalized to actin, 0 hours = 1). (C) OTUD5 protects chromatin regulators from proteasomal degradation in differentiating hESCs. Control or OTUD5-depleted hESCs or hESC subjected to neural conversion for 3 days were treated with the proteasome inhibitor MG132 for 4 hours followed by immunoblotting with indicated antibodies. (D) Chromatin regulator binding-deficient OTUD5ΔCterm does not support neural conversion. hESC cells expressing shRNA-resistant and doxycycline-inducible WT or chromatin regulator binding-deficient (ΔCterm) FLAGHAOTUD5 were generated. Cells were depleted of endogenous OTUD5 using shRNA as indicated, treated with or without doxycycline (DOX), and subjected to neural conversion for 6 days. Differentiation was monitored by immunoblotting using the indicated antibodies against hESC, CNS precursor, and neural crest markers. Note that anti-OTUD5 antibodies were raised against the C terminus of OTUD5 and do not recognize OTUD5ΔCterm. (E) Individual depletion of chromatin regulators, but not previously described OTUD5 substrates TRIM25 and TRAF3, partially phenocopies the aberrant neural conversion program observed upon OTUD5 reduction. hESCs were depleted of endogenous OTUD5 or indicated proteins using shRNAs, subjected to neural conversion for 6 days, and analyzed by qRT-PCR for CNS precursor markers (green) or neural crest markers (orange). Marker expression was normalized to shcontrol followed by hierarchical clustering. RPL27, endogenous control.
OTUD5 controls chromatin remodeling at enhancers driving ectodermal cell fate decisions
Given that OTUD5 uses its K48-specific deubiquitylation activity to control the stability of several chromatin regulators such as ARID1A and ARID1B during early ectodermal cell fate commitment, we hypothesized that the observed clinical and differentiation phenotypes underlie impaired chromatin dynamics. In line with this, ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) revealed that OTUD5 depletion caused loss of accessible chromatin at early stages of neural conversion, while accessibility was largely unaffected in self-renewing hESCs (Fig. 6, A and B, and fig. S9, A and B). This was accompanied by modest transcriptional changes (fig. S9, C and D), suggesting that differences in ATAC signal are not a consequence of failed differentiation but rather its initiating cause. Genes associated with chromatin regions exhibiting reduced accessibility at day 3 of neural conversion were enriched for GO terms involved in specification of CNS precursor and neural crest cells (Fig. 6C). Notably, more than half of the regions displaying lower accessibility were located at enhancers (Fig. 6D) and were enriched for neural and neural crest fate-promoting transcription factor motifs such as OTX2, SOX2, SOX3, SOX9, and SOX10 (fig. S10). Further supporting the idea that OTUD5 regulates cell fate commitment through stabilizing chromatin remodelers, we observed that OTUD5-regulated neural enhancers were often bound by SMARCA4 (63), a component of the BAF complex recruited by ARID1A/B (64) in neural progenitor cells (Fig. 6B). Among the ~600 genes associated with less accessible enhancers was the neuroectoderm cell fate–promoting transcription factor PAX6 and the neural crest and neuronal specification factor SEMA3A. These two cell fate regulators exhibited reduced mRNA expression upon OTUD5 depletion at these early stages of differentiation (Fig. 6E). Thus, OTUD5 controls neural cell fate commitment by regulating chromatin accessibility at neural- and neural crest–specific enhancers to enable activation of transcriptional networks that drive the differentiation program.
Fig. 6. OTUD5 is required for chromatin remodeling at enhancers driving neural and neural crest differentiation.
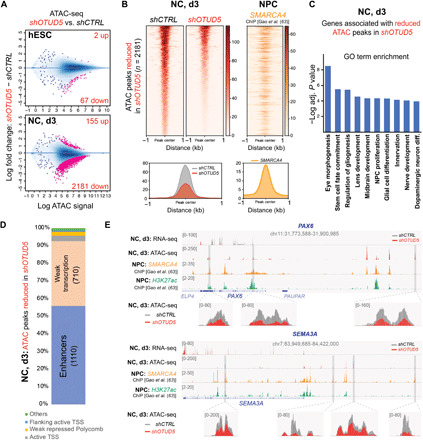
(A) Loss of OTUD5 leads to changes in chromatin accessibility specifically during differentiation. Changes in chromatin accessibility resulting from OTUD5 depletion in hESC and neural converted cells (NC, d3) are depicted as log2 fold change in ATAC-seq signal intensities at stringently identified peaks (IDR, 0.05). Numbers of statistically significant ATAC peaks (adjusted P value < 0.0001; pink dots) gained (up) or lost (down) upon OTUD5 depletion are indicated. In hESCs, there is a modest loss of ATAC signal upon shOTUD5 treatment compared to control, which is exacerbated during neural conversion of hESCs. (B) Averaged ATAC signal from shCTRL or shOTUD5 neural converted cells is plotted as a heatmap at 2181 peaks that lose accessibility upon shOTUD5 treatment. Average profile of ATAC signal in shCTRL and shOTUD5 cells is shown below. SMARCA4 ChIP-seq in neuronal precursor cells shows strong binding to regions with OTUD5-mediated reduction in chromatin accessibility, and peaks are centered at the differentially enriched ATAC-seq peaks. (C) OTUD5 is required for chromatin remodeling at genes promoting neural differentiation. ATAC-seq peaks significantly altered by shOTUD5 treatment in NC, day 3 were associated to genes using GREAT and subjected to GO term analysis. (D) OTUD5 is predominantly required for chromatin remodeling at enhancers. ATAC-seq regions with less enrichment in OTUD5-depleted differentiating cells were classified using ChromHMM genome functional annotation of H1-derived neural precursor cells. TSS, transcriptional start site. (E) Browser snapshots showing differences in transcription and chromatin accessibility between control (gray) and shOTUD5-treated (red) neural converted cells at two loci, PAX6 (top) and SEMA3A (bottom), which are enriched for H3K27ac and bound by SMARCA4.
DISCUSSION
In this study, guided by the application of genomic constraint scores, we discover LINKED syndrome, a multiple congenital anomaly disorder caused by hypomorphic variants in OTUD5. By using rare disease genetics and studying associated mutations, we identify an important physiological role for linkage-specific ubiquitin chain cleavage during embryonic development and aberrant degradation of chromatin regulators as a major disease mechanism underlying LINKED syndrome (Fig. 7). The K48 chain–specific cleavage activity of OTUD5 is required to stabilize the levels of a group of substrates (i.e., chromatin remodelers) during early stages of differentiation. We propose that in this manner, OTUD5 coordinates chromatin remodeling networks that promote the accessibility of enhancers to ensure transcriptional changes required for neuroectodermal cell fate commitment. Our study has important implications for and raises exciting questions concerning ubiquitin-dependent signaling during embryonic development, molecular mechanisms underlying chromatinopathies, and the use of genomic constraint metrics as a means to dissect molecular pathways critical for human health.
Fig. 7. OTUD5 controls developmental chromatin dynamics.
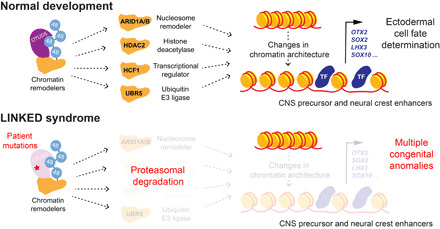
Model of how linkage-specific ubiquitin chain editing by OTUD5 controls development and is mis-regulated in disease. During normal early embryogenesis, OTUD5 uses its K48 linkage–specific deubiquitylation activity to target and stabilize several key chromatin regulators to coordinate chromatin remodeling events at CNS precursor and neural crest enhancers. This allows binding of lineage-promoting transcription factors (TF) to drive transcriptional networks required for neuroectodermal cell fate commitment. Hypomorphic patient mutations in OTUD5 result in dysregulation of this pathway and lead to a previously unrecognized multiple congenital anomaly disorder we name LINKED syndrome.
Signaling through linkage-specific ubiquitin chain cleavage of substrate groups during human development
Two E1 enzymes, ~40 E2 enzymes, and ~600 E3 ligases cooperate to generate a variety of ubiquitin signals on thousands of substrates to control virtually all cellular processes (3). The fact that the human genome only encodes ~100 DUBs is therefore unexpected, and it remains an outstanding question how this relatively small number of enzymes can control such a staggering complexity of ubiquitin signals. Our in vitro deubiquitylation and immunoprecipitation data (Figs. 2 and 4 and fig. S3) suggest that OTUD5 can recognize both the chromatin remodeler and the K48-ubiquitin chain, a mode of interaction that would endow OTUD5 with the ability to target a group of functionally related substrates carrying the same ubiquitin modification to drive differentiation. Other OTU DUBs such as OTUD7A and OTUD6B have or are expected to exhibit similar linkage-specific cleavage activities, and while their substrates have remained unknown, these enzymes have recently been linked to neurodevelopmental diseases (34–36). We hence propose that linkage-specific ubiquitin cleavage of substrate groups, as described here for OTUD5 and chromatin remodelers, will be a common signaling mode to coordinate embryonic differentiation. In addition to already described mechanisms of functional regulation and diversification of DUBs (23, 24), our results thus help to explain how the limited number of deubiquitylation enzymes in the human genome can efficiently regulate developmental ubiquitin signaling.
Our study shows that loss of OTUD5 activity results in aberrant neuroectodermal differentiation but has no obvious effects on maintenance of hESCs. This suggests that OTUD5-mediated stabilization of chromatin remodelers is subject to regulation at specific stages of stem cell differentiation. During neural conversion, we observe increased protein levels as well as the appearance of slower-migrating bands of OTUD5 (indicative of phosphorylation; Fig. 3, B and E). OTUD5 has previously been shown to be phosphorylated at multiple sites, including Ser177, which is essential for its activity (52). In addition, OTUD4, a related OTU DUB family member, can be switched from a K48 chain–specific to a K63 chain–specific enzyme by phosphorylation (65). Thus, determining the changes in levels and phosphorylation status of OTUD5 during neural conversion might reveal important insights into how OTUD5-dependent signaling is temporally regulated and embedded into the developmental program of the embryo.
We find that a select group of chromatin remodelers is degraded in the absence of OTUD5 only during differentiation and not in self-renewing hESCs (Fig. 5, A to C). This implies that both OTUD5 and the ubiquitin E3 ligase(s) it counteracts are activated at the same time during cell fate commitment. A possible function for such regulation could be to (i) protect a subpool of chromatin regulator molecules from degradation or (ii) allow for recycling of chromatin regulator subunits to ensure local chromatin remodeling at specific neuroectodermal enhancers. In the latter model, modification of the chromatin regulator with K48-ubiquitin chains would allow recognition by the ubiquitin-dependent ATPase p97/VCP that could extract it from chromatin (45, 66). OTUD5 would then protect these ubiquitylated regulators from proteasomal degradation by chain removal and allow for reuse of the regulator. Future experiments that will require identification of the cognate E3 ligase(s) may allow one to distinguish between these different models and elucidate the function of E3/OTUD5 co-activation during embryonic differentiation.
Another interesting question that arises from our study is how OTUD5 can discriminate between its cognate substrates that are decorated with either K48- or K63-linked ubiquitin chains. Linkage specificity in other OTU DUBs has been shown to rely on proper positioning of the proximal (lysine-donating) ubiquitin, which is achieved by either additional ubiquitin-binding domains or an S1′ ubiquitin-binding site on the OTU domain itself (29). OTUD5 harbors a UIM domain in its C terminus that has been shown to be required for K63-ubiquitin chain cleavage off TRAF3 (41), and here, we show that it is necessary for interaction with chromatin remodelers (Fig. 4D). It is therefore intriguing to speculate that OTUD5 uses its UIM domain to discriminate between its substrates by serving both as a ubiquitin-binding domain to position the proximal ubiquitin of K63 chains and as a binding site for chromatin regulators that carry K48-linked ubiquitin chains. In the latter case, an S1′ ubiquitin-binding site in or near the OTU domain around L352 (the residue that, when mutated, results in specific loss of K48-deubiquitylation activity) could serve to ensure K48-linkage specificity. Future biochemical and structural studies may allow one to delineate these different modes of recognition and cleavage and could provide important mechanistic insights to enable therapeutic targeting of OTUD5.
Implications for the molecular mechanisms underlying chromatinopathies
Coffin-Siris and Cornelia de Lange syndromes are genetic disorders caused by mutations in different chromatin remodelers. While exhibiting considerable phenotypic variability and locus heterogeneity, these syndromes share disease manifestations (67, 68). Here, we describe LINKED syndrome, which exhibits aspects both distinct from, and in common with, these two disorders (fig. S8). Through TUBE- and IP-based proteomics, proteasome inhibitor treatments, and CHX chases during neuroectodermal differentiation (Figs. 4 and 5 and fig. S5), we show that OTUD5 interacts with and targets ARID1A/B and HDAC2, proteins that are mutated in Coffin-Siris and Cornelia de Lange syndromes, respectively. Our findings link these proteins to a common neurodevelopmental pathway, thereby providing a molecular framework to explain the clinically overlapping features of these genetic disorders. In addition to neuroectodermal defects characterized in detail in this manuscript, patients with Coffin-Siris, Cornelia de Lange, and LINKED syndromes also exhibit phenotypes in tissues originating from endoderm and mesoderm. Thus, determining the cell-autonomous or nonautonomous functions of OTUD5 during the differentiation of other germ layers particularly in relation to the chromatin regulators mutated in Coffin-Siris and Cornelia de Lange syndromes will be an exciting area of research.
Conditional knockout studies in mice have revealed an important role for OTUD5 regulating both adaptive and innate immune responses (41, 53). However, none of our identified patients with LINKED syndrome have had recurrent infections, inflammation, or detectable hematopoietic defects. We believe that these phenotypes are likely masked by the severe embryonic and early developmental defects. Further genetic and biochemical studies may allow identification of OTUD5 alleles eliciting such immune phenotypes (e.g., those that would specifically affect K63-specific deubiquitylation activity and thus could potentially uncouple OTUD5’s functions during embryonic and hematopoietic development).
Genomic constraint–driven genotype-first approaches to dissect molecular pathways through human diseases
Identifying genetic causes of diseases is of paramount importance to understand mechanisms underlying human health. Historically, genetic studies have focused on sequencing select individuals with specific phenotypes followed by attempts to identify a genomic variant to explain their presentation. However, more recently, with the advent of large databases of exome and genome sequences from cohorts of healthy individuals, we now have metrics such as genomic constraint scores that allow for quantification of the tolerance of genes to loss-of-function and missense mutations in control populations (1). These tools facilitate not only identification of mutations in affected individuals but also depletion of mutations in unaffected individuals, both serving as a critical starting point for mechanistic investigations into physiologically relevant targets. In our work presented here, we take advantage of these metrics to delineate the importance of OTUD5 in human embryonic development (Fig. 1). This prioritization allowed for in-depth mechanistic studies to better characterize the specific biochemical defects associated with novel OTUD5 variants found in disease.
We believe that genomic constraints could be an invaluable first step for identifying genes critical for health and development in humans and envision a genotype-first approach of analyzing sequencing data agnostic to phenotype as a starting point for disease discovery and molecular characterization. For example, in addition to OTUD5, many other OTU DUB genes have highly restricted loss-of-function variation in healthy individuals that have never been linked to human disease (Fig. 1B). These encode the DUBs OTUD4, OTUB1, VCPIP, and ZRANB1/TRABID that exhibit various linkage-specific cleavage activities (29). We predict that targeted search for missense mutations in these genes in undiagnosed patients will likely allow identification of previously unknown developmental diseases and identify variants that might be useful in uncovering further functions and mechanisms of particular ubiquitin chain types during embryonic development.
While pLI and Z scores are powerful for identifying genes that are sensitive to haploinsufficiency, these tools do not completely capture intolerance toward other modes of autosomal dominant disease such as gain of function and dominant-negative and autosomal recessive inheritance. Thus, the genomic constraint–based approach presented here is currently best suited to explore genes sensitive to loss of a single allele as a means to understand and mechanistically dissect severe disease. Despite these minor limitations, current genomic intolerance scores are invaluable for identifying disease relevance of particular genetic variation, and development of novel metrics will only broaden this approach.
In summary, our study underscores the utility of genomic constraint metrics in prioritizing candidate disease variants, thus enabling the discovery of a developmental disease, LINKED syndrome. By studying this disease, we have identified linkage-specific deubiquitylation of substrate groups by OTUD5 as an essential signaling mode that coordinates chromatin remodeling during neuroectodermal cell fate commitment. We predict that similar genomic constraint–based approaches will be a common means for disease gene discovery and molecular dissection of embryonic pathways.
MATERIALS AND METHODS
Human subjects
Written informed consent was obtained from all individuals or family member legal representatives before exome sequencing. Consent was obtained for publication of photographs before inclusion in the study. Family 1, individuals 1 to 3, were counseled regarding the possible outcomes of exome sequencing and signed a consent form for research-based exome sequencing through the Johns Hopkins Hospital and the National Institutes of Health, which was approved by the National Institutes of Health Institutional Review Board. The rest of the participants were recruited through GeneMatcher (69). Individual 4 was consented for clinical exome sequencing through the Children’s Hospital of Orange County. Individual 5 was consented for clinical exome sequencing through Johns Hopkins Hospital. Individual 6 was consented for clinical and/or research-based exome sequencing through Shinshu University, Nagano, Japan. Individual 7 was consented for clinical and/or research-based exome sequencing through Radboud University, Nijmegen, The Netherlands. Individuals 8 and 9 were consented for clinical and/or research-based exome sequencing through Yokohama City University, Japan. Individual 10 was consented for research exome at the Tartu University Hospital, Estonia.
Exome and Sanger sequencing
Whole-exome sequencing and data analysis were performed as previously described. New candidate variants were filtered to remove those present in the gnomAD, 1000 Genomes Project, dbSNP, and ClinSeq databases and an in-house database with more than 1200 exomes, and variants were selected on the basis of autosomal recessive or X-linked recessive inheritance. For individual 3, trio-based exome sequencing was performed on genomic DNA isolated from amniocytes through Johns Hopkins Hospital and the National Institutes of Health. Novel variant in OTUD5 was identified using standard bioinformatics analysis. Sanger sequencing confirmed the presence of the OTUD5 variants in the trio and their absence in unaffected family members. Individual 4 had standard exome sequencing performed through the Children’s Hospital of Orange County, and a novel variant in OTUD5 was identified and Sanger confirmed. Individual 5 was consented for clinical exome sequencing through Johns Hopkins Hospital. Individual 6 was consented for clinical and/or research-based exome sequencing through Tokyo Medical and Dental University, Tokyo, Japan. Individual 7 was consented for clinical and/or research-based exome sequencing through Radboud University, Nijmegen, The Netherlands. All variants were confirmed using Sanger sequencing. All patients reported have no known definitive pathogenic variants identified in other genes causative for multiple congenital anomalies and developmental delay. All patients provided informed consent for exome sequencing and identifiable photographs. The approved study protocol for this work includes 94-HG-0105 and the Undiagnosed Diseases Network from the National Human Genome Research Institute.
Fibroblast/induced pluripotent cell lines
Dermal fibroblast cells derived from patients with OTUD5 or unrelated healthy donors were grown in DMEM (Dulbecco’s Modified Eagle Medium; Life Technologies) supplemented with 10% fetal calf serum (Gemini Bio-Products) and 1× antibiotics (Life Technologies). iPSCs were generated from individuals 2 and 3 and their unaffected carrier mother using a 3-week Sendai virus protocol previously described (70). Reprogramming efficiency was measured using both fluorescence-activated cell sorting analysis for pluripotency markers and teratoma formation in nude mice. Multiple clones were generated for each iPSC line and were used in specific experiments. Teratoma formation assays were performed on each clone, in quadruplicate, using bilateral gastrocnemius muscle injection in each mouse as described elsewhere (71). Slides were generated and stained using hematoxylin and eosin stain and quantified for ectodermal components using Adobe Illustrator.
Determination of skewed X-inactivation
The methylation status of the human androgen receptor (AR) gene at Xq12 was assessed to infer X chromosome inactivation in the heterozygous mother carrying the OTUD5 p.R274W mutation. One hundred nanograms of DNA isolated from peripheral blood was digested with the methylation-sensitive Hpa II enzyme (NEB, Ipswich, MA, USA), as originally described (72). Digested and undigested samples were then amplified by PCR with primers and protocol as previously described (73). The PCR products were separated by capillary electrophoresis on an ABI 3730xl DNA analyzer (Applied Biosystems) with the GeneScan 500 LIZ size standard (Applied Biosystems). Fragment analysis was performed with GeneMapper software (Applied Biosystems).
Mouse studies
Transgenic mice (C57BL/6J) were generated using CRISPR-Cas9 injection and electroporation after isolation of early embryos (74). For generating Otud5 knock-in mutations, we used a guide RNA (gRNA) p.Gly494Ser 5′-CACCCTGTGCACCAGGTCAG-3′, and for p.Leu352Pro, we used gRNA 5′-CCCCTGGCTTAAATGACGGT-3′ with repair templates including the specific missense mutation. After failing to identify viable pups with either indels or specific knock-in mutations, we began isolating day 12.5 embryos by microsurgery, imaging the embryo, and genotyping a small portion of the tail. As a control for injections, we either used saline injection or a nonessential gene used in the laboratory (Tbx21) 5′-CCCACTGTGCCCTACTACCG-3′. All mouse work included was approved by the National Human Genome Research Institute animal protocol.
Plasmids, shRNAs, and siRNAs
pDEST-FLAG-HA-USP5 and pDEST-FLAG-HA-OTUD5 were gifts from W. Harper (Addgene plasmids #22590 and #22610) (75). pRK5-HA-Ub-K48only and pRK5-HA-Ub-K63only were gifts from T. Dawson (Addgene plasmids #17605 and #1706). OTUD5 patient mutations (G494S, L352P, 161-164del, R274W, and D256N), point mutations (C224S or C224R), truncation mutations (ΔCterm = OTUD5 1-534, Cterm = OTUD5 534-571), and wobble mutations to make constructs resistant to shOTUD5#5 were introduced in this vector using the Q5 site-directed mutagenesis kit (E0554, NEB) following the manufacturer’s instructions. OTUD5 and OTUD5C224R were further subcloned into pKmyc using Bam HI and Not I sites (pKmyc-OTUD5 WT or C224R). For expression in hESCs, OTUD5 variants were cloned into pENTR1A or pENTR233 and recombined into pINDUCER20 (76). pLKO1-Puro Mission shRNA constructs targeting OTUD5 (#2: TRCN0000122275 and #5: TRCN0000233196), ARID1A (TRCN0000059092), ARID1B (TRCN00000420576), UBR5 (TRCN0000003411), HDAC2 (TRCN00000004819), HCFC1 (TRCN00000001625), TRAF3 (TRC00000034219), and TRIM25 (TRC0000003499) were purchased from Sigma-Aldrich. siRNA pools were purchased from Santa Cruz Biotechnology and Thermo Fisher Scientific.
Antibodies
The following antibodies were commercially purchased for immunoblotting and immunofluorescence microscopy: anti-OTUD5 (#20087S, clone D8Y2U, Cell Signaling, 1:1000 in IB), anti-PAX6 (#60433, clone D3A9V, Cell Signaling, 1:1000 in IB and 1:200 in IF), anti-TFAP2 (#2509, Cell Signaling, 1:1000 in IB and 1:200 in IF), anti-FOXG1 (ab18259, Abcam, 1:1000 in IB and 1:100 in IF), anti-ARID1A (#12354, clone D2A8U, Cell Signaling, 1:3000 in IB), anti-ARID1B (#65747, clone E1U7D, Cell Signaling, 1:1000 in IB), anti-UBR5 (#65344, clone D6O8Z, Cell Signaling, 1:1000 in IB), anti-HDAC2 (#5113, clone 3F3, Cell Signaling, 1:3000 in IB), anti-HCFC1 (#50708, Cell Signaling, 1:1000 in IB), anti-actin (#8691001, MP Biomedicals, 1:10,000 in IB), anti-SOX10 (#89356, Cell Signaling, 1:1000 in IB and 1:100 in IF), anti-TUJ1 (#5568, clone D71G9, Cell Signaling, 1:1000 in IB and 1:200 in IF), anti-NANOG (#4903, clone D73G4, Cell Signaling, 1:1000 in IB), anti-OCT4 (ac-8628, Santa Cruz Biotechnology, 1:1000 in IB), anti-OCT4 (#75463, clone D7O5G, Cell Signaling, 1:1000 in IB), anti-SNAIL2 (#9585, clone C19G7, Cell Signaling, 1:500 in IB), anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (#5174, clone D16H11, Cell Signaling, 1:10,000 in IB), anti-HA (hemagglutinin) (clone C29F4; Cell Signaling, 1:3000 in IB and 1:200 in IF), and anti-Flag (F1804, clone M2, Sigma-Aldrich, 1:2000 in IB). Anti-pOTUD5Ser177 antibodies were a gift from Genentech and were described previously (52).
Mammalian cell culture and transfections
HEK 293T cells were maintained in DMEM with 10% fetal bovine serum. Plasmid transfections of HEK 293T cells were carried out using PEI (polyethylenimine) siRNA transfections were carried out with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions using 10 nM for each siRNA. Cells were routinely tested for mycoplasma using the MycoAlert Mycoplasma Detection Kit from Lonza (LT07-118).
Pluripotent stem cell culture, lentiviral infections, and neural conversion
hES H1 cells were maintained under feeder-free conditions on Matrigel-coated plates (#354277, BD Biosciences) in mTeSR1 (#05871/05852, STEMCELL Technologies Inc.) and were routinely passaged with collagenase (#07909, STEMCELL Technologies Inc.). Cells were routinely tested for mycoplasma using the MycoAlert Mycoplasma Detection Kit from Lonza (LT07-118).
Lentiviruses were produced in 293T cells by cotransfection of lentiviral constructs with packaging plasmids (Addgene) for 48 to 72 hours. Transduction was carried out by infecting 2 × 105 hES H1 cells per well of a six-well plate with lentiviruses in the presence of Polybrene (6 μg/ml; Sigma-Aldrich) and 10 μM Y-27632 ROCK inhibitor. For transduction of lentiviruses carrying ectopic expression vectors, cells were centrifuged at 1000g at 30°C for 90 min. Media were replaced with 2 ml of mTESR1 containing 10 μM Y-27632 ROCK inhibitor. After 4 to 6 days of selection with appropriate antibiotic [puromycin (1 μg/ml) for pLKO1-puro-shRNA constructs and G418 (200 μg/ml) for pINDUCER20 constructs], hES H1 cells were analyzed and used in differentiation experiments.
Neural induction of hES H1 cells expressing different shRNA constructs was performed using STEMdiff Neural Induction Medium (#05831, STEMCELL Technologies Inc.) in combination with a monolayer culture method according to the manufacturer’s technical bulletin (#28044) and as previously described (54). In brief, single-cell suspensions were prepared by treatment of hES cells with Accutase, and 1.5 × 106 to 2.0 × 106 cells were seeded per well of a six-well plate in 4 ml of STEMdiff Neural Induction Medium supplemented with 10 μM Y-27632 ROCK inhibitor. Neural induction was performed for indicated time periods with daily medium change.
hES H1 rescue experiments
To rescue OTUD5-dependent phenotypes, hES H1 cells were stably transduced with pINDUCER-FLAGHAOTUD5 constructs (WT, C224S, L352P, or ΔC-term, containing wobble mutations that render them resistant to shOTUD5#5). Cells were selected and maintained with G418 (200 μg/ml) for 4 to 5 days. Cells were then transduced with control shRNAs or shRNAs targeting OTUD5 (shOTUD5#5) and selected and maintained with puromycin (1 μg/ml). For the rescue experiments, these cell lines were then treated in the absence or presence of doxycycline (1 μg/ml) and subjected to neural conversion for the indicated time periods. Cells were harvested for immunoblotting and RNA extraction.
Proteasome inhibitor treatment
To inhibit proteasome-mediated degradation of proteins, hES H1 cells and hES H1 cells undergoing neural conversion for 3 days were treated with the proteasome inhibitor MG132 (N-carbobenzyloxy-l-leucyl-l-leucyl-l-leucinal) at a concentration of 10 μM for 4 hours. After treatment, the cells were harvested by scraping in 1× phosphate-buffered saline (PBS) and centrifuged at 300g for 5 min. Cells were lysed in 2× urea sample buffer [150 mM tris (pH 6.5), 6 M urea, 6% SDS, 25% glycerol, and a few grains of bromophenol blue] followed by immunoblotting with indicated antibodies.
CHX chase assays
For CHX chase assays, control or OTUD5-depleted hES H1 cells and cells that had undergone neural induction for 3 days were treated with CHX (40 μg/ml) for 2, 4, and 8 hours. Cells were lysed in 2× urea sample buffer [150 mM tris (pH 6.5), 6 M urea, 6% SDS, 25% glycerol, and a few grains of bromophenol blue], sonicated, and analyzed by IB. For quantification, immunoblot signals for respective proteins were quantified using ImageJ (National Institutes of Health, http://rsbweb.nih.gov/ij/) and normalized to GAPDH or β-actin.
Quantitative real-time PCR analysis
For qRT-PCR analysis, total RNA was extracted and purified from cells using the NucleoSpin RNA kit (#740955, Macherey-Nagel) and transcribed into complementary DNA (cDNA) using the SuperScript IV First-Strand Synthesis System (#18091050, Thermo Fisher Scientific). Gene expression was quantified by PowerUp SYBR Green qPCR (#A25741, Thermo Fisher Scientific) on a CFX96 Real-Time System (Bio-Rad). Nonspecific signals caused by primer dimers were excluded by dissociation curve analysis and use of nontemplate controls. Loaded cDNA was normalized using RPL27 as an endogenous control. Gene-specific primers for qRT-PCR were designed by using the National Center for Biotechnology Information Primer-BLAST. Primer sequences can be found in table S5.
Cluster analysis
mRNA abundance was measured by qRT-PCR under different conditions. The datasets were plotted as a heatmap in Python using the Seaborn library. Hierarchical clustering of samples was performed using the standardized Euclidean method with average linkage.
Immunoprecipitations
HEK 293T cells were transiently transfected with WT FLAGHAOTUD5 or indicated variants and incubated for 48 hours at 37°C with 5% CO2. Cells were harvested by scraping in 1× PBS and centrifuged at 300g for 5 min. The cell pellets were either stored at −80°C or directly used for immunoprecipitation experiment. To detect OTUD5 interaction partners, HEK 293T expressing indicated FLAGHAOTUD5 variants (3 × 15-cm dishes per condition) were lysed in two pellet volumes of ice-cold lysis buffer [20 mM Hepes (pH 7.3) containing 110 mM potassium acetate, 2 mM magnesium acetate, 1 mM EGTA, 2 mM EDTA, 0.1% NP-40, 1× protease inhibitors (Roche), 1× PhosSTOP (Roche), and 2 mM phenanthroline]. Cells were sonicated, and the lysates were cleared by centrifugation at 20,000g for 25 min. To remove residual lipids, the supernatant was filtered through a 0.22-μm filter (Millex-GV). Subsequently, the lysates were quantified using the Pierce 660-nm reagent (Thermo Fisher Scientific, #22660), and an equal amount of lysates were incubated with anti-FLAG-M2 agarose (Sigma-Aldrich) for 2 hours at 4°C. Beads were then washed three times with lysis buffer and eluted in lysis buffer supplemented with 3xFLAG peptide (0.5 mg/ml; Sigma-Aldrich). Eluted proteins were precipitated by adding 20% trichloroacetic acid (TCA) followed by overnight incubation on ice. Protein pellets were washed three times with ice-cold 90% acetone in 0.01 M HCl, air-dried, and solubilized with 2× urea sample buffer followed by immunoblot analysis.
For in vitro deubiquitylation assays, OTUD5 and indicated variants were purified from HEK 293T cells (1 × 15-cm dishes per condition). Lysates were prepared and subjected to anti-FLAG immunoprecipitation as described above. Beads were washed twice with lysis buffer containing 1 M NaCl and three times with lysis buffer without NaCl, and OTUD5 was eluted from the beads with lysis buffer containing 10 mM dithiothreitol (DTT) and 3xFLAG peptide (0.5 mg/ml).
For mass spectrometry analysis, self-renewing or differentiating (neural conversion, 3 days) hES H1 cells or hES H1 cells expressing WT or catalytically inactive (C224S) FLAGHAOTUD5 were lysed and subjected to anti-FLAG immunoprecipitation as described above (5 × 15-cm dishes per condition). FLAG immunoprecipitates were further processed for multidimensional protein identification technology (MUDPIT) mass spectrometry as described below.
In vitro deubiquitylation assays
For in vitro deubiquitylation reactions, equal amounts of WT OTUD5 or indicated OTUD5 mutants (purified as described above) were incubated with 0.5 μM K48- or K63-tetra ubiquitin chains (Boston Biochem) in cleavage buffer [110 mM potassium acetate, 2 mM magnesium acetate, 1 mM EGTA, 20 mM Hepes (pH 7.3), 0.1% NP-40, 2 mM EDTA, and 10 mM DTT] at 30°C for different time periods (30, 60, 120 min). Reactions were stopped by an addition of equal amounts of 2× urea sample buffer.
Deubiquitylation of HDAC2 by OTUD5
HEK 293T cells were cotransfected with different combinations of HisHDAC2FLAG, HAUbiquitinK48only/HAUbiquitinK63only, and Myc-OTUD5 WT/Myc-OTUD5 C224R. After 48 hours of transfection, cells were treated with 20 μM MG132 for 4 hours. The cells were harvested by scraping in 1× PBS and centrifuged at 300g for 5 min. Harvested cells were lysed in 8 M urea lysis buffer [8 M urea, 20 mM imidazole, and 50 mM tris (pH 7.8)], sonicated, and centrifuged for 20 min at 20,000g. The lysates were then incubated with Ni-NTA (nitrilotriacetic acid) agarose beads for 1 hour. The beads were washed with urea lysis buffer three times and eluted with 2× urea sample buffer [150 mM tris (pH 6.5), 6 M urea, 6% SDS, 25% glycerol, and a few grains of bromophenol blue] followed by immunoblotting with indicated antibodies.
Ubiquitin linkage–specific TUBE pull downs from differentiating hES H1 cells
Fifteen-centimeter dishes (2×) of control or OTUD5-depleted hES H1 cells undergoing neural conversion for 1 day were treated with 20 μM MG132 for 4 hours, harvested by scraping in 1× PBS containing 10 μM PR619 (LifeSensors), and lysed in 100 mM tris-HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1% NP-40, 10 μM PR-619, 2 mM 1,10-phenantroline, and 1× protease inhibitors (Roche). Lysates were sonicated and cleared by centrifugation at 20,000g for 25 min. To remove residual lipids, the supernatant was filtered through a 0.22-μm filter (Millex-GV). Subsequently, the lysates were quantified using the Pierce 660-nm reagent (Thermo Fisher Scientific, #22660), and an equal amount of lysates were incubated with anti-K48 or anti-K63 TUBE magnetic beads (UM407M/UM404M, LifeSensors) for 2 hours at 4°C. Beads were then washed three times with lysis buffer and eluted in 2× urea sample buffer followed by immunoblotting with indicated antibodies.
Mass spectrometry to identify OTUD5 interactors
For mass spectrometry analysis, flag immunoprecipitates were prepared from self-renewing and differentiating hESCs as described above and precipitated with 20% TCA (Thermo Fisher Scientific) overnight. Proteins were resolubilized and denatured in 8 M urea (Thermo Fisher Scientific) and 100 mM tris (pH 8.5), followed by reduction with 5 mM TCEP [tris(2-carboxyethyl)phosphine, Sigma-Aldrich], alkylation with 10 mM iodoacetamide (Sigma-Aldrich), and overnight digestion with trypsin (0.5 mg/ml; Thermo Fisher Scientific). Samples were analyzed by MUDPIT mass spectrometry by the Vincent J. Coates Proteomics/Mass Spectrometry Laboratory at University of California Berkeley. High-confidence interactors of OTUD5 were defined as nuclear proteins only found in FLAGHAOTUD5WT/C224S and not in control immunoprecipitates.
TUBE-based mass spectrometry
TUBE-based mass spectrometry was performed by LifeSensors. The company provided the number of peptides detected, raw intensities, and calculated iBAQ (intensity-based absolute quantification) values. The data were further processed and analyzed using a Python script. Subsequent figures were produced using Matplotlib and Seaborn libraries. First, all the runs were normalized to each other using a sum normalization method, which we called the relative iBAQ values. This was done by taking the iBAQ values of each sample per run and dividing by the sum total of all iBAQ values for each sample per run.
We then used the minimum detection limit to fill any missing data. All zero/NaN values were replaced by the minimum relative iBAQ value in each individual sample. Afterward, the data were log-transformed before plotting. However, the sum-normalized values give a mixture of positive and negative numbers after log transformation, so all the data were multiplied by 1.0 × 107 to bring the smallest values above 1 in all the samples. The log-transformed data were plotted on scatterplots using Matplotlib and Seaborn. To enrich for proteins regulated in their ubiquitylation status in an OTUD5-dependent manner, we filtered for nuclear proteins with at least 13 unique peptides and identified in at least four of the six groups (depicted in gray in Fig. 4B) and, in addition, were found more than fivefold regulated upon OTUD5 depletion in TUBE pull downs from hESCs or hESC undergoing neural conversion for 1 or 3 days (depicted in light blue in Fig. 4B).
High-confidence OTUD5 substrate identification
To identify high-confidence OTUD5 substrates, we filtered for proteins that we found more than fivefold up-regulated in the TUBE-based mass spectrometry and to be physically interacting with OTUD5 in our MUDPIT mass spectrometry experiments. The list of these proteins was subjected to GO term analysis.
Immunofluorescence microscopy
For immunofluorescence analysis, hES H1 cells were seeded on Matrigel-coated coverslips using Accutase, fixed with 4% formaldehyde for 20 min, permeabilized with 0.5% Triton for 10 min, and stained with indicated antibodies and/or Hoechst 33342. Images were taken using a Nikon A1R+ MP microscope and processed using ImageJ.
ATAC-seq library preparation and downstream processing
ATAC-seq was performed using the OMNI-ATAC protocol as previously described (77). Briefly, cells were dissociated using Accutase, and 50,000 cells were subjected to the tagmentation reaction. Cells were first washed in resuspension buffer [10 mM tris-HCl (pH 8.0), 10 mM NaCl, and 3 mM MgCl2 in water], following which nuclei were isolated in 1 ml of lysis buffer [10 mM tris-HCl (pH 8.0), 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 0.1% Tween 20, and 0.01% digitonin in water] on ice for 3 min. Nuclei were rinsed once in wash buffer [10 mM tris-HCl (pH 8.0), 10 mM NaCl, 3 mM MgCl2, and 0.1% Tween 20], and tagmentation was carried out using 2.5 μl of Tn5 transposase (Illumina 15027865) for 30 min. Following tagmentation, DNA was purified using the Zymo DNA Clean and Concentrator kit. Libraries were prepared by PCR using Q5 High-Fidelity DNA polymerase (NEB) and using primers carrying Illumina Nextera i7 barcodes. First, gap filling was performed at 72°C for 5 min followed by 5 cycles of 98°C, 20 s; 63°C, 30 s; and 72°C, 1 min. After initial amplification, tubes were held on ice, while quantitative PCR was run on 1 μl of the preamplified library to determine additional number of cycles needed. Libraries were sequenced on HiSeq2500 using PE50.
Raw reads were processed using the ENCODE pipeline (encodeproject.org/atac-seq/) (78). Differentially accessible peaks were identified using the DiffBind (Stark and Brown, 2011, http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf).
For this comparative analysis, we used the set of ATAC peaks identified by the ENCODE analysis pipeline using the most restrictive approach [0.05 IDR (Irreproducibility Discovery Rate) of true replicates]. The default DESeq2 analysis using a threshold of 0.001 was used to define highly differential ATAC peaks. For each of the three comparisons shown, only the peaks identified in the two conditions being compared were considered. For visualization, bigwig files generated by the ENCODE pipeline that represent the P value signal of pooled true replicates were loaded into Integrated Genomics Viewer (IGV). Heatmaps and plot profiles were generated using the plotHeatmap and plotProfile function in DeepTools suite (79). To define the chromatin state of differentially enriched ATAC peaks, the ChromHMM model generated as part of the Roadmap consortium, using H1-derived NPC cells (80), was used. Briefly, ATAC peaks identified as differentially enriched were intersected with ChromHMM states not taking into account the size of intersect. Bedtools was used for intersections (81), and more than one state could be assigned to the same peak. Intersections with the quiescent state (characterized by not having enrichment of any chromatin mark) are not shown in the main figure but were assigned to 1066 peaks. Protein binding motifs enriched in differentially accessible enhancer peaks were identified using HOMER (82) by running the –size given and –mask parameters. Motifs with a P value lower than 1 × 10−12 were considered to be significantly enriched. Differentially accessible ATAC peaks were associated with genes using GREAT (83) using the default association tool. GO for biological processes was also carried out using GREAT. SMARCA4 and H3K27ac chromatin immunoprecipitation (ChIP)–seq data (GSE122631) were obtained from (63). ChIP-seq data were aligned to the human genome (hg38) using bowtie2 (84) and a MAPQ filter of 10. Duplicate reads were removed using Picard tools (http://broadinstitute.github.io/picard/). Enriched regions were called peaks using MACS2 (85) and corresponding input control. The two files of MACS2 peaks for each SMARCA4 ChIP-seq replicates were merged and used in bedtools to calculate the total intersection with differentially enriched peaks in shOTUD5 NC3 cells. Intersect size was not taken into account. For visualization in genome browser and heatmap, only one randomly selected replicate of SMARCA4 ChIP-seq is shown.
RNA sequencing
RNA from three replicates for cells treated with control shRNA or shRNA against OTUD5, at each stage of neural conversion, was isolated using TRIzol. After confirming that the RNA integration number for each sample was above 8, libraries were prepared using a TruSeq Stranded mRNA prep kit with PolyA purification and sequenced on HiSeq 2500 using a 1 × 50 single-read mode. RNA-seq analysis and identification of differentially expressed genes were performed using LCDB workflow (https://github.com/lcdb/lcdb-wf). For visualization, bigwig files created by the LCDB workflow were loaded onto IGV.
Supplementary Material
Acknowledgments
We thank the patients and their families for participating in research studies. We thank the NHLBI iPSC Core, the NHGRI Mouse Core, the NICHD Molecular Genomics Core, computational resources of the NIH HPC Biowulf cluster (hpc.nih.gov), and the NIDCR Imaging Core for excellent technical assistance and J. Mays for help with data analysis for teratoma studies. We also thank K. Walters, R. Youle, and M. Rape for critically reading this manuscript. We also acknowledge the members of the Undiagnosed Diseases Network (UDN), who are listed in alphabetical order in the Supplementary Materials accompanying this manuscript. Funding: This research was supported by the Intramural Research Program of the National Institutes of Dental and Craniofacial Research (NIDCR), the National Institute of Child Health and Development (NICHD), and the National Human Genome Research Institute (NHGRI), NIH. K.Õ. and S.P. were supported by Estonian Research Council grants PRG471, MOBTP175, and PUTJD827. Grants-in-Aid for Young Scientists (B) (17K17693) and Scientific Research (B) (JP19H03621) and (A) (JP17H01539) of the Japan Society for the Promotion of Science (JSPS) were provided to D.T.U., N.Mi., and N.Ma., respectively. This work was also supported by AMED under grant numbers JP20ek0109486, JP20dm0107090, JP20ek0109301, JP20ek0109348, and JP20kk020512 (to N.Ma.). Author contributions: D.B.B. conceived and designed the study, identified and saw patients with OTUD5, wrote the manuscript, and, together with M.A.B., performed most experiments. M.A.B. designed, performed, and interpreted stem cell differentiation, deubiquitylation, and immunoprecipitation experiments. A.J.A. analyzed and interpreted all mass spectrometry experiments, performed all immunofluorescence experiments, and performed and analyzed qPCR experiments. J.J.T. performed and analyzed ATAC-seq and RNA-seq experiments and wrote the manuscript. H.O. analyzed RNA-seq experiments. R.D. and A.M. analyzed ATAC-seq and RNA-seq experiments. D.T.U. performed and analyzed exome sequencing and methylation studies in P6-F4, and J.I. supervised the project. S.P. performed and analyzed exome sequencing in P10-F7. K.S., N.Mi., and N.Ma. performed exome sequencing in P8-F6. T.T., K.S., E.M., P.D., J.B., W.M., K.W.B., N.Mi., R.W., M.K., Y.N., S.O., T.K., N.Ma., M.W., I.T., K.Õ., and C.J.T. helped provide clinical information on patients reported in this study. I.A. conceived and designed the study and interpreted results. D.L.K. conceived and designed the study, interpreted results, and secured funding. P.P.R. designed, performed, and analyzed ATAC-seq and RNA-seq experiments, wrote the manuscript, and secured funding. A.W. conceived and designed the study, performed experiments, interpreted results, wrote the manuscript, and secured funding. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. RNA-seq and ATAC-seq datasets were deposited in the Gene Expression Omnibus (GSE160590). Additional data related to this paper may be requested from the corresponding author.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabe2116/DC1
REFERENCES AND NOTES
- 1.Claussnitzer M., Cho J. H., Collins R., Cox N. J., Dermitzakis E. T., Hurles M. E., Kathiresan S., Kenny E. E., Lindgren C. M., MacArthur D. G., North K. N., Plon S. E., Rehm H. L., Risch N., Rotimi C. N., Shendure J., Soranzo N., McCarthy M. I., A brief history of human disease genetics. Nature 577, 179–189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright C. F., FitzPatrick D. R., Firth H. V., Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 19, 253–268 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Komander D., Rape M., The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Strikoudis A., Guillamot M., Aifantis I., Regulation of stem cell function by protein ubiquitylation. EMBO Rep. 15, 365–382 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Werner A., Manford A. G., Rape M., Ubiquitin-dependent regulation of stem cell biology. Trends Cell Biol. 27, 568–579 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grabbe C., Husnjak K., Dikic I., The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 12, 295–307 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lydeard J. R., Schulman B. A., Harper J. W., Building and remodelling Cullin–RING E3 ubiquitin ligases. EMBO Rep. 14, 1050–1061 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oh E., Akopian D., Rape M., Principles of ubiquitin-dependent signaling. Annu. Rev. Cell Dev. Biol. 34, 137–162 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Di Fiore P. P., Polo S., Hofmann K., When ubiquitin meets ubiquitin receptors: A signalling connection. Nat. Rev. Mol. Cell Biol. 4, 491–497 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Haglund K., Dikic I., Ubiquitylation and cell signaling. EMBO J. 24, 3353–3359 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yau R., Rape M., The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Chau V., Tobias J., Bachmair A., Marriott D., Ecker D., Gonda D., Varshavsky A., A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 243, 1576–1583 (1989). [DOI] [PubMed] [Google Scholar]
- 13.Jin L., Williamson A., Banerjee S., Philipp I., Rape M., Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell 133, 653–665 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tokunaga F., Sakata S. I., Saeki Y., Satomi Y., Kirisako T., Kamei K., Nakagawa T., Kato M., Murata S., Yamaoka S., Yamamoto M., Akira S., Takao T., Tanaka K., Iwai K., Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol. 11, 123–132 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J. I., Chen Z. J., TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Meyer H. J., Rape M., Enhanced protein degradation by branched ubiquitin chains. Cell 157, 910–921 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yau R. G., Doerner K., Castellanos E. R., Haakonsen D. L., Werner A., Wang N., Yang X. W., Martinez-Martin N., Matsumoto M. L., Dixit V. M., Rape M., Assembly and function of heterotypic ubiquitin chains in cell-cycle and protein quality control. Cell 171, 918–933.e20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emmerich C. H., Ordureau A., Strickson S., Arthur J. S. C., Pedrioli P. G. A., Komander D., Cohen P., Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. U.S.A. 110, 15247–15252 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohtake F., Saeki Y., Ishido S., Kanno J., Tanaka K., The K48-K63 branched ubiquitin chain regulates NF-κB signaling. Mol. Cell 64, 251–266 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Komander D., Clague M. J., Urbe S., Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Newton K., Matsumoto M. L., Wertz I. E., Kirkpatrick D. S., Lill J. R., Tan J., Dugger D., Gordon N., Sidhu S. S., Fellouse F. A., Komuves L., French D. M., Ferrando R. E., Lam C., Compaan D., Yu C., Bosanac I., Hymowitz S. G., Kelley R. F., Dixit V. M., Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 134, 668–678 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Mevissen T. E. T., Komander D., Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 86, 159–192 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Sahtoe D. D., Sixma T. K., Layers of DUB regulation. Trends Biochem. Sci. 40, 456–467 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Leznicki P., Kulathu Y., Mechanisms of regulation and diversification of deubiquitylating enzyme function. J. Cell Sci. 130, 1997–2006 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Damgaard R. B., Walker J. A., Marco-Casanova P., Morgan N. V., Titheradge H. L., Elliott P. R., Hale D. M., Maher E. R., McKenzie A. N. J., Komander D., The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell 166, 1215–1230.e20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heideker J., Wertz I. E., DUBs, the regulation of cell identity and disease. Biochem. J. 465, 1–26 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Zhou Q., Yu X., Demirkaya E., Deuitch N., Stone D., Tsai W. L., Kuehn H. S., Wang H., Yang D., Park Y. H., Ombrello A. K., Blake M., Romeo T., Remmers E. F., Chae J. J., Mullikin J. C., Güzel F., Milner J. D., Boehm M., Rosenzweig S. D., Gadina M., Welch S. B., Özen S., Topaloglu R., Abinun M., Kastner D. L., Aksentijevich I., Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. U.S.A. 113, 10127–10132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clague M. J., Urbe S., Komander D., Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 20, 338–352 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Mevissen T. E. T., Hospenthal M. K., Geurink P. P., Elliott P. R., Akutsu M., Arnaudo N., Ekkebus R., Kulathu Y., Wauer T., Oualid F. E., Freund S. M. V., Ovaa H., Komander D., OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 154, 169–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Damgaard R. B., Elliott P. R., Swatek K. N., Maher E. R., Stepensky P., Elpeleg O., Komander D., Berkun Y., OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med. 11, e9324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Q., Wang H., Schwartz D. M., Stoffels M., Park Y. H., Zhang Y., Yang D., Demirkaya E., Takeuchi M., Tsai W. L., Lyons J. J., Yu X., Ouyang C., Chen C., Chin D. T., Zaal K., Chandrasekharappa S. C., P Hanson E., Yu Z., Mullikin J. C., Hasni S. A., Wertz I. E., Ombrello A. K., Stone D. L., Hoffmann P., Jones A., Barham B. K., Leavis H. L., van Royen-Kerkof A., Sibley C., Batu E. D., Gül A., Siegel R. M., Boehm M., Milner J. D., Ozen S., Gadina M., Chae J. J., Laxer R. M., Kastner D. L., Aksentijevich I., Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 48, 67–73 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Ligt J., Willemsen M. H., van Bon B. W. M., Kleefstra T., Yntema H. G., Kroes T., Vulto-van Silfhout A. T., Koolen D. A., de Vries P., Gilissen C., del Rosario M., Hoischen A., Scheffer H., de Vries B. B. A., Brunner H. G., Veltman J. A., Vissers L. E. L. M., Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Timal S., Hoischen A., Lehle L., Adamowicz M., Huijben K., Sykut-Cegielska J., Paprocka J., Jamroz E., van Spronsen F. J., Körner C., Gilissen C., Rodenburg R. J., Eidhof I., van den Heuvel L., Thiel C., Wevers R. A., Morava E., Veltman J., Lefeber D. J., Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum. Mol. Genet. 21, 4151–4161 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Uddin M., Unda B. K., Kwan V., Holzapfel N. T., White S. H., Chalil L., Woodbury-Smith M., Ho K. S., Harward E., Murtaza N., Dave B., Pellecchia G., D’Abate L., Nalpathamkalam T., Lamoureux S., Wei J., Speevak M., Stavropoulos J., Hope K. J., Doble B. W., Nielsen J., Wassman E. R., Scherer S. W., Singh K. K., OTUD7A regulates neurodevelopmental phenotypes in the 15q13.3 microdeletion syndrome. Am. J. Hum. Genet. 102, 278–295 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin J., Chen W., Chao E. S., Soriano S., Wang L., Wang W., Cummock S. E., Tao H., Pang K., Liu Z., Pereira F. A., Samaco R. C., Zoghbi H. Y., Xue M., Schaaf C. P., Otud7a Knockout mice recapitulate many neurological features of 15q13.3 microdeletion syndrome. Am. J. Hum. Genet. 102, 296–308 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santiago-Sim T., Burrage L. C., Ebstein F., Tokita M. J., Miller M., Bi W., Braxton A. A., Rosenfeld J. A., Shahrour M., Lehmann A., Cogné B., Küry S., Besnard T., Isidor B., Bézieau S., Hazart I., Nagakura H., Immken L. D. L., Littlejohn R. O., Roeder E., Kara B., Hardies K., Weckhuysen S., May P., Lemke J. R., Elpeleg O., Abu-Libdeh B., James K. N., Silhavy J. L., Issa M. Y., Zaki M. S., Gleeson J. G., Seavitt J. R., Dickinson M. E., Ljungberg M. C., Wells S., Johnson S. J., Teboul L., Eng C. M., Yang Y., Kloetzel P. M., Heaney J. D., Walkiewicz M. A., Afawi Z., Balling R., Barisic N., Baulac S., Craiu D., de Jonghe P., Guerrero-Lopez R., Guerrini R., Helbig I., Hjalgrim H., Jähn J., Klein K. M., Leguern E., Lerche H., Marini C., Muhle H., Rosenow F., Serratosa J., Sterbová K., Suls A., Moller R. S., Striano P., Weber Y., Zara F., Biallelic variants in OTUD6B cause an intellectual disability syndrome associated with seizures and dysmorphic features. Am. J. Hum. Genet. 100, 676–688 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The Deciphering Developmental Disorders Study , Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., O’Donnell-Luria A. H., Ware J. S., Hill A. J., Cummings B. B., Tukiainen T., Birnbaum D. P., Kosmicki J. A., Duncan L. E., Estrada K., Zhao F., Zou J., Pierce-Hoffman E., Berghout J., Cooper D. N., Deflaux N., DePristo M., Do R., Flannick J., Fromer M., Gauthier L., Goldstein J., Gupta N., Howrigan D., Kiezun A., Kurki M. I., Moonshine A. L., Natarajan P., Orozco L., Peloso G. M., Poplin R., Rivas M. A., Ruano-Rubio V., Rose S. A., Ruderfer D. M., Shakir K., Stenson P. D., Stevens C., Thomas B. P., Tiao G., Tusie-Luna M. T., Weisburd B., Won H.-H., Yu D., Altshuler D. M., Ardissino D., Boehnke M., Danesh J., Donnelly S., Elosua R., Florez J. C., Gabriel S. B., Getz G., Glatt S. J., Hultman C. M., Kathiresan S., Laakso M., McCarroll S., McCarthy M. I., McGovern D., McPherson R., Neale B. M., Palotie A., Purcell S. M., Saleheen D., Scharf J. M., Sklar P., Sullivan P. F., Tuomilehto J., Tsuang M. T., Watkins H. C., Wilson J. G., Daly M. J., MacArthur D. G.; Exome Aggregation Consortium , Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin H. C., Jones W. D., McIntyre R., Sanchez-Andrade G., Sanderson M., Stephenson J. D., Jones C. P., Handsaker J., Gallone G., Bruntraeger M., McRae J. F., Prigmore E., Short P., Niemi M., Kaplanis J., Radford E. J., Akawi N., Balasubramanian M., Dean J., Horton R., Hulbert A., Johnson D. S., Johnson K., Kumar D., Lynch S. A., Mehta S. G., Morton J., Parker M. J., Splitt M., Turnpenny P. D., Vasudevan P. C., Wright M., Bassett A., Gerety S. S., Wright C. F., FitzPatrick D. R., Firth H. V., Hurles M. E., Barrett J. C.; on behalf of the Deciphering Developmental Disorders Study , Quantifying the contribution of recessive coding variation to developmental disorders. Science 362, 1161–1164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karczewski K. J., Francioli L. C., Tiao G., Cummings B. B., Alföldi J., Wang Q., Collins R. L., Laricchia K. M., Ganna A., Birnbaum D. P., Gauthier L. D., Brand H., Solomonson M., Watts N. A., Rhodes D., Singer-Berk M., England E. M., Seaby E. G., Kosmicki J. A., Walters R. K., Tashman K., Farjoun Y., Banks E., Poterba T., Wang A., Seed C., Whiffin N., Chong J. X., Samocha K. E., Pierce-Hoffman E., Zappala Z., O’Donnell-Luria A. H., Minikel E. V., Weisburd B., Lek M., Ware J. S., Vittal C., Armean I. M., Bergelson L., Cibulskis K., Connolly K. M., Covarrubias M., Donnelly S., Ferriera S., Gabriel S., Gentry J., Gupta N., Jeandet T., Kaplan D., Llanwarne C., Munshi R., Novod S., Petrillo N., Roazen D., Ruano-Rubio V., Saltzman A., Schleicher M., Soto J., Tibbetts K., Tolonen C., Wade G., Talkowski M. E.; Genome Aggregation Database Consortium, Neale B. M., Daly M. J., MacArthur D. G., The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kayagaki N., Phung Q., Chan S., Chaudhari R., Quan C., O’Rourke K. M., Eby M., Pietras E., Cheng G., Bazan J. F., Zhang Z., Arnott D., Dixit V. M., DUBA: A deubiquitinase that regulates type I interferon production. Science 318, 1628–1632 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Gao J., Buckley S. M., Cimmino L., Guillamot M., Strikoudis A., Cang Y., Goff S. P., Aifantis I., The CUL4-DDB1 ubiquitin ligase complex controls adult and embryonic stem cell differentiation and homeostasis. eLife 4, e07539 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mena E. L., Kjolby R. A. S., Saxton R. A., Werner A., Lew B. G., Boyle J. M., Harland R., Rape M., Dimerization quality control ensures neuronal development and survival. Science 362, eaap8236 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Werner A., Iwasaki S., McGourty C. A., Medina-Ruiz S., Teerikorpi N., Fedrigo I., Ingolia N. T., Rape M., Cell-fate determination by ubiquitin-dependent regulation of translation. Nature 525, 523–527 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oh E., Mark K. G., Mocciaro A., Watson E. R., Prabu J. R., Cha D. D., Kampmann M., Gamarra N., Zhou C. Y., Rape M., Gene expression and cell identity controlled by anaphase-promoting complex. Nature 579, 136–140 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bustos F., Segarra-Fas A., Chaugule V. K., Brandenburg L., Branigan E., Toth R., Macartney T., Knebel A., Hay R. T., Walden H., Findlay G. M., RNF12 X-linked intellectual disability mutations disrupt E3 ligase activity and neural differentiation. Cell Rep. 23, 1599–1611 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaser R., Adusumalli S., Leng S. N., Sikic M., Ng P. C., SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Ioannidis N. M., Rothstein J. H., Pejaver V., Middha S., McDonnell S. K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D., Cannon-Albright L. A., Teerlink C. C., Stanford J. L., Isaacs W. B., Xu J., Cooney K. A., Lange E. M., Schleutker J., Carpten J. D., Powell I. J., Cussenot O., Cancel-Tassin G., Giles G. G., MacInnis R. J., Maier C., Hsieh C.-L., Wiklund F., Catalona W. J., Foulkes W. D., Mandal D., Eeles R. A., Kote-Jarai Z., Bustamante C. D., Schaid D. J., Hastie T., Ostrander E. A., Bailey-Wilson J. E., Radivojac P., Thibodeau S. N., Whittemore A. S., Sieh W., REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tate J. G., Bamford S., Jubb H. C., Sondka Z., Beare D. M., Bindal N., Boutselakis H., Cole C. G., Creatore C., Dawson E., Fish P., Harsha B., Hathaway C., Jupe S. C., Kok C. Y., Noble K., Ponting L., Ramshaw C. C., Rye C. E., Speedy H. E., Stefancsik R., Thompson S. L., Wang S., Ward S., Campbell P. J., Forbes S. A., COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cox B. J., Vollmer M., Tamplin O., Lu M., Biechele S., Gertsenstein M., van Campenhout C., Floss T., Kuhn R., Wurst W., Lickert H., Rossant J., Phenotypic annotation of the mouse X chromosome. Genome Res. 20, 1154–1164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Vivo A., Sanchez A., Yegres J., Kim J., Emly S., Kee Y., The OTUD5-UBR5 complex regulates FACT-mediated transcription at damaged chromatin. Nucleic Acids Res. 47, 729–746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang O. W., Ma X., Yin J. P., Flinders J., Maurer T., Kayagaki N., Phung Q., Bosanac I., Arnott D., Dixit V. M., Hymowitz S. G., Starovasnik M. A., Cochran A. G., Phosphorylation-dependent activity of the deubiquitinase DUBA. Nat. Struct. Mol. Biol. 19, 171–175 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Rutz S., Kayagaki N., Phung Q. T., Eidenschenk C., Noubade R., Wang X., Lesch J., Lu R., Newton K., Huang O. W., Cochran A. G., Vasser M., Fauber B. P., DeVoss J., Webster J., Diehl L., Modrusan Z., Kirkpatrick D. S., Lill J. R., Ouyang W., Dixit V. M., Deubiquitinase DUBA is a post-translational brake on interleukin-17 production in T cells. Nature 518, 417–421 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Chambers S. M., Fasano C. A., Papapetrou E. P., Tomishima M., Sadelain M., Studer L., Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shakiba N., Fahmy A., Jayakumaran G., McGibbon S., David L., Trcka D., Elbaz J., Puri M. C., Nagy A., van der Kooy D., Goyal S., Wrana J. L., Zandstra P. W., Cell competition during reprogramming gives rise to dominant clones. Science 364, eaan0925 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Huang L., Jolly L. A., Willis-Owen S., Gardner A., Kumar R., Douglas E., Shoubridge C., Wieczorek D., Tzschach A., Cohen M., Hackett A., Field M., Froyen G., Hu H., Haas S. A., Ropers H. H., Kalscheuer V. M., Corbett M. A., Gecz J., A noncoding, regulatory mutation implicates HCFC1 in nonsyndromic intellectual disability. Am. J. Hum. Genet. 91, 694–702 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jolly L. A., Nguyen L. S., Domingo D., Sun Y., Barry S., Hancarova M., Plevova P., Vlckova M., Havlovicova M., Kalscheuer V. M., Graziano C., Pippucci T., Bonora E., Sedlacek Z., Gecz J., HCFC1 loss-of-function mutations disrupt neuronal and neural progenitor cells of the developing brain. Hum. Mol. Genet. 24, 3335–3347 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Montgomery R. L., Hsieh J., Barbosa A. C., Richardson J. A., Olson E. N., Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc. Natl. Acad. Sci. U.S.A. 106, 7876–7881 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ronan J. L., Wu W., Crabtree G. R., From neural development to cognition: Unexpected roles for chromatin. Nat. Rev. Genet. 14, 347–359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wagner V. F., Hillman P. R., Britt A. D., Ray J. W., Farach L. S., A de novo HDAC2 variant in a patient with features consistent with Cornelia de Lange syndrome phenotype. Am. J. Med. Genet. A 179, 852–856 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Tsurusaki Y., Okamoto N., Ohashi H., Kosho T., Imai Y., Hibi-Ko Y., Kaname T., Naritomi K., Kawame H., Wakui K., Fukushima Y., Homma T., Kato M., Hiraki Y., Yamagata T., Yano S., Mizuno S., Sakazume S., Ishii T., Nagai T., Shiina M., Ogata K., Ohta T., Niikawa N., Miyatake S., Okada I., Mizuguchi T., Doi H., Saitsu H., Miyake N., Matsumoto N., Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 44, 376–378 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Li F., Sun Q., Liu K., Zhang L., Lin N., You K., Liu M., Kon N., Tian F., Mao Z., Li T., Tong T., Qin J., Gu W., Li D., Zhao W., OTUD5 cooperates with TRIM25 in transcriptional regulation and tumor progression via deubiquitination activity. Nat. Commun. 11, 4184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao F., Elliott N. J., Ho J., Sharp A., Shokhirev M. N., Hargreaves D. C., Heterozygous mutations in SMARCA2 reprogram the enhancer landscape by global retargeting of SMARCA4. Mol. Cell 75, 891–904.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chandler R. L., Brennan J., Schisler J. C., Serber D., Patterson C., Magnuson T., ARID1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol. Cell. Biol. 33, 265–280 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao Y., Mudge M. C., Soll J. M., Rodrigues R. B., Byrum A. K., Schwarzkopf E. A., Bradstreet T. R., Gygi S. P., Edelson B. T., Mosammaparast N., Mudge M. C., Soll J. M., Rodrigues R. B., Byrum A. K., Schwarzkopf E. A., Bradstreet T. R., Gygi S. P., Edelson B. T., Mosammaparast N., OTUD4 is a phospho-activated K63 deubiquitinase that regulates MyD88-dependent signaling. Mol. Cell 69, 505–516.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Franz A., Ackermann L., Hoppe T., Ring of change: CDC48/p97 drives protein dynamics at chromatin. Front. Genet. 7, 73 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.M. A. Deardorff, S. E. Noon, I. D. Krantz, Cornelia de Lange Syndrome, in GeneReviews®, M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, A. Amemiya, Eds. (University of Washington, 1993). [Google Scholar]
- 68.S. Schrier Vergano, G. Santen, D. Wieczorek, B. Wollnik, N. Matsumoto, M. A. Deardorff, Coffin-Siris Syndrome, in GeneReviews®, M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, A. Amemiya, Eds. (University of Washington, 1993). [PubMed] [Google Scholar]
- 69.Sobreira N., Schiettecatte F., Valle D., Hamosh A., GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 36, 928–930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beers J., Linask K. L., Chen J. A., Siniscalchi L. I., Lin Y., Zheng W., Rao M., Chen G., A cost-effective and efficient reprogramming platform for large-scale production of integration-free human induced pluripotent stem cells in chemically defined culture. Sci. Rep. 5, 11319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nelakanti R. V., Kooreman N. G., Wu J. C., Teratoma formation: A tool for monitoring pluripotency in stem cell research. Curr. Protoc. Stem Cell Biol. 32, 4A.8.1–4A.8.17 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Allen R. C., Zoghbi H. Y., Moseley A. B., Rosenblatt H. M., Belmont J. W., Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 51, 1229–1239 (1992). [PMC free article] [PubMed] [Google Scholar]
- 73.Kiedrowski L. A., Raca G., Laffin J. J., Nisler B. S., Leonhard K., McIntire E., Mongomery K. D., DNA methylation assay for X-chromosome inactivation in female human iPS cells. Stem Cell Rev. Rep. 7, 969–975 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Doe B., Brown E., Boroviak K., Generating CRISPR/Cas9-derived mutant mice by zygote cytoplasmic injection using an automatic microinjector. Methods Protoc. 1, 5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sowa M. E., Bennett E. J., Gygi S. P., Harper J. W., Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meerbrey K. L., Hu G., Kessler J. D., Roarty K., Li M. Z., Fang J. E., Herschkowitz J. I., Burrows A. E., Ciccia A., Sun T., Schmitt E. M., Bernardi R. J., Fu X., Bland C. S., Cooper T. A., Schiff R., Rosen J. M., Westbrook T. F., Elledge S. J., The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 3665–3670 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Corces M. R., Trevino A. E., Hamilton E. G., Greenside P. G., Sinnott-Armstrong N. A., Vesuna S., Satpathy A. T., Rubin A. J., Montine K. S., Wu B., Kathiria A., Cho S. W., Mumbach M. R., Carter A. C., Kasowski M., Orloff L. A., Risca V. I., Kundaje A., Khavari P. A., Montine T. J., Greenleaf W. J., Chang H. Y., An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.The ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramírez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y.-C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott G. N., Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J. M., Li W., Marra M. A., McManus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L.-H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J. A., Wang T., Kellis M., Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M., Bejerano G., GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nussbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabe2116/DC1