

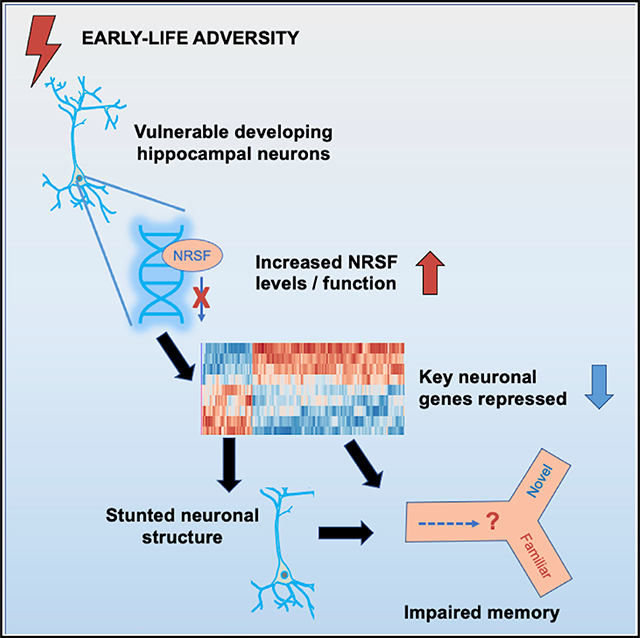
SUMMARY
Early-life adversity (ELA) is associated with lifelong memory deficits, yet the responsible mechanisms remain unclear. We impose ELA by rearing rat pups in simulated poverty, assess hippocampal memory, and probe changes in gene expression, their transcriptional regulation, and the consequent changes in hippocampal neuronal structure. ELA rats have poor hippocampal memory and stunted hippocampal pyramidal neurons associated with ~140 differentially expressed genes. Upstream regulators of the altered genes include glucocorticoid receptor and, unexpectedly, the transcription factor neuron-restrictive silencer factor (NRSF/REST). NRSF contributes critically to the memory deficits because blocking its function transiently following ELA rescues spatial memory and restores the dendritic arborization of hippocampal pyramidal neurons in ELA rats. Blocking NRSF function in vitro augments dendritic complexity of developing hippocampal neurons, suggesting that NRSF represses genes involved in neuronal maturation. These findings establish important, surprising contributions of NRSF to ELA-induced transcriptional programming that disrupts hippocampal maturation and memory function.
In Brief
Bolton et al. report enduring memory impairments after early-life adversity (ELA) associated with large-scale repression of key hippocampal genes contributing to neuronal maturation and neurotransmission. They identify glucocorticoid receptor (GR) and the repressive transcription factor neuron-restrictive silencer factor (NRSF) as candidate upstream regulators. Temporary block of NRSF chromatin-binding rescues hippocampal memory and neuronal structure.
Graphical Abstract
INTRODUCTION
Memory disorders affect tens of millions of individuals throughout the world (Guerchet et al., 2013; Prince et al., 2015). These disorders derive from a complex interplay of genetic and environmental factors (Klengel and Binder, 2015; Nestler and Hyman, 2010). Early postnatal life is a particularly sensitive period for the maturation of memory functions and the underlying neurons and brain circuits (Bale et al., 2010; Bath, 2020; Short and Baram, 2019), and adverse childhood experiences, such as poverty or neglect, are associated with cognitive deficits later in life (Kaplan et al., 2001; Nelson et al., 2007; Short et al., 2020). An enigmatic and crucial aspect of the consequences of early-life adversity (ELA) is their enduring and sometimes progressive nature (Brunson et al., 2005; Chen and Baram, 2016; Kaplan et al., 2001). How does a limited period of ELA provoke lifelong cognitive problems? Understanding the mechanisms of the enduring vulnerability to memory disorders following an early-life insult is fundamental for identifying new targets for prevention or intervention.
A potential mechanism for the long-lasting nature of cognitive changes involves enduring alterations in the expression of key neuronal genes that govern hippocampal neuron maturation, and these alterations may result from epigenetic/transcriptional processes (Rubin et al., 2014). Indeed, ELA promotes transcriptional changes in several relevant brain regions (Bale, 2015; Hunter and McEwen, 2013; Suderman et al., 2012) in both humans (Heim and Binder, 2012; Schwaiger et al., 2016; Suderman et al., 2014) and rodents (Deussing and Jakovcevski, 2017; Gray et al., 2018; Lucassen et al., 2013; McClelland et al., 2011a; Nestler, 2014; Peña et al., 2017; Ross et al., 2017; Russo et al., 2012; Szyf et al., 2016). A key master regulator of ELA-induced transcriptional changes is the receptor mediating many of the actions of the stress hormone corticosterone (cortisol in humans), i.e., glucocorticoid receptor (GR) (van Bodegom et al., 2017; Joëls and Baram, 2009; McEwen et al., 2016; Schmidt et al., 2013). GR acts as a transcription factor in concert with several interacting proteins (Binder, 2009; Ke et al., 2018; Kino, 2017; Klengel and Binder, 2015; Xu et al., 2017) to modulate gene expression enduringly and influence neuronal function (Daskalakis et al., 2015). Indeed, a key role for GR in mediating the effects of ELA on lifelong brain function has been suggested (Arnett et al., 2015; Chaudhury et al., 2014; McEwen et al., 2016) and addressed mechanistically (Arp et al., 2016; Lesuis et al., 2018; Loi et al., 2017). However, it remains unknown whether the complex and persistent ELA-provoked deficits in hippocampal structure (Dahmen et al., 2018; Molet et al., 2016a) and memory function (Brunson et al., 2005; Huot et al., 2002) are all attributable solely to the actions of GR.
Here we aimed to probe the transcriptional cascades leading to the serious consequences of ELA on spatial memory, a crucial function encoded in the dorsal hippocampus (Haettig et al., 2013; Maras et al., 2014). We imposed ELA by rearing rat pups in cages with limited bedding and nesting materials (LBN cages), which simulate resource scarcity and provoke aberrant maternal care (Gilles et al., 1996; Ivy et al., 2008; Molet et al., 2014). A week of exposure to this ELA (postnatal days 2–9) leads to persistent cognitive impairments later in life (Brunson et al., 2005; Molet et al., 2016a; Walker et al., 2017; Ivy et al., 2010; Bath et al., 2017; Short et al., 2020; Wang et al., 2011). Specifically, spatial memory deficits emerge in ELA rats during adolescence and are accompanied by structural changes in hippocampal neurons, including loss of dendrites, dendritic spines, and synapses (Ivy et al., 2010; Naninck et al., 2015; Molet et al., 2016a). We performed high-throughput mRNA sequencing (RNA-seq) and searched for potential ELA-induced “master regulators” that might orchestrate the observed disruption of gene expression programs in the hippocampus. We then targeted an unexpected candidate “driver” of the altered transcriptional programs and found that interfering with its function after the ELA period selectively rescued hippocampus-dependent memory function and hippocampal neuron maturation in ELA-experiencing rats.
RESULTS
ELA Provokes Spatial Memory Problems in Adult Rats
ELA leads to deficits in hippocampus-dependent memory, assessed via a variety of tests (Bath et al., 2017; Huot et al., 2002; Ivy et al., 2010; Molet et al., 2016a; Naninck et al., 2015; Short et al., 2020; Wang et al., 2013). Here we tested spatial memory using the object location task, because this test requires an intact dorsal hippocampus (Haettig et al., 2013; Maras et al., 2014) and involves little stress in itself (Bolton et al., 2017; Molet et al., 2016a). Memory of the location of an object was compromised in 2-month-old rats that had experienced ELA, apparent from a lower ratio of time spent exploring objects in novel versus familiar locations compared with controls (CTL; unpaired t test with Welch’s correction for unequal variance; t12 = 2.85, p = 0.02; Figure 1A). These deficits in spatial memory, in accord with prior findings using object location and the Morris water maze (Brunson et al., 2005; Ivy et al., 2010; Molet et al., 2016a), were not a result of anxiety or lack of motivation, because total exploration durations in both the training (Figure S1A) and the testing (Figure S1B) sessions failed to distinguish the groups (all p values > 0.1). In contrast with spatial memory, object recognition memory (Figure 1B), locomotion (Figure 1C), and anxiety-like behaviors were not influenced by ELA (Figure 1D; all p values > 0.3).
Figure 1. Early-Life Adversity (ELA) Provokes Selective Spatial Memory Deficits during Adulthood.

(A) Memory of object location, a measure of hippocampus-dependent spatial memory, was compromised in 2-month-old rats that had experienced ELA, spending a week early in life in cages with limited bedding and nesting materials. The memory deficits are apparent as a lower ratio of time spent exploring objects in novel versus familiar locations compared with littermates reared in control cages (p = 0.02; t test with Welch’s correction for unequal variance, n = 7/group).
(B) Object recognition memory, which is less dependent on the hippocampus, was not impaired by ELA.
(C and D) Neither locomotion (C) nor time spent in the center of an open field (D; a measure of anxiety-like behavior) was altered by ELA, suggesting that the effects on spatial memory were relatively specific. Data are mean ± SEM. *p < 0.05.
See also Figure S1.
Transcriptome-wide Changes in Adult Hippocampus after ELA
To determine the potential molecular mechanisms of the observed spatial memory problems, we performed high-throughput RNA-seq addressing the effect of ELA on dorsal hippocampal gene expression and found a subset of genes that were differentially expressed between the two conditions. Thirty-five genes were significantly upregulated and 107 were repressed in the dorsal hippocampus of male ELA rats (false discovery rate [FDR] < 0.05) (Figures 2A and B). When FDR < 0.1 was chosen as a cutoff, 63 genes were upregulated and 146 reduced (Figure 2B; Table S1). The high ratio of repressed genes over those that were upregulated is apparent from the heatmap (Figure 2C).
Figure 2. ELA Provokes Transcriptome-wide Changes in the Dorsal Hippocampus of Adult Rats.

(A) Overview of RNA-seq differential gene expression findings. Each bar represents the log2-fold change for the 142 differentially expressed genes in ELA versus control adult hippocampus. Genes were primarily repressed (107) rather than upregulated (35; STAR read alignment; FDR < 0.05). Error bars represent SEM.
(B) Scatterplot depicting the relation between transcript level for each gene (x axis) and the change in expression compared with the controls (y axis). Each gene is shown as a gray dot. Differentially regulated genes with FDR < 0.05 are shown in red, and those between 0.05 and 0.1 are shown in orange.
(C) Heatmap based on hierarchical clustering of Z scores for the 142 genes whose expression was significantly changed (FDR < 0.05) as a result of ELA. Plotted are gene-wise Z scores of genes and samples grouped by hierarchical clustering with the Euclidian distance metric.
(D) Gene Ontology identifies altered expression of specific pathways and gene families after ELA. Enriched terms were analyzed separately for significantly upregulated and downregulated genes (FDR < 0.05) using topGO and org.Rn.eg.db. Bar graphs represent Fisher’s exact test p values on a logarithmic scale.
(E and F) Validation of subgroups of repressed (E) and upregulated (F) genes identified by the RNA-seq of dorsal hippocampus samples. RT-PCR was performed on dorsal hippocampus samples of an independent rat cohort (n = 6 CTL and 5 ELA). Data are presented as mean ± SEM. *p < 0.05; #p ≤ 0.1 by one-sample t test.
Gene Ontology analysis of the differentially expressed genes identified significant distinctions between upregulated and downregulated gene clusters (Figure 2D). Among upregulated genes, enrichment of genes encoding lipid metabolism (Dgat2, Apoc1) and oxidative stress (Psap, Tmeff2, Ddit3/CHOP, Msrb1) was apparent, as well as genes involved in synaptogenesis (Tmem108/Retrolinkin, Sparcl1/Hevin). By contrast, the repressed genes were enriched in transcripts implicated in dendritic growth and axon guidance (e.g., cadherins, protocadherins, ephrin signaling; Table S2). Also enriched were genes encoding ion channels (Kcnn2, Kcnh7, Kcna4), ion channel auxiliary subunits (Cacng3/TARP-3, Shisa7/CKAMP59, Neto2, Pex5l/TRIP8b), and neurotransmitter receptors (Htr1b, Htr5b, Gabbr1, Gria4). Transcripts involved in intracellular transport and cytoskeletal dynamics (Sept6, Sept9, Asap1, Asap2, Arhgef26, Arhgap12) and in signal transduction were enriched among downregulated genes as well. Thus, the augmented and repressed genes clustered into functionally distinct categories that are linked to different aspects of neuronal function. Genes involved in neuronal differentiation and energy-demanding neuron-specific functions such as maintenance of membrane potential and neuronal firing were repressed, whereas genes contributing to cell metabolism and oxidative stress were augmented.
To corroborate the RNA-seq results, we obtained dorsal hippocampus samples from a separate cohort of rats and performed RT-PCR on subgroups of both repressed and upregulated genes, confirming their altered expression (Figures 2E and 2F; one-sample t test; repressed genes: Nell1: t[4] = 5.17, p = 0.007; Npas4: t[4] = 1.85, p = 0.1; Pex5l: t[4] = 3.42, p = 0.03; Rgs12: t[4] = 3.25, p = 0.03; upregulated genes: B3gat2: t[4] = 3.26, p = 0.03; Rspo3: t[4] = 2.06, p = 0.1; Sparcl1: t[4] = 3.20, p = 0.03; Tmem108; t[4] = 2.45, p = 0.07).
Targets of Two Transcription Factors Are Preferentially and Significantly Downregulated in Adult ELA Hippocampus
The data above demonstrated that hippocampal gene expression changes provoked by ELA were selective, suggesting that specific regulatory mechanisms were initiated by ELA to generate these potentially adaptive or compensatory changes. In addition, the high ratio of repressed genes over those that were upregulated (Figure 2) suggested that transcriptional repressors were involved. Using publicly available chromatin immunoprecipitation sequencing (ChIP-seq) data, we identified two candidate transcriptional regulators that might drive the differential gene expression after ELA (Figure 3A). Given the well-established effect of ELA on glucocorticoids (reviewed in van Bodegom et al., 2017), we first tested whether targets of the GR were over-represented among the differentially expressed genes using a ChIP-seq dataset from rat hippocampus (Polman et al., 2013). As might be expected after a stressful exposure such as ELA, we found enrichment in targets of the GR, which binds the stress hormone corticosterone (Figure 3B). GR, a transcription factor, is a well-established mediator of the epigenetic consequences of stress in general (Gray et al., 2017; Kino, 2017; Reul, 2014), as well as of early-life stress (Daskalakis et al., 2015; Klengel and Binder, 2015; Turecki and Meaney, 2016). As shown in the volcano plot (Figure 3B), among differentially regulated genes, three GR targets were upregulated, whereas 20 were repressed, in accord with the established functions of GR as both transcriptional enhancer and repressor (Kino, 2017) (Table S3). Notably, several of these genes were previously reported to be altered in the hippocampus after ELA or following adult stress (Gray et al., 2014).
Figure 3. Glucocorticoid Receptor (GR) and Neuron-Restrictive Silencer Factor (NRSF) Targets Are Enriched among Genes Enduringly Repressed after ELA.

(A) Tabular analysis of target enrichment demonstrates augmented representation of GR and NRSF/REST target genes, particularly among downregulated genes.
(B) A volcano plot showing p values for each gene plotted against fold changes in gene expression. Each gene is depicted as a gray dot. GR targets were assigned based on rat hippocampal data (Polman et al., 2013) and are shown as colored dots.
(C) A volcano plot highlighting NRSF targets, assigned based on Johnson et al. (2007), which employed cell lines, and shown as colored dots.
(D) A volcano plot highlighting NRSF targets, assigned based on rat ChIP-seq data from hippocampal progenitors (Mukherjee et al., 2016), and shown as colored dots. The significance cutoff p values corresponding to FDR of 0.05 and 0.1 are shown as dotted lines.
(E) Immunohistochemistry of NRSF in the dorsal hippocampus at P12 is shown in representative images from the CA1 of CTL and ELA rats. Bottom row shows single-cell resolution of NRSF protein levels within pyramidal neurons from the insets in images of the top row. Scale bar: 80 μm (top row); 20 μm (bottom row).
(F) The optical density of NRSF immunoreactivity was increased in the dorsal CA1 of P12 rats shortly following the ELA period (p = 0.01; unpaired t test). Data are mean ± SEM. *p < 0.05.
iQNP, induced quiescent neuronal progenitor; TAP, transit-amplifying progenitor. See also Tables S3 and S4.
To search for transcription factor targets among the differentially expressed genes in an unsupervised manner, we used the Enrichr tool (Kuleshov et al., 2016). Unexpectedly, enrichment analysis in the “ENCODE TF ChIP-seq 2015” database revealed neuron-restrictive silencer factor (NRSF, also known as REST) as the top hit based on ChIP-seq data across multiple tissues (Table S4); enrichment of NRSF targets was apparent for downregulated genes only and was confirmed in two independent ChIP-seq datasets (Johnson et al., 2007; Mukherjee et al., 2016) (Figures 3A, 3C, and 3D; Table S3). The selective enrichment of NRSF targets among genes downregulated by ELA is consistent with the repressive role of this transcription factor. Indeed, the canonical role of NRSF is to repress neuronal genes in non-neuronal tissues (Ballas et al., 2001; Johnson et al., 2007). To this end, numerous neuron-specific genes are endowed with an NRSF recognition site (NRSF-response element [NRSE]) and are sensitive to NRSF levels (McClelland et al., 2014). NRSF also crucially contributes to neuronal specification (Ballas et al., 2005; Abrajano et al., 2009; Gao et al., 2011). More recently, a role for NRSF in neuroplasticity after diverse early-life experiences has been emerging (Korosi et al., 2010; Patterson et al., 2017; Singh-Taylor et al., 2018). However, unlike the well-established role of GR in regulating gene expression following stress in several brain regions (Daskalakis et al., 2015; Gray et al., 2014), little is known about the potential role of NRSF in governing large-scale transcriptional programs in response to early-life insults. Adaptively, ELA-induced increase in NRSF function and the resultant repression of critical neuronal genes might reduce neuronal functions, such as synapse formation, maintenance of membrane potential, and action-potential firing, which are highly energy demanding, thus potentially enabling cell survival under adverse conditions (Mampay and Sheridan, 2019). Therefore, we tested whether NRSF contributed to ELA-induced hippocampal memory impairments and then probed the responsible cellular mechanisms.
Transiently Blocking NRSF Function Post Hoc Prevents Enduring Chromatin Changes and Rescues Spatial Memory in Adults that Had Experienced ELA
Prompted by the enriched representation of NRSF target genes among those repressed in ELA hippocampus, we measured the expression of this transcription factor in the hippocampus of control and ELA-experiencing rats. There was no evidence of significantly increased mRNA levels in the ELA group either shortly following the ELA epoch (1.008 ± 0.056 in controls and 1.110 ± 0.036 in ELA; p = 0.16; n = 6/group) or during adulthood (1.027 ± 0.1391 in controls [n = 4] and 1.167 ± 0.0599 in ELA [n = 5]; p = 0.35). This was not surprising, because augmented functional NRSF binding to the chromatin without significant changes in NRSF-mRNA expression has been reported following brain insults (McClelland et al., 2014; Rodenas-Ruano et al., 2012; Singh-Taylor et al., 2018). The hippocampus consists of numerous and diverse cell populations, such that a global assessment of mRNA expression might not enable detection of NRSF expression changes in the cell population in which functional changes related to augmented NRSF expression would be executed: dorsal CA1 pyramidal cells. Therefore, we next measured levels of NRSF-mRNA and protein with single-cell resolution using in situ hybridization and immunohistochemistry, respectively. We found no discernible changes in mRNA levels (data not shown). Remarkably, comparing protein expression specifically within the CA1 stratum pyramidale in the hippocampus of control and ELA rats at post-natal day 12, shortly after the end of the ELA period, was informative: ELA significantly increased NRSF protein in pyramidal neurons of the dorsal CA1 (t8 = 3.14, p = 0.01; Figures 3E and 3F).
To directly examine the potential role of NRSF-induced repression of key neuronal genes in the memory deficits provoked by ELA, we interfered with NRSF chromatin binding by infusing NRSF-blocking oligodeoxynucleotides (ODNs) into the lateral cerebral ventricles of control or ELA-experiencing rat pups during the 2 days following the ELA period (Figure S2). These ODNs, consisting of the stabilized sequence of the NRSF binding site (NRSE), enter brain cells and act as decoys, binding nuclear NRSF and preventing the interaction of this repressor with the chromatin (McClelland et al., 2011b, 2014; Patterson et al., 2017; Singh-Taylor et al., 2018) (Figures 4A and 5E). Random (scrambled [SCR]) ODNs were used as controls. We employed ChIP to quantify NRSF binding to the chromatin of several NRSF target genes immediately following the infusion but were unsuccessful: NRSF binding in all four groups of 12-day-old rats was <0.004% of input. We then reared the rats to adulthood and performed NRSF-ChIP analyses on the same groups (CTL+SCR, ELA+SCR, CTL+NRSE, and ELA+NRSE; n = 8/group). We tested whether ELA led to persistent increases of NRSF binding to key target genes in the adult hippocampus and whether this was prevented by NRSE-decoy ODNs. However, ChIP analysis of NRSF binding to several established targets of NRSF that were downregulated in the hippocampus of adult ELA-experiencing rats did not confirm this notion (data not shown). Therefore, we reasoned that the enduring repression of NRSF target genes might be a result of persistent changes to their chromatin that were initiated by transient NRSF binding. We looked specifically for augmented di-methylation of the histone 3 lysine 9 (H3K9me2) at NRSF target gene NRSEs, because this change was shown to follow transient NRSF binding in both the hippocampus and the hypothalamus (McClelland et al., 2011b; Singh-Taylor et al., 2018). We focused on the Npas4 gene because its promoter region is endowed with four putative NRSEs, and we performed ChIP on all four groups of adult rats. Two-way ANOVA revealed an increase in H3K9 di-methylation at the Npas4 promoter from the dorsal hippocampus of ELA+SCR rats compared with CTL+SCR, whereas blocking NRSF binding soon after the ELA period prevented this epigenetic mark from being laid down (significant main effects of ELA, F[1,20] = 6.61, p = 0.02, and of NRSE, F[1,20] = 4.97, p = 0.04; post hoc, p < 0.05; Figure 4B). Thus, the data support the notion that a transient binding of NRSF evolves to a persistent reduction in DNA accessibility via histone methylation-related changes of chromatin conformation.
Figure 4. Blocking NRSF Function Transiently after ELA Rescues Spatial Memory.

(A) A schematic of the mechanism by which the NRSE-ODN blocks NRSF function. The sequence of the synthetic, protected oligodeoxynucleotides (ODNs) replicates the recognition site for NRSF (NRSE). The ODNs, flooding the cells, bind available NRSF, acting as decoys and preventing NRSF binding to NRSE on genomic DNA (see also Figure 5E).
(B) H3K9 dimethylation at NRSE sites of the Npas4 gene was increased in adult dorsal hippocampus following ELA. Transiently blocking NRSF binding to chromatin with the use of NRSE-ODNs prevented this long-term ELA-provoked histone modification (significant main effects of ELA, F[1,20] = 6.61, p = 0.02, and of NRSE, F[1,20] = 4.97, p = 0.04; post hoc, p < 0.05).
(C) Memory for the prior location of a previously seen object was severely impaired in ELA rats receiving a random ODN sequence (Scrambled [SCR]) but was rescued in those receiving an NRSE-sequence decoy ODN to a level similar to that in control (CTL+SCR) rats. Two-way ANOVA demonstrated a significant interaction of the early-life experience and the type of treatment (F[1,26] = 16.44, p = 0.0004), and post hoc analyses identified a significant effect of the NRSF blockade intervention (p < 0.05).
(D) In an independent measure of spatial memory, ELA+SCR rats could not reliably discriminate between the novel arm and the familiar arm of the Y-maze, which they had explored 4 h earlier (see Y-maze schematic). However, NRSF blockade prevented this spatial memory deficit (significant ELA × NRSE interaction, F[1,25] = 7.26, p = 0.01, post hoc, p < 0.05). Representative heatmaps of the time spent in each area of the Y-maze are shown for each group, with the most time spent indicated with dark red and scaled through the least time spent represented with dark blue.
(E) Object recognition memory, which is less hippocampus-dependent, was unaltered by ELA or NRSF manipulation. Data in bar graphs of (C)–(E) are shown as the discrimination index: (time investigating novel location or object – time investigating familiar location or object)/(sum of time investigating both novel and familiar locations or objects).
(F–H) In contrast with the rescue of the impaired spatial memory, locomotion (open field test) (F), anxiety-like behaviors (elevated-plus maze) (G) and depressive-like behaviors (Porsolt forced-swim test) (H) were not affected by either the ELA or the ODN-NRSE treatment. Data are presented as mean ± SEM. *p < 0.05.
See also Figure S3.
Figure 5. ELA Induces an NRSF-Mediated Repression of Neuronal Maturation, and Blocking NRSF Enables Dendritic Arborization of Hippocampal Neurons.

(A) Sholl analyses of dorsal hippocampal pyramidal neurons in area CA1 of adult rats revealed that ELA stunted dendritic arborization (significant ELA × distance from soma interaction, F[38,494] = 1.90, p = 0.0013; post hoc, p < 0.05). Interfering with NRSF binding rescued neuronal dendritic complexity of ELA rats to control levels (significant NRSE × distance from soma interaction, F[38,494] = 2.46, p < 0.0001; post hoc, p < 0.05).
(B) A similar pattern of diminished dendritic complexity was observed in area CA3 of ELA rats (significant ELA × distance from soma interaction, F[54,702] = 1.60, p = 0.005; post hoc, p < 0.05).
(C) In the dentate gyrus, NRSE ODNs overall enhanced dendritic complexity (significant NRSE × distance from soma interaction, F[40,520] = 2.85, p < 0.0001), with no significant influence of ELA.
(D) Treatment of developing hippocampal neurons in vitro with NRSE ODNs, but not SCR ODNs, markedly increased dendritic complexity in pyramidal-like neurons (short/long axis ratio < 1.5; significant NRSE × distance from soma interaction, F[118,1593] = 1.79, p < 0.0001; post hoc, p < 0.05).
(E) NRSE ODNs labeled with BODIPY are observed inside the nuclei of cultured hippocampal neurons 40 h after a 2-h incubation with the ODNs, confirming that they are readily able to enter the cell and concentrate in the nucleus. Scale bar: 10 μm.
See also Figure S4.
We then tested adult ELA and control rats, with or without post hoc blockade of NRSF function, for memory performance. Memory for the location of a previously seen object was intact in CTL+SCR rats, which explored the object located to a novel area almost twice as long as the unmoved object (Figure 4C). There was a clear group effect on this function (significant ELA × NRSE interaction by two-way ANOVA; F[1,26] = 16.44, p = 0.0004). ELA led to profound deficits in spatial memory, as shown by a discrimination index near zero (Tukey’s post hoc test, ELA+SCR versus CTL+SCR, p < 0.05). Remarkably, blocking NRSF binding to the chromatin for a short period following the ELA experience restored memory up to control levels (post hoc, p < 0.05). We identified a significant reduction in spatial memory in CTL+NRSE rats (post hoc, p < 0.05) and excluded group differences in total exploration time of the two objects during the training or the testing sessions (Figure S3), or in locomotion (Figure 4F; all p values > 0.1).
To further probe the surprising rescue of ELA-induced enduring spatial memory deficits by a temporary interference with NRSF function, as well as the effect of NRSF blockade in controls, we conducted a second test that requires an intact hippocampus, the Y-maze (Conrad et al., 1996; Dellu et al., 1992). CTL+SCR rats performed well in this task (Figure 4D), whereas ELA+SCR rats exhibited marked memory deficits (significant ELA × NRSE interaction, F[1,25] = 7.26, p = 0.01; post hoc, p < 0.05). Blocking NRSF from binding to the chromatin completely prevented the ELA-induced spatial memory deficits (post hoc, p < 0.05). Notably, in the Y-maze, spatial memory of CTL+NRSE rats was intact, suggesting that NRSF blockade in controls may affect only the more rigorous hippocampus-dependent memory interrogated by the object location task.
The above data indicate that ELA generates profound deficits in spatial memory, and these are abrogated by blocking NRSF function. To test whether the influence of ELA and of NRSF blockade was selective to hippocampus-dependent memory, we evaluated rats using the object recognition test, which relies on broad limbic circuitry rather than the hippocampus specifically (Langston and Wood, 2010). In contrast with significant effects on object location and Y-maze performance, neither ELA nor blocking NRSF binding to chromatin influenced object recognition (Figure 4E; all p values > 0.3), because all groups exhibited intact object recognition memory.
We then delineated the spectrum of behavioral changes instigated by ELA and their response to transient interference with NRSF function. The use of standard tests of anxiety- and depressive-like behaviors demonstrated that neither ELA nor NRSF manipulations influenced any of these (open field: F[1,28] = 0.064, p = 0.94; elevated-plus maze: F[1,28] = 1.01, p = 0.32; Porsolt forced-swim: F[1,28] = 4.03, p = 0.054; Figures 4F–4H). These findings, in line with previous studies (Bolton et al., 2018; Brunson et al., 2005; Molet et al., 2016b), suggest that the impact of the type of ELA studied here on adult male rats is selective, with limited effects on measures of overt anxiety or learned helplessness.
Mechanisms of ELA-Induced Memory Deficits and Their Mitigation by Transient Interference with NRSF Functions
The enduring rescue of hippocampus-dependent memory by temporary interference with NRSF chromatin binding was striking. We reasoned that such an enduring effect must have resulted from the timing of the ELA, as well as of the NRSF-blocking intervention during a sensitive period for hippocampal neuronal maturation, including dendritic arborization and synapse formation. We first tested whether ELA led to a loss of CA1 pyramidal cells, a loss that might account for memory deficits. The number of pyramidal neurons in the stratum pyramidale of dorsal CA1 of the hippocampus was not altered by ELA (Figure S4), suggesting that the ELA-induced memory deficits were due to changes in the properties of these neurons, including the rewiring of hippocampal connectivity, rather than outright pyramidal cell loss. We then posited that ELA augmented NRSF-mediated repression of neuronal maturation, and blocking NRSF enabled expression of target neuronal genes during a period when their function was required for neuronal growth and the formation of mature dendritic trees. The latter is a prerequisite for normal neurotransmission and the execution of memory processes (Cline, 2001).
To test this possibility, we first analyzed the structure and dendritic arborization of hippocampal neurons in dorsal CA1, CA3, and the dentate gyrus and then employed in vitro methods to examine directly whether NRSF influenced dendritic length and arborization. Sholl analyses of dorsal hippocampal pyramidal neurons in area CA1 revealed a serious attenuation of dendritic arborization in ELA rats (significant ELA × distance from soma interaction, F[38,494] = 1.90, p = 0.0013; post hoc, p < 0.05; Figure 5A). Remarkably, the impoverished dendritic arborization was not diffuse or random. Rather, it was maximal at distances of ~180–360 μm from the cell body. This corresponds to stratum radiatum, a layer in which axons from area CA3 (Schaeffer collaterals) synapse onto CA1 neurons to carry out neurotransmission along the canonical hippocampal tri-synaptic pathway. Blocking NRSF function reversed the dendritic stunting of CA1 pyramidal cells by ELA (significant NRSE × distance from soma interaction, F[38,494] = 2.46, p < 0.0001; post hoc, p < 0.05). Consistent with the importance of the dendritic complexity of CA1 pyramidal cells in hippocampal function, a significant positive correlation (r[13] = 0.56, p < 0.05) was identified between the number of apical dendritic intersections at 320 μm away from the cell body (within the stratum radiatum) of an individual rat and the performance of the same rat in the hippocampus-dependent Y-maze task (i.e., the ratio of time spent in the novel versus familiar arms). Together, these data suggest that loss of neuronal structures mediating neurotransmission underlies ELA-induced memory deficits, and these structures were enduringly rescued by transient interference with NRSF function.
The structural changes induced by ELA were observed also in area CA3 pyramidal cells (significant ELA × distance from soma interaction, F[54,702] = 1.60, p = 0.005; post hoc, p < 0.05; Figure 5B). Notably, little effect of ELA was apparent in the apical dendritic arborization of the dentate gyrus granule cells (Figure 5C), which are largely born post-natally and mature later than the pyramidal cells (Bayer, 1980). Indeed, the interference with NRSF function, which took place following the ELA period, did influence apical dendritic arborization of the DG granule cells: NRSE ODNs overall enhanced dendritic complexity (significant NRSE × distance from soma interaction, F[40,520] = 2.85, p < 0.0001; Figure 5C). Together, the adult in vivo findings supported, but did not prove, an ELA-induced stunting of the differentiation of hippocampal neuron dendritic structure, a problem prevented by blocking NRSF actions. Therefore, to test directly whether NRSF stunts the development of hippocampal neuron dendritic trees, we cultured hippocampal neurons and applied the NRSF-blocking ODNs or random ODNs (SCR) onto the cultures (Andres et al., 2013; Noam et al., 2010; Patterson et al., 2017). Blocking the binding of NRSF to the chromatin, but not exposure to SCR ODNs, led to increased dendritic arborization in hippocampal pyramidal-like cells (short/long axis ratio < 1.5; significant NRSE × distance from soma interaction, F[118,1593] = 1.79, p < 0.0001; post hoc, p < 0.05; Figure 5D). The ability of the NRSE-ODNs to enter neurons and their nuclei was tested using ODNs rendered visible by the addition of the fluorescent dye boron-dipyrromethene (BODIPY) to the ODN molecules (Benniston and Copley, 2009; Hinkeldey et al., 2008; Patterson et al., 2017; Singh-Taylor et al., 2018). Figure 5E demonstrates that the ODNs readily enter the cell and appear to concentrate in neuronal nuclei.
DISCUSSION
Our principal findings are: (1) enduring impairments in hippocampus-dependent memory after a short period of ELA are associated with the repression of genes involved in neuronal maturation and function; (2) targets of GR and, unexpectedly, of the repressive transcription factor, NRSF, are enriched among downregulated hippocampal genes; (3) NRSF protein is increased in pyramidal neurons of the dorsal hippocampus shortly after ELA; (4) transient interference with the binding of NRSF to the chromatin selectively rescues hippocampal memory function and neuronal structure; and (5) NRSF directly represses dendritic arborization of hippocampal neurons. These discoveries identify an important, surprising contribution of NRSF to ELA-induced transcriptional processes that disrupt neuronal maturation and function. NRSF-mediated neuronal gene repression might be adaptive, promoting cell survival by reducing the high energetic cost of neuronal differentiation and activity.
Cognitive Deficits Resulting from ELA Are Accompanied by Major Changes in Dorsal Hippocampus Gene Expression
There is compelling human evidence for a major contribution of ELA to the burden of chronic cognitive disorders (Chen and Baram, 2016; Danese et al., 2017; Farah, 2018; Heckman, 2006; Kessler et al., 2010; Lupien et al., 2009; McLaughlin et al., 2019; Short and Baram, 2019), thus providing a strong impetus to uncover the responsible processes. We delineate here deficits in hippocampus-dependent spatial memory in adult rats that have experienced ELA, associated with significant and selective alteration of hippocampal gene expression profiles. These findings in an experimental system provide an opportunity to address key challenges to the understanding of the mechanisms of ELA-induced cognitive problems: What are the cellular processes triggered by ELA that result in memory deficits? How does a short period of adversity yield persistent cognitive dysfunction? And importantly, can we identify targets for prevention or intervention to mitigate the long-term consequences of ELA?
The observed memory disturbances in ELA rats were selective to spatial memory tasks that require an intact dorsal hippocampus (Chen et al., 2016; Haettig et al., 2013; Langston and Wood, 2010; Molet et al., 2016a). Dorsal hippocampus expression of genes encoding proteins linked to dendritic maturation and neurotransmission was reduced, plausibly contributing to the observed structural and functional impairments (Figures 1, 4, and 5). In addition, altered expression of several transcription factors was noted, which might maintain the persistent gene expression changes set in motion by the transient epoch of ELA (Table S1). For example, upregulated transcription factors included Tbr1 and Auts2, involved in cognitive disorders and autism (Gao et al., 2014; Huang et al., 2014), and downregulated transcription factors included Foxp1, Satb2, and Npas4, implicated in neuronal differentiation and neurotransmission (Bacon et al., 2015; Bloodgood et al., 2013; Britanova et al., 2008). Yet, even if these gene expression changes were to be causal, the large number of repressed and upregulated genes after ELA suggested that intervention at the level of individual genes might not be useful for ameliorating ELA-induced cognitive problems. Therefore, we searched instead for “upstream” regulators that might orchestrate ELA-induced transcriptional programs at a genomic scale and might thus serve as potential targets for intervention.
GR and NRSF Contribute to Gene Expression Profile Changes in ELA Hippocampus
In silico searches for potential transcriptional regulators of the ELA-induced changes in gene expression profiles identified two prominent candidates. First, as expected, we identified an enrichment in targets of GR, a transcriptional regulator that is sensitive to stress and the levels of corticosterone/cortisol (Kino, 2017). Surprisingly, we also discovered a highly significant enrichment of NRSF targets, specifically among the downregulated, but not upregulated, genes. This suggested that NRSF, a repressor that is generally considered to play a minor role in mature neurons, might also contribute to the mechanisms of ELA-induced memory problems. Of interest, the expression of both GR and NRSF is governed, in part, by a microRNA, miR-124 (Brennan et al., 2016; Conaco et al., 2006). Therefore, we analyzed the levels of this microRNA in the dorsal hippocampus of control and ELA-experiencing rat pups as a potential common upstream regulator of GR and NRSF. We found no evidence of a reduction of miR-124 levels, which would be expected to account for augmented GR and NRSF expression (CTL: 1.064 ± 0.146, n = 6; ELA: 1.482 ± 0.321, n = 5; p = 0.24). This is not surprising because, as noted above, enhanced repression of NRSF targets was the result of an increase in its protein levels selectively in hippocampal pyramidal cells and in chromatin binding, rather than augmented mRNA levels throughout the hippocampus.
NRSF is a zinc-finger transcription factor that binds to a conserved 21-bp target sequence (NRSE) and induces transcriptional repression in concert with a set of corepressors and chromatin-modifying enzymes (e.g., CoRest, Sin3a, MeCP2, HDACs). NRSF expression was originally described in non-neuronal tissues, where it suppresses neuron-specific genes (Ballas et al., 2001; Chen et al., 1998; Gao et al., 2011; Schoenherr and Anderson, 1995), indicating that many neuronal genes must carry NRSEs and are repressed by augmented NRSF levels (Ballas et al., 2005; McClelland et al., 2014). NRSF expression in mature neurons has been described, and the factor might be especially important for developing neurons, where expression of NRSF-regulated genes contributes to several aspects of maturation, including the development of excitatory synapses (Mandel et al., 2011). In this scenario, low levels of NRSF may be crucial for normal function, perhaps by constraining dendritic growth (Patterson et al., 2017; Singh-Taylor et al., 2018; Yang et al., 2012). This notion is supported by the current findings. First, blocking of NRSF function led to exuberant growth of dendritic trees of developing neurons both in vivo (Figure 5C) and in vitro (Figure 5D). Second, a transient block of NRSF function in immature control rats (post-natal days ~10–12) led to reduced memory function in the object location task (although not in the Y-maze, a less-taxing spatial memory task) during adulthood (Figures 4C and 4D). This pattern, observed also in other contexts, is consistent with a need for low levels of NRSF function for the maturation of the developing memory systems (Hsieh and Gage, 2004; Patterson et al., 2017). Indeed, dysregulation of NRSF-mediated gene silencing has recently been found to play a role in several pathological contexts, including neurodegeneration (Hwang and Zukin, 2018; Lu et al., 2014) and epilepsy (Delahaye-Duriez et al., 2016; McClelland et al., 2011b, 2014).
The Timing of ELA during Sensitive Developmental Periods Imprints NRSF-Dependent Structural Changes in the Dorsal Hippocampus
We identified here a specific pattern of hippocampal neuron structural stunting that reflects the developmental timing of the ELA and of the post hoc NRSF blocking. In CA1 pyramidal cells, the loss of dendritic arborization centered on the hippocampal stratum radiatum, a site of synaptic targets of axons from area CA3 and a pattern identified before (Brunson et al., 2005; Ivy et al., 2010; Patterson et al., 2017). These CA1 neurons, their dendrites, and their synapses actively develop during the first post-natal week in the rat (Tremblay et al., 1988; Tyzio et al., 1999), following the slightly earlier development of CA3 pyramidal cells. Both CA1 and CA3 neurons were affected by ELA, and it is likely that the arborization of these neurons requires the expression of NRSF target genes, because blocking NRSF function following ELA prevented dendritic impoverishment of these neurons. In contrast with pyramidal neurons, dentate gyrus granule cells are born and mature later (Bayer, 1980). Accordingly, the ELA-induced increase of NRSF-mediated repression did not impact granule cell dendritic structure. In contrast, blocking NRSF during the days that followed the ELA (well within the second post-natal week) led to exuberant dendritic arborization in these neurons.
The timing of the intervention should also be noted: we prevented NRSF binding to the chromatin early, immediately following the ELA period, a time point when NRSF protein levels were increased in pyramidal neurons of the dorsal hippocampus (Figures 3E and 3F). Although we were technically unable to demonstrate augmented binding of NRSF to the chromatin, a transient augmentation of its binding by diverse experiences was identified in both developing (Korosi et al., 2010; Singh-Taylor et al., 2018) and mature (McClelland et al., 2011b) brain. The transient binding of NRSF to one or multiple NRSEs led to the recruitment of co-factors such as MeCP2 and persistent changes in histone methylation state at the NRSE sites (e.g., H3K9me2), with likely consequent changes in chromatin conformation. These findings suggest that interference with NRSF binding to the chromatin during adulthood is unlikely to provide protection from the effects of ELA on memory. Whether GR might provide a therapeutic target in the adult remains unknown.
In summary, the findings described here establish a critical, unexpected contribution of NRSF to ELA-induced hippocampal transcriptional dysregulation, which disrupts neuronal maturation and leads to hippocampus-dependent memory deficits. From an evolutionary standpoint, both GR and NRSF may play an adaptive role: GR regulates a panoply of gene expression changes aiming to prepare for future adversity. In parallel, NRSF-mediated repression of neuron-specific genes should reduce the high metabolic demand involved in neurotransmission and maintenance of neuronal membrane potential, thus promoting cell survival. These transient developmental actions of NRSF suggest a window and a target for interventions aiming to mitigate the persistent cognitive deficits resulting from ELA.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tallie Z. Baram (tallie@uci.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession number for the RNA-seq data reported in this paper is GEO: GSE161498. This study did not generate code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experiments were performed in accordance with National Institutes of Health (NIH) guidelines and were approved by the UC Irvine animal care and use committee. All analyses were performed without knowledge of treatment group.
Animals
Primiparous time-pregnant Sprague Dawley dams gave birth in a temperature-controlled, quiet, and uncrowded vivarium and were maintained on 12-hr light/dark cycles and with unlimited access to chow and water. Pups were mixed among litters and adjusted to 12 per dam to obviate the potential confounding effect of genetic variables and litter size. After weaning at postnatal day (P)21, male rats were housed 2–3 per cage.
Chronic early-life adversity
Early-life adversity was induced using a previously described protocol (Avishai-Eliner et al., 2001; Gilles et al., 1996; Molet et al., 2014). Briefly, on P2, dams and pups randomly assigned to the early-adversity group were transferred to a cage fitted with a plastic-coated mesh platform sitting ~2.5 cm above the cage floor. Bedding was limited to sparsely cover the cage floor, and one-half of a paper towel was provided for nesting material, creating a limited bedding and nesting environment (LBN). Cages were maintained in a temperature-controlled room with a laminar flow to prevent ammonia accumulation. Control dams and pups resided in standard-bedded cages, containing ~0.33 cubic feet of corn cob bedding and one paper towel. Control and LBN cages were undisturbed during P2-P9. Maternal behaviors were monitored during the week of adversity as previously described (Ivy et al., 2008; Molet et al., 2016b). This experience generates significant chronic stress in the pups, measured by increased corticosterone levels and adrenal size, that both return to normal by adulthood (Brunson et al., 2005). The stress likely arises because of abnormal maternal behaviors provoked by the simulated poverty (Molet et al., 2014, 2016b). Notably, hypothermia and inanition are not observed (Bolton et al., 2019; Molet et al., 2014).
METHOD DETAILS
RNA preparation and sequencing
Dorsal hippocampi were obtained from 8-week-old male rats that were subjected to either control rearing conditions (n = 4) or ELA (n = 5), and did not undergo behavioral testing. RNA was isolated using the QIAGEN RNeasy Kit, according to the manufacturer’s instructions. Libraries were constructed using the Illumina TruSeq v2 kit with minor modifications. Briefly, 1 μg of total RNA was used as input, and poly(A) selection was carried out according to the protocol. Modifications to the TruSeq v2 protocol included a fragmentation time of 3 minutes. Additionally, 8 cycles of PCR were run for the enrichment of DNA fragments. Adapters were chosen according to protocol recommendations. Quantification of the libraries was accomplished using the KAPA qPCR kit for Illumina libraries. Sequencing was performed on an Illumina HiSeq 2500 system in rapid-run mode with paired-end 150 cycles and version 3 reagents. The libraries were clustered at 14 pM on the cBot machine using the Duo loading protocol. Pass-filter percentage of the overall 190 million paired-end reads was 94.2.
RNA-seq data analysis
About 20 million raw de-multiplexed reads were obtained for each sample and were mapped to the rat reference genome (rn6 genome assembly, Ensembl release 84) using STAR (Dobin et al., 2013), version 2.5.0., with ENCODE settings for long RNA-seq: –outFilterType BySJout–outFilterMultimapNmax 20–alignSJoverhangMin 8–alignSJDBoverhangMin 1–alignIntronMin 20–alignIntronMax 1000000–alignMatesGapMax 1000000. Gene models were obtained from: http://ftp.ensembl.org/pub/release-84/gtf/rattus_norvegicus. We then counted the number of reads corresponding to each mRNA using the htseq-count function of the Python program HTSeq (Anders et al., 2015) using only reads that uniquely mapped onto exons. The obtained read counts were normalized and analyzed for differential gene expression using the bioconductor package DESeq2 (Love et al., 2014). Statistical analyses employed the Benjamini-Hochberg correction to calculate false-discovery rates (FDR). Both FDR < 0.05 and < 0.1 were used for significance.
Gene ontology
Analyses of enriched gene ontology terms for significantly up- and downregulated genes employed the Bioconductor packages topGO and org.Rn.eg.db. The background gene set was defined as all genes expressed above the independent filtering threshold for low counts imposed by DESeq2 (mean normalized count > 33 for reads aligned with STAR). Over-representation was assessed using Fisher’s Exact Test. When redundant ontology terms were present, the most specific of the terms was plotted.
Analysis of transcription factor target enrichment
Analysis of enrichment for transcription factor targets relied on ChIP-seq data available from the Encyclopedia of DNA Elements (Dunham et al., 2012), using Enrichr for functional enrichment analysis (Kuleshov et al., 2016) (https://maayanlab.cloud/Enrichr/). We assigned a gene as a target based on published ChIP-seq datasets (Johnson et al., 2007; Polman et al., 2013), and tested for enrichment using Fisher’s Exact Test. Additional NRSF targets were assigned based on recently published rat ChIP-seq data from hippocampal progenitors (Mukherjee et al., 2016) (Figure 3).
qRT-PCR
Total RNA was isolated from the hippocampus of adult rats using the Direct-zol RNA purification kit (Zymo Research, Irvine, CA) and reverse-transcribed using the Transcriptor first strand cDNA synthesis kit (Roche) with oligo d(T) and random hexamer primers. Primer sequences are provided in Table S5. Samples were run in triplicate on a Roche Lightcycler 96 system, followed by relative quantification using the 2ˆ-ΔΔCt method.
NRSF Immunohistochemistry (IHC)
P12 male rats (n = 5/group) were euthanized with sodium pentobarbital and transcardially perfused with ice-cold phosphate-buffered saline (PBS; pH = 7.4) followed by 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH = 7.4). Brains were cryoprotected and frozen, then sectioned coronally into 20-μm-thick slices using a cryostat (1:5 series). Sections were subjected to NRSF-IHC using standard avidin-biotin complex methods, as described previously (Chen et al., 2001). Briefly, after several washes with PBS containing 0.3% Triton X-100 (PBS-T, pH 7.4), sections were treated with 0.3% H2O2/PBS for 30 min, then blocked with 5% normal goat serum (NGS) for 30 min to prevent non-specific binding. After rinsing, sections were incubated for 3 days at 4°C with rabbit anti-NRSF/REST antiserum (1:5,000, Bethyl Laboratories, Montgomery, TX) in PBS containing 1% BSA and washed in PBS-T (3 × 5 min). Sections were incubated with biotinylated goat-anti-rabbit IgG (1:400, Vector Laboratories, Burlingame, CA) for 2 hours at room temperature. After washing (3 × 5 min), sections were incubated with the avidin-biotin-peroxidase complex solution (1:200, Vector) for 3 hours, rinsed (3 × 5 min), and reacted with 3,3′-diaminobenzidine (DAB) containing H2O2 (Bioenno Tech, Santa Ana, CA) for 10 min.
Oligodeoxynucleotide administration in vivo
To block the binding of NRSF to its NRSE target sequence, we synthesized replicates of the NRSE as previously described (McClelland et al., 2011b), and introduced phosphothioate bond modifications into their backbone for stability. We administered these NRSE decoy oligodeoxynucleotides (NRSE-ODNs, 5′-GGAGCTGTCCACAGTTCTGAA-3′) into the lateral ventricles (i.c.v., see Figure S2; by using bregma demarcations, which are visible through the skin in a neonatal rat pup) of LBN and control rats. We used 0.25 nmol of ODNs diluted in 1 μL 0.25N NaCl per side, and administered the ODNs bilaterally twice, on P10 and on P11. As a control, we used scrambled (SCR) ODNs (5′-AGGTCGTACGTTAATCGTCGC-3′). Two outcome measures were investigated: First, we examined the efficacy of this intervention in blocking NRSF binding (target engagement) or H3K9me2 binding (a repressive histone modification) at the ODN dose used here. Then in a long-term experiment run on four cohorts, rats survived to adulthood and the effects of blocking NRSF function on outcome measures such as behavior were assessed. All experiments included four groups (CTL+SCR, CTL+NRSE, LBN+SCR, and LBN+NRSE, n = 8/group).
Chromatin Immunoprecipitation (ChIP)
We examined if NRSF binding to target genes is increased after early-life adversity, and if this is blocked by the NRSE-ODNs that were administered immediately after the LBN period. We investigated NRSF occupancy at its target sites on the first intron of neuronal PAS domain protein 4 (Npas4), a gene containing four NRSEs (McClelland et al., 2014; Singh-Taylor et al., 2018) that was repressed (p = 0.001; adjusted p = 0.08) in the RNA-seq screen. We also examined if the binding to Npas4 of H3K9me2, a known repressive histone modification, is altered by early-life adversity and NRSE-ODN treatment.
We employed a validated ChIP method (McClelland et al., 2011b, 2014; Singh-Taylor et al., 2018), with several modifications. Hippocampi were dissociated with an 18G needle and cross-linked with 1% formaldehyde for 10 min at room temperature in PBS. Neutralization of cross-linking was achieved with the addition of glycine. Pelleted tissue was dissociated in homogenization buffer (50 mM HEPES pH 8.0, 140 mM NaCl, 1 mM EDTA, 0.4% Igepel CA-630, 0.2% Triton X-100, and a cocktail of protease inhibitors), and then in Nuclear wash buffer (20 M mM Tris-Cl, pH 8.0, 0.15 M NaCl, and 1 mM EDTA), and centrifugation aided in nuclei collection. Nuclei were sonicated for 10 min using a Diagenode Bioruptor (Denville, NJ) to make genomic DNA of an average size of 500 bp. After cellular debris was removed, supernatant of homogenate was precleared overnight with Protein-A/G Beads (Santa Cruz, Dallas, TX) at 4°C, and then incubated with 5 μg of either control non-immune IgG (Cell Signaling, Danvers, MA) or anti-NRSF (Santa Cruz, Dallas, TX) or anti-Histone 3 Lysine 9 dimethyl antibody (Abcam, Cambridge, MA) overnight at 4°C in buffer containing 16.7 mM Tris-Cl, pH 8.0, 167 mM NaCl, 1.2 mM EDTA, 1.1% Triton X-100, and protease inhibitors. Protein A/G beads blocked with salmon sperm DNA (400 μg/ml) and BSA (400 μg/ml) were added to lysate for 2 hr. Beads were washed twice with the following buffers: low-salt wash buffer, high-salt wash buffer, LiCl wash buffer and TE, and then eluted using a buffer containing 2% SDS and 0.2 M sodium carbonate. After reversal of cross-linking at 65°C overnight, the bound DNA was purified and eluted using the MiniElute PCR purification kit (QIAGEN). Quantitative QCR (qPCR) amplification was done using SYBR green chemistry (Roche) on a Lightcycler 96 (Roche) with primers specific for NRSE regions of the Npas4 gene, or for H3K9me2 binding sites in the promoter region (±1kb TSS) of the Npas4 gene. NRSF or H3K9me2 binding was shown as a percentage of input after subtraction of non-specific binding to IgG-coupled beads. Primer sequences are provided in Table S6.
Object location memory task
We analyzed hippocampus-dependent memory using the object location memory task (n = 8/group; 2 cohorts), which consisted of two phases conducted over two days: a training session (on P62–65) and a testing phase (24 hr after training) (Molet et al., 2016a). All rats were handled daily for six days and habituated to the experimental apparatus for two days (10 min/day) prior to testing, and importantly, rats from each of the experimental groups (CTL+SCR, CTL+NRSE, LBN+SCR, LBN+NRSE) were run in parallel and at the same time of day. The experimental apparatus was a transparent plastic cage, coated with opaque paper and a visual cue (blue stripe) located on one side wall, and placed in a quiet, dimly lit testing room.
During the training session on day 1, rats were placed in the experimental apparatus with two identical objects (100-mL PYREX® beakers) and were allowed to explore the objects for 10 minutes. All objects were cleaned with 70% ethanol between trials. During the testing session on day 2, rats were presented with the two familiar objects from the training session with one object moved to a novel location, and were allowed to explore the two objects for 5 min. Both training and testing phases were video-recorded using an overhead camera, and the duration of exploration of each object (touching the object with the nose or sniffing with the nose < 2 cm from the object) as well as total object exploration was scored blindly without knowledge of the experimental groups. To assess location memory of the familiar object, exploration times for the novel (N) and familiar (F) objects were used to calculate a discrimination ratio (N/F) or a discrimination index (N-F/N+F).
Y-maze spatial memory task
As an independent test of hippocampus-dependent memory in two separate cohorts of animals (n = 4 per group per cohort, final n = 8/group; all groups represented and run in parallel), we employed the Y-maze task (Conrad et al., 1996; Dellu et al., 1992). All rats were handled daily for six days prior to testing. During training, 8-week-old rats were placed in the Y-maze apparatus (novel/familiar arms: 30 × 14.3 × 15 cm; home arm: 46 × 14.3 × 15 cm) with one arm blocked by an opaque plexi-glass partition. The rats were allowed to explore for 10 minutes. During the testing session 4 hr later, rats were placed in the Y-maze with both arms open to access for 5 minutes. In-between tests, the Y-maze apparatus was cleaned with 10% ethanol. Training and testing phases were video-recorded using an overhead camera, and the duration of exploration of each arm (starting and ending when the head of the rat crosses the arm threshold) were automatically analyzed by a computerized video tracking system (Noldus Ethovision). To assess spatial memory of the familiar arm, exploration times for the novel (N) and familiar (F) arms were used to calculate a discrimination index (N-F/N+F).
Object recognition memory task
The same rats that were tested in the Y-maze were also tested one week later in the object recognition memory task, which is not hippocampus-dependent (Langston and Wood, 2010; Molet et al., 2016a). Prior to testing, all rats were habituated to the experimental apparatus (same as that used for the object location memory task) for two days (10 min/day). During training, 9-week-old rats were placed in the experimental apparatus with two identical objects: a yellow radioactivity container or a brown glass bottle (objects were counterbalanced across groups), and they were allowed to explore the objects for 10 min. In-between trials, all objects were cleaned with 70% ethanol, and the experimental apparatus was cleaned with 10% ethanol. During the testing session 24 hr later, rats were placed in the experimental apparatus and allowed to explore two objects for 5 min. One object was the familiar object from the training session and the other, a novel object (yellow radioactivity container or brown glass bottle; which object was replaced was counterbalanced across groups). Training and testing phases were video-recorded using an overhead camera, and the duration of exploration of each object (touching the object with the nose or sniffing with the nose < 2 cm from the object) was scored without knowledge of the group. To assess recognition memory of the familiar object, exploration times for the novel (N) and familiar (F) objects were used to calculate a discrimination ratio (N/F) or a discrimination index (N-F/N+F).
Assessment of depressive- and anxiety-like behaviors
To analyze behaviors considered indicative of emotional states, we used the forced-swim test as a measure of depressive-like behavior, and the elevated-plus maze and open-field tests for anxiety-like behavior. For the latter tests, a computerized video tracking system (Noldus Ethovision) was used to calculate the time spent in the open “anxiogenic” regions of the apparatus, as well as the distance traveled. All tests were conducted in a quiet, empty and dimly lit room with no visual cues to distract the tested rat. All measurements and analyses were carried out without knowledge of treatment group.
Open field test:
The open field consisted of an open field box (100 × 100 cm) with black opaque walls and floor. Rats (P45–46) were placed in one corner of the open field, facing the wall at the start of the experiment. Each rat was allowed one 10-min trial and the box was cleaned with 10% ethanol after each trial. Duration of time spent in the center of the open field was used as an index of anxiety-like behavior, and total distance traveled was used a measure of locomotor activity.
Elevated-plus maze test:
The elevated-plus maze test consisted of two open arms (50 × 10 cm) and two cross-wise closed arms (50 × 10 × 40 cm; black opaque walls) with an open roof 50 cm above the floor. Rats (P48–51) were placed in the center of the maze, facing an enclosed arm at the start of the experiment. Each rat was allowed one 5-min trial in the maze, and the maze was wiped with 10% ethanol after each trial. The time on open arms was used as an index of anxiety-like behavior.
Porsolt forced-swim test:
The test consists of two sessions separated by 24 hr. The habituation session (Day 1), lasted 15 min. Rats (P50–52) were placed in a glass cylinder (20 cm in diameter and 60 cm high) containing water (23–25°C) filled to a depth of 45 cm. The test session occurred 24 hr later, and rats were placed in the cylinder for 5 min. Behavior was monitored using a video camera. The duration of immobility was scored and served as an indicator of depressive-like behaviors. Water was replaced and containers cleaned between trials.
Golgi method and Sholl analyses
Rats (~4 months old; n = 4–5/group from two separate cohorts) were sacrificed via rapid decapitation, at least 1 month after the object recognition memory test. Brains were immediately removed and immersed in impregnation solution (superGolgi Kit; Bioenno Lifesciences, Santa Ana, CA) for 10–11 days at room temperature in the dark, then sectioned into 200-μm coronal slices and mounted on gelatin-coated slides. After drying overnight, slices were stained according to the manufacturer’s instructions (superGolgi Kit; Bioenno). After washing and drying the slides thoroughly, sections were dehydrated with 100% EtOH and xylenes and coverslipped with Permount. Golgi analyses were conducted without knowledge of treatment groups at the same level in dorsal hippocampus for all animals and groups. Sholl analyses of dorsal hippocampal CA1 and CA3 cells were completed on fully impregnated pyramidal neurons (CA1: n = 103 neurons total, ~6 neurons/animal; CA3: n = 97 neurons total, ~6 neurons/animal). In the dentate gyrus (DG), granule cells were selected for analysis based on their structure (small soma with traceable apical processes and few basal processes) and their location. Cells were counted in both the upper and lower blades of the hilus of the dorsal hippocampus per animal (n = 151 neurons total, ~6 neurons/blade/animal) using ImageJ software (U.S. National Institutes of Health, Bethesda, MD).
Hippocampal neuron culture experiments
Dissociated hippocampal primary cultures were prepared from P0 Sprague-Dawley rats as previously described (Noam et al., 2010). Briefly, hippocampi were quickly dissected, meninges removed, and tissue incubated for 30 min in buffered salt solution containing 10 units/ml papain (Worthington). After removal of the papain, cells were mechanically triturated and plated at a density of 400–600 cells/mm2 on 12-mm coverslips that were precoated with poly-D-lysine (Sigma). Cultures were initially maintained in Neurobasal Medium with B-27 supplement (Invitrogen) at 36°C and 5% CO2. 3–4 hr after plating, half of the culture medium was replaced with a Neurobasal Medium/B-27-based medium that was preconditioned for 24 hr by 1–2 week-old non-neuronal cell cultures prepared from P3–4 rat cortices. Cultures were subsequently refreshed every 3–4 days with the conditioned medium. On the third day in vitro (DIV 3), 1 μM cytosine-arabinoside (Sigma, St. Louis, MO) was added to the culture medium to inhibit glial proliferation.
To examine the role of NRSF in constraining dendritic growth and arborization, cultured hippocampal neurons were treated with SCR or NRSE ODNs (0.1 μM) or normal medium (naive group) on DIV 7. New ODNs were added to the cultures each time they were refreshed. Cultures were collected on DIV 21 by fixing them with fresh ice-cold 4% PFA (Fisher) and affixing coverslips to precoated microscope slices with mounting media (Southern Biotech, Birmingham, AL). Sholl analyses were performed on presumed pyramidal cells (long/short radius < 1.5).
For ODN visualization experiments, DIV 17 hippocampal neurons in culture were exposed to 0.5 μM BODIPY-linked NRSE ODNs. BODIPY is a small, fluorescent molecule that allows for visualization of the ODN without structurally interfering with its transportation across membranes (Benniston and Copley, 2009; Hinkeldey et al., 2008). Cells were incubated with the tagged ODNs for 2 hr, then washed with culture medium. At 40 hr post-wash, cells were imaged live using a 40x objective and a CCD monochrome 12-bit camera (Retiga 2000R; Qimaging), acquired with NIS-Elements-D software (Nikon).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical considerations and methods of analysis
Differences after ELA (i.e., object location and objection recognition memory; distance traveled and time spent in center in open field) were assessed using unpaired t-tests, with Welch’s correction for unequal variance as necessary, or one-sample t-tests (i.e., qPCR validation). Differences after ELA and NRSF intervention (i.e., object location and object recognition memory, Y-maze, forced-swim, elevated-plus maze, and open-field tests, as well as NRSF and H3K9me2 ChIP, were analyzed by two-way ANOVA (ELA X NRSE) followed by Tukey’s post hoc tests. Sholl analysis results were assessed by three-way ANOVA (ELA X NRSE X Distance from Soma) for in vivo data or one-way ANOVA for in vitro data, followed by Tukey’s post hoc tests. Significance levels were set at 0.05, and data are presented as mean ± SEM. Grubbs’ test was used to remove statistical outliers from the data, and technical outliers were removed if rats failed to explore objects or arms of the Y-maze sufficiently during training or testing (i.e., < 20 s). Statistical analyses were performed using GraphPad Prism 6.0 software (GraphPad, San Diego, CA, USA). All experiments were assessed blindly without prior knowledge of the experimental group.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-REST/NRSF IHC antibody | Bethyl Laboratories | Cat# IHC-00141; RRID: AB_2285179 |
| Anti-Histone H3 (dimethyl K9) antibody | Abcam | Cat# ab1220; RRID: AB_449854 |
| Anti-NRSF (H-290) ChIP antibody | Santa Cruz | Cat# sc-25398; RRID: AB_2179625 |
| Critical Commercial Assays | ||
| superGolgi Kit | Bioenno Lifesciences | Cat# 003010 |
| Deposited Data | ||
| Raw and analyzed RNA-seq data from hippocampus | This paper | GEO: GSE161498 |
| GR ChIP-seq dataset from hippocampus | (Polman et al., 2013) | GEO: GSE84202 |
| NRSF ChIP-seq dataset from cell line | (Johnson et al., 2007) | GEO: GSE13047 |
| NRSF ChIP-seq dataset from hippocampal progenitors | (Mukherjee et al., 2016) | GEO: GSE70695 |
| Experimental Models: Organisms/Strains | ||
| Rat: Hsd:Sprague-Dawley SD | Envigo | Product code #002 |
| Oligonucleotides | ||
| NRSE-ODNs: 5′-GGAGCTGTCCACA GTTCTGAA-3′ | Sigma-Aldrich | N/A |
| SCR-ODNs: 5′-AGGTCGTACGTTA ATCGTCGC-3′ | Sigma-Aldrich | N/A |
| Primers for qRT-PCR (see Table S5) | IDT | N/A |
| Npas4 Primers for ChIP (see Table S6) | IDT | N/A |
| Software and Algorithms | ||
| Rat genome (rn6) and annotation | Ensembl release 84 | N/A |
| STAR RNA-seq read aligner, v2.5.0 | Dobin et al., 2013 | N/A |
| Python program HTSeq | Anders et al., 2015 | N/A |
| Bioconductor package DESeq2 | Love et al., 2014 | N/A |
| Bioconductor package org.Rn.eg.db | https://bioconductor.org/packages/release/data/annotation/html/org.Rn.eg.db.html | N/A |
| Bioconductor package topGO | https://bioconductor.org/packages/release/bioc/html/topGO.html | N/A |
| Enrichr (https://maayanlab.doud/Enrichr/) | Kuleshov et al., 2016 | N/A |
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
Highlights.
Hippocampus-dependent memory is impaired long term by early-life adversity (ELA)
Large-scale repression of key neuronal genes and neuronal stunting are found
Target genes of GR and of the repressor NRSF/REST are enriched
Transient block of NRSF function prevents ELA-induced memory and neuronal deficits
ACKNOWLEDGMENTS
This work was supported by the NIH (grants P50 MH096889, NS28912, MH73136, and K99 MH120327) and the Hewitt Foundation for Biomedical Research (United States). We thank the high-throughput genomics facility at UC Irvine for library preparation and RNA-seq. We also thank Erin Jackson, Stephanie Min Law, and Jennifer Daglian for excellent technical assistance.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108511.
REFERENCES
- Abrajano JJ, Qureshi IA, Gokhan S, Zheng D, Bergman A, and Mehler MF (2009). REST and CoREST modulate neuronal subtype specification, maturation and maintenance. PLoS ONE 4, e7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres AL, Regev L, Phi L, Seese RR, Chen Y, Gall CM, and Baram TZ (2013). NMDA receptor activation and calpain contribute to disruption of dendritic spines by the stress neuropeptide CRH. J. Neurosci. 33, 16945–16960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett MG, Pan MS, Doak W, Cyr PEP, Muglia LM, and Muglia LJ (2015). The role of glucocorticoid receptor-dependent activity in the amygdala central nucleus and reversibility of early-life stress programmed behavior. Transl. Psychiatry 5, e542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arp JM, Ter Horst JP, Loi M, den Blaauwen J, Bangert E, Fernández G, Joëls M, Oitzl MS, and Krugers HJ (2016). Blocking glucocorticoid receptors at adolescent age prevents enhanced freezing between repeated cue-exposures after conditioned fear in adult mice raised under chronic early life stress. Neurobiol. Learn. Mem. 133, 30–38. [DOI] [PubMed] [Google Scholar]
- Avishai-Eliner S, Eghbal-Ahmadi M, Tabachnik E, Brunson KL, and Baram TZ (2001). Down-regulation of hypothalamic corticotropin-releasing hormone messenger ribonucleic acid (mRNA) precedes early-life experience-induced changes in hippocampal glucocorticoid receptor mRNA. Endocrinology 142, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon C, Schneider M, Le Magueresse C, Froehlich H, Sticht C, Gluch C, Monyer H, and Rappold GA (2015). Brain-specific Foxp1 deletion impairs neuronal development and causes autistic-like behaviour. Mol. Psychiatry 20, 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL (2015). Epigenetic and transgenerational reprogramming of brain development. Nat. Rev. Neurosci. 16, 332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, Nemeroff CB, Reyes TM, Simerly RB, Susser ES, and Nestler EJ (2010). Early life programming and neurodevelopmental disorders. Biol. Psychiatry 68, 314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, et al. (2001). Regulation of neuronal traits by a novel transcriptional complex. Neuron 31, 353–365. [DOI] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, and Mandel G (2005). REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121, 645–657. [DOI] [PubMed] [Google Scholar]
- Bath KG (2020). Synthesizing Views to Understand Sex Differences in Response to Early Life Adversity. Trends Neurosci. 43, 300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bath KG, Nitenson AS, Lichtman E, Lopez C, Chen W, Gallo M, Goodwill H, and Manzano-Nieves G (2017). Early life stress leads to developmental and sex selective effects on performance in a novel object placement task. Neurobiol. Stress 7, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA (1980). Development of the hippocampal region in the rat. II. Morphogenesis during embryonic and early postnatal life. J. Comp. Neurol. 190, 115–134. [DOI] [PubMed] [Google Scholar]
- Benniston AC, and Copley G (2009). Lighting the way ahead with boron dipyrromethene (Bodipy) dyes. Phys. Chem. Chem. Phys. 11, 4124–4131. [DOI] [PubMed] [Google Scholar]
- Binder EB (2009). The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 34 (Suppl 1), S186–S195. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sharma N, Browne HA, Trepman AZ, and Greenberg ME (2013). The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 503, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton JL, Molet J, Ivy A, and Baram TZ (2017). New insights into early-life stress and behavioral outcomes. Curr. Opin. Behav. Sci. 14, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton JL, Molet J, Regev L, Chen Y, Rismanchi N, Haddad E, Yang DZ, Obenaus A, and Baram TZ (2018). Anhedonia Following Early-Life Adversity Involves Aberrant Interaction of Reward and Anxiety Circuits and Is Reversed by Partial Silencing of Amygdala Corticotropin-Releasing Hormone Gene. Biol. Psychiatry 83, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton JL, Short AK, Simeone KA, Daglian J, and Baram TZ (2019). Programming of Stress-Sensitive Neurons and Circuits by Early-Life Experiences. Front. Behav. Neurosci. 13, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan GP, Dey D, Chen Y, Patterson KP, Magnetta EJ, Hall AM, Dube CM, Mei YT, and Baram TZ (2016). Dual and Opposing Roles of MicroRNA-124 in Epilepsy Are Mediated through Inflammatory and NRSF-Dependent Gene Networks. Cell Rep. 14, 2402–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, et al. (2008). Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron 57, 378–392. [DOI] [PubMed] [Google Scholar]
- Brunson KL, Kramár E, Lin B, Chen Y, Colgin LL, Yanagihara TK, Lynch G, and Baram TZ (2005). Mechanisms of late-onset cognitive decline after early-life stress. J. Neurosci. 25, 9328–9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S, Aurbach EL, Sharma V, Blandino P Jr., Turner CA, Watson SJ, and Akil H (2014). FGF2 is a target and a trigger of epigenetic mechanisms associated with differences in emotionality: partnership with H3K9me3. Proc. Natl. Acad. Sci. USA 111, 11834–11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, and Baram TZ (2016). Toward understanding how early-life stress reprograms cognitive and emotional brain networks. Neuropsychopharmacology 41, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z-F, Paquette AJ, and Anderson DJ (1998). NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 20, 136–142. [DOI] [PubMed] [Google Scholar]
- Chen Y, Bender RA, Frotscher M, and Baram TZ (2001). Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J. Neurosci. 21, 7171–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Molet J, Lauterborn JC, Trieu BH, Bolton JL, Patterson KP, Gall CM, Lynch G, and Baram TZ (2016). Converging, synergistic actions of multiple stress hormones mediate enduring memory impairments after acute simultaneous stresses. J. Neurosci. 36, 11295–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline HT (2001). Dendritic arbor development and synaptogenesis. Curr. Opin. Neurobiol. 11, 118–126. [DOI] [PubMed] [Google Scholar]
- Conaco C, Otto S, Han J-J, and Mandel G (2006). Reciprocal actions of REST and a microRNA promote neuronal identity. Proc. Natl. Acad. Sci. USA 103, 2422–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad CD, Galea LAM, Kuroda Y, and McEwen BS (1996). Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav. Neurosci. 110, 1321–1334. [DOI] [PubMed] [Google Scholar]
- Dahmen B, Puetz VB, Scharke W, von Polier GG, Herpertz-Dahlmann B, and Konrad K (2018). Effects of Early-Life Adversity on Hippocampal Structures and Associated HPA Axis Functions. Dev. Neurosci. 40, 13–22. [DOI] [PubMed] [Google Scholar]
- Danese A, Moffitt TE, Arseneault L, Bleiberg BA, Dinardo PB, Gandelman SB, Houts R, Ambler A, Fisher HL, Poulton R, and Caspi A (2017). The origins of cognitive deficits in victimized children: Implications for neuroscientists and clinicians. Am. J. Psychiatry 174, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daskalakis NP, De Kloet ER, Yehuda R, Malaspina D, and Kranz TM (2015). Early Life Stress Effects on Glucocorticoid-BDNF Interplay in the Hippocampus. Front. Mol. Neurosci. 8, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delahaye-Duriez A, Srivastava P, Shkura K, Langley SR, Laaniste L, Moreno-Moral A, Danis B, Mazzuferi M, Foerch P, Gazina EV, et al. (2016). Rare and common epilepsies converge on a shared gene regulatory network providing opportunities for novel antiepileptic drug discovery. Genome Biol. 17, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellu F, Mayo W, Cherkaoui J, Le Moal M, and Simon H (1992). A two-trial memory task with automated recording: study in young and aged rats. Brain Res. 588, 132–139. [DOI] [PubMed] [Google Scholar]
- Deussing JM, and Jakovcevski M (2017). Histone modifications in major depressive disorder and related rodent models. In Neuroepigenomics in Aging and Disease Advances in Experimental Medicine and Biology, Delgado-Morales R, ed. (Springer; ), pp. 169–183. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. ; ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah MJ (2018). Socioeconomic status and the brain: prospects for neuroscience-informed policy. Nat. Rev. Neurosci. 19, 428–438. [DOI] [PubMed] [Google Scholar]
- Gao Z, Ure K, Ding P, Nashaat M, Yuan L, Ma J, Hammer RE, and Hsieh J (2011). The master negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J. Neurosci. 31, 9772–9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Lee P, Stafford JM, von Schimmelmann M, Schaefer A, and Reinberg D (2014). An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles EE, Schultz L, and Baram TZ (1996). Abnormal corticosterone regulation in an immature rat model of continuous chronic stress. Pediatr. Neurol. 15, 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JD, Rubin TG, Hunter RG, and McEwen BS (2014). Hippocampal gene expression changes underlying stress sensitization and recovery. Mol. Psychiatry 19, 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JD, Rubin TG, Kogan JF, Marrocco J, Weidmann J, Lindkvist S, Lee FS, Schmidt EF, and McEwen BS (2018). Translational profiling of stress-induced neuroplasticity in the CA3 pyramidal neurons of BDNF Val66-Met mice. Mol. Psychiatry 23, 904–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JD, Kogan JF, Marrocco J, and McEwen BS (2017). Genomic and epigenomic mechanisms of glucocorticoids in the brain. Nat. Rev. Endocrinol. 13, 661–673. [DOI] [PubMed] [Google Scholar]
- Guerchet M, Prina M, and Prince M (2013). Policy Brief for Heads of Government: The Global Impact of Dementia 2013–2050 (Alzheimer’s Disease International). https://www.alzint.org/u/2020/08/GlobalImpactDementia2013.pdf.
- Haettig J, Sun Y, Wood MA, and Xu X (2013). Cell-type specific inactivation of hippocampal CA1 disrupts location-dependent object recognition in the mouse. Learn. Mem. 20, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman JJ (2006). Skill formation and the economics of investing in disadvantaged children. Science 312, 1900–1902. [DOI] [PubMed] [Google Scholar]
- Heim C, and Binder EB (2012). Current research trends in early life stress and depression: review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp. Neurol. 233, 102–111. [DOI] [PubMed] [Google Scholar]
- Hinkeldey B, Schmitt A, and Jung G (2008). Comparative photostability studies of BODIPY and fluorescein dyes by using fluorescence correlation spectroscopy. ChemPhysChem 9, 2019–2027. [DOI] [PubMed] [Google Scholar]
- Hsieh J, and Gage FH (2004). Epigenetic control of neural stem cell fate. Curr. Opin. Genet. Dev. 14, 461–469. [DOI] [PubMed] [Google Scholar]
- Huang T-N, Chuang H-C, Chou W-H, Chen C-Y, Wang H-F, Chou S-J, and Hsueh Y-P (2014). Tbr1 haploinsufficiency impairs amygdalar axonal projections and results in cognitive abnormality. Nat. Neurosci. 17, 240–247. [DOI] [PubMed] [Google Scholar]
- Hunter RG, and McEwen BS (2013). Stress and anxiety across the lifespan: structural plasticity and epigenetic regulation. Epigenomics 5, 177–194. [DOI] [PubMed] [Google Scholar]
- Huot RL, Plotsky PM, Lenox RH, and McNamara RK (2002). Neonatal maternal separation reduces hippocampal mossy fiber density in adult Long Evans rats. Brain Res. 950, 52–63. [DOI] [PubMed] [Google Scholar]
- Hwang JY, and Zukin RS (2018). REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 48, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy ASS, Brunson KLL, Sandman C, and Baram TZZ (2008). Dysfunctional nurturing behavior in rat dams with limited access to nesting material: a clinically relevant model for early-life stress. Neuroscience 154, 1132–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy AS, Rex CS, Chen Y, Dubé C, Maras PM, Grigoriadis DE, Gall CM, Lynch G, and Baram TZ (2010). Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J. Neurosci. 30, 13005–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, and Baram TZ (2009). The neuro-symphony of stress. Nat. Rev. Neurosci. 10, 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, and Wold B (2007). Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 316, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Kaplan GA, Turrell G, Lynch JW, Everson SA, Helkala E-LL, and Salonen JT (2001). Childhood socioeconomic position and cognitive function in adulthood. Int. J. Epidemiol. 30, 256–263. [DOI] [PubMed] [Google Scholar]
- Ke X, Fu Q, Majnik A, Cohen S, Liu Q, and Lane R (2018). Adverse early life environment induces anxiety-like behavior and increases expression of FKBP5 mRNA splice variants in mouse brain. Physiol. Genomics 50, 973–981. [DOI] [PubMed] [Google Scholar]
- Kessler RC, McLaughlin KA, Green JG, Gruber MJ, Sampson NA, Zaslavsky AM, Aguilar-Gaxiola S, Alhamzawi AO, Alonso J, Angermeyer M, et al. (2010). Childhood adversities and adult psychopathology in the WHO World Mental Health Surveys. Br. J. Psychiatry 197, 378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino T (2017). Glucocorticoid receptor. In Endotext K. Feingold, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland HJ, and Kaltsas G, et al. , eds. (MDText.com). https://www.ncbi.nlm.nih.gov/books/NBK279171/.
- Klengel T, and Binder EB (2015). Epigenetics of Stress-Related Psychiatric Disorders and Gene 3 Environment Interactions. Neuron 86, 1343–1357. [DOI] [PubMed] [Google Scholar]
- Korosi A, Shanabrough M, McClelland S, Liu Z-W, Borok E, Gao X-B, Horvath TL, and Baram TZ (2010). Early-life experience reduces excitation to stress-responsive hypothalamic neurons and reprograms the expression of corticotropin-releasing hormone. J. Neurosci. 30, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston RF, and Wood ER (2010). Associative recognition and the hippocampus: differential effects of hippocampal lesions on object-place, object-context and object-place-context memory. Hippocampus 20, 1139–1153. [DOI] [PubMed] [Google Scholar]
- Lesuis SL, Weggen S, Baches S, Lucassen PJ, and Krugers HJ (2018). Targeting glucocorticoid receptors prevents the effects of early life stress on amyloid pathology and cognitive performance in APP/PS1 mice. Transl. Psychiatry 8, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi M, Sarabdjitsingh RA, Tsouli A, Trinh S, Arp M, Krugers HJ, Karst H, van den Bos R, and Joëls M (2017). Transient Prepubertal Mifepristone Treatment Normalizes Deficits in Contextual Memory and Neuronal Activity of Adult Male Rats Exposed to Maternal Deprivation. eNeuro 4, ENEURO.0253–17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang T-H, Kim H-M, Drake D, Liu XS, et al. (2014). REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucassen PJ, Naninck EFGG, van Goudoever JB, Fitzsimons C, Joels M, and Korosi A (2013). Perinatal programming of adult hippocampal structure and function; emerging roles of stress, nutrition and epigenetics. Trends Neurosci. 36, 621–631. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, and Heim C (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 10, 434–445. [DOI] [PubMed] [Google Scholar]
- Mampay M, and Sheridan GK (2019). REST: An epigenetic regulator of neuronal stress responses in the young and ageing brain. Front. Neuroendocrinol. 53, 100744. [DOI] [PubMed] [Google Scholar]
- Mandel G, Fiondella CG, Covey MV, Lu DD, Loturco JJ, and Ballas N (2011). Repressor element 1 silencing transcription factor (REST) controls radial migration and temporal neuronal specification during neocortical development. Proc. Natl. Acad. Sci. USA 108, 16789–16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maras PM, Molet J, Chen Y, Rice C, Ji SG, Solodkin A, and Baram TZ (2014). Preferential loss of dorsal-hippocampus synapses underlies memory impairments provoked by short, multimodal stress. Mol. Psychiatry 19, 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland S, Korosi A, Cope J, Ivy A, and Baram TZ (2011a). Emerging roles of epigenetic mechanisms in the enduring effects of early-life stress and experience on learning and memory. Neurobiol. Learn. Mem. 96, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland S, Flynn C, Dubé C, Richichi C, Zha Q, Ghestem A, Esclapez M, Bernard C, and Baram TZ (2011b). Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann. Neurol. 70, 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland S, Brennan GP, Dubé C, Rajpara S, Iyer S, Richichi C, Bernard C, and Baram TZ (2014). The transcription factor NRSF contributes to epileptogenesis by selective repression of a subset of target genes. eLife 3, e01267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Nasca C, and Gray JD (2016). Stress Effects on Neuronal Structure: Hippocampus, Amygdala, and Prefrontal Cortex. Neuropsychopharmacology 41, 3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin KA, Weissman D, and Bitrán D (2019). Childhood Adversity and Neural Development: A Systematic Review. Annu. Rev. Dev. Psychol. 1, 277–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molet J, Maras PM, Avishai-Eliner S, and Baram TZ (2014). Naturalistic rodent models of chronic early-life stress. Dev. Psychobiol. 56, 1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molet J, Maras PM, Kinney-Lang E, Harris NG, Rashid F, Ivy AS, Solodkin A, Obenaus A, and Baram TZ (2016a). MRI uncovers disrupted hippocampal microstructure that underlies memory impairments after early-life adversity. Hippocampus 26, 1618–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molet J, Heins K, Zhuo X, Mei YT, Regev L, Baram TZ, and Stern H (2016b). Fragmentation and high entropy of neonatal experience predict adolescent emotional outcome. Transl. Psychiatry 6, e702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Brulet R, Zhang L, and Hsieh J (2016). REST regulation of gene networks in adult neural stem cells. Nat. Commun. 7, 13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naninck EFGGG, Hoeijmakers L, Kakava-Georgiadou N, Meesters A, Lazic SE, Lucassen PJ, and Korosi A (2015). Chronic early life stress alters developmental and adult neurogenesis and impairs cognitive function in mice. Hippocampus 25, 309–328. [DOI] [PubMed] [Google Scholar]
- Nelson CA 3rd, Zeanah CH, Fox NA, Marshall PJ, Smyke AT, and Guthrie D (2007). Cognitive recovery in socially deprived young children: the Bucharest Early Intervention Project. Science 318, 1937–1940. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2014). Epigenetic mechanisms of depression. JAMA Psychiatry 71, 454–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, and Hyman SE (2010). Animal models of neuropsychiatric disorders. Nat. Neurosci. 13, 1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noam Y, Zha Q, Phan L, Wu RL, Chetkovich DM, Wadman WJ, and Baram TZ (2010). Trafficking and surface expression of hyperpolarization-activated cyclic nucleotide-gated channels in hippocampal neurons. J. Biol. Chem. 285, 14724–14736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KP, Barry JM, Curran MM, Singh-Taylor A, Brennan G, Rismanchi N, Page M, Noam Y, Holmes GL, and Baram TZ (2017). Enduring Memory Impairments Provoked by Developmental Febrile Seizures Are Mediated by Functional and Structural Effects of Neuronal Restrictive Silencing Factor. J. Neurosci. 37, 3799–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña CJ, Kronman HG, Walker DM, Cates HM, Bagot RC, Purushothaman I, Issler O, Eddie Loh YH, Leong T, Kiraly DD, et al. (2017). Early life stress confers lifelong stress susceptibility in mice via ventral tegmental area OTX2. Science 356, 1185–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman JAE, de Kloet ER, and Datson NA (2013). Two populations of glucocorticoid receptor-binding sites in the male rat hippocampal genome. Endocrinology 154, 1832–1844. [DOI] [PubMed] [Google Scholar]
- Prince M, Wimo A, Guerchet M, Gemma-Claire Ali M, Wu Y-T, Prina M, Yee Chan K, and Xia Z (2015). World Alzheimer Report 2015: The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends (Alzheimer’s Disease International). https://www.alzint.org/u/WorldAlzheimerReport2015.pdf.
- Reul JMHM (2014). Making memories of stressful events: a journey along epigenetic, gene transcription, and signaling pathways. Front. Psychiatry 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenas-Ruano A, Chávez AE, Cossio MJ, Castillo PE, and Zukin RS (2012). REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat. Neurosci. 15, 1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross DA, Arbuckle MR, Travis MJ, Dwyer JB, van Schalkwyk GI, and Ressler KJ (2017). An Integrated Neuroscience Perspective on Formulation and Treatment Planning for Posttraumatic Stress Disorder: An Educational Review. JAMA Psychiatry 74, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin TG, Gray JD, and McEwen BS (2014). Experience and the everchanging brain: what the transcriptome can reveal. BioEssays 36, 1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Murrough JW, Han M-HH, Charney DS, and Nestler EJ (2012). Neurobiology of resilience. Nat. Neurosci. 15, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MV, Abraham WC, Maroun M, Stork O, and Richter-Levin G (2013). Stress-induced metaplasticity: from synapses to behavior. Neuroscience 250, 112–120. [DOI] [PubMed] [Google Scholar]
- Schoenherr CJ, and Anderson DJ (1995). The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science 267, 1360–1363. [DOI] [PubMed] [Google Scholar]
- Schwaiger M, Grinberg M, Moser D, Zang JCS, Heinrichs M, Hengstler JG, Rahnenführer J, Cole S, and Kumsta R (2016). Altered Stress-Induced Regulation of Genes in Monocytes in Adults with a History of Childhood Adversity. Neuropsychopharmacology 41, 2530–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short AK, and Baram TZ (2019). Early-life adversity and neurological disease: age-old questions and novel answers. Nat. Rev. Neurol. 15, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short AK, Maras PM, Pham AL, Ivy AS, and Baram TZ (2020). Blocking CRH receptors in adults mitigates age-related memory impairments provoked by early-life adversity. Neuropsychopharmacology 45, 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Taylor A, Molet J, Jiang S, Korosi A, Bolton JL, Noam Y, Simeone K, Cope J, Chen Y, Mortazavi A, and Baram TZ (2018). NRSF-dependent epigenetic mechanisms contribute to programming of stress-sensitive neurons by neonatal experience, promoting resilience. Mol. Psychiatry 23, 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suderman M, McGowan PO, Sasaki A, Huang TCT, Hallett MT, Meaney MJ, Turecki G, and Szyf M (2012). Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc. Natl. Acad. Sci. USA 109 (Suppl 2), 17266–17272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suderman M, Borghol N, Pappas JJ, Pinto Pereira SM, Pembrey M, Hertzman C, Power C, and Szyf M (2014). Childhood abuse is associated with methylation of multiple loci in adult DNA. BMC Med. Genomics 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyf M, Tang Y-Y, Hill KG, and Musci R (2016). The dynamic epigenome and its implications for behavioral interventions: a role for epigenetics to inform disorder prevention and health promotion. Transl. Behav. Med. 6, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay E, Roisin MP, Represa A, Charriaut-Marlangue C, and Ben-Ari Y (1988). Transient increased density of NMDA binding sites in the developing rat hippocampus. Brain Res. 461, 393–396. [DOI] [PubMed] [Google Scholar]
- Turecki G, and Meaney MJ (2016). Effects of the Social Environment and Stress on Glucocorticoid Receptor Gene Methylation: A Systematic Review. Biol. Psychiatry 79, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Represa A, Jorquera I, Ben-Ari Y, Gozlan H, and Aniksztejn L (1999). The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. J. Neurosci. 19, 10372–10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bodegom M, Homberg JR, and Henckens MJAG (2017). Modulation of the Hypothalamic-Pituitary-Adrenal Axis by Early Life Stress Exposure. Front. Cell. Neurosci. 11, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker C-D, Bath KG, Joels M, Korosi A, Larauche M, Lucassen PJ, Morris MJ, Raineki C, Roth TL, Sullivan RM, et al. (2017). Chronic early life stress induced by limited bedding and nesting (LBN) material in rodents: critical considerations of methodology, outcomes and translational potential. Stress 20, 421–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X-D, Rammes G, Kraev I, Wolf M, Liebl C, Scharf SH, Rice CJ, Wurst W, Holsboer F, Deussing JM, et al. (2011). Forebrain CRF1 modulates early-life stress-programmed cognitive deficits. J. Neurosci. 31, 13625–13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD, Su YA, Wagner KV, Avrabos C, Scharf SH, Hartmann J, Wolf M, Liebl C, Kühne C, Wurst W, et al. (2013). Nectin-3 links CRHR1 signaling to stress-induced memory deficits and spine loss. Nat. Neurosci. 16, 706–713. [DOI] [PubMed] [Google Scholar]
- Xu J, Wang R, Liu Y, Liu D, Jiang H, and Pan F (2017). FKBP5 and specific microRNAs via glucocorticoid receptor in the basolateral amygdala involved in the susceptibility to depressive disorder in early adolescent stressed rats. J. Psychiatr. Res. 95, 102–113. [DOI] [PubMed] [Google Scholar]
- Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, Wang EP, Marshall-Walker CA, Barry BJ, Murn J, et al. (2012). Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 151, 1097–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is GEO: GSE161498. This study did not generate code.