

Abstract
Background
Previous studies have reported that mitochondrial dysfunction participates in the pathological process of osteoarthritis (OA). However, studies that improve mitochondrial function are rare in OA. Mitochondrial transfer from mesenchymal stem cells (MSCs) to OA chondrocytes might be a cell-based therapy for the improvement of mitochondrial function to prevent cartilage degeneration. This study aimed to determine whether MSCs can donate mitochondria and protect the mitochondrial function and therefore reduce cartilage degeneration.
Methods
Bone-marrow-derived mesenchymal stromal cells (BM-MSCs) were harvested from the marrow cavities of femurs and tibia in young rats. OA chondrocytes were gathered from the femoral and tibial plateau in old OA model rats. BM-MSCs and OA chondrocytes were co-cultured and mitochondrial transfer from BM-MSCs to chondrocytes was identified. Chondrocytes with mitochondria transferred from BM-MSCs were selected by fluorescence-activated cell sorting. Mitochondrial function of these cells, including mitochondrial membrane potential (Δψm), the activity of mitochondrial respiratory chain (MRC) enzymes, and adenosine triphosphate (ATP) content were quantified and compared to OA chondrocytes without mitochondrial transfer. Chondrocytes proliferation, apoptosis, and secretion ability were also analyzed between the two groups.
Results
Mitochondrial transfer was found from BM-MSCs to OA chondrocytes. Chondrocytes with mitochondrial from MSCs (MSCs + OA group) showed increased mitochondrial membrane potential compared with OA chondrocytes without mitochondria transfer (OA group) (1.79 ± 0.19 vs. 0.71 ± 0.12, t = 10.42, P < 0.0001). The activity of MRC enzymes, including MRC complex I, II, III, and citrate synthase was also improved (P < 0.05). The content of ATP in MSCs + OA group was significantly higher than that in OA group (161.90 ± 13.49 vs. 87.62 ± 11.07 nmol/mg, t = 8.515, P < 0.0001). Meanwhile, we observed decreased cell apoptosis (7.09% ± 0.68% vs.15.89% ± 1.30%, t = 13.39, P < 0.0001) and increased relative secretion of type II collagen (2.01 ± 0.14 vs.1.06 ± 0.11, t = 9.141, P = 0.0008) and proteoglycan protein (2.08 ± 0.20 vs. 0.97 ± 0.12, t = 8.227, P = 0.0012) in MSCs + OA group, contrasted with OA group.
Conclusions
Mitochondrial transfer from BM-MSCs provided protection for OA chondrocytes against mitochondrial dysfunction and degeneration through improving mitochondrial function, cell proliferation, and inhibiting apoptosis in chondrocytes. This finding may offer a new therapeutic direction for OA.
Keywords: Mitochondrial transfer, Osteoarthritis, Mitochondrial dysfunction, Chondrocyte, BM-MSCs
Introduction
Osteoarthritis (OA) is a common aging-associated degenerative joint disease attributed to degeneration of articular cartilage, joint pain, and functional impairment.[1–3] As the sole functional cells present in mature cartilage, the chondrocytes regulate plenty of physiological processes, including the synthesis, deposition, and modification of the extracellular matrix.[2] However, the molecular mechanism of OA remains poorly known. Recent studies reported mitochondrial dysfunction in OA chondrocytes.[1,4] An ex vivo study analyzing the mitochondrial respiratory chain (MRC) displayed decreased activity of complexes I, II, and III in OA chondrocytes compared to normal chondrocytes.[5] Further studies showed that mitochondrial functions, such as adenosine triphosphate (ATP) synthesis and MRC activity, were affected in OA chondrocytes.[2] Our previous study also discovered decreased MRC activity and reduced mitochondrial membrane potential (Δψm) in OA chondrocytes.[4]
Mitochondrial dysfunction affects several pathways involved in OA pathology, including chondrocyte apoptosis, oxidative stress, cytokine-induced chondrocyte inflammation, and calcification of the cartilage matrix.[2,6,7] Therefore, enhancing mitochondrial activity may be a therapeutic alternative for OA patients. Mitochondrial transfer from donor cells into injured cells has been proposed as a cell-based therapy for the improvement of mitochondrial activity.[8–10] However, studies that regulate or improve mitochondrial function are not currently mentioned in OA.
Therefore, our study investigated whether mitochondrial transfer from mesenchymal stem cells (MSCs) to OA chondrocytes exist. And whether the mitochondrial transfer could improve the mitochondrial function of OA chondrocytes and attenuate cartilage degeneration.
Methods
Ethical approval
This study was approved by the Institutional Ethical Committee for Animal Care and Use of Peking University First Hospital (No. 201866). All animals were provided by the Laboratory Animal Centre of Peking University First Hospital, and this study was also performed in the Laboratory Animal Centre of Peking University First Hospital.
Bone-marrow-derived mesenchymal stromal cells (BM-MSCs)
According to the protocol previously described, BM-MSCs were harvested from 2-week old male rats and cultured.[11] In brief, rats femurs and tibia were cut off and the marrow cavities were flushed using sterile phosphate buffer saline in aseptic condition. The harvested cell suspension was filtered and isolated. BM-MSCs were cultured with Dulbecco modified Eagle medium (DMEM) (Hyclone, Ge Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS) (Hyclone, GE Healthcare Life Sciences) in a 37°C incubator with 5% CO2 atmosphere, 3rd to 5th passage cells were used in subsequent studies.
OA chondrocytes
OA animal model was established according to the protocol previously described.[12] Briefly, 8-week old rats were anesthetized by pentobarbital (5 mg/kg, intraperitoneal injection), bilateral anterior cruciate ligament and medial meniscus were resected to induce the pathogenesis of OA. The rats were sacrificed eight weeks later. Cartilage samples were gathered from the femoral and tibial plateau, and chondrocytes were obtained using the modified Klagsbrun method. After cut into small pieces (1 mm3), the cartilage was incubated with 0.4% Hank balanced salt solution (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China) for 2 h. Further digestion was done with sterile 0.2% collagenase (Nanjing KeyGen Biotech Co., Ltd.) for another 5 h. Then, cell suspension was filtered two times, and isolated chondrocytes were cultured in DMEM (Hyclone), mixed with 10% FBS (Hyclone) and 1% penicillin-streptomycin (Nanjing KeyGenvBiotech Co., Ltd.) for 24 h. The cells were incubated in a humidified atmosphere at 37°C, containing 5% CO2. For cell synchronization, chondrocytes were cultured by serum starvation for 24 h.
Co-culture of OA chondrocytes and BM-MSCs
Mitochondrial targeting green fluorescence protein (lentiviral-mediated mitochondrial-specific fragment fused with green fluorescence protein, LV-Mito-GFP, Cat. No. Cyto102-PA-1, System Biosciences, Palo Alto, CA, USA) was transfected. Briefly, 5 × 105 BM-MSCs were seeded before infection, 24 h later, 1 mL growth medium with LV-Mito-GFP was added to cells and incubated for 16 h. LV-Mito-GFP vector, encoding a mitochondrial cytochrome oxidase subunit VIII and a fusion of GFP, will allow us to distinguish mitochondrial trafficking. Chondrocytes were labeled with CellTrace Violet. Labeled OA chondrocytes and labeled MSCs were co-cultured for 24 h at a ratio of 1:1. The co-cultured cells were harvested and the chondrocytes merged labeled mitochondrial targeting green fluorescence protein were selected by flow cytometry (BD FACSAria III, BD Biosciences, Franklin Lakes, NJ, USA). For contrast, labeled chondrocytes which were not co-cultured with BM-MSCs were also selected.
Transmission electron microscope (TEM) observation of chondrocytes
Chondrocytes were prepared as a monoplast suspension of >1 × 105 cells/mL and were fixed by glutaraldehyde for 24 h and then osmic acid for 2 h. The chondrocytes were embedded by resin after dehydration and stained with uranium acetate and lead citrate. The morphology of the chondrocytes, including organelles, the number and shape of mitochondria, the electron density in the outer membrane, and the cristae of the inner membrane was then observed.
Mitochondrial membrane potential (Δψm) measurement of chondrocytes
OA chondrocytes with (MSCs + OA group) or without (OA group) merged labeled mitochondrial targeting green fluorescence protein were harvested. After preparing a monoplast suspension with density more than 1 × 105 cells/mL, cells were stained with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine (JC-1) at 37°C for 20 min, according to the operation instruction of the Δψm assay kit (Sigma-Aldrich, St Louis Missouri, USA). JC-1 is a carbocyclic lipophilic fluorescent dye which is sensitive to membrane potential, it can form polymers in the mitochondrial matrix of normal membrane potential cells and exhibit bright red fluorescence emission. In apoptotic cells, however, the membrane potential decreased, JC-1 forms as monomer and exhibits green fluorescence emission. Flow cytometry was performed to measure the Δψm within 30 min after staining. The JC-1 monomer was detected under excitation wavelength at 488 nm and emission wavelength at 530 nm, and the JC-1 polymer was observed using excitation wavelength at 490 nm and emission wavelength at 590 nm. Data were analyzed using FlowJo (version 7.6.5, BD Biosciences).
Identification of respiratory chain complex activity and ATP content
In order to investigate if the mitochondrial transfer from BM-MSCs could improve the mitochondrial function of OA chondrocytes, quantitative analysis of the MRC enzymes’ activities, including MRC complexes I, II, III, and citrate synthase were determined. Quantification of the ATP was also detected. OA chondrocytes of two groups were prepared as a monoplast suspension with a density of more than 1 × 107 cells/mL. According to the protocol of mitochondrial extraction kit (Nanjing KeyGen Biotech Co., Ltd.), mitochondria were extracted. The activities of the mitochondrial enzymes and the content of ATP were calculated using the initial velocity equation for pseudo-first-order reactions at 25°C.
Measurement of cell proliferation and apoptosis
Cellular proliferation of chondrocytes was assessed with Cell Counting Kit-8 (Applygen Technologies, Inc., Beijing, China). Optical density values of the cells between the two groups were measured and compared on days 1, 3, 5, and 7. For apoptosis, chondrocytes were stained by propidium iodide and Annexin V-enhanced green fluorescent protein at 37°C for 15 min, then cells were assessed by flow cytometry (BD Biosciences) with Annexin V-fluorescein isothiocyanate kit (Nanjing KeyGen Biotech Co., Ltd.). Data collected were then analyzed with FlowJo (version 7.6.5, BD Biosciences).
Quantitative analysis of type II collagen and proteoglycan protein
Total protein in chondrocytes was extracted by the total protein extraction kit (Nanjing KeyGen Biotech Co., Ltd.). The quantification of type II collagen and proteoglycan was determined via Western blotting. Briefly, the concentration was measured with Bio-Rad Protein Assay Kit (Bio-Rad Laboratories, Hercules, CA, USA) by running on 10% polyacrylamide gel. Polyvinylidene fluoride membranes with protein were blocked and incubated with the primary antibodies, collagen II (ab34712, Abcam, Cambridge, UK), proteoglycan (ab2501, Abcam), and glyceraldehyde-3-phosphate dehydrogenase (ab128915, Abcam), overnight at 4°C. Then, rabbit anti-mouse secondary antibody was incubated at 37°C for 1 h.
Statistical analysis
Statistical analysis was operated with SPSS software (version 23.0; IBM Corp., New York, USA). All data were reported as the mean ± standard deviation. Student's t test with subsequent Bonferroni correction was adapted to compare results from two groups, with P < 0.05 considered statistically significant.
Results
Mitochondrial transfer from BM-MSCs to OA chondrocytes
MSCs with LV-Mito-GFP were successfully built to reveal the mitochondrial transfer [Figure 1A]. OA chondrocytes were labeled with CellTrace Violet [Figure 1B]. OA chondrocytes (violet) and BM-MSCs (green) can be distinguished. After 24 h co-culture of these two kinds of cells, Mito-GFP was observed in many violet fluorescence-positive cells [Figure 1C], indicating that mitochondria from MSCs had translocated to chondrocytes.
Figure 1.
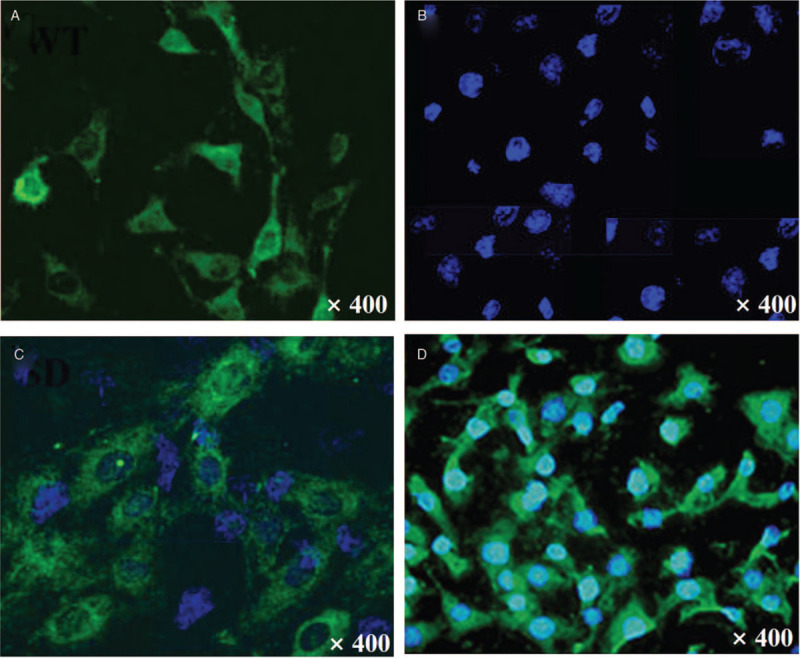
Intercellular mitochondrial transfer from BM-MSCs to OA chondrocytes was detected by confocal microscope. (A) BM-MSCs labeled with green LV-Mito-GFP. (B) OA chondrocytes labeled with CellTrace Violet. (C) Mito-GFP was observed in many violet fluorescence-positive OA chondrocytes after 24 h co-culturing of violet-labeled OA chondrocytes and Mito-GFP-labeled MSCs. (D) Selected violet fluorescence-positive cells transferred with Mito-GFP. Original magnification, ×400. BM-MSCs: Bone-marrow-derived mesenchymal stromal cells; LV-Mito-GFP: Lentiviral-mediated mitochondrial-specific fragment fused with green fluorescence protein; OA: Osteoarthritis.
TEM observation of chondrocytes
Chondrocytes of MSCs + OA group were selected by flow cytometry [Figure 1D] and chondrocytes not co-cultured (OA group) were selected for observation by TEM. In MSCs + OA group, various organelles were visible, including the endoplasmic reticulum, the Golgi apparatus, and the number of mitochondria was much more than OA chondrocytes. The shape of mitochondria was oblong. In the outer membrane, the electron density was continuous and high, and in the inner membrane, the cristae was regularly arranged [Figure 2A]. However, in OA group, the mitochondria were more swollen, and its shape was more round rather than oblong. In the outer membrane, the electron density became discontinuous and asymmetrical, while in the inner membrane, the cristae exhibited an irregular pattern, or even cannot be detected. [Figure 2B].
Figure 2.

Transmission electron microscope and Δψm results. (A) Transmission electron microscope results in MSCs + OA group. Organelles were visible in the cytoplasm, including a large number of mitochondria, the endoplasmic reticulum, and the Golgi apparatus. The shape of mitochondria was oblong. The electron density in the outer membrane was continuous and high, the cristae in the inner membrane was regularly arranged. Scale bar = 0.5 μm. (B) Transmission electron microscope results in OA group. The number of organelles in the cytoplasm was decreased compared with MSC + OA group. The mitochondria were more swollen, the electron density was discontinuous and the structure of the cristae was disarrayed (arrow). (C and D) Immunofluorescence staining of Δψm in OA and MSCs + OA groups, respectively. Original magnification, ×1000. (E) The ratios of red/green fluorescent signal in OA group and MSCs + OA group were 0.71 ± 0.12 and 1.79 ± 0.19, respectively (∗P < 0.01, OA group vs. MSCs + OA group). Δψm: Mitochondrial membrane potential; MSCs: Mesenchymal stromal cells; OA: Osteoarthritis.
Δψm measurement of chondrocytes
Compared with OA group, the red fluorescence in MSCs + OA group was increased, while the green fluorescence was decreased [Figure 2C and 2D]. The ratio of red/green fluorescent signal in MSCs + OA group was 1.79 ± 0.19, whereas the signal in OA group was 0.71 ± 0.12. Importantly, the differences between MSCs + OA and OA groups showed statistically significant (t = 10.42, P < 0.01) [Figure 2E].
Detection of respiratory chain complex activity and ATP content.
The activities of the MRC complexes I, II, III, and citrate synthase in MSCs + OA group were remarkably increased compared with OA group (69.11 ± 16.69 vs. 30.61 ± 6.24 nmol·min−1·mg−1, t = 3.174, P = 0.013; 36.67 ± 5.65 vs. 12.52 ± 2.96 nmol·min−1·mg−1, t = 6.323, P = 0.0002; 118.74 ± 9.50 vs. 47.61 ± 8.94 nmol·min−1·mg−1, t = 6.523, P = 0.0002; and 196.40 ± 26.18 vs. 115.00 ± 15.37 nmol·min−1·mg−1, t = 5.362, P = 0.0007, respectively).
The content of ATP in OA group was 87.62 ± 11.07 nmol/mg. Whereas in MSCs + OA group, it was 161.90 ± 13.49 nmol/mg, which was significantly increased compared with OA group (t = 8.515, P < 0.01). The present results proposed that mitochondrial transfer from BM-MSCs was able to improve mitochondrial function of OA chondrocytes.
Cellular viability assay
Cell proliferation assay revealed that the proliferative ability of MSCs + OA group was significantly increased compared with OA group on days 3, 5, 7 (all P < 0.05) [Figure 3A]. Meanwhile, the total apoptosis rate in OA group was 15.89% ± 1.30% (The early and late apoptosis rates were 1.54% ± 0.91% and 14.35% ± 1.25%, respectively) [Figure 3B]. In MSCs + OA group, the early, late, and total apoptosis rates were 1.85% ± 0.71%, 5.24% ± 0.99%, and 7.09% ± 0.68%, respectively [Figure 3C]. Significant difference in total apoptosis rates can be detected between two groups (t = 13.39, P < 0.01) [Figure 3D]. These results suggested that improved mitochondrial function was able to enhance cell proliferation and decrease the apoptosis rate of OA chondrocytes.
Figure 3.

Cellular viability results. (A) Cell proliferation assay showed that the proliferative ability of MSCs + OA group was significantly increased compared with OA group on days 3, 5, and 7 (∗P < 0.05, vs. OA group). (B) In OA group, the early, late, and total apoptosis rates were 1.54% ± 0.91%, 14.35% ± 1.25%, and 15.89% ± 1.30%, respectively. (C) In MSCs + OA group, the early, late, and total apoptosis were 1.85% ± 0.71%, 5.24% ± 0.99%, and 7.09% ± 0.68%, respectively. (D) There was a significant difference in total apoptosis between the two groups (∗P < 0.01, vs. OA group). MSCs: Mesenchymal stromal cells; OA: Osteoarthritis.
Quantitative detection of type II collagen and proteoglycan protein
Type II collagen and proteoglycan protein levels were significantly elevated in MSCs + OA group than those in OA group determined (2.01 ± 0.14 vs. 1.06 ± 0.11, t = 9.141, P = 0.0008; and 2.08 ± 0.20 vs. 0.97 ± 0.12, t = 8.227, P = 0.0012, respectively) [Figure 4].
Figure 4.
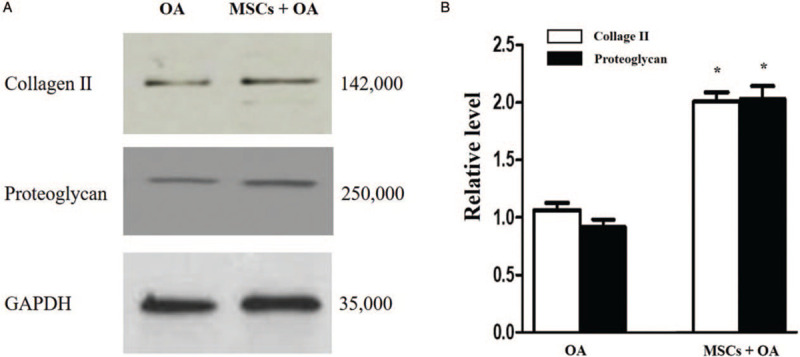
Relative quantitative detection of type II collagen and proteoglycan protein in two groups. (A) Western blotting results revealed the level of type II collagen and proteoglycan protein. (B) The relative levels of collagen II and proteoglycan in OA group and MSCs + OA group (∗P < 0.01 vs. the corresponding relative protein expression level in the OA group). GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; MSCs: Mesenchymal stromal cells; OA: Osteoarthritis.
Discussion
As a progressive joint degenerative disease, OA may often lead to physical disability. Mitochondrial dysfunction may influence the pathogenesis of OA and the function of chondrocytes. Cillero-Pastor et al[13] reported that inhibiting complex III activity using antimycin-A (a specific MRC inhibitor) could increase the apoptosis in human chondrocytes. Several studies have identified that specific MRC inhibitors suppressed the secretion of collagen and proteoglycans in human chondrocytes.[14–16] So, improving mitochondrial function may be a therapeutic alternative for OA. MSCs have been recommended as a valuable cell source in the therapy of OA, which exert therapeutic functions mainly via anti-inflammatory actions, paracrine, and cartilage repair.[17–19] Demonstrated as an important mechanism of MSCs therapy, the mitochondrial transfer is associated with the rescue of aerobic respiration and restoration of mitochondria function in the recipient cells.[8,9,20,21] It played a critical role during disease healing processes, such as cerebral injury, cardiac myopathies, lung diseases, and acute respiratory disorders.[20–24] However, the potential mechanism of mitochondrial function improvement in OA through MSCs therapy is not currently included. In our study, we show for the first time that mitochondrial transfer can occur from BM-MSCs to OA chondrocytes in vitro. These results also revealed that functional mitochondrial transfer from MSCs can directly protect chondrocytes against mitochondrial dysfunction and reverses states of OA chondrocytes, which is a critical mechanism in MSC therapy.
TEM imaging provides the visualization of mitochondrial morphological features. The morphology and number of the cristae in the mitochondrial inner membrane are associated with cell metabolism. Ma et al[25] revealed that the loss of normal mitochondrial morphology may be associated with the dysfunction of the MRC, including the uncovering of the mitochondrial permeability transition pore, the decrease of Δψm and the collapse of the outer membrane. In our study, we found more normal mitochondria in MSCs + OA groups compared with OA chondrocytes. This result suggested that MSCs can transfer healthy mitochondria to OA chondrocytes, which can hence improve the mitochondrial dysfunction of OA chondrocytes.
Mitochondrial dysfunction participates in the process of chondrocyte apoptosis, while numerous preclinical studies indicated that chondrocyte apoptosis can be reversed by mitochondrial modulation.[26] Previous studies proved that mitochondrial transfer could increase ATP content and enhance gene and protein expression relating to the tricarboxylic acid cycle, thus protect against cell injury and apoptosis.[9,11] Here, we also exhibited that mitochondrial transfer from MSCs to OA chondrocytes increased the activity of the complexes of MRC, mitochondrial membrane potential, and ATP content. Meanwhile, we indicated that mitochondrial transfer was an important strategy in preventing the chondrocytes from mitochondrion-driven apoptosis. In our study, cell proliferation increased by about 98% in the 7th day, while total apoptosis rates decreased by about 55%, secretion level of type II collagen and proteoglycan protein improved about 1-fold in MSCs + OA group. This finding confirmed that mitochondrial transfer from MSCs to OA chondrocytes can significantly increase the mitochondrial function and ameliorate degeneration of OA chondrocytes.
There are several limitations in this study. First, the relative roles of paracrine secretion vs. mitochondrial transfer in the BM-MSCs protective effect were not mentioned. Jiang et al[21] indicated that paracrine action and mitochondrial transfer were two independent processes, which collaborated to MSC-mediated protection. Second, the potential mechanisms of mitochondrial transfer from BM-MSCs to chondrocytes in our study were not elucidated. Many studies have indicated that various mechanisms including tunnel tube formation, gap junctions, microvesicle formation, cell fusion, and other modes of transfer are involved in mitochondrial transfer.[8,20,21] Further studies are needed in terms of mitochondrial transfer from MSCs to chondrocytes.
In conclusion, our study reveals that mitochondrial transfer from BM-MSCs might protect OA chondrocytes against mitochondrial dysfunction and cartilage degeneration through improving mitochondrial function, cell proliferation, and inhibiting apoptosis in chondrocytes. This finding may offer a new therapeutic direction for OA. Further studies are needed in the mechanism of mitochondrial transfer from MSCs to chondrocytes.
Funding
This work was supported by a grant from the Scientific Research Seed Fund of Peking University First Hospital (No. 2018SF020).
Conflicts of interest
None.
Footnotes
How to cite this article: Wang R, Maimaitijuma T, Ma YY, Jiao Y, Cao YP. Mitochondrial transfer from bone-marrow-derived mesenchymal stromal cells to chondrocytes protects against cartilage degenerative mitochondrial dysfunction in rats chondrocytes. Chin Med J 2021;134:212–218. doi: 10.1097/CM9.0000000000001057
References
- 1.Tang Q, Zheng G, Feng Z, Chen Y, Lou Y, Wang C, et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis 2017; 8:e3081.doi: 10.1038/cddis.2017.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol 2011; 7:161–169. doi: 10.1038/nrrheum.2010.213. [DOI] [PubMed] [Google Scholar]
- 3.Li X, Yang X, Maimaitijuma T, Cao XY, Jiao Y, Wu H, et al. Plant homeodomain finger protein 23 inhibits autophagy and promotes apoptosis of chondrocytes in osteoarthritis. Chin Med J 2019; 132:2581–2587. doi: 10.1097/CM9.0000000000000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu H, Li Z, Cao Y, Cui Y, Yang X, Meng Z, et al. Effect of chondrocyte mitochondrial dysfunction on cartilage degeneration: a possible pathway for osteoarthritis pathology at the subcellular level. Mol Med Rep 2019; 20:3308–3316. doi: 10.3892/mmr.2019.10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maneiro E, Martín MA, de Andres MC, López-Armada MJ, Fernández-Sueiro JL, del Hoyo P, et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum 2003; 48:700–708. doi: 10.1002/art.10837. [DOI] [PubMed] [Google Scholar]
- 6.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003; 552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruiz-Romero C, Calamia V, Mateos J, Carreira V, Martinez-Gomariz M, Fernandez M, et al. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics 2009; 8:172–189. doi: 10.1074/mcp.M800292-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinha P, Islam MN, Bhattacharya S, Bhattacharya J. Intercellular mitochondrial transfer: bioenergetic crosstalk between cells. Curr Opin Genet Dev 2016; 38:97–101. doi: 10.1016/j.gde.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun C, Liu X, Wang B, Wang Z, Liu Y, Di C, et al. Endocytosis-mediated mitochondrial transplantation: transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 2019; 9:3595–3607. doi: 10.7150/thno.33100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayakawa K, Bruzzese M, Chou SH, Ning M, Ji X, Lo EH. Extracellular mitochondria for therapy and diagnosis in acute central nervous system injury. JAMA Neurol 2018; 75:119–122. doi: 10.1001/jamaneurol.2017.3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 2012; 18:759–765. doi: 10.1038/nm.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martínez-Calleja A, Cruz R, Miranda-Sánchez M, Fragoso-Soriano R, Vega-López MA, Kouri JB. Latexin expression correlated with mineralization of articular cartilage during progression of post-traumatic osteoarthritis in a rat model. Histol Histopathol 2020; 35:269–278. doi: 10.14670/HH-18-151. [DOI] [PubMed] [Google Scholar]
- 13.Cillero-Pastor B, Caramés B, Lires-Deán M, Vaamonde-García C, Blanco FJ, López-Armada MJ. Mitochondrial dysfunction activates cyclooxygenase 2 expression in cultured normal human chondrocytes. Arthritis Rheum 2008; 58:2409–2419. doi: 10.1002/art.23644. [DOI] [PubMed] [Google Scholar]
- 14.López-Armada MJ, Caramés B, Martín MA, Cillero-Pastor B, Lires-Dean M, Fuentes-Boquete I, et al. Mitochondrial activity is modulated by TNFα and IL-1β in normal human chondrocyte cells. Osteoarthritis Cartilage 2006; 14:1011–1022. doi: 10.1016/j.joca.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Johnson K, Jung A, Murphy A, Andreyev A, Dykens J, Terkeltaub R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum 2000; 43:1560–1570. doi: 10.1002/1529-0131(200007)43:7<1560::aid-anr21>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 16.Tomita M, Sato EF, Nishikawa M, Yamano Y, Inoue M. Nitric oxide regulates mitochondrial respiration and functions of articular chondrocytes. Arthritis Rheum 2001; 44:96–104. doi: 10.1002/1529-0131(200101)44:1<96::aid-anr13>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 17.Desando G, Giavaresi G, Cavallo C, Bartolotti I, Sartoni F, Nicoli Aldini N, et al. Autologous bone marrow concentrate in a sheep model of osteoarthritis: new perspectives for cartilage and meniscus repair. Tissue Eng Part C Methods 2016; 22:608–619. doi: 10.1089/ten.TEC.2016.0033. [DOI] [PubMed] [Google Scholar]
- 18.Shapiro SA, Kazmerchak SE, Heckman MG, Zubair AC, O’Connor MI. A prospective, single-blind, placebo-controlled trial of bone marrow aspirate concentrate for knee osteoarthritis. Am J Sports Med 2017; 45:82–90. doi: 10.1177/0363546516662455. [DOI] [PubMed] [Google Scholar]
- 19.Whitehouse MR, Howells NR, Parry MC, Austin E, Kafienah W, Brady K, et al. Repair of torn avascular meniscal cartilage using undifferentiated autologous mesenchymal stem cells: from in vitro optimization to a first-in-human study. Stem Cells Transl Med 2017; 6:1237–1248. doi: 10.1002/sctm.16-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paliwal S, Chaudhuri R, Agrawal A, Mohanty S. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J Biomed Sci 2018; 25:31.doi: 10.1186/s12929-018-0429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang D, Gao F, Zhang Y, Wong DS, Li Q, Tse HF, et al. Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis 2016; 7:e2467.doi: 10.1038/cddis.2016.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA 2006; 103:1283–1288. doi: 10.1073/pnas.0510511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Yu Z, Jiang D, Liang X, Liao S, Zhang Z, et al. iPSC-MSCs with high intrinsic MIRO1 and sensitivity to TNF-alpha yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Reports 2016; 7:749–763. doi: 10.1016/j.stemcr.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Gerdes HH. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ 2015; 22:1181–1191. doi: 10.1038/cdd.2014.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma Q, Fang H, Shang W, Liu L, Xu Z, Ye T, et al. Superoxide flashes: early mitochondrial signals for oxidative stress-induced apoptosis. J Biol Chem 2011; 286:27573–27581. doi: 10.1074/jbc.M111.241794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HA, Blanco FJ. Cell death and apoptosis in osteoarthritic cartilage. Curr Drug Targets 2007; 8:333–345. doi: 10.2174/138945007779940025. [DOI] [PubMed] [Google Scholar]