

Abstract
Smart drugs, such as antibody-drug conjugates, for targeted therapy rely on the ability to deliver a warhead to the desired location and to achieve activation at the same site. Thus, designing a smart drug often requires proper linker chemistry for tethering the warhead with a vehicle in such a way that either allows the active drug to retain its potency while being tethered or ensures release and thus activation at the desired location. Recent years have seen much progress in the design of new linker activation strategies. Herein, we review the recent development of chemical strategies used for linking the warhead with a delivery vehicle for preferential cleavage at the desired sites.
Keywords: Linker chemistry, Drug delivery, Targeted delivery, Triggered release, Antibody-drug conjugates, Click and release
1. Introduction
In the world of drug discovery and development, the concept of “smart drugs” broadly refers to those that can selectively affect disease-relevant target(s). However, to a certain degree, essentially all drugs are already selective toward the desired target(s) whether they are enzyme inhibitors, receptor ligands, ion channel blockers, inhibitors of protein-protein interaction, or even genotoxins used for cancer chemotherapy, among others. They would not have made through the various milestones of drug discovery and developmental processes if they were not sufficiently selective for the intended target(s). However, once administered into the human body, the highly complex living system often leads to many unexpected off-target effects. Thus, various additional approaches have been studied and employed to further enhance the level of selectivity to ensure efficacy with a minimal level of off-target effects. Broadly speaking, synthetic lethality, conjugation of a drug to a targeting moiety (e.g., antibody, receptor ligand), selective activation of a prodrug at the desired site, suicide inhibitors, and selective trapping of a drug at the desired site are all approaches aimed at adding an extra layer of selectivity to a drug that normally relies on single-target selectivity.1 Among all these approaches, conjugation of a drug to a targeting moiety is arguably the most commonly used method to further enhance selectivity. Antibody-drug conjugates (ADCs) are excellent examples.2–6 There are other ligands that have been extensively used for targeting purposes, including folate,7–9 integrin ligands,10–12 ligands for the prostate-specific antigen,13–15 to name a few. Ideally, such conjugates can achieve two things: enrichment at the desired site because of the targeting vector and selective release/activation of the parent drug molecule at the site of action. The second aspect is where the linker chemistry comes into play. Earlier efforts often rely on linker chemistry that offers limited selectivity in its cleavage, resulting in premature drug release or inadequate release. In recent years, there has been some very impressive progress made in the selective activation of a prodrug. The selective cleavage of such a linker affords a second layer of selectivity; much of this has been achieved through the innovative use of linker chemistry and triggering mechanism(s) for activation. In discussing the specific linker chemistry, there are two key components: the chemistry needed to allow the linker to be removed and the triggering mechanism. There are many different permutations and combinations of the “trigger” and “linker” components. It should also be noted that there have been several recent reviews about the application of chemical linkers in various examples of prodrugs.16–18 Herein, we do not strive to be comprehensive and do not want to unnecessarily duplicate what has already been elegantly described in these reviews. Instead, we will use examples to show innovative combinations of linker chemistry and activation mechanism to allow for improved performance of targeting strategies.
2. pH-sensitive linkers
There are two commonly seen scenarios that allow for the selective activation of pH-sensitive linkers, both of which are developed for targeting cancer. First, it is well-known that tumor tissues generally have lower pH (6–7) than normal tissues (pH 7.4), largely due to elevated glycolysis known as the Warburg effect and poor lymphatic drainage. Second, in an endocytic or exocytic process, pH inside the endosome and lysosome generally drops to about 5.5~6.0 and 4.5–5.0, respectively.19 Therefore, pH-sensitive linkers take advantage of this pH difference to trigger selective prodrug release in tumor tissue or release the payload of a targeted therapeutic conjugate through an endocytosis or exocytosis process.
In most of the reported pH-sensitive linker applications, the enrichment effect is largely achieved by the targeting moiety. The linker itself should be stable enough to avoid premature release and sensitive enough to ensure efficient release upon being taken up through endocytosis. There have been extensive reviews of this area,20–23 and yet it is well-recognized that the choice of suitable linker chemistry in the context of the intended disease target(s) is mostly based on intuition.24 On the other hand, the pH difference between endosome and extracellular space is significant (ΔpH ≈ 2); it would be easier to achieve selective release by taking advantage of this mechanism than to utilize the minor difference between cancerous and normal tissues. In this context, we focus on the chemistry that allows for pH-dependent hydrolysis profiles; and also in the discussion of linker chemistry, we would like to introduce the ratio between the release rates at lower and neutral pH as an indicator for selectivity, as suggested by Wagner el al.24 If data is available, release rates at various pH will also be included for comparison.
2.1. Imine/hydrazone/oxime linkers
The chemistry of acid-catalyzed hydrolysis of oxime and hydrazone has been extensively used in pH-sensitive linker strategies, especially in the delivery of anticancer drugs through the use of nanocarriers,25 polymer-based carriers,19 and ADCs.26,27 For discussing the various design principles, it is important to understand the basics of the hydrolysis chemistry and reaction rate, especially in comparative studies of the hydrolytic cleavage of oxime and hydrazone (C1=N1-X). In a 2008 study using NMR in a deuterated buffer, it was found that the attack by a water molecule on C1 is the rate-determining step and is subject to acid catalysis through protonation of the N1 nitrogen atom (Figure 1A).28 Tethering an electron-withdrawing X group reduces the propensity for N1 to be protonated, leading to an overall decrease in the hydrolysis rate under physiological conditions. Therefore, the hydrolytic stability at pH 7.0 is ranked in the order of trialkylhydrazonium ion (no detectable hydrolysis) >> oxime >> acyl hydrazone > primary hydrazone > secondary hydrazone > imine (Table 1).
Figure 1.

Mechanism of hydrolytic cleavage of hydrazone/oxime linkers.
Table 1.
Summary of hydrolysis kinetics of different hydrazones in deuterated buffer.
| Entry | Structure | t1/2 at pD 7.0 | t1/2 at pD 6.0 | t1/2 at pD 5.0 | Ratio |
|
|---|---|---|---|---|---|---|
| kpD6.0/kpD7.0 | kpD5.0/kpD7.0 | |||||
| 1 | 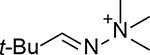 |
No hydrolysis in 22 d | Not determined | 10.3% hydrolysis in 17 d | Not applicable | Not applicable |
| 2 | 25 d | 4.4 ± 0.3 d | 15.7 ± 0.4 h | 5.7 | 38.2 | |
| 3 | 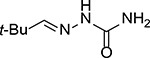 |
3.8 ± 0.5 h | 36 ± 2 min | 8.5 ± 0.4 min | 6.3 | 26 |
| 4 |  |
2.0 ± 0.2 h | 21.4 ± 0.8 min | 2.4 ± 0.4 min | 5.6 | 50 |
| 5 | 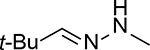 |
1.0 ± 0.1 h | 24.5 ± 0.6 min | 9 ± 1 min | 2.4 | 6.6 |
| 6 | 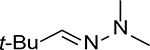 |
32 ± 3 min | 11.3 ± 0.2 min | 7.4 ± 0.5 min | 2.8 | 4.3 |
For a pH-sensitive linker, it is very important to understand the effect of pH on the linker stability. It can be seen from Table 1 that all these linkers are pH-sensitive to some degree. However, the quaternary ammonium hydrazone (Entry 1) is too stable to be used as a linker at pH 5. The remaining five all show substantial reaction rate differences between pH 7 and 5. Among all these, the acyl hydrazone linker (Entry 4) stands out as very unique. It is more resistant to hydrolysis than alkylhydrazones at neutral pH but is more labile at lower pH (pH 5). Such properties of the acylhydrazone linker presumably reflect a delicate overall balance between the need for electrophilicity of the imine carbon and the ability for an acid to catalyze the hydrolysis reaction at pH 5 through protonation. The stability profile makes acylhydrazone an excellent pH-responsive linker for anticancer drug delivery (Entry 4). Indeed, the half-life of drug release at ~pH 5.0 for various acyl hydrazine-linked doxorubicin ADCs was determined to be as short as 2.4 min. In contrast, the half-life was more than 2.0 h at pH 7.0.20,29,30 For its unique pH sensitivity, the acylhydrazone linker was employed in the first generation of ADCs, including cBR96-Dox ADC (Figure 2A). Only a small amount of pre-mature release of doxorubicin in the serum was found with cBR96-Dox ADC.31 In phase I clinical trials, cBR96-Dox did not show myelosuppression and alopecia side-effects, which are commonly associated with doxorubicin. Such results indicate that premature Dox release was not pronounced. The most common side effect of the cBR96-Dox conjugate was upper GI toxicity, which could be attenuated by pretreating with anti-acid drug omeprazole.32 However, cBR96-Dox was discontinued after observing promising efficacy in phase II clinical trials,33,34 presumably due to corporate business decisions.
Figure 2.

Schematic view of gemtuzumab ozogamicin showing the AcBut-acyl hydrazone linker.
For developing ADCs with improved effectiveness, extremely cytotoxic pay loads such as calicheamicin (sub-nM cellular IC50) are often used. This requires reliable linkers with improved stability under physiological conditions to avoid premature release of the payload. Therefore, in the development of ADC targeting CD33+ myeloid leukemia, a series of aliphatic and aromatic aldehydes and ketones were assessed as part of the acylhydrazone linker to search optimal results.35 Such aldehydes/ketones also contain a carboxylic acid group for conjugation with the antibody as the targeting moiety. Among these acyl hydrazone linkers, the one formed between 4-(4-acetylphenoxy) butanoic acid (AcBut) and acyl hydrazone (Figure 2B) showed the highest pH selectivity. The resulting ADC gemtzumab ozogamicin (gem-ozo) was shown to be stable with only 6% hydrolysis at pH 7.4 after 24 h at 37 °C, and yet was able to release 97% of tethered calicheamicin after 24 h at 37 °C and pH 4.5, which corresponds to the lysosomal pH. Such results are consistent with the stabilization of the acyl hydrazone linker by the p-alkyloxyphenyl group, which increases the electron density of the hydrazone carbon (as C1 in Figure 1), leading to its decreased propensity to be attacked by a nucleophilic water molecule. In-vitro cytotoxicity of the conjugate showed 78000-fold selectivity to CD33+ HL-60 leukemia cells (IC50 = 0.46 pg cal/mL, equivalent of the weight of calicheamicin) versus CD33− Raji human Burkitt lymphoma cell (IC50 = 35.7 ng cal/mL). In an HL-60 xenograft mouse model, the gem-ozo treatment group showed complete tumor regression by day 28 with a dosage of 50 μg/kg (calicheamicin equivalent) for a total of 3 doses.35 In 2000, gem-ozo became the first ADC approved by the FDA for human use under the brand name of Mylotarg.
In 2010, Mylotarg was voluntarily withdrawn from the U.S. and European markets due to an elevated fatality rate compared to the control group in a phase-3 clinical trial. There were different opinions about the elevated fatality rate. Some retrospective reviews suggested that the toxicity was due to payload release in the blood circulation.36–38 On the other hand, there are others suggesting that the side effect should be attributed to target (CD33) selection, not the linker.40,41,42 Even in the context of these disagreement, the issues was not about the pH-sensitive nature of the linker chemistry. As a matter of fact, a CD22-targeting ADC, inotuzumab ozogamicin (Besponsa®, launched in 2017), utilizes the same hybridized AcBut-acylhydrazone linker, which led to a half-life of 12.3 days, demonstrating stability. The ADC was also shown to be well tolerated.43 Furthermore, FDA approved Mylotarg again in 2017 as an orphan drug for the treatment of CD33-positive acute myeloid leukemia, after Pfizer revised the dosing regimen and confirmed the efficacy, which out-weighs the harmful effects.44
With the aim of further improving the hydrazone linker, Zheng et al. developed a safety-catch strategy by combining an acyl hydrazone linker with a proteolytic bridge to form a peptide-bridged twin-acylhydrazone (PTA) linker (Figure 3). A protease substrate was used to join two acyl hydrazone linkers derived from a wishbone-like bis-aldehyde spacer.45 As a proof of concept, two peptides, GGPLGLAGG and GGFLGG were assessed individually. The first is a substrate for extracellular matrix metalloproteinase 2 (MMP-2) and the second for chymotrypsin. In model studies, a fluorophore (Oregon Green 488)-quencher (Dabcyl) pair was used to examine efficiency of the protease-mediated cleavage. As such, cleavage of the linker and the subsequent hydrolysis of the hydrazone moiety would separate the quencher from the fluorophore, leading to increased fluorescence. When incubated at both pH 7.4 and 4.0 for 24 h, there was no appreciable increase of fluorescent intensity, indicating the chemical stability of the linker. The study attributed this chemical stability of the embedded hydrazone linkers to the cooperative effect brought upon by the spatial proximity of the two hydrazone linkers, which facilitates the reformation of acylhydrazone upon partial hydrolysis. On the other hand, incubation with the corresponding MMP-2 or chymotrypsin for 2 h followed by incubation in buffer for 24 h led to about 50% fluorescent recovery at pH 4.0 and about 10% fluorescent recovery at pH 7.4. The PTA linker with the same fluorescent reporter system was then conjugated to an RGD targeting moiety for studies in U87 cells, which highly express the αvβ3 integrin, a receptor for RGD. Fluorescence increase was observed by confocal microscopy after 24 h incubation. Then the same system was used to deliver a cytotoxic payload to cancer cells. The PTA linker was used to link the RGD targeting moiety and cytotoxic monomethyl auristatin E (MMAE, IC50 = 0.75 nM in U87 cell). MMP-2, chymotrypsin, or lysosomal cathepsin peptide substrates were separately employed as the peptide bridge. The conjugated MMAE with different peptide bridges showed cytotoxicity (IC50) of 61.9 nM (MMP-2 substrate bridge), 140.9 nM (chymotrypsin substrate bridge), and 623.1 nM (cathepsin substrate bridge) respectively in U87 cells. One thing worth noting is that MMP-2 and chymotrypsin are extracellular enzymes. Thus, cleavage of the peptide bridge before the entire delivery system gets into the lysosome is a prerequisite. This safety-catch mechanism offers the advantage of payload release only under the combined conditions of an acidic environment and the presence of an appropriate protease. Even though the release profile in the in-vitro studies led to some promising results, the release rate was considerably slower and less efficient than the conventional acyl hydrazone linker. Thus, this two-stage release mechanism should be further investigated in animal models to assess its applicability.
Figure 3.

Proteolytic unlocking of twin-acylhydrazone linkers for lysosomal acid-triggered release of anticancer drugs.
Compared with the hydrazone linker, another example is the oxime linker. However, the release rate for an oxime linker formed with terephthalaldehyde used in a PEG-Dox micelle drug delivery system was shown to be slow at pH 5.0 (t1/2 ≈ 15 h) and yet the overall release ratio at pH 7.4 is high (≈ 20% after 12 h).46 Such results indicate a smaller stability margin than the acyl hydrazone linker. It should be noted that many of the reported oxime linkers were actual imine chemistry, which is less stable at physiological pH than oxime, presenting an even greater degree of premature release.47,48
Overall, there are some very useful and interesting linker chemistries in the categories of hydrazone, oxime, and imine. However, it is necessary to understand the detailed chemistry before choosing a specific linker for a particular application.
2.2. Phosphoramidate linkers
Besides imine chemistry, phosphoramidate chemistry has also been used in pH-sensitive linker design with tunable release rates.49 One such design takes advantage of general-acid catalysis enabled by an appropriately positioned pyridinyl group (Figure 4). Specifically, the pKa of a protonated pyridine is slightly below physiological pH, allowing the pyridine to remain in the largely unprotonated form at pH 7.4. However, at a lower pH, the protonation of the pyridinyl nitrogen allows for general acid catalysis as a way to facilitate the hydrolytic cleavage of the phosphoramidate linker. As a result, both the pKa values of the amine in the payload part and the pyridinyl group in the linker segment affect the release rate. The trend for the hydrolysis rate of the various phosphoramidate analogs (n = 2) at pH 5.5 was determined as follow: 1a (t1/2: 1.64 h) > 1b (t1/2: 2.13 h) > 1c (t1/2: 16.2 h) > 1d (t1/2: 31.5 h). The position of the neighboring protonated pyridinium nitrogen also influences the hydrolysis rate through intramolecular hydrogen bond formation. For example, in the hydrolysis tests of various 2-pyridinyl phosphate conjugates, it was found that the release rate for compound 1e (n = 1) was very fast and the half-life was reported as t1/2 = 0 h. Presumably, hydrolysis happened instantly after mixing with the aqueous buffer. Hydrolysis of compound 1a (n = 2) at pH 5.5 was much slower with t1/2 of 1.64 h. Such results suggest that the formation of an 8-membered ring in the transition state is more favorable than the formation of a 9-membered ring in the process of accomplishing general-acid catalysis. Along a similar line, for compounds with R1 and R2 being phenylethyl and methyl groups respectively, changing the pyridine nitrogen from an ortho- (reported as t1/2 = 0 h) to a meta-position (t1/2 = 4.33 h), led to an increase in half-life at pH 5.5. The pKa of the amine is inversely related to linker stability. With the pyridine moiety being the same, an aliphatic amine has a shorter half-life than an aryl amine (e.g., 1a vs. 1b, 1c vs. 1d, 1e vs. 1f). The overall design of the phosphoramidate linker chemistry cleverly takes advantage of general-acid catalysis and entropic control to achieve selectivity between different pHs.
Figure 4.

Phosphoramidate linker chemistry.
2.3. Acetal-based linkers
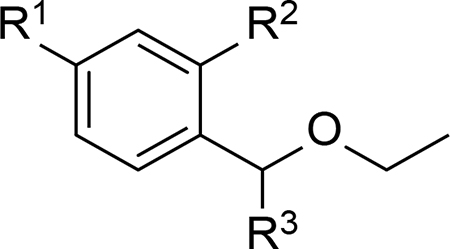
As an acid-labile protecting group of the aldehyde group, acetal can readily undergo hydrolysis under acidic conditions (Figure 5). Wagner et al. assessed several simple benzaldehyde acetals for their hydrolysis profiles and applicability as pH-sensitive linkers.24 It was found that the best selectivity and efficiency were achieved with ethoxybenzyl acetal derivatives (Table 2). Therefore, R1 and R3 could be further modified to prepare pH-sensitive linkers. Indeed, the imidazole derivative represented in Entry 4 of Table 2 was successfully utilized to develop the NEBI linker as discussed later.
Figure 5.

Acid-catalyzed hydrolysis of acetals.
Table 2.
Summary of hydrolysis kinetics of different acetals
 |
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 |
t1/2 (h) |
Ratio (kpH 5.5/kpH 7.4 ) | |
| pH 5.5 | pH 7.4 | |||||
| 1 | H | H | OEt | 0.8 | 12.2 | 15.3 |
| 2 | OMe | H | OEt | 0.1 | 5.3 | 53 |
| 3 | H | OMe | OEt | 0.1 | 19.9 | 199 |
| 4 | H | OMe |  |
1.1 | 28.8 | 31.7 |
In the same study, they also compared the release profiles of the spiro-di-orthoester linker (also known as SpiDo linker),50 acylalkylhydrazone linker, and the acetal linker by constructing a FRET-based fluorescent reporting system consisting of a tetramethylrhodamine (TAMRA) fluorophore, a black hole quencher (BHQ-2), and the linker that joins them together (Figure 6, the same approach was also reported in another publication from the same group51). By monitoring the fluorescent changes at different pH, the study revealed that the spiro diorthoester conjugate 14 gave the highest pH-dependent selectivity and efficiency. It was consistent in achieving instant and complete hydrolysis below pH 3.0, whereas 1 h and 4.5 h were needed to achieve complete hydrolysis at pH 4.0 and 5.0, respectively. At pH 6.0 and 7.0, it showed 65% and 24% of hydrolysis after 15 h. Therefore, the spiro diorthoester linker seemed to be more sensitive to low pH compared with the acylalkylhydrazone linker.
Figure 6.

Fluorescent probes used to assess the hydrolysis profile.
2.4. An N-ethoxybenzylimidazole (NEBI) linker
The N-ethoxybenzylimidazole (NEBI) moiety was used by Yang et al. as a tunable pH-sensitive linker52 and was applied later in the targeted delivery of indenoisoquinoline to cancer via the folate receptor (Figure 7).53 It was based on the hydrolysis of “aminol” ether. In this case, the prodrug used an imidazole nitrogen to form the aminol ether. After protonation of the imidazole nitrogen under acidic conditions, the linker would spontaneously collapse to release the imidazole-containing parent drug. By varying the substitution (R group) on the phenyl ring, the release rate is tunable. For example, installing an electron-donating groups (EDG) such as a methoxyl group on the benzaldehyde ring led to a prodrug with a t1/2 = 0.6 h at pH 5.5, whereas an electron-withdrawing group (EWG) such as nitro group at the same position resulted in t1/2 = 6900 h at pH 5.5. In addition, the half-life at pH 7.4 was found to be about 10-fold greater than that at pH 5.5, independent of the substitution group. This phenomenon is desirable for the rational design of linkers with somewhat predictable pH-dependency and release efficiency. Targeted delivery and release of the payload in 12 were verified through in-vitro studies using folate receptor-positive KB cells, which showed IC50 of 60 μM. In contrast, the IC50 of the same conjugate in folate receptor knockdown KB cells was 655 μM. Further, a compound with an uncleavable poly(ethyleneglycol) (PEG) linker showed an IC50 of greater than 250 μM in folate receptor-positive KB cells. The uptake of the conjugate was examined with fluorescence microscopy, which showed co-localization of the conjugate and Lysotracker blue (a lysosome fluorescent dye) in the KB cells. The results suggest that the pH-sensitive NEBI linker is needed for the conjugate to take effect through folate-receptor mediated endocytosis. However, the high IC50 values for the prodrugs suggest there is room for improvement.
Figure 7.

Targeted delivery of indenoisoquinoline to cancer via the folate receptor.
2.5. Maleic acid-derived linkers
Maleic acid derivatives (including cis-aconityl group) have been used as linkers based on the intramolecular cyclization of maleoyl amide at pH less than the pKa of the free carboxylic acid group (Figure 8). This linker chemistry was first reported in 1981 with a highly desirable pH-sensitive release profile, allowing complete release within 3 hours at pH 4.0 while remaining stable at pH 7.0 without any detectable release for up to 96 h. It should be noted that earlier research often uses cis-aconitic anhydride, which could lead to regioisomers of the final conjugate. In addition, conjugation using cis-aconitric acid often leads to decarboxylation and/or trans-isomerization.54 This issue has been addressed by Fletcher et al.55 by using protected 2-(5-aminopentyl)-maleoyl acid instead of cis-aconitic anhydride to form the linker between a sugar moiety and a lipid dioleoylphosphatidylethanolamine (DOPE). This was used to develop a pH-sensitive phospholipid for potential use in drug and gene delivery (Figure 9). The linker precursor 21 can be synthesized from Boc-ε-aminocaproic acid via a four-step procedure and can be introduced to the conjugate via a coupling reaction. Deprotection by tetra-n-butylammonium hydroxide (TBAH) leads to in situ cyclization product 25. Further hydrolysis allows for the reopening of the imide to form the acid-labile linker conjugate. However, the formation of α- and β-substituted regioisomers (26 and 27) in an equal ratio seems inevitable.
Figure 8.

Maleic acid-derived linkers.
Figure 9.

DOPE as a pH-sensitive gene-delivery vector.
The release mechanism of the maleic amide linker remains speculative. Zhang et al. summarized this aspect based on experimental findings and literature reports (Figure 10).56 At pH below the pKa of the acid, 29 is the dominant specie, leading to at least three possible pathways for subsequent reactions. Pathway A was considered to be the main mechanism by Zhang and co-authors. It was proposed to involve intramolecular proton transfer from the carboxylic acid to the amide group followed by ring closure, leading to the tetrahedral intermediate 30. The subsequent collapse of the tetrahedral intermediate leads to the release of the drug and maleic anhydride. In determining the rate-limiting step of this pathway, Kirby et al. conducted detailed studies in the 1970s.57 It was found that the rate of hydrolysis was independent of the pH from pH 0 to 2. At pH higher than 2, reaction rate decreases with increasing pH. At pH above the pKa, there was no detectable hydrolysis of the amide. The intramolecular proton transfer that leads to the formation of the tetrahedral intermediate 30 was found to be the key step that gave this unique hydrolysis profile of the maleamic acid derivatives (including phthalamic amide). The pH dependence of the reaction indicates there is a component of specific-acid catalysis.57 However, because there is a second proton-transfer step in Pathway A, general-acid catalysis also plays a role. In accordance with the findings by Kirby et al.,57 Zhang et al.56 also found that double substitution on maleamic acid led to a 700-fold increase in the amide hydrolysis rate. It was presumably because the two alkyl groups impose entropic advantages for cyclization. The rate-limiting step for the hydrolysis was confirmed to be the breakdown of 30.57 However, in the case of the maleamic acid substitution being an isopropyl group, the proton transfer process was found to become the rate-determining step, leading to strong general-acid catalysis.58 There have not been thorough studies as to whether the reaction is under kinetic or thermodynamic control. In Pathway B, attack by the amide oxygen leads to the isomaleimide 31, which can undergo hydrolysis to 30. Subsequent collapse of the tetrahedral intermediate to release the drug can follow a similar pathway as that of Pathway A. Pathway C does not directly lead to drug release. However, it is possible that all reactions are fully reversible under the conditions used and on a practical timescale. In one example,56 a methyl substituted maleamic acid derivative was used as the linker to tether glutathione (GSH) to doxorubicin (Dox) (Figure 11). Due to the polar nature of the GSH moiety, the conjugate was not cell-permeable and thus was inactive. The linker is able to respond to minor changes in the pH of the tumor microenvironment and release Dox on site. The cleavage ratio between pH 6.0 and 7.0 was found to be about 7 (70% Dox release at pH 6.0 vs. 10% release at pH 7.0 at 5 h). This selectivity is higher than the previously discussed spiro diorthoester linker with the pH 6/7 cleavage ratio being around 4 at 5 h. This advantage reaffirms why the old maleamic acid linker strategy is still being actively studied in the field.
Figure 10.

Possible release mechanisms of the maleamic acid linker.
Figure 11.

Prodrug with the di-substitute maleamic acid linker.
Due to the free amino group (pKa = 8.2) in Dox, it remains in the protonated form in the slightly acidic microenvironment of the tumor tissue, which might impede the cellular uptake of Dox and reduce its effectiveness. For example, in ES-2 ovarian cancer cells, the IC50 of free Dox is higher (1.6 ± 0.1 μM) at pH 6.7 (extracellular pH) and lower (0.7 ± 0.1 μM) at pH 7.4. The pH-sensitive nature of the maleamic acid linker mitigates this pH-dependency for Dox. For example, in ES-2 ovarian cancer cells, conjugate 33 was shown to have an IC50 of 2.1 ± 0.3 μM at pH 6.7 and 3.2 ± 0.4 μM at pH 7.4. Although 33 is about 2-fold less potent than free Dox, this prodrug mitigates pH dependency of Dox and may benefit Dox chemotherapy by increasing Dox concentration in the tumor tissue and circumvent the issue of the undesirable pH-profile of Dox. However, in employing this strategy, the issue of disulfide stability in vivo (see disulfide linker part for detail) should be emphasized and further studied.
To summarize, pH-sensitive linkers respond to pH changes in the microenvironment and undergo acid-catalyzed cleavage. To target endosome/lysosome with a relatively low pH, acylhydrazone linkers can achieve fast and selective release. To target solid tumor with only a slight drop in pH (to 6.0–7.0), it might be desirable to use a spiro-di-orthoester linker and/or a maleamic acid linker, which offers the advantages of being sensitive to subtle pH changes. Even with all the progress, new chemistry for pH-sensitive linkers with tunable release rate is still highly desirable for the development of prodrugs including ADC and gene-delivery.
3. Protease-sensitive linkers
One way to achieve targeted delivery is to take advantage of the differential expression levels of enzymes at the desired sited of action. Along this line, protease is often considered as a useful trigger. Thus, a linker of an appropriately designed peptide sequence should offer the chance of selective cleavage at the site of action (Figure 12A). There have been examples of using proteases and others for this purpose.59 Examples include cathepsin B overexpression in invasive and metastatic cancer phenotypes60 and aberrant levels of kallikrein peptidase family (KLK1~15) in certain tumor types. Among them, the well-characterized KLK3, also known as the prostate-specific antigen (PSA), has been used to for designing prodrugs to treat prostate cancer with the first example dating back to 1998.61 Table 3 summarizes some examples.
Figure 12.

Two types of peptide linkers.
Table 3.
Prodrugs with a peptide linker.
| Entry | Name | Target tissue | Protease trigger | Peptide linker sequence | Conjugate moiety | Payload drug | Ref |
|---|---|---|---|---|---|---|---|
| 1 | NTD | Cancer | Cathepsin B | GFLG | Tat cell penetration peptide | Doxorubicin (multiple) | 62 |
| 2 | PEG-vcTNF-α | Cancer | Cathepsin B | VCit | PEG | TNF-α | 63 |
| 3 | BMS936561 | Cancer | Cathepsin B | VCit | CD70 mAb | MED-A (duocarmycin derivative) | 64 |
| 4 | Dendrimer-GEM | Breast cancer | Cathepsin B | GFLG | PEG dendrimer | Gemcitabine | 65 |
| 5 | DEVD-Cysteamide-EMCS-doxorubicin | Cancer | Caspase-3 | RDEVD | none | Doxorubicin (modified) | 66 |
| 6 | uPA-cPPP | Cancer | Urokinase plasminogen activator (uPA) | GSGRSAG | Cyclopeptide template c-KAAPGAKAPG | Photosensitizer Pheophorbide A | 67 |
| 7 | GKAFRR-L12ADT | Prostate cancer | KLK2 | GKAFRR | none | Thapsigargin derivative L12ADT | 68 |
| 8 | Z-GP-EPI | Epithelia tumor | Fibroblast activation protein-α (FAPα) | Cbz-GP | none | Epirubicin | 69 |
| 9 | rTLM-PEG | Cancer | MMP-2 | PLGLAG | PEG | Cell-penetrating peptide-trichosanthin | 69 |
| 10 | BSD352 | Prostate cancer | KLK3 (PSA) | HSSKLQ | Tat cell penetration peptide | BH3 domain, anti-VEGF peptide (SP5.2), anti-bFGF peptide (DG2) | 70 |
In some cases, directly tethering a peptide linker serves the purpose (Figure 12A). In other cases, directly linking the drug to a peptide may lead to interference with the proteolytic cleavage due to steric hindrance.71 Therefore, especially in ADCs, a small self-immolation spacer such as p-aminobenzyloxycarbonyl (PABC) is often used to form a hybridized peptide linker (Figure 12B). One example of using a cleavable peptide linker to tether a cytotoxic payload with a monoclonal antibody (mAb) is the FDA-approved ADC brentuximab vedotin (Adcetris).72 It uses a valine-citrulline (Val-Cit) moiety, a cathepsin B substrate, as the peptide linker along with the PABC spacer. Upon antigen-targeted delivery and internalization into cancer cells, cathepsin hydrolyzes the amide bond between Cit and the PABC spacer, leading to the subsequent 1,6-elimination to release the cytotoxic monomethyl auristatin E (MMAE). To study the stability of the linker as well as the entire conjugate, 2, 4, or 8 equivalents of the MMAE payloads were attached to one monoclonal antibody cAC10 through the Val-Cit linker.73 It was found that a higher loading degree did not alter the antigen-binding profile or the linker stability but decreased the stability of the conjugated mAb. The 4-payloads species showed a drug release half-life comparable to that of the parental cAC10 mAb (t1/2 about 14 days in mice). This was considered desirable for extending the ADC exposure time to the tumor and also for reducing binding competition between the ADC and the unmodified mAb.73,74 It was reported that the in-vivo half-life of this linker was around 144 h (6 days) in mice and 230 h (9.6 days) in cynomolgus monkey, which was significantly more stable than the acylhydrazone linker. Such results suggest that this linker would be highly stable in human. In 2011, phase II trials of this ADC produced a response rate (the percentage of patients whose cancer shrunk or disappeared after treatment) of 75% in patients with Hodgkin lymphoma (n = 102) and 87% in patients with anaplastic large cell lymphoma (n = 30).72 Subsequently, this drug was granted accelerated approval for the treatment of relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma.75,76
In addition to PABC, several other self-immolative spacers (Figure 13) have also been employed.77–79 Before designing prodrugs with such linkers, several factors should be considered, including proteolytic cleavage kinetics of the amide bond between the peptide and self-immolative spacer, cyclization or elimination release rate of the spacer, and the chemical stability the ester/amide bond between the spacer and the drug. For example, by using a phenylacetamide moiety as the substrate of penicillin G acylase (PGA) and 7-hydroxyl coumarin as a fluorescent reporter, a study tested various self-immolative linkers to compare the enzymatic release kinetics (Figure 13).80 Upon treating with PGA, the combined PABC-cyclization spacer 38 was found to give the fastest cleavage rate, leading to complete release within 10 min. On the other hand, the release rate for the compound (37) with a single PABC spacer was slower than that of 38 with a half-life of 6 min (complete release expected to be at least 36 min). One explanation is that the extended linker 38 is a better substrate for the protease. Further the elimination of CO2 from the carbamate linker offers an additional thermodynamic driving force for the linker cleavage. Other more sophisticated designs (39 and 40) with a dual-cyclization spacer gave slower release kinetics than 37 with about one-fourth of the drug released within 30 min. The slower release of the payload can be attributed to the cyclization reactions involving the thiol group attacking the carbamate. The single cyclization linker in 41 failed to release the fluorophore, indicating the inability for PGA to hydrolyze the N-methylated phenacetamide bond.
Figure 13.

Comparison of self-immolation spacers.
Substrate structure is an important determinant of the release kinetics in ADC design. One can tune the release rate by selecting from different substrates for the same protease. For example, a study using PEG-Onc112 (an anti-bacterial peptide) conjugates examined 20 different trypsin-like protease substrates as the peptide linker.81 It was found that the LVRPLVPRLVPR linker was the most susceptible toward serum degradation with a half-life of 1.5 h whereas the ADRG linker was the most stable with a half-life of about 42 h.
Dubowchik and co-workers conducted a comprehensive study on the enzymatic release profiles of cathepsin B substrates as the peptide linker.76 They compared ten different DOX conjugates of dipeptide substrates with/without the PABC spacer (N-Cbz-dipeptide-(PABC)-Doxorubicin). In the in-vitro enzymatic release assay, both Phe-Lys and Val-Lys with a PABC linker showed fast release with t1/2 of around 8 min. However, the same dipeptide without the PABC spacer showed no hydrolysis over 6 h. A valine-citrulline (Val-Cit) linker showed t1/2 = 240 min with a PABC spacer, but no hydrolysis was found without the PABC spacer. An Ala-Lys-PABC linker gave a t1/2 of 60 min, while other dipeptides such as Phe-Cit, Leu-Cit, Ile-Cit, Trp-Cit, Phe-Arg(NO2), and Phe-Arg(Ts) generally gave t1/2 > 500 min even with a PABC spacer. It was further demonstrated that the Phe-Lys-PABC and Val-Cit-PABC linkers were cleaved in rat liver lysosomes with t1/2 of 29 min but were stable in human plasma.
It should be noted that in many cases, metabolic kinetics of a peptide linker could vary among difference species due to changes in the expression and distribution of the various isozymes. This factor should be taken into consideration in the development of prodrugs or ADCs using a peptide-based linker. For example, the most widely used Val-Cit linker was found to be stable in human plasma but labile in mouse plasma. It turned out that mouse blood contains an extracellular carboxylesterase 1c (Ces1c) that can cleave this linker.82 This phenomenon often poses challenges in the design and preclinical evaluation of ADCs that rely on enzyme-mediated activation, and further states the importance of examining the issue of human relevance early on.
Another key issue is substrate specificity for the target isoenzyme and interference by isozymes of similar substrate specificity. A study showed that deletion or silencing the CTSB gene, which encoded cathepsin B, did not decrease the potency of ADC with the Val-Cit linker between mAb and the cytotoxic MMAE payload. Subsequently, cathepsins K/L/S were also found to be able to cleave the Val-Cit linker.83 Such results suggest that the preferential expression of cathepsin B or other specific proteases in cancer may not necessarily increase the therapeutic index of ADCs with a peptide linker. Therefore, one should be very careful in extrapolating from results from biochemical and cell culture results to animal models.
4. β-D-Glucuronidase-sensitive linkers
β-D-Glucuronidase84 is a non-circulating enzyme overexpressed in some tumor types.85,86 Therefore, it is also an enzyme that one can take advantage of in designing cancer-targeting strategies. For the design of prodrugs, parent drugs are usually linked to β-glucuronide through a self-immolative linker, such as a substituted para-hydroxylbenzyl carbamate. The introduction of the glucuronic acid moiety makes the parent drug more specific since the glucuronide-linked prodrug only gets hydrolyzed in the lysosome by the available β-D-glucuronidase. It should be noted that the cleavage of the β-glucuronide glycosidic bond by β-glucuronidase automatically triggers a 1,6-elimination of the substituted para-hydroxylbenzyl spacer thereby releasing the free cytotoxic drug.
In 2006, Jeffrey et al. first reported the use of glucuronic acid as a linker in ADCs87 to take advantage of the presence of lysosomal β-glucuronidase for drug activation. They reported mAb conjugates of 43a and 43b (Figure 14). The IC50 of the conjugate between anti-CD30 mAb cAC10 and MMAE (43a) was determined to be 0.06 nM in CD30+ line Karpas 299. In comparison, the nonbinding CD70 mAb-MMAE conjugate did not show any cytotoxicity at up to 30 nM. The drug-linker conjugate is inactive in the systemic circulation as the linker cleavage only happens in the presence of β-glucuronidase, which resides in the lysosome.84 Specifically, to test the stability of the linker itself in rat plasma, the reactive maleimide of 42b was reacted with excess DTT to afford dihydro-42b, which was then incubated in rat plasma for 7 days. It was found that 89% of the drug-linker conjugate dihydro-42b was still intact by LC-MS analysis. This puts the half-life of the linker at about 81 days as compared to 6.25 days for the conjugate between valine-citrulline (Val-Cit) and monomethyl auristatin F (MMAF). Then, the efficacy of 43a was further evaluated in nude mice with subcutaneous Karpas 299 ALCL tumors. Mice treated with a single dose of 0.5, 1.0, or 3 mg/kg of 43a on day 14 post tumor implantation all showed much-decreased tumor size (from ~100 mm3 to 0 mm3) after 110 days. In comparison, the tumor of non-treated mice developed very fast, reaching the size of 100 mm3 to ~1000 mm3 in one month after tumor implantation. The maximum-tolerated dose of 43a was determined to be approximately 100 mg/kg, giving a therapeutic index of more than 200. Incidentally, the hydrophilicity of the β-glucuronide moiety also helps to enhance the solubility of the prodrug and reduces aggregation when used as an ADC linker. Jeffrey’s group also demonstrated that β-glucuronide-linked conjugates showed less than 5% aggregation as compared to 80% aggregation for the comparable peptide-linked conjugates.88 Hence, the use of this water-soluble β-glucuronide linker represents an excellent strategy for targeted delivery of drugs to tumor cells.89 The excellent stability and desirable release profile of the glucuronide moiety significantly adds to the value of its use as a linker for prodrugs and ADCs. Over the years, β-glucuronide has been widely applied in ADC designs along with cytotoxic agents such as duocarmycin, auristatins, maytansines, γ-calicheamicin, cyclopamine, and irciniastatin A.62,83,89–93
Figure 14.

Mechanism of cleavage of beta-glucuronide linkers.
5. Thiol-sensitive linkers
In biological systems, free thiol species such as glutathione (GSH) generally serve as buffers to maintain cellular redox homeostasis.94–95 It should be noted that the intracellular concentration of GSH is in the range of 1–10 mM. However, in plasma and other extracellular fluids, the concentration is at micromolar levels. Tumor cells were found to contain a higher level of GSH than normal cells.96 Thiol-sensitive linkers, commonly disulfide bridges, have several advantages such as good biocompatibility, ease of synthesis, and a high release ratio. Taking advantage of all these features, disulfide linkers have been broadly explored in prodrug development. For targeted delivery, a disulfide linkage is generally used as a connection between the payload and the targeting moiety.
For example, Perez and coworkers reported a conjugate of folate and doxorubicin by a disulfide bond.97 Folate was used in targeting the high-affinity folate receptor, which is commonly over-expressed in cancer cells. A disulfide linker was installed with two carboxylic groups at its two ends, which was used to tether Dox and folate by forming amide bonds. Upon conjugation, the fluorescence of Dox was quenched due to its proximity to the folate moiety. In the presence of intracellular GSH or other reducing agents, the disulfide bridge can be cleaved through thiol exchange reactions, leading to the release of Dox and folate. Although the released Dox moiety (Dox-SH) is still a modified version, its cytotoxicity and fluorescence are kept at the same level as Dox itself. For example, when 5 mM of the prodrug was treated with 5 mM GSH in PBS, a five-fold increase of the fluorescence from Dox was observed within 3 h. After 3 h, almost 100% recovery of the Dox-SH was observed with HPLC. To compare the cytotoxicity of Dox, Dox-SH, Dox-S-S-Folate, and Dox-C-C-Folate (a non-cleavable conjugate), A549 (folate receptor positive) and MCF-7 (folate receptor negative) cell lines were used for the cell culture studies. A similar time-dependent decrease in viability for A549 cells was observed from Dox-, Dox-SH- and Dox-S-S-Folate-treated groups. After 48 h with 1.2 μM of each individual drug, almost 90% of the cells were dead from these groups. However, the non-cleavable conjugate, Dox-C-C-Folate, only caused 5% cell death at the same time point. Towards MCF-7 cells, only Dox and Dox-SH maintained the same potency, while Dox-S-S-Folate and Dox-C-C-Folate failed to induce cell death, presumably due to the absence of folate receptor-mediated internalization. Dose-dependent experiments were also conducted on the A549 cell line, and the IC50 values of Dox and Dox-S-S-Folate were determined to be 2.03 μM and 1.27 μM, respectively. These results support the feasibility of the design strategy. Since the improvement in potency for Dox-S-S-folate over Dox alone was small, it is hard to analyze the true effect of folate conjugation in enhancing potency.
Mitochondria are known to be the energy producer and play a critical regulatory role in apoptosis in mammalian cells. Many human diseases are related to mitochondrial dysfunctions. Therefore, targeting and delivery of drugs to mitochondria have attracted widespread interest. Kelly and coworkers developed a mitochondrial delivery system by employing conjugates of a mitochondria-penetrating peptide (MPP) and a payload using a disulfide bond.98 In the intracellular environment, the concentration of GSH is much higher than that in plasma, which may cause cleavage of the disulfide bond before entering the mitochondria. Therefore, chemical modifications were applied to increase the stability of the disulfide bond. As a model test, a fluorophore (TAMRA)-quencher (BHQ-2) pair was linked through a disulfide bond. This pair was further tethered to a mitochondria penetrating peptide (Figure 15). The alkyl group attached to the disulfide was modified with substitutions that impose steric hindrance to provide stabilization effects. The breakage of the disulfide bond can lead to an increase in fluorescent intensity from the fluorophore due to detachment from the quencher. Such a reporter system was used to study the cleavage kinetics of different disulfide linkers in PBS and in K562 cell culture. In the presence of 0.5 mM dithiothreitol in PBS, the unsubstituted analog 46 was fully cleaved within 10 min, while the mono- and disubstituted analogs (47 and 48) required more than 45 min for cleavage. In K562 cell culture, 10% cleavage from the unsubstituted analog was immediately observed after the treatment and the maximum release ratio (around 80%) was achieved in 20 h. The mono- and di-substituted linkers shown a much lower initial cleavage (less than 10%), and the maximal release (around 70%) took about 48 h. For the studies in living cells, during the incubation with different substituted disulfide linkers, fluorescent images were taken at various time points (0.5, 8 and 24 h). Compared to non-and di-substituted linkers, the conjugate with the mono-substituted linker in 47 exhibited low pre-incubation cleavage and efficient release within 24 h, and therefore was chosen for further development of the delivery system. Luminespib, an HSP90 inhibitor, was selected as the cargo, which was connected to the mitochondria-penetrating peptide through a mono-substituted disulfide linker. By design, the disulfide bond reduction is to be followed by the self-immolation of the thiol-carbonate, which can lead to the release of Luminespib. The localization of the delivery vehicle in mitochondria was affirmed by a fluorescently labeled analog. However, no quantitative data to assess the release efficiency in cells was reported. Compared to a low level of toxicity from the non-cleavable analog, the cleavable compound showed a time-dependent increase in toxicity against K562 cells over 48 h. Through the assessment of the mode of cell death, mitochondrial effects and mitochondrial depolarization, its toxicity was deduced to be due to mitochondrial swelling-mediated apoptosis. This thiol-sensitive linker has also been successfully used in the targeted delivery of per-acetylated N-azidoacetylmannosamine (Ac4ManNAz) to tumor cells. Ac4ManNAz incubation can lead to azidomannose incorporation into cell surface sialic acid. The azido group can serve as a handle for further labeling for imaging and immune therapy applications.99 In K562 cells, it took twice as much time for the mono- and disubstituted analogs to complete the cleavage as compared to the unsubstituted one. By combining this monosubstituted disulfide linker system and a mitochondria-penetrating peptide, mitochondrial delivery of an HSP90 inhibitor was achieved.
Figure 15.

Modifications on the disulfide linker to improve stability.
In the field of ADCs, the disulfide bond is also used to link a mAb with various cytotoxic agents. For example, Sliwkowski and coworkers reported a series of Trastuzumab-Maytansinoid (DM) conjugates, by using a disulfide or a thioether bond as the linker.100 As shown in Figure 16, disulfide linkers were constructed with different numbers of methyl groups (49-52) and a thioether linker 53 was also developed. All conjugates showed improved potency on HER2 -amplified breast cancer cell lines BT-474 and SK-BR-3 as compared to free DM. The IC50 of compound 50 (Figure 16) decreased to 0.24 nM and 2.71 nM compared to 1.00 nM and 4.52 nM of DM in SK-BR-3 and BT-474 cell lines, respectively. In normal or HER2-negative cell lines such as MCF-7 and MDA-MB-468, the cytotoxicity of the conjugate decreased. For example, in MCF-7 cell lines, free DM showed an IC50 of 2.92 nM, while the IC50 of the disulfide-linked prodrugs 50 and 53 was over 100 nM. Pharmacokinetic analyses of the various conjugates were conducted in nude mice. The least hindered disulfide conjugate (compound 49) was more easily degraded in plasma compared to other conjugates, and the ADC level was undetectable after three days. With increasing steric hindrance on the linker, the ADC clearance time in serum increased accordingly. On day three, 60% of conjugate 50 and 20% of conjugate 51 were degraded. The most hindered disulfide-containing analog 52 maintained 70% of the total conjugate concentration after seven days, which was similar to the result of the non-reducible thioether linker. An MMTV-HER2 Fo5 mammary tumor transplant (trastuzumab-resistant) model was used for in-vivo efficacy studies. By measuring the tumor volume after treatment with ADCs of different linkers, a positive correlation between the linker stability and the anti-tumor activity was obtained. The ADCS with the most stable disulfide linker and the non-reducible thioether linker exhibited potent efficacy. The safety profiles indicated that conjugate 50 with a less stable disulfide linker led to a significant decrease in mouse body weight within 5 days of treatment, indicating significant gross toxicity. In contrast, the conjugate with a non-reducible linker only induced a slight decrease in body weight and was thus chosen for further experiments. Thus, the stability of the linker most likely affects the systemic toxicity of the trastuzumab conjugate. Judged by the favorable efficacy, pharmacokinetic, and safety profile, ADC 53 with a non-cleavable thioether linker was chosen to be further evaluated in the clinical study.100 It is interesting to note that in this case, it is a non-cleavable linker that afforded the desired properties. Such results further indicate the need to examine each specific case and application very carefully in optimizing the conjugates.
Figure 16.

Application of the disulfide linker in ADCs.
Overall, thiol-sensitive linkers have been extensively studied, and have played important roles in the development of ADCs. However, in using thiol-sensitive linkers, it is important to balance the issue of stability during circulation and facile release inside cancer cells. This is a challenging task and needs to be carefully analyzed for each application.
6. ROS sensitive linkers
Reactive oxygen species (ROS) include free radicals, singlet oxygen (1O2), hydrogen peroxide (H2O2), and ions such as superoxide (O2−), hypochlorite (OCl−), etc. ROS has been shown to play a major role in various cellular signaling processes and in turn affects diverse cellular events such as proliferation, differentiation, regulation of immune responses, etc. The oxidative environment generated by ROS is also capable of causing post-translational modifications of various proteins by oxidizing the thiol group in cysteine to reactive forms such as sulfenic acid (-SOH). Further reaction of the -SOH group with nearby a cysteine leads to disulfide bond formation and/or result in changes in the structure and function of various proteins.101 Many pathogenic conditions, including cancer, are characterized by high concentrations of ROS.102 Drug delivery systems can take advantage of the high ROS concentration in cancer cells to achieve targeted drug delivery.103 Linkers containing boronate/boronic ester, sulfide, selenide/telluride thioether, and ferrocene are commonly used to exploit the presence of ROS for targeted drug delivery. Several very recent studies in this area are summarized below.
6.1. Aryl boronic acid and boronate-based linkers
Boronic acid and boronate groups have been widely used in designing ROS-sensitive prodrugs to deliver anticancer drugs such as SAHA, SN-38, nitrogen mustard, NO donors, etc. Boronic acids have been used in FDA-approved human medicine and the end product of boronic acid oxidation, boric acid, is an ingredient in vegetables and plants. The selectivity of this group towards ROS makes it a great choice for preparing ROS-sensitive prodrugs.104
Recently, Xue and co-workers designed boronic acid- and boronate-based prodrugs of 5-fluorouracil (5-FU).105 5-FU is generally used to treat solid tumors of the breast, stomach, colon, and pancreas. Though 5-FU acts as a potent anticancer agent, it suffers from major side effects such as myelosuppression, gastrointestinal toxicity, and central neurotoxicity.106,107 The more pressing problem with 5-FU is its metabolic instability and its conversion to cardio- and neurotoxic α-fluoro-β-alanine (FBAL) through catabolism initiated by dihydropyrimidine dehydrogenase (DPD). Only 1 to 3% of the original dose of 5-FU mediates the cytotoxic effects on tumor cells and normal tissues through anabolic actions.107 To overcome such issues, a para-boronate benzyl group was introduced to impede metabolic degradation by DPD at the N1 position of 5-FU. The arylboronate group was used as the ROS-sensitive trigger for drug activation upon exposure to H2O2 via a phenol intermediate. Instability of the phenolic intermediate would result in spontaneous disintegration to give 5-FU and a reactive quinone methide byproduct that is quickly hydrolyzed in aqueous media to yield nontoxic 4-(hydroxymethyl)phenol. Specifically, compounds 54a and 54b (100 μM) (Figure 17) were activated efficiently in the presence of 5 equivalents of H2O2 as monitored using RP-UPLC-MS. Prodrug 54a was completely converted to 5-FU within 5 minutes, while it took 250 minutes for 54b to complete the conversion. The ability for such prodrugs to inhibit growth against 60 cancer cell lines (the Developmental Therapeutics Program of NCI) was studied. Incubation with 54a (50 μM) for 48 h led to growth inhibition of over 50% for most of the cancer cell lines tested. The boronate prodrug 54a showed better inhibition percentages (92% in lung NCI-H522, 96% in melanoma MDA-MB-435, 95% in ovarian OVCAR-3 and 81 and 87% respectively in breast MCF-7 and MDA-MB-468 cancer cell lines) in comparison to the boronic acid prodrug 54b (44% in lung NCI-H522, 65% in melanoma MDA-MB-435, 30% in ovarian OVCAR-3 and 33 and 22% respectively in breast MCF-7 and MDA-MB-468 cell lines). The difference in activity was attributed to the improved permeability of the boronate prodrug. A comparative study of the prodrugs was performed on breast cancer cell line MCF-7 and normal MEC1 cells. Compound 54a (70% inhibition at 50 μM) showed less potent anti-proliferative activity in comparison to 5-FU (70% inhibition at 10 μM); compound 54b (∼65% inhibition at 100 μM) was found to have a weaker effect in comparison to 54a. Presumably, the difference was again due to the difference in permeability through the membrane. The toxicities of both prodrugs 54a and 54b to MEC1 normal cells (cell viability of > 95%) at up to 100 μM were found to be reduced in comparison to 5-FU (cell viability of ∼50% at 1μM conc.; ∼10% at 100 μM conc.). Though the design principle seems to suggest selectivity of the boronic acid-based prodrugs, the concentrations used in all these in-vitro experiments were on the high side. Prodrug 54a was tested in wild-type C57BL/6 male mice for safety. 5-FU killed all the mice on the twelfth day at a dose of 100 mg/kg, while prodrug 54a at the same dose did not cause death or any abnormalities in the mice. Such results showed that 54a had an improved safety profile when compared to 5-FU and also showed reduced toxicity in the normal tissues.
Figure 17.

ROS activated prodrugs.
In summary, aryl boronic acids and esters are known for their relative stability and selective reactivity towards H2O2. Boronic acid and ester prodrugs as well as the boric acid byproduct formed after the release of the payload are known to be tolerated in human.108 All these factors, along with the relatively fast in-vitro kinetics in the presence of physiologically relevant H2O2 levels, make boronic acid/esters good structural moieties for use as a ROS-sensitive trigger.109 Though the examples listed are technically not linkers, the concept of prodrug activation is similar.
6.2. Thioketal-based linkers
Thioketals are another class of ROS-sensitive linkers, which can be cleaved by various ROS species to give the corresponding thiol 57 and acetone (Figure 18).110 In 2012, Xia and coworkers developed a ROS-responsive thioketal-based cationic polymeric system for targeted gene transfection in prostate cancer cells.111 The polymer was synthesized by Michael addition-based polymerization between thioketal 58 and TFA-protected N, N’-bis(2-aminoethyl)-1,3-propanediamin 59, followed by deprotection using TFA to yield poly(amino thioketal) (PATK, 60) (Figure 18). The degradation kinetics of these polymers were studied in the presence of ROS using 1H NMR. Specifically, PATK was dissolved in D2O containing H2O2 and a trace amount of CuCl2 to induce radical production. The degradation rate for the polymer was found to be proportional to the H2O2 concentration. The half-life of PATK was found to be 20 h in the presence of 100 mM H2O2 and 11 h in the presence of 200 mM H2O2. DNA was efficiently complexed by PATK, which was confirmed by the size and the surface charge on the polyplex. A comparative study of gene transfection efficiency by PATK in ROS-rich prostate cancer LNCaP cells and non-cancerous CHO cells was performed. eGFP expression of the different types of cells incubated with PATK complexing eGFP encoding plasmid DNA was found to be higher for LNCAP (32% transfection rate) in comparison to CHO cells (25% transfection rate). This polymeric system was further made cancer cell-selective by conjugating it with a GRP78 binding peptide (GRP78 protein is overexpressed in various tumor cells). DNA/GRP78P-PATK showed higher transfection (normalized change of about 2.2 folds) and cellular uptake (∼ 3.5 folds) in comparison to DNA/GRP78P free-PATK (transfection ∼1.0 fold and cellular uptake of ∼ 1.2 folds).
Figure 18.

Synthesis of ROS-responsive PATK.
Given the relatively slow degradation profile of the thioketal linker under physiologically relevant ROS concentrations in tumor, there is the question of whether there is a way to provide an extra boost to ROS generation at the site of action. Along this line, Wang and co-workers utilized such a thioketal linker to reduce off-site drug leakage in their nanoparticle design.112 In this design, a ROS-sensitive thioketal linker (TK) was used to link a polyphosphoester with anticancer drug doxorubicin to form PPE-TK-DOX. This drug-loaded polymer was assembled into nanoparticle formulations with or without embedded chlorin e6 (Ce6), namely PPE-TK-DOX NP and Ce6@PPE-TK-DOX NP, respectively. Here, Ce6 is a photosensitizer which could generate ROS upon red light irradiation. Therefore, 660 nm red light was designed as a trigger to release DOX by inducing ROS and hence cleaving the thioketal linker. Such a design was meant to reduce premature release of DOX in the blood circulation and to afford light-controlled drug release. In an in-vitro buffer-based study, 57% Dox was released from Ce6@PPE-TK-DOX NP upon exposure to red light, while less than 10% of the drug was released from the plain mixture of Ce6 and PPE-TK-DOX nanoparticles (Ce6+PPE-TK-DOX NP). Further, no DOX release was observed in the absence of light exposure. Then the fluorescent intensity of MDA-MB-231 cancer cells after drug exposure was analyzed using flow cytometry. It was found that treatment with Ce6@PPE-TK-DOX nanoparticles in the presence of red light gave much higher intracellular DOX fluorescence in comparison to that of the non-illuminated controls. The cells incubated with a mixture of Ce6+PPE-TK-DOX NP showed no significant DOX fluorescence signal. However, the total intracellular DOX content was found to be similar (∼0.2 μg/ 106 cells) by HPLC for both formulations with and without light. Although the reason was not completely clear, the authors attributed the lower fluorescence in the control groups to fluorescence quenching caused by the encapsulation of the DOX in the nanoparticles. Based on this assumption, it might indicate that the cellular uptake of the DOX nanoparticles in both formulations is similar, but light induced ROS generation was probably the reason for the enhanced intracellular release of the free DOX. Further, the particles were examined in mouse models with implanted MDA-MD-231 cells. It was found that in a 16-day experiment, twice a week treatment with Ce6@PPE-TK-DOX NP at a Dox-equivalent dosage of 5.0 mg/kg followed by red light exposure on the tumor site (660 nm laser at 0.1W/cm2 for 30 min) resulted in significantly reduced tumor size compared with the control groups including Ce6@PPE-TK-DOX NP without light exposure, plain mixture of Ce6 and PPE-TK-DOX NP with or without light exposure, Free DOX, and Free Ce6. Thus, Ce6@PPE-TK-DOX NP was shown to allow controlled release the drug at tumor site by using light exposure to initiate ROS generation. Further, thioketal-based drug delivery systems are widely reported in ROS triggered drug release114 and in hybridized PDT-chemotherapy studies.114 The stability of the thioketal bond under physiological conditions and its triggered cleavage only in the presence of ROS species could help to reduce off-target release of the cytotoxic drug. Thus, such properties make the thioketal linker a good choice for PDT-based chemotherapy. However, the slow release of this linker under conventional conditions might be a reason why it is not generally utilized in small molecule-based drug delivery systems.
6.3. Thioether-based linkers
Thioethers have been widely used in biomedical research as an ROS-sensitive group. In the presence of an oxidative species, the hydrophobic thioether moiety is converted to a more hydrophilic sulfoxide or sulfone. Once these polymeric systems are exposed to the ROS trigger, the payload is released due to destabilization resulting from this change of polymer properties.115 In 2015, Zhu and co-workers reported the design of a type of H2O2-responsive polymeric micelles. The system consists of two parts. On the outside part is a polymer HPG-2S-SN38 formed by tethering anticancer drug 7-ethyl-10-hydroxy-camptothecin (SN38) with hyperbranched polyglycerol (HPG) via an ROS-sensitive thioether linker.116 This ROS sensitive polymer was then self-assembled into nanomicelles with cinnamaldehyde (CA) being encapsulated in the core. CA is said to have antitumor effects by inducing ROS generation in the mitochondria of cancer cells.117 However, CA’s clinical application is hampered by its low water solubility, low bioavailability, low stability and lack of selectivity toward tumor cells.116,117 It was said that encapsulation of CA in nanoparticle would not only potentially address these drawbacks, but also further increase ROS generation through CA release and thus facilitate linker cleavage to release SN38. In the presence of 1 mM H2O2, in-vitro studies of triggered release of CA from CA-loaded HPG-2S-SN38 micelles showed 70% CA and 60% SN38 release in 12 h. In contrast, in the absence of H2O2, only less than 20% SN38 and less than 30% CA were released in the first 48 h of incubation. The release of conjugated SN38 was found to be slower than that of CA from the micelles. It was assumed that breaking the covalent linkage between HPG and SN38 was slower than the dissociation of CA from the destabilized micelles. Based on the H2O2 responsiveness of the nanomicelle system, the in-vitro cytotoxic effect was studied in MCF-7, HN-4, and HeLa cells. No cytotoxicity was observed with the unloaded pure HPG micelles in tumor cells at a concentration of 2 mg/mL after 48 h. The physical mixture of CA and SN38 showed more potent anticancer activity than free SN38, free CA, and HPG-2S-S38, highlighting the synergistic or additive effect of the two drugs. Furthermore, the CA loaded HPG-2S-SN38 nanomicelles showed more potent activity compared to the physical mixture. The authors reasoned that this additive or synergistic anticancer activity could be due to the higher intracellular uptake and endosomal escape of the nanomicelles. As a result, it was concluded that this nanomicelle system improved the solubility and stability of both drugs. The results were further interpreted as to indicate CA’s ability to generate ROS, which in turn facilitates drug release from the nanomicelles. Aside from aforementioned examples, thioether linkers have also found applications in various dual-response-based systems such as thermal and ROS responsive prodrugs118 that could increase the oxidative response of these prodrugs as well as NPs that are able to generate exogenous ROS at the tumor site and thus accelerate payload release.119
In summary, several pathological states have now been shown to be associated with a high ROS level. Drug delivery systems based on this particular stimulus are thus of importance to the field of drug delivery. For a ROS-sensitive drug delivery system to work efficiently, factors such as the type of linker used, its sensitivity to different types of ROS, the position of the linker in the delivery system and the toxicity of the byproducts play a significant role in affecting the viability of the drug delivery approach. ROS-sensitive linkers such as thioether utilize the solubility switch concept (from hydrophobic to hydrophilic) for release of payloads while other linkers such as boronates and thioketals are cleavage-based linkers. A major challenge in this field is to avoid premature leakage of the drug to normal cells containing low levels of ROS and thus to effectively deliver drugs to the desired site containing increased levels of ROS. Since different disease states may involve various elevated levels of ROS, the choice of linker used should be based on the application in hand.
7. Pre-targeting strategy-based linkers
As mentioned above, ADC is a widely used strategy for targeted delivery, especially in cancer therapy. Despite the enormous success of this strategy, there are still areas where improvements are needed. First, the limited number of cell surface receptors (~105 receptors per cell) and the low target-background ratio of mAb usually results in a low concentration of the cytotoxic drug at the desired site.120,121 Second, it is difficult to achieve an optimal balance between the needed stability of the linker during circulation and cleavability at the desired site. To overcome such limitations, bioorthogonal chemistry-based122–125 pre-targeting strategies have been used by using a click reaction to trigger release. Specifically, the nontoxic targeting moiety can be conjugated with one click-handle such as an azido group. This conjugate can be administered to achieve accumulation at the desired site. This constitutes the pre-targeting step. Then, a prodrug conjugated with a trigger such as trans-cyclooctene (TCO) is administered. One important aspect of such a design is the need for the bimolecular click reaction to set up the cleavage of the linker that tethers the drug to the targeting moiety. There has been some very significant development in this area in recent years.126–128
One strategy that has been used for pre-targeting delivery is the “click and release” reaction between an azido group and TCO. A cytotoxic drug can be caged in its inactive form, and activated by a reagent from a click-reaction pair such as azido-TCO, leading to drug release.128,129 A targeting ligand can be installed to direct the activator to the desired location for release of the cytotoxic drug (Figure 19).
Figure 19.

Mechanism of using azido and TCO as click-release tool.
Specifically, as shown in Figure 19, the reaction between the azido group of 61 and TCO would form an unstable 1, 2, 3-triazoline compound 62, leading to the release of molecular nitrogen and formation of aldimine 63. The latter can be hydrolyzed to afford aldehyde 67 and amine 64. 1,6-Elimination of 64 would leads to the release of payload 65. Coumarin prodrug 61a and TCO-OH were used to demonstrate the proof of concept. The second-order rate constant of the 1,3-dipolar cycloaddition between 61a and the equatorial and axial isomers of TCO-OH was determined to be 0.017 M−1S−1 ± 0.003 (equatorial isomer of TCO-OH) and 0.027 M−1S−1 ± 0.006 (axial isomers of TCO-OH) respectively in CH3CN/PBS (1:1, 37 °C). The subsequent 1,6-elimination of the PABC intermediate 65a was very fast; it is faster than what the NMR time scale allows. For 100 μM of Dox prodrug 61c, after 4 h incubation in serum/PBS (1:1) with 500 μM TCO-OH (equatorial isomer), the yield of drug release was determined to be 51%, and the cytotoxicity of 61c (IC50 = 0.96 μM) was found to be comparable to that of its parent drug Dox (0.71 μM). However, the azido prodrug was shown to have stability issues, leading to a recovery yield of only 68% after 24 h incubation in mouse serum/PBS (1:1). To ensure sufficient drug release, much faster kinetics of the bimolecular reaction was needed. The introduction of an electron-withdrawing group such as fluoro substitution is known to facilitate such 1,3-dipolar cycloaddition.130 Thus a tetrafluoro-substituted aryl azide was studied and was shown to have a second-order rate constant of 0.110± 0.036 M−1S−1 in CH3CN/PBS (1:1, 37 °C) with TCO-OH, compared to 0.017 ± 0.003 M−1S−1 for the counterpart without a fluoro substitution. Although the fluoro-substitution accelerated the cycloaddition reaction, it led to a slower overall payload release rate by stabilizing the intermediate aldimine. The chemical feasibility of this targeted delivery approach has been demonstrated. However, further application of this strategy is still hindered by the relative sluggish reaction kinetics, and the application of such chemistry in pre-targeting delivery in vivo has not yet been reported. In addition, electron-deficient aryl azides have an increased tendency to undergo decomposition and nitrene-based side reactions, which could pose a problem for future applications.131
To achieve the goal of pre-targeting-based delivery, fast reaction kinetics are needed for drug release. The reaction between TCO and tetrazine was found to possess fast kinetics, and the reaction rates for this reaction can be tuned by modification of the tetrazine part. In 2013, Robillard and co-workers designed a prodrug activation system based on this reaction (Figure 20).132,133 A 1, 4-dihydropyridazine intermediate 69 was formed after reaction between TCO-drug conjugate and tetrazine; further elimination led to the release of the payload. The release yield was about 79% from the TCO-Dox conjugate (25 μM) after incubation of 16 min at 37 °C with 10 equivalences of tetrazine 68b or 68c. Further investigation into the relationship between reaction kinetics and substitutions on the tetrazine moiety was reported by Chen and co-workers in 2016.134 It was found that substitutions by an EWG on tetrazine accelerated the cycloaddition step, but lowered the rate of the decaging reaction. In contrast, EDGs had the opposite effect. To balance these two effects, a series of unsymmetric tetrazines bearing both an EWG at the 3-position and an alkyl group at the 6-position were synthesized, leading to an optimized decaging efficiency of >90 % with 50 μM 68d within 4 min.
Figure 20.

Mechanism of the click-release reaction between tetrazine and TCO.
After demonstrating the promising release profiles by this click-based design in vitro, Robillard and co-workers further applied this strategy in ADC by conjugating the mAb CC49 against anti-tumor-associated glycoprotein-72 (TAG72) and Dox via TCO (Figure 21).133 Circulation of CC49-conjugated TCO 73 was similar to the native CC49, suggesting the compatible nature of the conjugation chemistry used. Dox release was then tested from conjugate 73 in mice bearing colon carcinoma xenografts. A 5 mg/kg dose of 73 achieved a tumor uptake of 30–40% of injected dose per gram (ID/g) 30 h post-injection. In this case, conjugate 73 was administered first to allow for accumulation in cancer cells for the pre-targeting purpose and a tetrazine was applied as an activator to trigger the drug release as shown in the previous mechanism. The extra conjugate 73 was removed 24 h after ADC administration by an albumin-based clearing agent comprising a liver-directing galactose moiety (designed for fast clearance) and tetrazine 68a (designed for click reaction with extra TCO in blood without causing any drug release) to prevent any off-target Dox release in blood. However, the fast clearance of activator 68b or 68c would lead to an incomplete on-tumor reaction between TCO and activator. Thus, activators 74 and 75 conjugated with 10 kDa dextran were designed to increase the clearance time. Complete tumor blockage was finally achieved without any acute toxicity after iv administration of 74 (10 × dosage, 0.335 mmol tetrazine/kg) and 75 (1 × dosage, 35 μmol tetrazine/kg) in mice.
Figure 21.

Targeting delivery by mAb-TCO-Dox conjugates and tetrazine activators.
The pre-targeting strategy utilizing a click reaction offers excellent complementation of the traditional linker chemistry applied in prodrug activation. The bioorthogonality of click reactions provides selectivity for linker activation/cleavage and thus is superior to the traditional linker chemistry in some specific situations of prodrug activation. However, the development of bioorthogonal reaction-based pre-targeting strategy is still in its infancy and has many limitations. One major challenge is the difficulty in drug administration as it is hard to control the pharmacokinetic (PK) profiles of two components and the stability issues of such click partners may present extra difficulty in this field. Further, the extra pre-targeting components in circulation would consume activators and it must be removed by clearing agents. Overall, this is a very innovative and promising approach with a few remaining issues to work out.
8. Enrichment-triggered drug release
The idea of targeted delivery constitutes essentially two steps: enrichment and release/activation of the drug moiety. If one can take advantage of the enrichment factor to trigger drug activation, it would allow for maximal efficiency in targeted drug delivery while minimizing pre-mature release and potential side effects. Along this line, Wang and co-workers designed an “enrichment-triggered drug release” strategy.135 The rationale for this design is to take advantage of the concentration-dependent nature of a bimolecular reaction to achieve enrichment-triggered release. By conjugating with the same targeting moiety, the concentrations of two partners of a bioorthogonal reaction can be enhanced greatly at the desired sites. This should lead to a corresponding reaction rate increase to drive the release of the drug. As proof of concept work, two systems using a triphenyl phosphonium (TPP) moiety as the mitochondria-targeting group were designed.
This strategy was first examined using a bimolecular carbon monoxide (CO) prodrug system by conjugating TPP with exo-BCN and cyclopentadienone, respectively (Figure 22A). Briefly, carbon monoxide is a known endogenous signaling molecule with strong cytoprotective and anti-inflammatory effects, among others. One key obstacle in developing CO-based therapeutics is the difficulty in its controlled delivery because inhalation of gaseous CO does not allow easy control of quantity and risk factors.136,137 Therefore, there has been much interest in developing CO-releasing molecules (CO-RMs)137–139 and recently CO prodrugs for controlled delivery via oral or parenteral administration.126,140 Aiming for developing controlled CO delivery methods, an enrichment-triggered method was used to target the mitochondrion, which is the site of action for CO. Briefly, an inverse-electron demand reaction between a substituted cyclopentadienone 77 and a strained alkyne 78 was used for the generation of a norbornadien-7-one intermediate, which is known to undergo a facile cheletropic extrusion reaction to release CO and a fluorescent byproduct, 79.140 Specifically, compounds 67b and 68b, each with a TPP moiety for mitochondrion-targeting, were prepared. The design was based on prior literature reports that TPP-conjugation enables enrichment in mitochondria by up to 1000-fold.141 Imaging studies of compounds 77b and 78b at 5 μM in RAW 264.7 cells showed intense fluorescence of 79 after incubation for 4 h, while the control group without TPP conjugation (77a and 78a) at the same concentration did not lead to any fluorescence increase. Such results are consistent with the original design in that a second-order rate constant of 0.25 M−1S−1 for the reaction is expected to lead to a t1/2 being over 100 h at 10 μM. However, even a modest 50-fold enrichment would reduce the t1/2 to around 2.2 h, which should enable CO release within a time frame that allows for anti-inflammatory effects.142, 143 The prodrug approach was examined in both cell culture and animal models for their effect on suppressing inflammation and offering cytoprotection. For example, when tested in an acetaminophen-induced liver injury model, the EC50 in suppressing ALT (a liver injury marker) level was less than 0.4 mg/kg, while similar effects with other CO prodrugs were only seen at about 15 mg/kg.128,143,144 The same strategy was used for the delivery of doxorubicin by using a click pair of tetrazine 80 and strained alkyne 81 for the click-based coupling (Figure 22B).145 The second-order reaction constant can be tuned from 0.0075 M−1s−1 to 2 M−1s−1 by modification of the alkyne and tetrazine components, allowing for easy control of the reaction in such a way that minimizes the click reaction before enrichment and enhances the bimolecular reaction rate to the point that enables doxorubicin release within a short period of time. Between the two regioisomeric products of the click reaction, only 83 but not 82 is expected to undergo the subsequent intramolecular lactonization to release doxorubicin and to form byproduct 84. In cell culture studies, the EC50 of the click pairs was 1.2 μM, very similar to that of doxorubicin itself (1.0 μM), while the control prodrug without TPP conjugation did not show cytotoxicity at a much higher concentration (> 100 μM). These two studies both demonstrate the general feasibility of the “enrichment triggered drug release” approach. The same idea can be applicable in ADC. However, some additional work in applying this method in enriching the components on cell surface is needed to demonstrate general applicability if cell surface biomarkers are the targets.
Figure 22.

Two examples of enrichment-triggered drug release.
9. CONCLUDING REMARKS
A wide range of drug conjugates/prodrugs with various triggering mechanisms including activation by endogenous stimuli have been reported. In these applications, chemical linkers have enabled the drugs to be “smarter” by further improving targeting selectivity through preferential activation at the desired site of action. However, each approach still has its own problems. For example, the mitochondria-targeted enrichment-triggered release example from our lab relies on conjugation with a triphenylphosphonium (TPP) moiety, which is large, hydrophobic, and may have toxicity problems depending on the application.141,146–148 In other cases, the needs for improvement may include stability and toxicity of the click partners, reaction kinetics, and side reactions with other components commonly encountered in a biological system. It is also worth noting that some very interesting work in developing new bioorthogonal reactions has been reported.149–152 Though these new reactions may not have been used in targeted drug delivery, they offer additional opportunities in this area of research. We hope that the publication of this review will provide researchers a good reference for choosing a suitable chemical linker and chemical strategies for their prodrug design, and more importantly will stimulate more research into overcoming obstacles that this field still faces in developing smart drugs for clinical applications.
ACKNOWLEDGMENTS
We thank the Center for Diagnostic and Therapeutics for providing a CDT fellowship to Abiodun Anifowose, the Molecular Basis of Disease Program for providing MBD fellowships to Zhixiang Pan and Manjusha R. Choudhury, and the Brains and Behavior program for fellowships to Bingchen Yu and Zhengnan Yuan. Work in the lab of Binghe Wang on drug delivery is partially supported by the National Institutes of Health (DK119202). We also thank the Georgia Research Alliance for its general financial support through an Eminent Scholar endowment for research in the lab of Binghe Wang. Partial financial support from a Georgia Research Alliance Eminent Scholar endowment (Binghe Wang), GSU Brains and Behaviors Fellowship (Zhengnan Yuan and Bingchen Yu), the Molecular Basis of Disease Program (Manjusha R. Choudhury and Zhixiang Pan), the Center for Diagnostics and Therapeutics (Abiodun Anifowose), and GSU internal resources is gratefully acknowledged. CO-related research is also partially supported by a grant from the National Institutes of Health (R01DK119202).
Bio-sketches of the authors
 Xiaoxiao Yang received his Ph.D. degree in medicinal chemistry in 2008 from Chinese Academy of Medical Sciences (CAMS), Peking Union Medical College (Beijing, China). After that he started his research work in the Institute of Materia Medica, CAMS as an assistant research scientist and then associate research scientist. In 2017, he joined Dr. Binghe Wang’ group as a postdoc at Georgia State University. His current research interests lie in the areas of medicinal chemistry to develop new therapeutic and diagnostic agents for cancer, and also to develop carbon monoxide prodrug as a therapeutic agent.
Xiaoxiao Yang received his Ph.D. degree in medicinal chemistry in 2008 from Chinese Academy of Medical Sciences (CAMS), Peking Union Medical College (Beijing, China). After that he started his research work in the Institute of Materia Medica, CAMS as an assistant research scientist and then associate research scientist. In 2017, he joined Dr. Binghe Wang’ group as a postdoc at Georgia State University. His current research interests lie in the areas of medicinal chemistry to develop new therapeutic and diagnostic agents for cancer, and also to develop carbon monoxide prodrug as a therapeutic agent.
 Zhixiang Pan received his bachelor’s degree in pharmacy in 2014 from Anhui Medical University (Hefei, China). In the same year, he joined Dr. Binghe Wang’s group as a research assistant. During his PhD study, his research was focused on the development of organic carbon monoxide prodrugs and drug delivery. He has received his PhD degree in May, 2019 and now serves as a senior research scientist in WuxiApptec, Shanghai.
Zhixiang Pan received his bachelor’s degree in pharmacy in 2014 from Anhui Medical University (Hefei, China). In the same year, he joined Dr. Binghe Wang’s group as a research assistant. During his PhD study, his research was focused on the development of organic carbon monoxide prodrugs and drug delivery. He has received his PhD degree in May, 2019 and now serves as a senior research scientist in WuxiApptec, Shanghai.
 Manjusha Roy Choudhury is currently pursuing her PhD at Georgia State University under Dr. Binghe Wang. She completed her Bachelor’s in Chemistry from St. Stephens, Delhi University, India followed by a Masters in Chemistry from University of Manchester, UK. She is currently working on bacterial membrane sensitizers and also towards developing novel mitochondria targeting small molecules.
Manjusha Roy Choudhury is currently pursuing her PhD at Georgia State University under Dr. Binghe Wang. She completed her Bachelor’s in Chemistry from St. Stephens, Delhi University, India followed by a Masters in Chemistry from University of Manchester, UK. She is currently working on bacterial membrane sensitizers and also towards developing novel mitochondria targeting small molecules.
 Zhengnan Yuan received his B.S. degree at Capital Normal University (2015). He is currently pursuing his Ph.D. degree in chemistry under Dr. Binghe Wang’s supervision at Georgia State University. He is currently working on developing prodrug strategies for drug delivery.
Zhengnan Yuan received his B.S. degree at Capital Normal University (2015). He is currently pursuing his Ph.D. degree in chemistry under Dr. Binghe Wang’s supervision at Georgia State University. He is currently working on developing prodrug strategies for drug delivery.
 Abiodun Anifowose received his bachelor’s degree from the University of Lagos, Nigeria (2008) and obtained his Ph.D. degree in Chemistry from Georgia State University (2019). He is currently working as a chemist in a peptide API manufacturing company in South Carolina.
Abiodun Anifowose received his bachelor’s degree from the University of Lagos, Nigeria (2008) and obtained his Ph.D. degree in Chemistry from Georgia State University (2019). He is currently working as a chemist in a peptide API manufacturing company in South Carolina.
 Bingchen Yu received his B.S. degree at Shandong University (2014) and his Ph.D. degree in chemistry at Georgia State University in 2018. He worked as a postdoctoral fellow in Dr. Binghe Wang’s lab during 2019 and is now working as a postdoctoral fellow in Dr. Lei Wang’s lab in University of California, San Francisco. His research focus on sulfur-related prodrug design, new antimicrobial drug development and protein function modification utilizing genetic code expansion tools.
Bingchen Yu received his B.S. degree at Shandong University (2014) and his Ph.D. degree in chemistry at Georgia State University in 2018. He worked as a postdoctoral fellow in Dr. Binghe Wang’s lab during 2019 and is now working as a postdoctoral fellow in Dr. Lei Wang’s lab in University of California, San Francisco. His research focus on sulfur-related prodrug design, new antimicrobial drug development and protein function modification utilizing genetic code expansion tools.
 Wenyi Wang joined Dr. Binghe Wang’s group at Georgia State University in 2013 and received her Ph.D. degree in Organic Chemistry in 2018. During graduate study, her research mainly focused on prodrug design and synthesis of anticancer drugs and gasotransmitters. After graduation, she entered the pharmaceutical industry and is now working as a research scientist in the field of small molecule formulation development.
Wenyi Wang joined Dr. Binghe Wang’s group at Georgia State University in 2013 and received her Ph.D. degree in Organic Chemistry in 2018. During graduate study, her research mainly focused on prodrug design and synthesis of anticancer drugs and gasotransmitters. After graduation, she entered the pharmaceutical industry and is now working as a research scientist in the field of small molecule formulation development.
 Binghe Wang is Regents’ Professor of Chemistry and Georgia Research Alliance Eminent Scholar in Drug Discovery at Georgia State University. He is the Founding Director of the Center for Diagnostics and Therapeutics, and the founding serial editor of “Wiley Series in Drug Discovery and Development.” He also serves on the editorial boards of numerous journals. His research interests are in the areas of drug discovery, drug delivery, and new diagnostics. Dr. Wang, together with his students and postdocs, has published over 290 papers and edited several books covering the same general areas.
Binghe Wang is Regents’ Professor of Chemistry and Georgia Research Alliance Eminent Scholar in Drug Discovery at Georgia State University. He is the Founding Director of the Center for Diagnostics and Therapeutics, and the founding serial editor of “Wiley Series in Drug Discovery and Development.” He also serves on the editorial boards of numerous journals. His research interests are in the areas of drug discovery, drug delivery, and new diagnostics. Dr. Wang, together with his students and postdocs, has published over 290 papers and edited several books covering the same general areas.
REFERENCES
- 1.Ji X, Zhou C, Ji K, Aghoghovbia R, Pan Z, Chittavong V, Ke B, Wang B. Click and release: a chemical strategy toward developing gasotransmitter prodrugs by using an intramolecular Diels-Alder reaction. Angew Chem Int Ed Engl 2016;55:15846–15851. [DOI] [PubMed] [Google Scholar]
- 2.Diamantis N, Banerji U. Antibody-drug conjugates-an emerging class of cancer treatment. Brit J Cancer 2016;114(4):362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sievers EL, Senter PD. Antibody-Drug conjugates in cancer therapy. Annu Rev Med 2013;64:15–29. [DOI] [PubMed] [Google Scholar]
- 4.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov 2017;16:315–337. [DOI] [PubMed] [Google Scholar]
- 5.Tsuchikama K, An Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell 2018;9:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuchikama K Novel chemical linkers for next-generation antibody-drug conjugates(ADCs). Yakugaku Zasshi 2019;139(2):209–219. [DOI] [PubMed] [Google Scholar]
- 7.Ceborska M Folate appended cyclodextrins for drug, DNA, and siRNA delivery. Eur J Pharm Biopharm 2017;120:133–145. [DOI] [PubMed] [Google Scholar]
- 8.Carron PM, Crowley A, O’Shea D, McCann M, Howe O, Hunt M, Devereux M. Targeting the folate receptor: improving efficacy in inorganic medicinal chemistry. Curr Med Chem 2018;25:2675–2708. [DOI] [PubMed] [Google Scholar]
- 9.Lu YJ, Low PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Deliver Rev 2012;64:342–352. [DOI] [PubMed] [Google Scholar]
- 10.Juliano RL, Ming X, Nakagawa O, Xu RZ, Yoo H. Integrin targeted delivery of gene therapeutics. Theranostics 2011;1:211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arosio D, Casagrande C. Advancement in integrin facilitated drug delivery. Adv Drug Deliver Rev 2016;97:111–143. [DOI] [PubMed] [Google Scholar]
- 12.Arosio D, Manzoni L, Corno C, Perego P. Integrin-targeted peptide- and peptidomimetic-drug conjugates for the treatment of tumors. Recent Pat Anticancer Drug Discov 2017;12:148–168. [DOI] [PubMed] [Google Scholar]
- 13.Barve A, Jin W, Cheng K. Prostate cancer relevant antigens and enzymes for targeted drug delivery. J Control Release 2014;187:118–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mangadlao JD, Wang X, McCleese C, Escamilla M, Ramamurthy G, Wang Z, Govande M, Basilion JP, Burda C. Prostate-specific membrane antigen targeted gold nanoparticles for theranostics of prostate cancer. ACS Nano 2018;12:3714–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lv Q, Yang J, Zhang R, Yang Z, Yang Z, Wang Y, Xu Y, He Z. Prostate-specific membrane antigen targeted therapy of prostate cancer using a DUPA-paclitaxel conjugate. Mol Pharm 2018;15:1842–1852. [DOI] [PubMed] [Google Scholar]
- 16.Frigerio M, Kyle AF. The chemical design and synthesis of linkers used in antibody drug conjugates. Curr Top Med Chem 2017;17(32):3393–3424. [DOI] [PubMed] [Google Scholar]
- 17.Jain N, Smith SW, Ghone S, Tomczuk B. Current ADC linker chemistry. Pharm Res 2015;32(11):3526–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leriche G, Chisholm L, Wagner A. Cleavable linkers in chemical biology. Bioorgan Med Chem 2012;20(2):571–582. [DOI] [PubMed] [Google Scholar]
- 19.Sonawane SJ, Kalhapure RS, Govender T. Hydrazone linkages in pH responsive drug delivery systems. Eur J Pharm Sci 2017;99:45–65. [DOI] [PubMed] [Google Scholar]
- 20.Tong R, Tang L, Ma L, Tu C, Baumgartner R, Cheng J. Smart chemistry in polymeric nanomedicine. Chem Soc Rev 2014;43(20):6982–7012. [DOI] [PubMed] [Google Scholar]
- 21.Hassanzadeh P, Atyabi F, Dinarvand R. Linkers: The key elements for the creation of efficient nanotherapeutics. J Control Release 2018;270:260–267. [DOI] [PubMed] [Google Scholar]
- 22.Singh SK, Luisi DL, Pak RH. Antibody-drug conjugates: design, formulation and physicochemical stability. Pharm Res 2015;32(11):3541–3571. [DOI] [PubMed] [Google Scholar]
- 23.He X, Li J, An S, Jiang C. pH-sensitive drug-delivery systems for tumor targeting. Ther Deliv 2013;4(12):1499–1510. [DOI] [PubMed] [Google Scholar]
- 24.Jacques SA, Leriche G, Mosser M, Nothisen M, Muller CD, Remy JS, Wagner A. From solution to in-cell study of the chemical reactivity of acid sensitive functional groups: a rational approach towards improved cleavable linkers for biospecific endosomal release. Org Biomol Chem 2016;14(21):4794–4803. [DOI] [PubMed] [Google Scholar]
- 25.Kanamala M, Wilson WR, Yang MM, Palmer BD, Wu ZM. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: A review. Biomaterials 2016;85:152–167. [DOI] [PubMed] [Google Scholar]
- 26.Singh R, Erickson HK. Antibody-cytotoxic agent conjugates: preparation and characterization. Methods Mol Biol 2009;525:445–467. [DOI] [PubMed] [Google Scholar]
- 27.Kumaresan PR, Luo J, Song A, Marik J, Lam KS. Evaluation of ketone-oxime method for developing therapeutic on-demand cleavable immunoconjugates. Bioconjug Chem 2008;19(6):1313–1318. [DOI] [PubMed] [Google Scholar]
- 28.Kalia J, Raines RT. Hydrolytic stability of hydrazones and oximes. Angew Chem Int Ed 2008;47(39):7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patil R, Portilla-Arias J, Ding H, Konda B, Rekechenetskiy A, Inoue S, Black KL, Holler E, Ljubimova JY. Cellular delivery of doxorubicin via pH-controlled hydrazone linkage using multifunctional nano vehicle based on poly(beta-l-malic acid). Int J Mol Sci 2012;13(9):11681–11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruan Y, Wang L, Zhao Y, Yao Y, Chen S, Li J, Guo H, Ming C, Chen S, Gong F, Chen G. Carbon monoxide potently prevents ischemia-induced high-mobility group box 1 translocation and release and protects against lethal renal ischemia-reperfusion injury. Kidney Int 2014;86(3):525–537. [DOI] [PubMed] [Google Scholar]
- 31.Saleh MN, Sugarman S, Murray J, Ostroff JB, Healey D, Jones D, Daniel CR, LeBherz D, Brewer H, Onetto N, LoBuglio AF. Phase I trial of the anti-Lewis Y drug immunoconjugate BR96-doxorubicin in patients with lewis Y-expressing epithelial tumors. J Clin Oncol 2000;18(11):2282–2292. [DOI] [PubMed] [Google Scholar]
- 32.Saleh MN, Sugarman S, Murray J, Ostroff JB, Healey D, Jones D, Daniel CR, LeBherz D, Brewer H, Onetto N, LoBuglio AF. Phase I trial of the anti-Lewis Y drug immunoconjugate BR96-doxorubicin in patients with lewis Y-expressing epithelial tumors. J Clin Oncol 2000;18(11):2282–2292. [DOI] [PubMed] [Google Scholar]
- 33.Tolcher AW, Sugarman S, Gelmon KA, Cohen R, Saleh M, Isaacs C, Young L, Healey D, Onetto N, Slichenmyer W. Randomized phase II study of BR96-doxorubicin conjugate in patients with metastatic breast cancer. J Clin Oncol 1999;17(2):478–484. [DOI] [PubMed] [Google Scholar]
- 34.Ross HJ, Rudin CM, Hart LL, Swanson PM, Rarick MU, Figlin RA, Jacobs AD, Miller DM. Randomized phase II study of SGN-15 (CBR96-doxorubicin immunoconjugate) with docetaxel in patients with advanced or metastatic non-small cell lung cancer (NSCLC). J Clin Oncol 2004;22(14_suppl):7039–7039. [Google Scholar]
- 35.Hamann PR, Hinman LM, Hollander I, Beyer CF, Lindh D, Holcomb R, Hallett W, Tsou HR, Upeslacis J, Shochat D, Mountain A, Flowers DA, Bernstein I. Gemtuzumab Ozogamicin, A potent and selective anti-CD33 antibody−calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem 2002;13(1):47–58. [DOI] [PubMed] [Google Scholar]
- 36.Tsuchikama K, An Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell 2018;9(1):33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu J, Jiang F, Lu A, Zhang G. Linkers having a crucial role in antibody-drug conjugates. Int J Mol Sci 2016;17(4):561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroeder GM, Vite GD, Borzilleri RM. Antibody-drug conjugates: current status and future directions. Drug Discov Today 2014;19(7):869–881. [DOI] [PubMed] [Google Scholar]
- 39.Boghaert ER, Khandke KM, Sridharan L, Dougher M, DiJoseph JF, Kunz A, Hamann PR, Moran J, Chaudhary I, Damle NK. Determination of pharmacokinetic values of calicheamicin-antibody conjugates in mice by plasmon resonance analysis of small (5 microl) blood samples. Cancer Chemother Pharmacol 2008;61(6):1027–1035. [DOI] [PubMed] [Google Scholar]
- 40.van Der Velden VH, te Marvelde JG, Hoogeveen PG, Bernstein ID, Houtsmuller AB, Berger MS, van Dongen JJ. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: in vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001;97(10):3197–3204. [DOI] [PubMed] [Google Scholar]
- 41.Ricart AD. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin Cancer Res 2011;17(20):6417–6427. [DOI] [PubMed] [Google Scholar]
- 42.Rajvanshi P, Shulman HM, Sievers EL, McDonald GB. Hepatic sinusoidal obstruction after gemtuzumab ozogamicin (Mylotarg) therapy. Blood 2002;99(7):2310–2314. [DOI] [PubMed] [Google Scholar]
- 43.Paci A, Desnoyer A, Delahousse J, Blondel L, Maritaz C, Chaput N, Mir O, Broutin S. Pharmacokinetic/pharmacodynamic relationship of therapeutic monoclonal antibodies used in oncology: Part 1, monoclonal antibodies, antibody-drug conjugates and bispecific T-cell engagers. Eur J Cancer 2020;128:107–118. [DOI] [PubMed] [Google Scholar]
- 44.Appelbaum FR, Bernstein ID. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood 2017;130(22):2373–2376. [DOI] [PubMed] [Google Scholar]
- 45.Zheng Y, Ren J, Wu Y, Meng X, Zhao Y, Wu C. proteolytic unlocking of ultrastable twin-acylhydrazone linkers for lysosomal acid-triggered release of anticancer drugs. Bioconjug Chem 2017;28(10):2620–2626. [DOI] [PubMed] [Google Scholar]
- 46.Jin Y, Song L, Su Y, Zhu L, Pang Y, Qiu F, Tong G, Yan D, Zhu B, Zhu X. Oxime linkage: a robust tool for the design of pH-sensitive polymeric drug carriers. Biomacromolecules 2011;12(10):3460–3468. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Yang C, Wang W, Liu J, Liu Q, Huang F, Chu L, Gao H, Li C, Kong D, Liu Q, Liu J. Co-delivery of doxorubicin and curcumin by pH-sensitive prodrug nanoparticle for combination therapy of cancer. Sci Rep 2016;6:21225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu W, Ding J, Xiao C, Li L, Zhuang X, Chen X. Versatile preparation of intracellular-acidity-sensitive oxime-linked polysaccharide-doxorubicin conjugate for malignancy therapeutic. Biomaterials 2015;54:72–86. [DOI] [PubMed] [Google Scholar]
- 49.Choy CJ, Geruntho JJ, Davis AL, Berkman CE. Tunable pH-sensitive linker for controlled release. Bioconjug Chem 2016;27(3):824–830. [DOI] [PubMed] [Google Scholar]
- 50.Deslongchamps P, Dory YL, Li S. The relative rate of hydrolysis of a series of acyclic and six-membered cyclic acetals, ketals, orthoesters, and orthocarbonates. Tetrahedron 2000;56(22):3533–3537. [Google Scholar]
- 51.Leriche G, Nothisen M, Baumlin N, Muller CD, Bagnard D, Remy JS, Jacques SA, Wagner A. Spiro Diorthoester (SpiDo), a human plasma stable acid-sensitive cleavable linker for lysosomal release. Bioconjug Chem 2015;26(8):1461–1465. [DOI] [PubMed] [Google Scholar]
- 52.Kong SD, Luong A, Manorek G, Howell SB, Yang J. Acidic hydrolysis of N-ethoxybenzylimidazoles (NEBIs): Potential applications as pH-sensitive linkers for drug delivery. Bioconjug Chem 2007;18(2):293–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao Y, Yang J. Development of a folate receptor (FR)-targeted indenoisoquinoline using a pH-sensitive N-ethoxybenzylimidazole (NEBI) bifunctional cross-linker. Bioconjug Chem 2014;25(5):873–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dinand E, Zloh M, Brocchini S. Competitive reactions during amine addition to cis-aconityl anhydride. Aust J Chem 2002;55(7):467–474. [Google Scholar]
- 55.Fletcher S, Jorgensen MR, Miller AD. Facile preparation of an orthogonally protected, pH-sensitive, bioconjugate linker for therapeutic applications. Org Lett 2004;6(23):4245–4248. [DOI] [PubMed] [Google Scholar]
- 56.Zhang A, Yao L, An M. Reversing the undesirable pH-profile of doxorubicin via activation of a di-substituted maleamic acid prodrug at tumor acidity. Chem Commun 2017;53(95):12826–12829. [DOI] [PubMed] [Google Scholar]
- 57.Kirby AJ, Lancaster PW. Structure and efficiency in intramolecular and enzymic catalysis. Catalysis of amide hydrolysis by the carboxy-group of substituted maleamic acids. J Chem Soc, Perkin Trans 2 1972(9):1206–1214. [Google Scholar]
- 58.Aldersley MF, Kirby AJ, Lancaster PW. Rate-determining proton transfer in intramolecular catalysis of amide hydrolysis by the carboxylic acid group. J Chem Soc, Chem Comm 1972(9):570–571. [Google Scholar]
- 59.Andresen TL, Thompson DH, Kaasgaard T. Enzyme-triggered nanomedicine: Drug release strategies in cancer therapy (Invited Review). Mol Membr Biol 2010;27(7):353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gondi CS, Rao JS. Cathepsin B as a cancer target. Expert Opin Ther Targets 2013;17(3):281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Denmeade SR, Nagy A, Gao J, Lilja H, Schally AV, Isaacs JT. Enzymatic activation of a doxorubicin-peptide prodrug by prostate-specific antigen. Cancer Res 1998;58(12):2537–2540. [PubMed] [Google Scholar]
- 62.Chen Z, Zhang P, Cheetham AG, Moon JH, Moxley JW Jr., Lin YA, Cui H. Controlled release of free doxorubicin from peptide-drug conjugates by drug loading. J Control Release 2014;191:123–130. [DOI] [PubMed] [Google Scholar]
- 63.Dai C, Fu Y, Li B, Wang Y, Zhang X, Wang J, Zhang Q. Linkage with cathepsin B-sensitive dipeptide promotes the in vitro and in vivo anticancer activity of PEGylated tumor necrosis factor-alpha (TNF-alpha) against murine fibrosarcoma. Sci China Life Sci 2011;54(2):128–138. [DOI] [PubMed] [Google Scholar]
- 64.Wang H, Rangan VS, Sung MC, Passmore D, Kempe T, Wang X, Thevanayagam L, Pan C, Rao C, Srinivasan M, Zhang Q, Gangwar S, Deshpande S, Cardarelli P, Marathe P, Yang Z. Pharmacokinetic characterization of BMS-936561, an anti-CD70 antibody-drug conjugate, in preclinical animal species and prediction of its pharmacokinetics in humans. Biopharm Drug Dispos 2016;37(2):93–106. [DOI] [PubMed] [Google Scholar]
- 65.Zhang C, Pan D, Li J, Hu J, Bains A, Guys N, Zhu H, Li X, Luo K, Gong Q, Gu Z. Enzyme-responsive peptide dendrimer-gemcitabine conjugate as a controlled-release drug delivery vehicle with enhanced antitumor efficacy. Acta Biomater 2017;55:153–162. [DOI] [PubMed] [Google Scholar]
- 66.Chung SW, Lee BS, Choi J, Kim SW, Kim IS, Kim SY, Byun Y. Optimization of a stable linker involved DEVD peptide-doxorubicin conjugate that is activated upon radiation-induced caspase-3-mediated apoptosis. J Med Chem 2015;58(16):6435–6447. [DOI] [PubMed] [Google Scholar]
- 67.Bouilloux J, Yuschenko O, Dereka B, Boso G, Zbinden H, Vauthey E, Babic A, Lange N. Cyclopeptidic photosensitizer prodrugs as proteolytically triggered drug delivery systems of pheophorbide A: part I - self-quenched prodrugs. Photochem Photobiol Sci 2018. [DOI] [PubMed] [Google Scholar]
- 68.Janssen S, Jakobsen CM, Rosen DM, Ricklis RM, Reineke U, Christensen SB, Lilja H, Denmeade SR. Screening a combinatorial peptide library to develop a human glandular kallikrein 2–activated prodrug as targeted therapy for prostate cancer. Mol Cancer Ther 2004;3(11):1439–1450. [PubMed] [Google Scholar]
- 69.Chen Y, Zhang M, Jin H, Tang Y, Wang H, Xu Q, Li Y, Li F, Huang Y. Intein-mediated site-specific synthesis of tumor-targeting protein delivery system: Turning PEG dilemma into prodrug-like feature. Biomaterials 2017;116:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li B, Zhang L, Zhang Z, Long M, Ren J, Lin F, Wang X, Wei J, Dong K, Zhang H. Synergistic tumor growth-inhibitory effect of the prostate-specific antigen-activated fusion peptide BSD352 for prostate cancer therapy. Anticancer Drugs 2011;22(3):213–222. [DOI] [PubMed] [Google Scholar]
- 71.Richard JA, Meyer Y, Jolivel V, Massonneau M, Dumeunier R, Vaudry D, Vaudry H, Renard PY, Romieu A. Latent fluorophores based on a self-immolative linker strategy and suitable for protease sensing. Bioconjug Chem 2008;19(8):1707–1718. [DOI] [PubMed] [Google Scholar]
- 72.Katz J, Janik JE, Younes A. Brentuximab Vedotin (SGN-35). Clin Cancer Res 2011;17(20):6428–6436. [DOI] [PubMed] [Google Scholar]
- 73.Sanderson RJ, Hering MA, James SF, Sun MMC, Doronina SO, Siadak AW, Senter PD, Wahl AF. In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate. Clin Cancer Res 2005;11(2):843–852. [PubMed] [Google Scholar]
- 74.Bornstein GG. Antibody Drug Conjugates: Preclinical Considerations. AAPS J 2015;17(3):525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mizuno M, Nozaki M, Morine N, Suzuki N, Nishikawa K, Morgan BP, Matsuo S. A protein toxin from the sea anemone Phyllodiscus semoni targets the kidney and causes a severe renal injury with predominant glomerular endothelial damage. Am J Pathol 2007;171(2):402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dubowchik GM, Firestone RA, Padilla L, Willner D, Hofstead SJ, Mosure K, Knipe JO, Lasch SJ, Trail PA. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug Chem 2002;13(4):855–869. [DOI] [PubMed] [Google Scholar]
- 77.Kumar SK, Williams SA, Isaacs JT, Denmeade SR, Khan SR. Modulating paclitaxel bioavailability for targeting prostate cancer. Bioorgan Med Chem 2007;15(14):4973–4984. [DOI] [PubMed] [Google Scholar]
- 78.Meyer Y, Richard JA, Massonneau M, Renard PY, Romieu A. Development of a new nonpeptidic self-immolative spacer. Application to the design of protease sensing fluorogenic probes. Org Lett 2008;10(8):1517–1520. [DOI] [PubMed] [Google Scholar]
- 79.Meyer Y, Richard JA, Massonneau M, Renard PY, Romieu A. Development of a new nonpeptidic self-immolative spacer. Application to the design of protease sensing fluorogenic probes. Org Lett 2008;10(8):1517–1520. [DOI] [PubMed] [Google Scholar]
- 80.Meyer Y, Richard JA, Delest B, Noack P, Renard PY, Romieu A. A comparative study of the self-immolation of para-aminobenzylalcohol and hemithioaminal-based linkers in the context of protease-sensitive fluorogenic probes. Org Biomol Chem 2010;8(8):1777–1780. [DOI] [PubMed] [Google Scholar]
- 81.Böttger R, Knappe D, Hoffmann R. Readily adaptable release kinetics of prodrugs using protease-dependent reversible PEGylation. J Control Release 2016;230:88–94. [DOI] [PubMed] [Google Scholar]
- 82.Anami Y, Yamazaki CM, Xiong W, Gui X, Zhang N, An Z, Tsuchikama K. Glutamic acid-valine-citrulline linkers ensure stability and efficacy of antibody-drug conjugates in mice. Nat Commun 2018;9(1):2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Caculitan NG, Chuh JD, Ma Y, Zhang DL, Kozak KR, Liu YC, Pillow TH, Sadowsky J, Cheung TK, Phung Q, Haley B, Lee BC, Akita RW, Sliwkowski MX, Polson AG. Cathepsin B is dispensable for cellular processing of cathepsin b-cleavable antibody-drug conjugates. Cancer Res 2017;77(24):7027–7037. [DOI] [PubMed] [Google Scholar]
- 84.Di YY, Ji SP, Wolf P, Krol ES, Alcorn J. Enterolactone glucuronide and beta-glucuronidase in antibody directed enzyme prodrug therapy for targeted prostate cancer cell treatment. AAPS Pharmscitech 2017;18(6):2336–2345. [DOI] [PubMed] [Google Scholar]
- 85.Albin N, Massaad L, Toussaint C, Mathieu MC, Morizet J, Parise O, Gouyette A, Chabot GG. Main Drug-Metabolizing Enzyme-Systems in Human Breast-Tumors and Peritumoral Tissues. Cancer Res 1993;53(15):3541–3546. [PubMed] [Google Scholar]
- 86.de Graaf M, Boven E, Scheeren HW, Haisma HJ, Pinedo HM. Beta-glucuronidase-mediated drug release. Curr Pharm Design 2002;8(15):1391–1403. [DOI] [PubMed] [Google Scholar]
- 87.Jeffrey SC, Andreyka JB, Bernhardt SX, Kissler KM, Kline T, Lenox JS, Moser RF, Nguyen MT, Okeley NM, Stone IJ, Zhang XQ, Senter PD. Development and properties of beta-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconjug Chem 2006;17(3):831–840. [DOI] [PubMed] [Google Scholar]
- 88.Burke PJ, Senter PD, Meyer DW, Miyamoto JB, Anderson M, Toki BE, Manikumar G, Wani MC, Kroll DJ, Jeffrey SC. Design, synthesis, and biological evaluation of antibody−drug conjugates comprised of potent camptothecin analogues. Bioconjugate Chem 2009;20(6):1242–1250. [DOI] [PubMed] [Google Scholar]
- 89.Jeffrey SC, De Brabander J, Miyamoto J, Senter PD. Expanded utility of the beta-glucuronide Linker: ADCs that deliver phenolic cytotoxic agents. ACS Med Chem Lett 2010;1(6):277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Renoux B, Legigan T, Bensalma S, Chadeneau C, Muller JM, Papot S. A new cyclopamine glucuronide prodrug with improved kinetics of drug release. Org Biomol Chem 2011;9(24):8459–8464. [DOI] [PubMed] [Google Scholar]
- 91.Doronina S, Toki B, Torgov M, Mendelsohn B, Cerveny C, Chace D, DeBlanc R, Gearing R, Bovee T, Siegall C, Francisco J, Wahl A, Meyer D, Senter P. Erratum: Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol 2003;21(8):778–784. [DOI] [PubMed] [Google Scholar]
- 92.Govindan S, Cardillo T, Sharkey R, Tat F, Gold D, Goldenberg D. Milatuzumab-SN-38 conjugates for the treatment of CD74 cancers. Mol Cancer Ther 2013;12(6):968–978. [DOI] [PubMed] [Google Scholar]
- 93.Kolakowski RV, Haelsig KT, Emmerton KK, Leiske CI, Miyamoto JB, Cochran JH, Lyon RP, Senter PD, Jeffrey SC. the methylene alkoxy carbamate self-immolative unit: utilization for the targeted delivery of alcohol-containing payloads with antibody-drug conjugates. Angew Chem Int Ed 2016;55(28):7948–7951. [DOI] [PubMed] [Google Scholar]
- 94.Aquilano K, Baldelli S, Ciriolo MR. Glutathione: new roles in redox signaling for an old antioxidant. Front Pharmacol 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Townsend D, Tew K, Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother 2003;57(3–4):145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Godwin A, Meister A, Odwyer PJ, Huang CS, Hamilton TC, Anderson ME. High-resistance to cisplatin in human ovarian-cancer cell-lines is associated with marked increase of glutathione synthesis. Proc. Natl Acad Sci USA 1992;89(7):3070–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Santra S, Kaittanis C, Santiesteban OJ, Perez JM. Cell-Specific, Activatable, and theranostic prodrug for dual-targeted cancer imaging and therapy. J Am Chem Soc 2011;133(41):16680–16688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lei EK, Kelley SO. Delivery and release of small-molecule probes in mitochondria using traceless linkers. J Am Chem Soc 2017;139(28):9455–9458. [DOI] [PubMed] [Google Scholar]
- 99.Li S, Yu B, Wang J, Zheng Y, Zhang H, Walker M, Yuan Z, Zhu H, Zhang J, Wang P, Wang B. Biomarker-Based Metabolic Labeling for Redirected and Enhanced Immune Response. Acs Chem Biol 2018;13(6):1686–1694. [DOI] [PubMed] [Google Scholar]
- 100.Lewis Phillips G, Li G, Dugger D, Crocker L, Parsons K, Mai E, Blättler W, Lambert J, Chari R, Lutz R, Wong W, Jacobson F, Koeppen H, Schwall R, Kenkare-Mitra S, Spencer S, Sliwkowski M. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody–cytotoxic drug conjugate. Cancer research 2008;68(22):9280–9290. [DOI] [PubMed] [Google Scholar]
- 101.Ray P, Huang B, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012;24(5):981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liou G, Storz P. Reactive oxygen species in cancer. Free Radical Res 2010;44(5):479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saravanakumar G, Kim J, Kim W. Reactive-oxygen-species-responsive drug delivery systems: promises and challenges. Adv Sci 2017;4(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Peng X, Gandhi V. ROS-activated anticancer prodrugs: a new strategy for tumor-specific damage. Ther Deliv 2012;3(7):823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ai Y, Obianom ON, Kuser M, Li Y, Shu Y, Xue FT. Enhanced tumor selectivity of 5-fluorouracil using a reactive oxygen species-activated prodrug approach. ACS Med Chem Lett 2019;10(1):127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heggie GD, Sommadossi JP, Cross DS, Huster WJ, Diasio RB. Clinical pharmacokinetics of 5-fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res 1987;47(8):2203–2206. [PubMed] [Google Scholar]
- 107.Miura K, Kinouchi M, Ishida K, Fujibuchi W, Naitoh T, Ogawa H, Ando T, Yazaki N, Watanabe K, Haneda S, Shibata C, Sasaki I. 5-FU metabolism in cancer and orally-administrable 5-FU drugs. Cancers (Basel) 2010;2(3):1717–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kuang Y, Balakrishnan K, Gandhi V, Peng X. Hydrogen peroxide inducible DNA cross-linking agents: targeted anticancer prodrugs. J Am Chem Soc 2011;133(48):19278–19281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Peng X, Gandhi V. ROS-activated anticancer prodrugs: a new strategy for tumor-specific damage. Therapeutic Delivery 2012;3(7):823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu QH, He CL, Xiao CS, Chen XS. Reactive oxygen species (ROS) responsive polymers for biomedical applications. Macromol Biosci 2016;16(5):635–646. [DOI] [PubMed] [Google Scholar]
- 111.Shim MS, Xia Y. A reactive oxygen species (ROS)-responsive polymer for safe, efficient, and targeted gene delivery in cancer cells. Angew Chem Int Ed 2013;52(27):6926–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pei P, Sun C, Tao W, Li J, Yang X, Wang J. ROS-sensitive thioketal-linked polyphosphoester-doxorubicin conjugate for precise phototriggered locoregional chemotherapy. Biomaterials 2019;188:74–82. [DOI] [PubMed] [Google Scholar]
- 113.Ling X, Zhang S, Shao P, Wang P, Ma X, Bai M. Synthesis of a reactive oxygen species responsive heterobifunctional thioketal linker. Tetrahedron Lett 2015;56(37):5242–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang X, Meng G, Zhang S, Liu X. A Reactive 1O2 - responsive combined treatment system of photodynamic and chemotherapy for cancer. Sci Rep 2016;6:29911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Taresco V, Alexander C, Singh N, Pearce AK. Stimuli-responsive prodrug chemistries for drug delivery. Adv Ther 2018;1(4). [Google Scholar]
- 116.Liu B, Wang DL, Liu YK, Zhang Q, Meng LL, Chi HR, Shi JN, Li GL, Li JC, Zhu XY. Hydrogen peroxide-responsive anticancer hyperbranched polymer micelles for enhanced cell apoptosis. Polym Chem-Uk 2015;6(18):3460–3471. [Google Scholar]
- 117.Noh J, Kwon B, Han E, Park M, Yang W, Cho W, Yoo W, Khang G, Lee D. Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nat Commun 2015;6:6907. [DOI] [PubMed] [Google Scholar]
- 118.Tang M, Hu P, Zheng Q, Tirelli N, Yang X, Wang Z, Wang Y, Tang Q, He Y. Polymeric micelles with dual thermal and reactive oxygen species (ROS)-responsiveness for inflammatory cancer cell delivery. J Nanobiotechnology 2017;15(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Qiao Z, Zhao W, Cong Y, Zhang D, Hu Z, Duan Z, Wang H. Self-assembled ROS-sensitive polymer–peptide therapeutics incorporating built-in reporters for evaluation of treatment efficacy. Biomacromolecules 2016;17(5):1643–1652. [DOI] [PubMed] [Google Scholar]
- 120.Devaraj NK, Upadhyay R, Hatin JB, Hilderbrand SA, Weissleder R. Fast and sensitive pretargeted labeling of cancer cells through a tetrazine/trans-cyclooctene cycloaddition. Angew Chem Int Ed 2009;48(38):7013–7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rossin R, van Duijnhoven SM, Lappchen T, van den Bosch SM, Robillard MS. Trans-cyclooctene tag with improved properties for tumor pretargeting with the diels-alder reaction. Mol Pharm 2014;11(9):3090–3096. [DOI] [PubMed] [Google Scholar]
- 122.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl Acad Sci USA 2007;104(43):16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Best MD. Click chemistry and bioorthogonal reactions: unprecedented selectivity in the labeling of biological molecules. Biochem 2009;48(28):6571–6584. [DOI] [PubMed] [Google Scholar]
- 124.Thirumurugan P, Matosiuk D, Jozwiak K. Click chemistry for drug development and diverse chemical-biology applications. Chem Rev 2013;113(7):4905–4979. [DOI] [PubMed] [Google Scholar]
- 125.Wright MH, Sieber SA. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat Prod Rep 2016;33(5):681–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ji X, Pan Z, Yu B, De La Cruz L, Zheng Y, Ke B, Wang B. Click and release: bioorthogonal approaches to “on-demand” activation of prodrugs. Chem Soc Rev 2019;48(4):1077–1094. [DOI] [PubMed] [Google Scholar]
- 127.de Geus M, ten Hoeve W, Hudson P, Janssen H, Khasanov A, van Onzen A, Robillard MS, Rossin R, Steenbergen EJ, Versteegen RM, Wessels HJ, Wu J. Chemically cleavable antibody-drug conjugates: drug release in one click. Synlett 2018;29(18):A180–A184. [Google Scholar]
- 128.Wang P, Yao L, Zhou L, Liu Y, Chen M, Wu H, Chang R, Li Y, Zhou M, Fang X, Yu T, Jiang L, Huang Z. carbon monoxide improves neurologic outcomes by mitochondrial biogenesis after global cerebral ischemia induced by cardiac arrest in rats. Int J Biol Sci 2016;12:1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Matikonda S, Orsi D, Staudacher V, Jenkins I, Fiedler F, Chen J, Gamble A. Bioorthogonal prodrug activation driven by a strain-promoted 1,3-dipolar cycloaddition. Chem Sci 2015;6(2):1212–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sustmann P, Trill D. Substituent effects in 1,3-dipolar cycloadditions of phenyl azide. Angew Chem Int Ed 1972;11(9):838. [Google Scholar]
- 131.Peng H, Dornevil K, Draganov A, Chen W, Dai C, Nelson W, Liu A, Wang B. An unexpected copper catalyzed ‘reduction’ of an arylazide to amine through the formation of a nitrene intermediate. Tetrahedron 2013;69(25):5079–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Versteegen R, Rossin R, ten Hoeve W, Janssen H, Robillard M. Click to Release: Instantaneous Doxorubicin Elimination upon Tetrazine Ligation. Angew Chem Int Ed 2013;52(52):14112–14116. [DOI] [PubMed] [Google Scholar]
- 133.Rossin R, van Duijnhoven S, Ten Hoeve W, Janssen H, Kleijn L, Hoeben F, Versteegen R, Robillard M. Triggered drug release from an antibody-drug conjugate using fast “click-to-release” chemistry in mice. Bioconjug Chem 2016;27(7):1697–1706. [DOI] [PubMed] [Google Scholar]
- 134.Fan X, Ge Y, Lin F, Yang Y, Zhang G, Ngai W, Lin Z, Zheng S, Wang J, Zhao J, Li J, Chen P. Optimized tetrazine derivatives for rapid bioorthogonal decaging in living cells. Angew Chem Int Ed 2016;55(45):14046–14050. [DOI] [PubMed] [Google Scholar]
- 135.Zheng Y, Ji X, Yu B, Ji K, Gallo D, Csizmadia E, Zhu M, Choudhury M, De La Cruz L, Chittavong V, Pan Z, Yuan Z, Otterbein LE, Wang B. Enrichment-triggered prodrug activation demonstrated through mitochondria-targeted delivery of doxorubicin and carbon monoxide. Nat Chem 2018(10):787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ji X, Damera K, Zheng Y, Yu B, Otterbein LE, Wang B. Toward carbon monoxide–based therapeutics: critical drug delivery and developability issues. J Pharm Sci 2017;105(2):406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov 2010;9(9):728–743. [DOI] [PubMed] [Google Scholar]
- 138.Soboleva T, Berreau LM. 3-hydroxyflavones and 3-hydroxy-4-oxoquinolines as carbon monoxide-releasing molecules. Molecules 2019;24(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Motterlini R, Haas B, Foresti R. Emerging concepts on the anti-inflammatory actions of carbon monoxide-releasing molecules (CO-RMs). Med Gas Res 2012;2(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ji X, Wang B. Strategies toward organic carbon monoxide prodrugs. Acc Chem Res 2018;51(6):1377–1385. [DOI] [PubMed] [Google Scholar]
- 141.Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M, Kalyanaraman B. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev 2017;117(15):10043–10120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ji X, Zhou C, Ji K, Aghoghovbia R, Pan Z, Chittavong V, Ke B, Wang B. Click and Release: A Chemical Strategy toward Developing Gasotransmitter Prodrugs by Using an Intramolecular Diels-Alder Reaction. Angew Chem Int Ed Engl 2016;55:15846–15851. [DOI] [PubMed] [Google Scholar]
- 143.Ji X, Pan Z, Li C, Kang T, De La Cruz LKC, Yang L, Yuan Z, Ke B, Wang B. Esterase-Sensitive and pH-Controlled Carbon Monoxide Prodrugs for Treating Systemic Inflammation. J Med Chem 2019;62(6):3163–3168. [DOI] [PubMed] [Google Scholar]
- 144.Correa-Costa M, Gallo D, Csizmadia E, Gomperts E, Lieberum J, Hauser C, Ji X, Wang B, Camara NOS, Robson SC, Otterbein LE. Carbon monoxide protects the kidney through the central circadian clock and CD39. Proc Natl Acad Sci USA 2018;115(10):E2302–E2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chen W, Wang D, Dai C, Hamelberg D, Wang B. Clicking 1,2,4,5-tetrazine and cyclooctynes with tunable reaction rates. Chem Commun (Camb) 2012;48(12):1736–1738. [DOI] [PubMed] [Google Scholar]
- 146.Isales G, Hipszer R, Raftery T, Chen A, Stapleton H, Volz D. Triphenyl phosphate-induced developmental toxicity in zebrafish: Potential role of the retinoic acid receptor. Aquat Toxicol 2015;161:221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Trnka J, Elkalaf M, Andel M. Lipophilic triphenylphosphonium cations inhibit mitochondrial electron transport chain and induce mitochondrial proton leak. Plos One 2015;10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Elkalaf M, Tuma P, Weiszenstein M, Polak J, Trnka J. Mitochondrial Probe Methyltriphenylphosphonium (TPMP) Inhibits the Krebs Cycle Enzyme 2-Oxoglutarate Dehydrogenase. Plos One 2016;11(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chio TI, Gu H, Mukherjee K, Tumey LN, Bane SL. Site-Specific Bioconjugation and multi-bioorthogonal labeling via rapid formation of a boron-nitrogen heterocycle. Bioconjug Chem 2019;30(5):1554–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tu J, Xu M, Parvez S, Peterson RT, Franzini RM. Bioorthogonal Removal of 3-isocyanopropyl groups enables the controlled release of fluorophores and drugs in vivo. J Am Chem Soc 2018;140(27):8410–8414. [DOI] [PubMed] [Google Scholar]
- 151.Yao Q, Lin F, Fan X, Wang Y, Liu Y, Liu Z, Jiang X, Chen P, Gao Y. Synergistic enzymatic and bioorthogonal reactions for selective prodrug activation in living systems. Nat Commun 2018;9(1):5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sabatino V, Rebelein JG, Ward TR. “Close-to-release”: spontaneous bioorthogonal uncaging resulting from ring-closing metathesis. J Am Chem Soc 2019;141(43):17048–17052. [DOI] [PMC free article] [PubMed] [Google Scholar]