

Summary
KRAS is a frequent oncogenic driver in solid tumors, including non-small cell lung cancer (NSCLC). It was previously thought to be an “undruggable” target due to the lack of deep binding pockets for specific small-molecule inhibitors. A better understanding of the mechanisms that drive KRAS transformation, improved KRAS-targeted drugs, and immunological approaches that aim at yielding immune responses against KRAS neoantigens have sparked a race for approved therapies. Few treatments are available for KRAS mutant NSCLC patients, and several approaches are being tested in clinicals trials to fill this void. Here, we review promising therapeutics tested for KRAS mutant NSCLC.
Keywords: non-small cell lung cancer, KRAS, targeted therapy, immunotherapy, clinical trials, personalized medicine
Graphical Abstract
Highlights
KRAS, an oncogenic driver in lung cancer, is known as an “undruggable” target
Different strategies are under investigation to indirectly and directly target KRAS
Clinical trials of small-molecule inhibitors report promising interim results
In this review, Salgia et al. discusses KRAS, a prominent oncogenic driver in lung cancer that is notoriously difficult to target. Studies have shown preclinical rationale in leveraging neoantigens and inhibiting KRAS signaling pathways in KRAS-mutant cancers. Recent advancements in clinical trials have demonstrated encouraging results with small-molecule inhibitors.
Introduction
Mutations in Kirsten rat sarcoma viral oncogene homolog (KRAS) are one of the most common genomic alterations identified in solid tumors, especially lung cancer, colorectal cancer, and pancreatic ductal adenocarcinoma (Figure 1). These cancers rely on continued activation and signaling of KRAS, therefore making it an ideal therapeutic target. Unfortunately, therapeutic approaches specifically targeting KRAS, such as attempts to inhibit KRAS membrane localization with farnesyltransferase inhibitors, failed in clinical trials and have thus far not been successful. In non-small lung cancer (NSCLC), the current treatment for KRAS mutant patients relies on chemotherapy, with an average overall survival (OS) of 22 months, which is less than desired.1 Since previous efforts failed to specifically target KRAS, researchers began to indirectly target KRAS through its role in various signaling pathways. Unfortunately, targeting KRAS dependencies in NSCLC has highlighted the difficulty in the effective inhibition of indirect targeting of KRAS.2,3 In addition, downstream pathway blockade of rapidly accelerated fibrosarcoma kinase/mitogen-activated protein kinase/phosphatidylinositol 3-kinase (RAF/MEK/PI3K) with inhibitors has not yet been proven to be effective as shown by the negative results of the Selumetinib Evaluation as Combination Therapy-1 (SELECT-1) trial of selumetinib combined with docetaxel.4, 5, 6 Small-molecule inhibitors have been challenging to develop since mutant KRAS has a very high affinity for guanosine triphosphate (GTP) and the catalytic sites are small and tough to target. Analyses of the KRAS subpopulation in landmark clinical trials in NSCLC have provided information on response (overall response rate of 17%–18%) and survival (median OS of 3 months) of this patient cohort for approved therapeutics,7, 8, 9 but there is still a lack of effective treatments for mutant KRAS. Consequently, the search for therapies that successfully target frequent KRAS mutations has continued due to its large incidence in various cancers.
Figure 1.

Frequency of KRAS mutations in relation to NRAS and HRAS mutation in various malignancies. Uterine CS, uterine carcinosarcoma; AML, acute lymphoblastic leukemia; DLBC, diffuse B cell lymphoma; pRCC, papillary renal cell carcinoma.
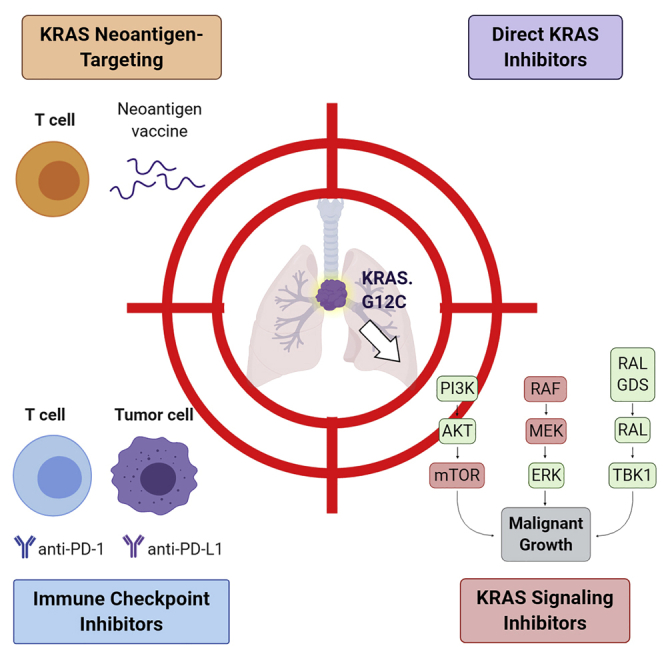
In <5 years, the availability of novel therapeutics has transformed the landscape of KRAS treatments, and a target that was once considered “undruggable” now has several therapeutic inhibitors undergoing clinical trials.10, 11, 12 There are essentially four novel approaches to target KRAS that can be divided into targeted therapies and immunotherapies, potentially in combination with one another:1 a novel class of direct KRAS inhibitors that specifically inhibit the G12C mutations through direct interaction with the cysteine residue, as well as a somewhat broader approach using a pan-KRAS inhibitor that does not attempt to inhibit specific mutations;2 novel signaling inhibitors that can inhibit chronic activation of KRAS-dependent pathways;3 immune checkpoint inhibitors (ICIs) that can be used in combination with other approaches;4 and approaches that take advantage of the neoantigen feature of mutated KRAS protein and attempt to improve the T cell response. Therefore, we present in this review recent advances in therapeutic options from a clinical standpoint, to understand the mechanisms of these inhibitors and potential future considerations regarding KRAS and therapeutic efficacy in NSCLC.
Mutated KRAS is a prominent oncogene in lung cancer
The intracellular guanine nucleotide-binding protein (G protein) KRAS is a member of the small GTPases RAS gene family that also includes NRAS and HRAS. KRAS encodes for six exons, resulting in two major splice variants, KRAS4A and KRAS4B, which differ in the C-terminal sequence. This region is required for membrane localization and contains a dual membrane-targeting motif in KRAS4A, in contrast to KRAS4B, which only contains one farnelysation site (Figure 2A). Since KRAS4B is the dominant form and KRAS4A is only marginally expressed in normal cells, it was long thought that the splicing of KRAS was of little consequence. In tumors, the overall expression of KRAS4A can be greatly elevated.13 This is of interest since KRAS4A has been reported to be required for the formation of chemically induced lung cancer in mice.14 Also, active KRAS4A functions as a regulator of hexokinase 1, which depends on its palmitoylation and colocalization with the metabolic enzyme on the outer mitochondrial membrane, thus effectively modulating glucose metabolism.15 Most mutations in KRAS affect codons 12, 13, 61, and 146; however, codon 146 is rarely altered in some cancers, such as NSCLC. Oncogenic KRAS missense mutations result in the high-affinity binding of GTP and loss of GTPase activity, thereby leaving KRAS in an “on” state and deregulating various signaling pathways that rely on active RAS (Figure 2B).16,17 More recent work suggests that another potential mechanism of oncogenic KRAS activation (A146T mutant and possibly also V14I and G12D) involves the acceleration of intrinsic and guanine nucleotide exchange factor-induced nucleotide exchange.18 The most frequent KRAS mutations in NSCLC are G12C, with almost half of all cases, followed by G12V and G12D (Figure 2C).19,20 Thus, lung cancer cells express mutations in both KRAS4A and KRAS4B.
Figure 2.

Structure and function of KRAS
(A) The KRAS gene consists of 6 exons, wherein exon −1 and part of exon 1 are non-coding. The mature protein exists in 2 major splice variants, KRAS4A and KRAS4B (top). KRAS4A and KRAS4B differ in the C-terminal membrane-targeting region. KRAS4A contains a single palmitoylation and farnesylation site, while KRAS4B contains 1 farnesylation and a putative serine phosphorylation site (bottom).
(B) RAS proteins are cycled from an active form (ON) through GTP binding facilitated by a guanine nucleotide-exchange factor (GEF) to an inactive form (OFF) by GTP hydrolysis through RAS itself, catalyzed by a GTPase-activating protein (GAP). In oncogenic RAS (e.g., with a G12 mutation, G12∗), this process is disrupted, and RAS remains active.
(C) Common KRAS mutations in lung adenocarcinoma by specific subtypes.
Lung cancer is the number one cause of cancer-related death worldwide and accounts for ~228,820 new cases and 140,730 deaths in 2020.21 NSCLC is the most frequent lung cancer subtype, and patients who are diagnosed with metastatic disease have reported 5-year OS rates of only 6%.22 With ~25% to 30% of cases, KRAS mutations are represented in a large portion of NSCLC patients,23 but effective therapies against KRAS have yet to be approved by the US Food and Drug Administration (FDA). The oncogenic mutations that drive NSCLC include not only KRAS but also epidermal growth factor receptor (EGFR) (17% in NSCLC), anaplastic lymphoma kinase (ALK) (7% in NSCLC), mesenchymal epithelial transition kinase (MET) (3% in NSCLC), and others, which are generally mutually exclusive.24, 25, 26, 27 It should be noted that there are ethnic differences in the frequency of KRAS mutations in NSCLC, with a higher incidence rate of KRAS mutations in White NSCLC patients as compared to Black or east Asian patients.28, 29, 30, 31 However, these differences are not known to affect the potential efficacy of novel KRAS therapeutics. Numerous targeted therapies against these actionable mutations have been developed and tyrosine kinase inhibitors (TKIs) targeting EGFR, ALK, or MET are already standard of care. In fact, a poor prognosis and a shorter median survival are reported for patients with KRAS mutant NSCLC versus patients with actionable mutations coupled with approved personalized therapies (2.41 versus 3.5 years, p < 0.001).32 These data demonstrate the impact of targeted therapies on response and OS in NSCLC cases caused by a specific driver mutation. Fortunately, several KRAS-targeted therapies are under investigation in clinical trials. Interestingly, many of the oncogenes in NSCLC signal through KRAS, and some of the approaches developed against active KRAS may also be applicable in these patients.
KRAS mutations and clinical characteristics
In past studies, KRAS mutations have been reported as a prognostic indicator of poor OS in NSCLC,33, 34, 35 while other studies have demonstrated no difference in response to chemotherapy/targeted therapy or OS.36, 37, 38 Although its true prognostication is somewhat unclear due to the complexity of KRAS-driven tumors, several analyses have noted a predictive value of KRAS, indicating poor prognosis.39,40 For example, a retrospective analysis of 482 lung adenocarcinoma cases at a single institution characterized the incidence of KRAS and its association with age, gender, race, and smoking history.41 The study determined that NSCLC patients who are positive for KRAS mutations are typically White and have a history of cigarette smoking. They also reported that age, gender, or the number of pack-years was not associated with KRAS mutation incidence. Interestingly, never smokers were more likely to have G12A transition mutations, while former or current smokers were more likely to report G12T or G12C transversion mutations,41 which was recapitulated in other studies.19,42,43 Another study by Dogan et al.19 examined 3,026 lung adenocarcinoma cases for clinical correlations of common driver mutations EGFR and KRAS. They reported a high frequency of KRAS G12C in women compared to men (p = 0.007).
Reports have been inconsistent in determining whether treatment response, progression-free survival (PFS), and OS differ based on the KRAS mutation subtype. KRAS mutation subtypes have been reported to rely on different signaling pathways, with G12C and G12V activating the Ral signaling pathway, while KRAS G12C tumors use PI3K and MEK signaling pathways.44 However, the putative molecular mechanisms that regulate these pathway switches are unclear, and the results have not been independently validated. Similarly, in vitro studies have demonstrated that KRAS point mutations have variable responses to chemotherapy,45 but other analyses found no significant differences in response to or survival in chemotherapy or immunotherapy.7 In an analysis of 179 surgically resected lung adenocarcinoma patients, KRAS G12C was found to be associated with worse disease-free survival (DFS) and OS as compared to G12D (hazard ratio [HR] 2.81, p = 0.035) or G12A (HR 5.99, p = 0.016).46 However, this was not true when compared to G12V (HR 1.62, p = 0.259). An analysis of 43 KRAS mutant NSCLC patients who were enrolled in the BATTLE-1 (Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination) clinical trial determined that KRAS mutation variants G12C and G12V were significantly associated with worse median PFS compared to other KRAS variants or wild-type KRAS (1.84 versus 3.35 versus 1.95 months, respectively, p = 0.046).44 In contrast to these findings, a large retrospective study evaluated 677 patients with KRAS-mutated metastatic NSCLC and discovered no significant difference between KRAS mutation subtypes.47 The overall data suggest that differences among mutation subtypes are rather small.
KRAS and other transforming pathways in NSCLC
The mutational landscape in NSCLC is complex, and despite the mutational activation of KRAS, one can distinguish KRAS-dependent or KRAS-independent groups of KRAS mutant NSCLC through their gene expression signatures. KRAS-dependent tumors, which rely on the continued activation of mutant KRAS through downstream signaling pathways, are correlated with a well-differentiated epithelial signature. Meanwhile, KRAS-independent tumors, which have decreased KRAS coupling to downstream signaling pathways, are correlated with an epithelial-to-mesenchymal transition (EMT) signature, a key process in cancer cell invasion, migration, and drug resistance.48,49 These findings suggest that KRAS-dependent and -independent NSCLCs should be therapeutically approached as distinct subtypes since they are driven by different signaling cascades.50 Although KRAS amplification can occur concurrently with EGFR amplification,51 KRAS mutations tend to occur independently of EGFR mutations in NSCLC and do not respond to EGFR TKIs, as highlighted by their contrasting clinical characteristics.52, 53, 54, 55 Like other important NSCLC driver oncogenes such as EGFR and ALK, co-mutations with KRAS are quite frequent.56 Research has established that the existence of KRAS and its specific co-mutation partner signifies distinct biological subtypes and variable responses to therapeutic treatments.57 For example, KRAS was found to occur often with other important oncogenes such as TP53 (40%), STK11/LKB1 (32%), and CDKN2A (19.8%).58 The study demonstrated that with each of these common co-mutation partners, the downstream signaling effects diverge, leading to variances in genetic events, tumor differentiation, and gene expression, suggesting a difference in response to therapies. It also indicated a worse prognosis in KRAS mutant NSCLC patients with STK11/LKB1 or CDKN2A alterations compared to patients with TP53 alterations. The prognostic role of KRAS and TP53 co-occurrence as an indicator of worse PFS remains controversial.59,60 A study on co-occurring mutations in KRAS mutant NSCLC confirmed a high prevalence of TP53 and STK11 but also noted KEAP1/NFE2L2 incidence (27%).61 KEAP1 and KRAS co-mutated tumors were associated with shorter OS after the initiation of immunotherapy. Skoulidis et al.62 found that KRAS mutant NSCLC patients with STK11/LKB1 co-mutations stimulated resistance to anti-programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) antibodies. Of 174 KRAS mutant lung adenocarcinomas, most of which were treated with immunotherapy, 31% expressed STK11/LKB1 alterations, while the rest either expressed TP53 or no major co-mutation. Objective response rates (ORRs) were calculated, and patients expressing both KRAS and STK11/LKB1 alterations demonstrated an ORR of 7.4%. This was significant when compared with KRAS/TP53 or KRAS alone cohorts (7.4% versus 35.7% versus 28.6%, respectively; p < 0.001). The authors confirmed this finding by analyzing the KRAS subgroup ORR to nivolumab in the CheckMate-057 trial (0% in KRAS/STK11 patients versus 57.1% in KRAS/TP53 patients versus 18.2% in KRAS alone; p = 0.047).63 The median PFS (1.8 versus 3.0 versus 2.7 months; p = 0.0018) and OS (6.4 versus 16.0 versus 16.1 months; p = 0.0045) of these patients were also similarly affected.
Targeting KRAS dependencies in lung cancer
Since KRAS mutant tumors survive and proliferate via major signaling pathways often involving MEK, PI3K-AKT, and mammalian target of rapamycin (mTOR), inhibition of their signaling mechanisms should ideally result in response and tumor regression. In the search for signaling mechanisms that are required for mutant KRAS transformation in NSCLC, several genes have been identified via synthetic lethal screenings, including B-cell lymphoma-extra large (BCL-XL), TANK-binding kinase 1 (TBK1), and cyclin-dependent kinase 4 (CDK4), and targeting their expression resulted in the death of mutant KRAS cells in vitro.64 Corcoran et al.65 demonstrated that the dual inhibition of BCL-XL and MEK triggered significant apoptosis and tumor regression in KRAS mutant xenograft models. Based on preclinical data, an ongoing clinical trial is investigating the safety and tolerability of combining trametinib (MEK1/2 inhibitor) and navitoclax (BCL-XL/BCL-2 inhibitor) in advanced or metastatic solid tumors (NCT02079740). Barbie et al.66 identified TBK1, a gene involved in the Ral-GEF signaling pathway essential for mutant KRAS tumors, as a synthetic lethal gene in preclinical models. Subsequent research revealed that TBK1 enhances tumor survival by activating CCL5 and interleukin-6 in aggressive KRAS-mutated lung adenocarcinoma mouse models.67 A phase IB study of momelotinib (JAK2 and TBK1 inhibitor) combined with trametinib (MEK1/2 inhibitor) in previously treated, refractory, or metastatic KRAS mutant NSCLCs2 of 21 patients showed no ORR in any patient, and 12 patients (57.1%) had a disease control rate (DCR) of 8 weeks (90% confidence interval [CI], 37.2%–75.5%). The median PFS and median OS were 3.6 months (90% CI, 2.2–5.6 months) and 7.4 months (90% CI, 4.0–15.3 months), respectively. However, the maximum plasma concentrations of momelotinib failed to inhibit TKB1 in patients, emphasizing the need for the development of more potent and selective inhibitors. Thus, whereas the addition of momelotinib demonstrated no clinical improvement over trametinib monotherapy in KRAS mutant NSCLC, these results do not answer the question of whether TKB1 targeting could have therapeutic benefits in vivo.
Another synthetic lethal screening revealed that loss of the CDK4, involved in cell-cycle progression, resulted in cell deterioration and tumor inhibition of mutant KRAS cells in mouse models.68 Although CDK inhibitors demonstrate major drug toxicities and limited clinical activity as monotherapy, several clinical trials have been conducted to elucidate the role of CDK inhibitors in KRAS mutant lung cancer. A phase I trial examining abemaciclib (CDK4/6 inhibitor) confirmed encouraging clinical activity in patients with KRAS mutant versus KRAS wild-type NSCLC tumors.69 Recently, the team conducting the phase III JUNIPER study (A Study of Abemaciclib [LY2835219] in Participants With Previously Treated KRAS Mutated Lung Cancer), examining abemaciclib in previously treated mutant KRAS NSCLC, presented its findings, expressing that the trial did not reach its primary endpoint of OS.3 Four hundred fifty-three previously treated KRAS mutant NSCLC patients were randomized 3:2 to receive abemaciclib (n = 270) or erlotinib (n = 183). Although abemaciclib did not improved median OS compared to erlotinib (7.4 versus 7.9 months, respectively), it did demonstrate an improvement in median PFS (3.6 versus 1.9 months; HR [95% CI]: 0.58 [0.47–0.72], p < 0.001) as well as a significantly higher ORR (8.9% versus 2.7%, p = 0.01). Palbociclib (CDK4/6 inhibitor) is under investigation in the National Lung Matrix Trial (NCT02664935) for patients diagnosed with NSCLC with a CDK4/6 genomic alteration. The RAS signaling pathway shows some level of redundancy or cross-activation, and single synthetic lethal targets may not be sufficient to block transformation by mutant KRAS in vivo. These studies highlight the difficulty in achieving effective target inhibition of synthetic lethal partners of KRAS in patients. Recent preclinical data have demonstrated that inhibitors against KRAS-dependent pathway targets, including mTOR and insulin-like growth factor 1, combined with KRAS G12C inhibitors instead of MEK inhibitors, significantly improved the efficacy and selectivity of blocking downstream MEK/ERK signaling in KRAS G12C mutant tumor cells in vitro and in mouse models.70 Effective treatment of KRAS tumors could involve a combination approach of direct KRAS targeting and indirect targeting through synthetic lethality of downstream effectors.
Targeting KRAS-regulated pathways
KRAS is involved in various downstream signaling pathways that could allow for the indirect targeting of KRAS. Mutations in KRAS result in dysregulated signaling of different cascades, including MET overexpression, the RAF/MEK/ERK, the PI3K/AKT/mTOR, the RHO/FAK, and the Ral/GEF pathways.71 Targeting patients with KRAS mutant cancers via downstream pathways as a strategy to affect KRAS has been examined in clinical trials (Table 1) with average response rates of <20%,72 as compared to EGFR or ALK response rates of 60% to 80% when treated with TKIs.73,74 These differential outcomes can be explained considering that EGFR and ALK can be targeted with specific inhibitors, while direct inhibition of KRAS has been made possible only recently.
Table 1.
Select completed trials investigating inhibitors against KRAS-regulated pathway targets in KRAS-mutant NSCLC
| Therapeutic drug | Target | Trial | Activity/results in KRAS subgroup | Toxicity |
|---|---|---|---|---|
| TVB-2640 | FASN | NCT02223247: A Phase 1, First-In-Human Study of Escalating Doses of Oral TVB-2640 in Patients With Solid Tumors75 | • 11/18 patients demonstrated stable disease for a minimum of 19 weeks | common related AEs include alopecia (41%), palmar-plantar erythrodysesthesia (47%), and decreased appetite (13%) |
| Sorafenib | RAF | NCT00863746: A Phase III, Multicenter, Placebo-Controlled Trial of Sorafenib (BAY43-9006) in Patients With Relapsed or Refractory Advanced Predominantly Non-Squamous Non-Small Cell Lung Cancer (NSCLC) After 2 or 3 Previous Treatment Regimens for Advanced Disease76 | • median OS sorafenib 6.4 mo versus placebo 5.1 mo | common related AEs include skin toxicities (40.5%), fatigue (36.1%), and diarrhea (35.8%) |
| • median PFS sorafenib 2.6 mo versus placebo 1.7 mo | ||||
| • ORR sorafenib 2.9% versus placebo 0% | ||||
| Selumetinib + docetaxel | MEK | NCT01933932: A Phase III, Double-Blind, Randomized, Placebo-Controlled Study to Assess the Efficacy and Safety of Selumetinib (AZD6244; ARRY-142886) (Hyd-Sulfate) in Combination With Docetaxel, in Patients Receiving Second Line Treatment for KRAS Mutation-Positive Locally Advanced or Metastatic Non Small Cell Lung Cancer (Stage IIIB–IV) (SELECT 1)6 | • median OS selumetinibc+cdocetaxel 8.7 months versus placeboc+cdocetaxel 7.9 mo | common related AEs in the selumetinibc+cdocetaxel group were diarrhea (61%), nausea (38%), rash (34%), and peripheral edema (30%) |
| • median PFS selumetinibc+cdocetaxel 3.9 months versus placeboc+cdocetaxel 2.8 mo | ||||
| • ORR selumetinibc+cdocetaxel 20.1% versus placeboc+cdocetaxel 13.7% | ||||
| Ridaforolimus | mTOR | NCT00818675: A Randomized Discontinuation Phase II Trial of Ridaforolimus in Non-Small Cell Lung Cancer (NSCLC) Patients With KRAS Mutations5 | • median OS ridaforolimus 18 mo versus placebo 5 mo | common related AEs include fatigue (10%), mucositis/stomatitis (10%), pneumonia (10%), dyspnea (9%), diarrhea (6%), and hyperglycemia (6%) |
| • median PFS ridaforolimus 4 mo versus placebo 2 mo | ||||
| • ORR at 8 weeks ridaforolimus 1% |
See also https://clinicaltrials.gov/. AEs, adverse events; FASN, fatty acid synthase; KRAS, Kirsten rat sarcoma viral oncogene homolog; MEK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; RAF, rapidly accelerated fibrosarcoma kinase.
Fatty acid synthase (FASN)
It was previously shown that KRAS induces fatty acid synthesis by activating lipogenesis and ERK2 kinase activity. Also, inhibition of one of the FASN subunits with cerulenin in KRAS mutant lung adenocarcinoma cell lines showed a significant decrease in cell growth.77 The small-molecule drug TVB-2640, an orally bioavailable and first-in-class inhibitor of FASN, was studied as monotherapy or with paclitaxel in a phase I clinical trial of NSCLC patients.78 Preliminary results from the phase I trial showed promising clinical activity in KRAS mutant NSCLC.75,79,80 Of the 31 NSCLC patients included in the study, 18 were KRAS mutant. Six patients on monotherapy TVB-2640 and 5 patients on the combination regimen achieved stable disease for at least 19 and 23 weeks, respectively. KRAS mutant NSCLC patients achieved a longer PFS on TVB-2640 monotherapy, with 60% of KRAS mutant patients still on trial at 12 weeks versus 0% of KRAS wild-type patients. A phase II clinical trial investigating TVB-2640 is under way in KRAS mutant NSCLC (NCT03808558).
RAF
RAF inhibition was examined in preclinical and clinical models to determine potential efficacy against KRAS. BRAF inhibitors vemurafenib and dabrafenib are contraindicated for use in KRAS tumors since they stimulate tumorigenesis through binding to BRAF, also known as the MAPK paradox.81 Thus, targeting other members of the RAF family was explored in KRAS-mutated NSCLC. Preclinical data demonstrated the inhibition of cell growth in KRAS mutant NSCLC cells when treated with sorafenib, an oral multikinase inhibitor directed at RAF and similar transmembrane receptors.4 Although promising, clinical models did not demonstrate great efficacy when using sorafenib. A phase II clinical trial investigated sorafenib in KRAS-mutated stage IIIB/IV NSCLC patients that progressed on a previous line of platinum chemotherapy with a primary endpoint of the DCR.82 Fifty-seven patients treated with sorafenib demonstrated an observed DCR of 52.6%, a median PFS of 2.3 months, and a median OS of 5.3 months. Grade III/IV adverse events (AEs) secondary to sorafenib, including skin and gastrointestinal toxicities, were reported in 10 patients (17.5%). This modest clinical activity warranted further investigation and resulted in a phase III multicenter, placebo-controlled clinical trial in relapsed or refractory non-squamous NSCLC patients treated with monotherapy sorafenib after progression on at least two treatment lines.76 Although OS was comparable, PFS was significantly longer in the KRAS mutant subpopulation as compared to the KRAS wild-type group when treated with sorafenib. Common AEs included skin toxicities (40.5%), fatigue (36.1%), and diarrhea (35.8%). Finally, in the BATTLE-1 trial, although sorafenib demonstrated better DCR in KRAS mutant patients compared with all of the other TKIs (61% versus 32%), this was not significant (p = 0.11).83
MEK
Clinical trials targeting the KRAS effector MEK with CI-1040 or PD-0325901 in various solid tumor cancers demonstrated insufficient antitumor activity.84 Selumetinib, a potent, selective MEK inhibitor, was shown to inhibit tumor growth and proliferation in KRAS mutant xenograft models.85 This was significantly enhanced when combined with cytotoxic chemotherapy docetaxel or irinotecan. Several phase II clinical trials were initiated to examine selumetinib versus single-agent chemotherapy agents. Selumetinib versus pemetrexed was first evaluated in advanced NSCLC patients who progressed on first-line treatment and no clinical benefit over the standard of care was found.86 Based on the previous preclinical study, the second trial investigated selumetinib with docetaxel in previously treated KRAS mutant NSCLC. Eighty-seven patients were enrolled in the trial and were randomly assigned to receive selumetinib and docetaxel (n = 44) or placebo and docetaxel (n = 43).87 Adding selumetinib to docetaxel demonstrated promising clinical activity in terms of ORR and PFS. However, the phase III SELECT-1 trial of selumetinib and docetaxel in KRAS mutant NSCLC failed to demonstrate improved PFS versus docetaxel alone.6 Of the 510 patients who were randomized to receive treatment, 505 actually received treatment with either selumetinib and docetaxel (n = 251) or placebo and docetaxel (n = 254). Median OS (8.7 versus 7.9 months, p = 0.64) and median PFS (3.9 versus 2.8 months, p = 0.44) were not significant in the selumetinib and docetaxel group compared to the placebo and docetaxel group. The trial reported an ORR of 20.1% in the combination regimen and 13.7% in the placebo and docetaxel cohort (p = 0.05); however, the addition of selumetinib to docetaxel ultimately did not improve OS or PFS. A phase II clinical trial examined monotherapy selumetinib or in combination with erlotinib in previously treated advanced NSCLC patients.88 Although the ORR improved in the mutant KRAS combination cohort (0% for selumetinib alone versus 10% for combination), combination therapy led to increased AEs. Trametinib, a selective allosteric MEK inhibitor, has also been examined in clinical trials and demonstrated results comparable to those of selumetinib.89, 90, 91 Several trials investigating MEK inhibitors combined with other therapies are ongoing (NCT02964689, NCT01859026, NCT03170206, NCT03299088, and NCT02185690).
PI3K/AKT/mTOR pathway
Although KRAS alterations can occur with PI3K alterations, research has shown that PI3K inhibitors as monotherapy are ineffective. Recently, a preclinical study reported that combination treatment with KRAS G12C inhibitor ARS1620 and a PI3K inhibitor overcame resistance in patient-derived xenograft models resistant to the KRAS inhibitor.92 mTOR inhibitor ridaforolimus was investigated in a phase II clinical trial in KRAS mutant advanced NSCLC.5 Seventy-nine patients were enrolled and randomized 1:1 to receive ridaforolimus or placebo. Although the trial investigators reported a low ORR (1%) and no significant benefit in OS, they reported prolonged median PFS in patients treated with ridaforolimus (4 versus 2 months, p = 0.013, HR 0.36). Common grade 3/4 AEs were fatigue (10%), mucositis/stomatitis (10%), pneumonia (10%), dyspnea (9%), diarrhea (6%), and hyperglycemia (6%). Overall, preclinical and clinical data on targeting this pathway in KRAS mutant tumors suggest that combination treatment with other inhibitors could enhance therapeutic efficacy.
KRAS inhibitors
Since RAS proteins must localize to the plasma membrane to be functionally active, it was long thought that blocking KRAS farnesylation would be beneficial. The majority of the effort was focused on developing farnesyltransferase inhibitors (FTIs) due to lipid post-translational modifications catalyzed by these enzymes that are required for the oncogenic activity of KRAS.93 Unfortunately, clinical trial testing of this class of inhibitors yielded poor results.94, 95, 96, 97 The development of novel inhibitors that directly target KRAS is expected to transform the therapeutic landscape of cancers affected by this oncogenic driver. Although none of these inhibitors are FDA approved, several promising drugs have entered clinical trials (Table 2). Ideally, efforts began in developing a small-molecule inhibitor targeted at the KRAS-binding site to prevent GTP-KRAS interactions, but they have failed primarily due to the high affinity of GTP for KRAS and the lack of suitable binding pockets for small-molecule drugs that could interfere with RAS function. Thus, the focus has shifted away from approaching KRAS inhibition as “one-size-fits-all” and instead to approaching it from many sides. Drugs including the pan-KRAS inhibitor BBP-454 (BridgeBio Pharma) may change the paradigm of KRAS as an undruggable target as it is designed to bind to previously unrecognized binding pockets.98,99 It is in the preclinical phase and data have not yet been formally reported. Also, interest in targeting KRAS G12C arose when it was discovered that KRAS had an allosteric regulatory site that could lead to inhibition.100 Preliminary research on targeting the allosteric site revealed that it was mutation specific to KRAS G12C.101, 102, 103 Thus, various pharmaceutical companies have begun to develop KRAS G12C-specific inhibitors, many of which are being explored in ongoing clinical trials, as noted in the following four subsections.
Table 2.
Select clinical trials in KRAS mutant cancers
| Therapeutic drug | Target | Trial | Primary objective(s) |
|---|---|---|---|
| AMG 510 | KRAS | NCT03600883: A Phase I/II, Open-label Study Evaluating the Safety, Tolerability, Pharmacokinetics, and Efficacy of AMG 510 in Subjects With Solid Tumors With a KRAS p.G12C Mutation and AMG 510 Combination Therapy in Subjects with Advanced NSCLC with KRAS p.G12C Mutation (CodeBreak 100) | evaluate the safety and tolerability of AMG 510, estimate the MTD and/or a recommended phase II dose, ORR assessed by RECIST 1.1 criteria of AMG 510 as monotherapy |
| G12C | |||
| MRTX849 | KRAS | NCT03785249: Phase 1/2 Multiple Expansion Cohort Trial of MRTX849 in Patients with Advanced Solid Tumors with KRAS G12C mutation KRYSTAL-1 | characterize the safety of MRTX849 and evaluate the pharmacokinetics of the drug, ORR assessed by RECIST 1.1 criteria of MRTX849 as monotherapy |
| G12C | |||
| JNJ-74699157/ ARS-3248 | KRAS | NCT04006301: A First-in-Human Study of the Safety, Pharmacokinetics, Pharmacodynamics, and Preliminary Antitumor Activity of JNJ-74699157 in Participants with Advanced Solid Tumors Harboring the KRAS G12C Mutation | determine the MTD and RP2D of JNJ-74699157, determine the safety and preliminary antitumor activity of JNJ-74699157 |
| G12C | |||
| BI 1701963 | Pan-KRAS | NCT04111458: A Phase I Open-label Dose Escalation Trial of BI 1701963 as Monotherapy and in Combination With Trametinib in Patients With KRAS Mutated Advanced or Metastatic Solid Tumors | determine the MTD and RP2D of BI 1701963 as monotherapy and in combination with trametinib |
| mRNA-5671 (cancer vaccine) | KRAS G12C/ G12D/ G13D/ G12V | NCT03948763: A Phase 1, Open-Label, Multicenter Study to Assess the Safety and Tolerability of mRNA-5671/V941 as a Monotherapy and in Combination With Pembrolizumab in Participants With KRAS Mutant Advanced or Metastatic Non-Small Cell Lung Cancer, Colorectal Cancer or Pancreatic Adenocarcinoma | determine the safety and tolerability and establish a preliminary RP2D of mRNA-5671/V941 as a monotherapy and in combination with pembrolizumab infusion |
| KRAS-targeted long peptide vaccine | KRAS | NCT04117087: Pooled Mutant KRAS-Targeted Long Peptide Vaccine Combined With Nivolumab and Ipilimumab for Patients With Resected MMR-p Colorectal and Pancreatic Cancer | evaluate the safety and tolerability of the KRAS-based peptide vaccine, determine the drug-related toxicities, determine the fold change in interferon-producing mutant-KRAS-specific CD8 and CD4 T cells at 16 weeks |
| mDC3/8-KRAS vaccine | KRAS G12C/ G12D/ G12R/ G12V | NCT03592888: Pilot Study of Mature Dendritic Cell Vaccination Against Mutated KRAS in Patients With Resectable Pancreatic Cancer | evaluate the safety and tolerability of the KRAS mature dendritic cell vaccine |
| Mutant KRAS G12V-specific TCR transduced autologous T cells | KRAS G12V | NCT04146298: Clinical Trial Evaluating the Safety and Activity of Mutant KRAS G12V-specific TCR Transduced T Cell Therapy for Advanced Pancreatic Cancer | evaluate the safety and tolerability of the KRAS G12V-specific TCR transduced T cell therapy; ORR assessed by RECIST 1.1 criteria |
| Anti-KRAS G12D murine TCR peripheral blood lymphocytes | KRAS G12D | NCT03745326: A Phase I/II Study Administering Peripheral Blood Lymphocytes Transduced With a Murine T-Cell Receptor Recognizing the G12D Variant of Mutated RAS in HLA-A∗11:01 Patients | evaluate the safety and tolerability of anti-KRAS G12D murine TCR peripheral blood lymphocytes, ORR assessed by RECIST 1.1 criteria |
| Anti-KRAS G12V murine TCR peripheral blood lymphocytes | KRAS G12V | NCT03190941: A Phase I/II Study Administering Peripheral Blood Lymphocytes Transduced With a Murine T-Cell Receptor Recognizing the G12V Variant of Mutated RAS in HLA-A∗11:01 Patients | evaluate the safety and tolerability of anti-KRAS G12V murine TCR peripheral blood lymphocytes, ORR assessed by RECIST 1.1 criteria |
See also https://clinicaltrials.gov/. MMR-p, mismatch repair protein; MTD, maximum tolerated dose; RP2D, the highest dose with acceptable toxicity, usually defined as the dose level producing ~20% of dose-limiting toxicity; TCR, T cell receptor.
BI 1701963 (Boehringer Ingelheim)
BI 1701963 is a pan-KRAS inhibitor that interferes with KRAS binding to SOS1, a guanine nucleotide exchange factor essential in activating KRAS. By targeting and binding to SOS1, RAS activation is disrupted through the inhibition of the RAS-SOS1 interaction. Thus, due to its design, it can target mutated KRAS beyond the G12C mutation. The drug demonstrated promising antitumor activity against KRAS, and it was advanced into an ongoing phase I clinical trial as monotherapy or in combination with trametinib (NCT04111458).
AMG 510/sotorasib (Amgen)
The KRAS G12C inhibitor AMG 510/sotorasib was reported in preclinical studies to inhibit KRAS signaling and tumor growth in cell lines and patient-derived xenograft models.104 Furthermore, the drug enhanced in vitro antitumor activity and tumor immune infiltration in immunocompetent mouse models when combined with targeted therapy/chemotherapy and immunotherapy, respectively. The G12C inhibitor is being investigated in an ongoing phase I/II clinical trial in advanced KRAS mutant solid tumor patients (NCT03600883).105 The trial also contains an arm examining AMG 510 in combination with anti-PD-1/PD-L1 antibodies. The primary endpoints include the safety and tolerability of the drug as well as ORR. Preliminary data from the first 29 patients enrolled in the trial were presented at ESMO 2019.106 Ten of the 29 patients were diagnosed with NSCLC, 5 patients of whom exhibited a partial response, 4 exhibited stable disease, and 1 patient progressed. Based on the preliminary data, no disease-limiting toxicities were noted, and the majority of patients demonstrated good clinical activity. Recently, the final results from the phase I trial showed a median PFS of 6.3 months and a median duration of response of 10.9 months in their NSCLC subgroup (n = 59).10 Nineteen NSCLC patients (32.2%) had a confirmed ORR and 52 patients (88.1%) demonstrated disease control. At the first restaging scan at 6 weeks, 42 patients (71%) showed a decrease in tumor burden, with a median time to response of 1.4 months. The trial confirmed that AMG 510 exhibits antitumor efficacy in pretreated KRAS G12C mutant NSCLC, consistent with previous preliminary results.
JNJ-74699157/ARS-3248 (Janssen Biotech and Wellspring Biosciences)
Preclinical data allowed Nagasaka et al.11 to move the KRAS G12C inhibitor into a phase I clinical trial in advanced solid tumors harboring KRAS G12C mutations (NCT04006301).
MRTX849/adagrasib (Mirati Therapeutics)
Similar to its competitor drugs, MRTX849/adagrasib, a KRAS G12C inhibitor, exhibited significant inhibition of KRAS signaling and tumor growth in vivo, as well as tumor regression in mutant KRAS G12C cell lines and patient-derived xenograft models.107 KRAS G12C mutant patients are enrolling in the expansion cohort of the KRYSTAL (A Phase I/II Multiple Expansion Cohort Trial of MRTX849 in Patients With Advanced Solid Tumors With KRAS G12C Mutation) phase I/II clinical trial (NCT03785249). Preliminary data from the trial demonstrated great antitumor activity in 6 pretreated metastatic NSCLC patients with a >30% tumor decrease in half of those patients after 6 weeks.12
As the KRAS G12C trials continue, future directions will focus on strategies to prolong PFS, overcome treatment resistance by developing combination therapies, and develop more KRAS inhibitors to target other mutations. Recent preclinical work has attempted to elucidate how to maximize KRAS G12C mutant cell response to targeted therapy.108 It has been previously reported that while KRAS G12C cells fluctuate between active and inactive forms, KRAS G12C inhibitors only bind to its inactive state, or the GDP-bound state, and leave the cell in its inactivated form.109 The same study showed that in KRAS G12C mutant cell lines, suppressing the nucleotide exchange activity that allows cells to switch from active to inactive enhanced the inhibition of the drug. Researchers have discovered that this limitation allows tumor cells to undergo adaptive changes after the inhibitor is introduced to the tumor environment to become insusceptible to the drug.108 Instead of relying on the drug-inhibited MEK pathway to proliferate, these adapted cells now use EGFR and aurora kinase downstream signaling cascades as an escape mechanism to remain in their active form. Another preclinical study demonstrated similar results of feedback reactivation of the RAS pathway via receptor tyrosine kinase (RTK)-mediated activation of non-mutant RAS.110 Mainardi et al.111 found that Src homology region 2 (SHP2), a cytoplasmic Src homology 2 domain containing protein tyrosine phosphatase that regulates several cellular processes, is necessary for KRAS mutant tumor cell growth in in vivo models of NSCLC. The authors also discovered that the inhibition of SHP2 promoted a senescence response in KRAS mutant NSCLC cells, suggesting a preclinical rationale for targeting SHP2 in KRAS mutant tumors.111 Interestingly, a recent study reported that combined inhibition of KRAS and SHP2 effectively suppressed feedback reactivation of the RAS pathway and produced significant tumor regression in xenograft models.110 These data help elucidate why the majority of KRAS G12C patients in these clinical trials tend to exhibit partial responses to treatment versus complete radiographic response. Escape mechanisms such as these need to be addressed through the development of combination targeted therapies to create a more durable response to KRAS G12C inhibitors.
ICIs and mutant KRAS
Prior research has shown that PD-L1 expression is associated with KRAS alterations in KRAS NSCLC cell lines and patient tumor tissue.112 Also, mutated KRAS contributes to an immunosuppressive environment through phenotypic conversion of T cells113 and is able to upregulate PD-L1 expression in preclinical lung adenocarcinoma models through MEK/ERK signaling,112,114 suggesting a role for active KRAS in immunoediting of NSCLC. In lung adenocarcinoma tumor cells, PD-L1 expression variation correlates with KRAS, TP53, and STK11 alterations.115 Furthermore, KRAS alterations measured in the blood by liquid biopsy next-generation sequencing was associated with favorable clinical responses to ICIs.116 Consistent with previous reports,8,117 a recent preclinical study found a strong connection between KRAS mutation and immune markers associated with responses to ICI treatment, such as inflammatory tumor microenvironment, increased tumor-infiltrating lymphocytes, and high tumor mutation burden.118 These results indicate that mutant KRAS NSCLC patients in particular would gain clinical benefit from ICI treatment.
Immunotherapy with ICIs is already a lead standard-of-care treatment in NSCLC.63,119, 120, 121, 122, 123 A subgroup analysis of the KRAS NSCLC population in the KEYNOTE-042 clinical trial was performed.124 The results showed clinical benefits in patients treated with pembrolizumab monotherapy versus chemotherapy, with an ORR of 56.7% versus 18.0% in patients with any KRAS mutation (n = 69) and an ORR of 66.7% versus 23.5% in patients with KRAS G12C mutations (n = 29), respectively. The median PFS was comparable between any KRAS mutation and KRAS G12C. KRAS wild-type patients (n = 232) had a significantly smaller ORR (29.1% versus 21.0%) and observed no differences in median PFS between each arm (6 months for each treatment). A subgroup analysis of NSCLC patients treated with PD-1 inhibitors identified KRAS and STK11 co-mutations as negative predictors of response to ICIs.62 The median PFS was significantly shorter in the KRAS-STK11 co-mutation group when compared to the KRAS-TP53 co-mutation group or the KRAS-only group (1.8 versus 3.0 and 2.7 months, respectively, p = 0.0018).
Overall, these clinical studies report clinical activity in KRAS mutant NSCLC patients treated with ICIs; however, further analysis is necessary to confirm a distinct benefit of KRAS mutations and immunotherapy treatment. Preclinical data have shown that KRAS G12C inhibitor AMG 510 promotes a pro-inflammatory tumor microenvironment, suggesting potential synergy between KRAS G12C inhibitors and ICIs.104 This study also combined AMG 510 and anti-PD-1 therapy in mouse models, which produced long-term T cell responses against KRAS mutant tumor cells, providing preclinical rationale for the combination treatment of KRAS inhibitors and PD-1 blockade. There is an ongoing phase I/II study of AMG 510 treatment in KRAS G12C mutant tumors with an experimental arm to evaluate the combination therapy of AMG 510 and anti-PD-1/PD-L1 monoclonal antibodies (NCT03600883).
Targeting KRAS neoantigens
KRAS mutations create new epitopes that can be recognized by the immune system as neoantigens. The goal is to either elicit a T cell response against these neoantigens through vaccination or through adoptive T cell therapy, which are under investigation in several clinical trials (Table 2). mRNA-5671, a cancer vaccine against KRAS G12C, G12D, G13D, and G12V (from Moderna Therapeutics and Merck), is the only mRNA vaccine currently being considered. This vaccine is encoded as a neoantigen concatemer in a single RNA molecule, and once administered, is expected to be optimally presented on HLA-A11:01 and/or HLA-C∗08:02 receptors. Preclinical data reported an enhanced CD8+ T cell response against KRAS antigens after KRAS mRNA vaccination.125 A phase I clinical trial examining mRNA-5671 with or without pembrolizumab is recruiting advanced or metastatic KRAS mutant NSCLC patients (NCT03948763). In resected pancreatic ductal adenocarcinoma, an ongoing pilot study is recruiting eligible patients to undergo treatment with a vaccine composed of autologous dendritic cells pulsed with mutant KRAS peptides corresponding to the KRAS mutational variant of each patient, including G12C, G12D, G12R, and G12V (NCT03592888). A different strategy uses pooled KRAS neoantigen peptide-based vaccines in a clinical trial in combination with nivolumab and ipilimumab in patients with resected mismatch repair-proficient colorectal cancer and resected pancreatic cancer (NCT04117087). Preclinical reports in humanized mouse models demonstrated a strong induction of cytotoxic and T cell helper response against mutations through KRAS-targeted long peptide vaccinations.126 Another strategy is to infuse in patients autologous T cells transduced with mutant KRAS-specific T cell receptors (TCRs). Preclinical data showed that KRAS mutant-specific TCRs inhibited tumor growth in immunodeficient mouse models.127 A single case report demonstrated specific tumor response when using four different T cell clonotypes from a patient who specifically targeted KRAS G12D.128 In another case of successful adoptive T cell therapy, four different TCRs were identified within the tumor-infiltrating T cell population, recognizing a nonamer and a decamer neoantigen specific to KRAS G12D.129 There is a phase I/II clinical trial evaluating the safety and tolerability of KRAS G12V-specific TCR transduced T cell therapy in advanced pancreatic cancer (NCT04146298). There are also several ongoing phase I/II clinical trials examining gene transfer with murine TCRs that are equipped to recognize KRAS mutational variants such as G12D (NCT03745326) and G12V (NCT03190941) in KRAS mutant cancer patients that are HLA-A11:01, based on preclinical studies of murine T cells against epitopes in KRAS G12D/G12V mutant cells produced from HLA-A11:01 transgenic mice.127 Even though some of the therapeutics are initially not tested in lung cancers, these approaches are expected to have more general applicability. It is also important to note that these therapeutic approaches require meaningful MHC1-peptide display of the KRAS neoantigens, which has not been demonstrated by physical detection methods, and immunoediting may select against this.
Conclusions
NSCLC is a heterogenous disease, initially subclassified histologically and currently classified by molecular markers as well. We have made strides against EGFR mutations, ALK translocations, and other genomic alterations in lung cancer. The ICIs have also led to enhanced OS in certain tumors. KRAS mutations in lung cancer have been very difficult to treat. Recent developments in therapeutics have led to the resurgence of KRAS as one of the most promising oncogenic drivers with therapeutic potential in NSCLC, and efforts to directly target KRAS oncoproteins are becoming clinically relevant. For many years, KRAS had built a reputation as being “undruggable,” but several ongoing clinical trials are redefining the efficacy of different direct and indirect KRAS inhibitors through appropriate patient stratification and identification of subgroup mutations. Other treatment strategies including the inhibition of specific pathway genes such as MEK, RAF, and others are also under clinical investigation. Encouraging preliminary response and safety results for the direct allosteric irreversible inhibition of KRAS G12C has sparked a therapeutic race for FDA approval. This also triggered the development of a number of novel pan-KRAS inhibitors that interact with active KRAS independent of mutational status. KRAS inhibitors targeting specific mutations such as AMG 510 and MRTX849 are the most promising therapeutic approaches, and the biggest challenge that could prevent them from having a lasting impact in the clinic is therapeutic resistance caused by resistance mutations, phenotypic switching, and tumor plasticity, which has been noted in other targeted therapies such as EGFR and ALK. The role of ICIs in the treatment of mutant KRAS cancers is also under consideration and may enhance or may be enhanced by targeted therapy approaches. KRAS is an early mutational event in the development of NSCLC,46,130, 131, 132 and it would therefore be interesting to see whether immunological approaches that target the mutant KRAS neoantigen have the capacity to efficiently eradicate cancer stem cells to cause a durable response. Specific KRAS inhibition has been difficult to accomplish due to the small size of the protein and a surface area with few deep pockets for drug interaction, but numerous agents under investigation in preclinical and clinical models have overcome this challenge by using the distinct alterations of KRAS mutant tumors to provide targeted therapeutics that may finally provide significantly improved survival for this patient group.
Acknowledgments
The work was supported by the National Cancer Institute of the National Institutes of Health under award nos. P30CA033572, U54CA209978, R01CA247471, and R01CA218545.
Author contributions
All of the authors contributed to the manuscript.
Declaration of interests
R.S. reports speakers’ bureau participation with AstraZeneca and Merck and is an advisory board member for Janssen and Novartis. R.P., I.M., A.N., and M.S. declare no competing interests.
References
- 1.Awad M.M., Gadgeel S.M., Borghaei H., Patnaik A., Yang J.C.-H., Powell S.F., Gentzler R.D., Martins R.G., Stevenson J.P., Altan M. Long-Term Overall Survival From KEYNOTE-021 Cohort G: Pemetrexed and Carboplatin With or Without Pembrolizumab as First-Line Therapy for Advanced Nonsquamous NSCLC. J. Thorac. Oncol. 2020 doi: 10.1016/j.jtho.2020.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Barbie D.A., Spira A., Kelly K., Humeniuk R., Kawashima J., Kong S., Koczywas M. Phase 1B Study of Momelotinib Combined With Trametinib in Metastatic, Kirsten Rat Sarcoma Viral Oncogene Homolog-Mutated Non-Small-Cell Lung Cancer After Platinum-Based Chemotherapy Treatment Failure. Clin. Lung Cancer. 2018;19:e853–e859. doi: 10.1016/j.cllc.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldman J.W., Mazieres J., Barlesi F., Koczywas M., Dragnev K.H., Göksel T., Cortot A.B., Girard N., Wesseler C., Bischoff H. A randomized phase 3 study of abemaciclib versus erlotinib in previously treated patients with stage IV NSCLC with KRAS mutation: JUNIPER. J. Clin. Oncol. 2018;36:9025. doi: 10.3389/fonc.2020.578756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takezawa K., Okamoto I., Yonesaka K., Hatashita E., Yamada Y., Fukuoka M., Nakagawa K. Sorafenib inhibits non-small cell lung cancer cell growth by targeting B-RAF in KRAS wild-type cells and C-RAF in KRAS mutant cells. Cancer Res. 2009;69:6515–6521. doi: 10.1158/0008-5472.CAN-09-1076. [DOI] [PubMed] [Google Scholar]
- 5.Riely G.J., Brahmer J.R., Planchard D., Crinò L., Doebele R.C., Lopez L.A.M., Gettinger S.N., Schumann C., Li X., Atkins B.M. A randomized discontinuation phase II trial of ridaforolimus in non-small cell lung cancer (NSCLC) patients with KRAS mutations. J. Clin. Oncol. 2012;30:7531. [Google Scholar]
- 6.Jänne P.A., van den Heuvel M.M., Barlesi F., Cobo M., Mazieres J., Crinò L., Orlov S., Blackhall F., Wolf J., Garrido P. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA. 2017;317:1844–1853. doi: 10.1001/jama.2017.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeanson A., Tomasini P., Souquet-Bressand M., Brandone N., Boucekine M., Grangeon M., Chaleat S., Khobta N., Milia J., Mhanna L. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC) J. Thorac. Oncol. 2019;14:1095–1101. doi: 10.1016/j.jtho.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 8.Dong Z.-Y., Zhong W.-Z., Zhang X.-C., Su J., Xie Z., Liu S.-Y., Tu H.-Y., Chen H.-J., Sun Y.-L., Zhou Q. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017;23:3012–3024. doi: 10.1158/1078-0432.CCR-16-2554. [DOI] [PubMed] [Google Scholar]
- 9.Passiglia F., Cappuzzo F., Alabiso O., Bettini A.C., Bidoli P., Chiari R., Defferrari C., Delmonte A., Finocchiaro G., Francini G. Efficacy of nivolumab in pre-treated non-small-cell lung cancer patients harbouring KRAS mutations. Br. J. Cancer. 2019;120:57–62. doi: 10.1038/s41416-018-0234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong D.S., Fakih M.G., Strickler J.H., Desai J., Durm G.A., Shapiro G.I., Falchook G.S., Price T.J., Sacher A., Denlinger C.S. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020;383:1207–1217. doi: 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagasaka M., Li Y., Sukari A., Ou S.I., Al-Hallak M.N., Azmi A.S. KRAS G12C game of thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 2020;84:101974. doi: 10.1016/j.ctrv.2020.101974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jänne P., Papadopoulous K., Ou S., Rybkin I., Johnson M. Paper presented at AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, Boston, MA, October 28. 2019. A phase 1 clinical trial evaluating the pharmacokinetics (PK), safety, and clinical activity of MRTX849, a mutant-selective small molecule KRAS G12C inhibitor, in advanced solid tumors.https://www.mirati.com/wp-content/uploads/2019/10/AACR-NCI-EORTC-Clinical-Data-Presentation_Janne_October-2019-1-1.pdf [Google Scholar]
- 13.Tsai F.D., Lopes M.S., Zhou M., Court H., Ponce O., Fiordalisi J.J., Gierut J.J., Cox A.D., Haigis K.M., Philips M.R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. USA. 2015;112:779–784. doi: 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.To M.D., Wong C.E., Karnezis A.N., Del Rosario R., Di Lauro R., Balmain A. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat. Genet. 2008;40:1240–1244. doi: 10.1038/ng.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amendola C.R., Mahaffey J.P., Parker S.J., Ahearn I.M., Chen W.C., Zhou M., Court H., Shi J., Mendoza S.L., Morten M.J. KRAS4A directly regulates hexokinase 1. Nature. 2019;576:482–486. doi: 10.1038/s41586-019-1832-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riely G.J., Marks J., Pao W. KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 2009;6:201–205. doi: 10.1513/pats.200809-107LC. [DOI] [PubMed] [Google Scholar]
- 17.Malumbres M., Barbacid M. RAS oncogenes: the first 30 years. Nat. Rev. Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 18.Poulin E.J., Bera A.K., Lu J., Lin Y.J., Strasser S.D., Paulo J.A., Huang T.Q., Morales C., Yan W., Cook J. Tissue-Specific Oncogenic Activity of KRASA146T. Cancer Discov. 2019;9:738–755. doi: 10.1158/2159-8290.CD-18-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dogan S., Shen R., Ang D.C., Johnson M.L., D’Angelo S.P., Paik P.K., Brzostowski E.B., Riely G.J., Kris M.G., Zakowski M.F., Ladanyi M. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. 2012;18:6169–6177. doi: 10.1158/1078-0432.CCR-11-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Osta B., Behera M., Kim S., Berry L.D., Sica G., Pillai R.N., Owonikoko T.K., Kris M.G., Johnson B.E., Kwiatkowski D.J. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2019;14:876–889. doi: 10.1016/j.jtho.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 22.ASCO . 2020. Lung Cancer – Non-Small Cell: Statistics.https://www.cancer.net/cancer-types/lung-cancer-non-small-cell/statistics#:~:text=The%205%2Dyear%20survival%20rate%20for%20all%20people%20with%20all,for%20small%20cell%20lung%20cancer [Google Scholar]
- 23.Wood K., Hensing T., Malik R., Salgia R. Prognostic and Predictive Value in KRAS in Non-Small-Cell Lung Cancer: A Review. JAMA Oncol. 2016;2:805–812. doi: 10.1001/jamaoncol.2016.0405. [DOI] [PubMed] [Google Scholar]
- 24.Kazandjian D., Blumenthal G.M., Chen H.-Y., He K., Patel M., Justice R., Keegan P., Pazdur R. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist. 2014;19:e5–e11. doi: 10.1634/theoncologist.2014-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khozin S., Blumenthal G.M., Jiang X., He K., Boyd K., Murgo A., Justice R., Keegan P., Pazdur R. U.S. Food and Drug Administration approval summary: erlotinib for the first-line treatment of metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. Oncologist. 2014;19:774–779. doi: 10.1634/theoncologist.2014-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yi L., Fan J., Qian R., Luo P., Zhang J. Efficacy and safety of osimertinib in treating EGFR-mutated advanced NSCLC: a meta-analysis. Int. J. Cancer. 2019;145:284–294. doi: 10.1002/ijc.32097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oncology Times Brigatinib Granted Accelerated Approval for NSCLC. Oncology Times. 2017;39:39. [Google Scholar]
- 28.Soh J., Toyooka S., Matsuo K., Yamamoto H., Wistuba I.I., Lam S., Fong K.M., Gazdar A.F., Miyoshi S. Ethnicity affects EGFR and KRAS gene alterations of lung adenocarcinoma. Oncol. Lett. 2015;10:1775–1782. doi: 10.3892/ol.2015.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Y., Ren Y., Fang Z., Li C., Fang R., Gao B., Han X., Tian W., Pao W., Chen H., Ji H. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J. Clin. Oncol. 2010;28:4616–4620. doi: 10.1200/JCO.2010.29.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou W., Christiani D.C. East meets West: ethnic differences in epidemiology and clinical behaviors of lung cancer between East Asians and Caucasians. Chin. J. Cancer. 2011;30:287–292. doi: 10.5732/cjc.011.10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reinersman J.M., Johnson M.L., Riely G.J., Chitale D.A., Nicastri A.D., Soff G.A., Schwartz A.G., Sima C.S., Ayalew G., Lau C. Frequency of EGFR and KRAS mutations in lung adenocarcinomas in African Americans. J. Thorac. Oncol. 2011;6:28–31. doi: 10.1097/JTO.0b013e3181fb4fe2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kris M.G., Johnson B.E., Berry L.D., Kwiatkowski D.J., Iafrate A.J., Wistuba I.I., Varella-Garcia M., Franklin W.A., Aronson S.L., Su P.-F. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311:1998–2006. doi: 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slebos R.J., Kibbelaar R.E., Dalesio O., Kooistra A., Stam J., Meijer C.J., Wagenaar S.S., Vanderschueren R.G., van Zandwijk N., Mooi W.J. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N. Engl. J. Med. 1990;323:561–565. doi: 10.1056/NEJM199008303230902. [DOI] [PubMed] [Google Scholar]
- 34.Mascaux C., Iannino N., Martin B., Paesmans M., Berghmans T., Dusart M., Haller A., Lothaire P., Meert A.P., Noel S. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br. J. Cancer. 2005;92:131–139. doi: 10.1038/sj.bjc.6602258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meng D., Yuan M., Li X., Chen L., Yang J., Zhao X., Ma W., Xin J. Prognostic value of K-RAS mutations in patients with non-small cell lung cancer: a systematic review with meta-analysis. Lung Cancer. 2013;81:1–10. doi: 10.1016/j.lungcan.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 36.Brady A.K., McNeill J.D., Judy B., Bauml J., Evans T.L., Cohen R.B., Langer C., Vachani A., Aggarwal C. Survival outcome according to KRAS mutation status in newly diagnosed patients with stage IV non-small cell lung cancer treated with platinum doublet chemotherapy. Oncotarget. 2015;6:30287–30294. doi: 10.18632/oncotarget.4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mellema W.W., Dingemans A.M., Thunnissen E., Snijders P.J., Derks J., Heideman D.A., Van Suylen R., Smit E.F. KRAS mutations in advanced nonsquamous non-small-cell lung cancer patients treated with first-line platinum-based chemotherapy have no predictive value. J. Thorac. Oncol. 2013;8:1190–1195. doi: 10.1097/JTO.0b013e318298764e. [DOI] [PubMed] [Google Scholar]
- 38.Rulli E., Marabese M., Torri V., Farina G., Veronese S., Bettini A., Longo F., Moscetti L., Ganzinelli M., Lauricella C., TAILOR Trialists Value of KRAS as prognostic or predictive marker in NSCLC: results from the TAILOR trial. Ann. Oncol. 2015;26:2079–2084. doi: 10.1093/annonc/mdv318. [DOI] [PubMed] [Google Scholar]
- 39.Hames M.L., Chen H., Iams W., Aston J., Lovly C.M., Horn L. Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung cancer. Lung Cancer. 2016;92:29–34. doi: 10.1016/j.lungcan.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marabese M., Ganzinelli M., Garassino M.C., Shepherd F.A., Piva S., Caiola E., Macerelli M., Bettini A., Lauricella C., Floriani I. KRAS mutations affect prognosis of non-small-cell lung cancer patients treated with first-line platinum containing chemotherapy. Oncotarget. 2015;6:34014–34022. doi: 10.18632/oncotarget.5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riely G.J., Kris M.G., Rosenbaum D., Marks J., Li A., Chitale D.A., Nafa K., Riedel E.R., Hsu M., Pao W. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res. 2008;14:5731–5734. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Govindan R., Ding L., Griffith M., Subramanian J., Dees N.D., Kanchi K.L., Maher C.A., Fulton R., Fulton L., Wallis J. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imielinski M., Berger A.H., Hammerman P.S., Hernandez B., Pugh T.J., Hodis E., Cho J., Suh J., Capelletti M., Sivachenko A. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ihle N.T., Byers L.A., Kim E.S., Saintigny P., Lee J.J., Blumenschein G.R., Tsao A., Liu S., Larsen J.E., Wang J. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J. Natl. Cancer Inst. 2012;104:228–239. doi: 10.1093/jnci/djr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garassino M.C., Marabese M., Rusconi P., Rulli E., Martelli O., Farina G., Scanni A., Broggini M. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann. Oncol. 2011;22:235–237. doi: 10.1093/annonc/mdq680. [DOI] [PubMed] [Google Scholar]
- 46.Nadal E., Chen G., Prensner J.R., Shiratsuchi H., Sam C., Zhao L., Kalemkerian G.P., Brenner D., Lin J., Reddy R.M. KRAS-G12C mutation is associated with poor outcome in surgically resected lung adenocarcinoma. J. Thorac. Oncol. 2014;9:1513–1522. doi: 10.1097/JTO.0000000000000305. [DOI] [PubMed] [Google Scholar]
- 47.Yu H.A., Sima C.S., Shen R., Kass S., Gainor J., Shaw A., Hames M., Iams W., Aston J., Lovly C.M. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J. Thorac. Oncol. 2015;10:431–437. doi: 10.1097/JTO.0000000000000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalluri R., Weinberg R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mohanty A., Nam A., Pozhitkov A., Bhattacharya S., Yang L., Nathan A., Wu X., Srivastava S., Mambetsariev I., Nelson M. A Non-genetic Mechanism for Chemoresistance in Lung Cancer: The Role of Integrin β4/Paxillin Axis. bioRxiv. 2019 doi: 10.1101/781807. [DOI] [Google Scholar]
- 50.Yang H., Liang S.Q., Schmid R.A., Peng R.W. New Horizons in KRAS-Mutant Lung Cancer: Dawn After Darkness. Front. Oncol. 2019;9:953. doi: 10.3389/fonc.2019.00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roper N., Brown A.-L., Wei J.S., Pack S., Trindade C., Kim C., Restifo O., Gao S., Sindiri S., Mehrabadi F. Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell Rep. Med. 2020;1:100007. doi: 10.1016/j.xcrm.2020.100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kosaka T., Yatabe Y., Endoh H., Kuwano H., Takahashi T., Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919–8923. doi: 10.1158/0008-5472.CAN-04-2818. [DOI] [PubMed] [Google Scholar]
- 53.Pao W., Wang T.Y., Riely G.J., Miller V.A., Pan Q., Ladanyi M., Zakowski M.F., Heelan R.T., Kris M.G., Varmus H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shigematsu H., Lin L., Takahashi T., Nomura M., Suzuki M., Wistuba I.I., Fong K.M., Lee H., Toyooka S., Shimizu N. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 55.Tam I.Y., Chung L.P., Suen W.S., Wang E., Wong M.C., Ho K.K., Lam W.K., Chiu S.W., Girard L., Minna J.D. Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin. Cancer Res. 2006;12:1647–1653. doi: 10.1158/1078-0432.CCR-05-1981. [DOI] [PubMed] [Google Scholar]
- 56.Skoulidis F., Heymach J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer. 2019;19:495–509. doi: 10.1038/s41568-019-0179-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Skoulidis F., Byers L.A., Diao L., Papadimitrakopoulou V.A., Tong P., Izzo J., Behrens C., Kadara H., Parra E.R., Canales J.R. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860–877. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adderley H., Blackhall F.H., Lindsay C.R. KRAS-mutant non-small cell lung cancer: converging small molecules and immune checkpoint inhibition. EBioMedicine. 2019;41:711–716. doi: 10.1016/j.ebiom.2019.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shepherd F.A., Lacas B., Le Teuff G., Hainaut P., Jänne P.A., Pignon J.P., Le Chevalier T., Seymour L., Douillard J.Y., Graziano S., LACE-Bio Collaborative Group Pooled Analysis of the Prognostic and Predictive Effects of TP53 Comutation Status Combined With KRAS or EGFR Mutation in Early-Stage Resected Non-Small-Cell Lung Cancer in Four Trials of Adjuvant Chemotherapy. J. Clin. Oncol. 2017;35:2018–2027. doi: 10.1200/JCO.2016.71.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao J., Han Y., Li J., Chai R., Bai C. Prognostic value of KRAS/TP53/PIK3CA in non-small cell lung cancer. Oncol. Lett. 2019;17:3233–3240. doi: 10.3892/ol.2019.10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arbour K.C., Jordan E., Kim H.R., Dienstag J., Yu H.A., Sanchez-Vega F., Lito P., Berger M., Solit D.B., Hellmann M. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018;24:334–340. doi: 10.1158/1078-0432.CCR-17-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skoulidis F., Goldberg M.E., Greenawalt D.M., Hellmann M.D., Awad M.M., Gainor J.F., Schrock A.B., Hartmaier R.J., Trabucco S.E., Gay L. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018;8:822–835. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borghaei H., Paz-Ares L., Horn L., Spigel D.R., Steins M., Ready N.E., Chow L.Q., Vokes E.E., Felip E., Holgado E. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaelin W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 65.Corcoran R.B., Cheng K.A., Hata A.N., Faber A.C., Ebi H., Coffee E.M., Greninger P., Brown R.D., Godfrey J.T., Cohoon T.J. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–128. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu Z., Aref A.R., Cohoon T.J., Barbie T.U., Imamura Y., Yang S., Moody S.E., Shen R.R., Schinzel A.C., Thai T.C. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4:452–465. doi: 10.1158/2159-8290.CD-13-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Puyol M., Martín A., Dubus P., Mulero F., Pizcueta P., Khan G., Guerra C., Santamaría D., Barbacid M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 69.Patnaik A., Rosen L.S., Tolaney S.M., Tolcher A.W., Goldman J.W., Gandhi L., Papadopoulos K.P., Beeram M., Rasco D.W., Hilton J.F. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016;6:740–753. doi: 10.1158/2159-8290.CD-16-0095. [DOI] [PubMed] [Google Scholar]
- 70.Molina-Arcas M., Moore C., Rana S., van Maldegem F., Mugarza E., Romero-Clavijo P., Herbert E., Horswell S., Li L.-S., Janes M.R. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med. 2019;11:eaaw7999. doi: 10.1126/scitranslmed.aaw7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan T.L., Amzallag A., Bagni R., Yi M., Afghani S., Burgan W., Fer N., Strathern L.A., Powell K., Smith B. Differential Effector Engagement by Oncogenic KRAS. Cell Rep. 2018;22:1889–1902. doi: 10.1016/j.celrep.2018.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stinchcombe T.E., Johnson G.L. MEK inhibition in non-small cell lung cancer. Lung Cancer. 2014;86:121–125. doi: 10.1016/j.lungcan.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chong C.R., Jänne P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shaw A.T., Kim D.W., Nakagawa K., Seto T., Crinó L., Ahn M.J., De Pas T., Besse B., Solomon B.J., Blackhall F. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 75.Falchook G., Patel M., Infante J., Arkenau H.-T., Dean E., Brenner A., Borazanci E., Lopez J., Moore K., Schmid P. Abstract CT153: First in human study of the first-in-class fatty acid synthase (FASN) inhibitor TVB-2640. Cancer Res. 2017;77:CT153. [Google Scholar]
- 76.Paz-Ares L., Hirsh V., Zhang L., de Marinis F., Yang J.C., Wakelee H.A., Seto T., Wu Y.L., Novello S., Juhász E. Monotherapy Administration of Sorafenib in Patients With Non-Small Cell Lung Cancer (MISSION) Trial: A Phase III, Multicenter, Placebo-Controlled Trial of Sorafenib in Patients with Relapsed or Refractory Predominantly Nonsquamous Non-Small-Cell Lung Cancer after 2 or 3 Previous Treatment Regimens. J. Thorac. Oncol. 2015;10:1745–1753. doi: 10.1097/JTO.0000000000000693. [DOI] [PubMed] [Google Scholar]
- 77.Gouw A.M., Eberlin L.S., Margulis K., Sullivan D.K., Toal G.G., Tong L., Zare R.N., Felsher D.W. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc. Natl. Acad. Sci. USA. 2017;114:4300–4305. doi: 10.1073/pnas.1617709114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bartolacci C., Padanad M., Andreani C., Melegari M., Rindhe S., George K., Frankel A., McDonald J., Scaglioni P.P. Fatty Acid Synthase Is a Therapeutic Target in Mutant KRAS Lung Cancer. J. Thorac. Oncol. 2017;12:S1538. [Google Scholar]
- 79.O’Farrell M., Heuer T., Grimmer K., Crowley R., Waszczuk J., Fridlib M., Ventura R., Rubio C., Lai J., Buckley D. Abstract LB-214: FASN inhibitor TVB-2640 shows pharmacodynamic effect and evidence of clinical activity in KRAS-mutant NSCLC patients in a phase I study. Cancer Res. 2016;76:LB-214. [Google Scholar]
- 80.Dean E.J., Falchook G.S., Patel M.R., Brenner A.J., Infante J.R., Arkenau H.-T., Borazanci E.H., Lopez J.S., Pant S., Schmid P. Preliminary activity in the first in human study of the first-in-class fatty acid synthase (FASN) inhibitor, TVB-2640. J. Clin. Oncol. 2016;34:2512. doi: 10.1016/j.eclinm.2021.100797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heidorn S.J., Milagre C., Whittaker S., Nourry A., Niculescu-Duvas I., Dhomen N., Hussain J., Reis-Filho J.S., Springer C.J., Pritchard C., Marais R. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dingemans A.-M.C., Mellema W.W., Groen H.J.M., van Wijk A., Burgers S.A., Kunst P.W.A., Thunnissen E., Heideman D.A.M., Smit E.F. A phase II study of sorafenib in patients with platinum-pretreated, advanced (Stage IIIb or IV) non-small cell lung cancer with a KRAS mutation. Clin. Cancer Res. 2013;19:743–751. doi: 10.1158/1078-0432.CCR-12-1779. [DOI] [PubMed] [Google Scholar]
- 83.Kim E.S., Herbst R.S., Wistuba I.I., Lee J.J., Blumenschein G.R., Jr., Tsao A., Stewart D.J., Hicks M.E., Erasmus J., Jr., Gupta S. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rinehart J., Adjei A.A., Lorusso P.M., Waterhouse D., Hecht J.R., Natale R.B., Hamid O., Varterasian M., Asbury P., Kaldjian E.P. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 2004;22:4456–4462. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 85.Davies B.R., Logie A., McKay J.S., Martin P., Steele S., Jenkins R., Cockerill M., Cartlidge S., Smith P.D. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol. Cancer Ther. 2007;6:2209–2219. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 86.Hainsworth J.D., Cebotaru C.L., Kanarev V., Ciuleanu T.E., Damyanov D., Stella P., Ganchev H., Pover G., Morris C., Tzekova V. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J. Thorac. Oncol. 2010;5:1630–1636. doi: 10.1097/JTO.0b013e3181e8b3a3. [DOI] [PubMed] [Google Scholar]
- 87.Jänne P.A., Shaw A.T., Pereira J.R., Jeannin G., Vansteenkiste J., Barrios C., Franke F.A., Grinsted L., Zazulina V., Smith P. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 88.Carter C.A., Rajan A., Keen C., Szabo E., Khozin S., Thomas A., Brzezniak C., Guha U., Doyle L.A., Steinberg S.M. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Ann. Oncol. 2016;27:693–699. doi: 10.1093/annonc/mdw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blumenschein G.R., Jr., Smit E.F., Planchard D., Kim D.W., Cadranel J., De Pas T., Dunphy F., Udud K., Ahn M.J., Hanna N.H. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC) Ann. Oncol. 2015;26:894–901. doi: 10.1093/annonc/mdv072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gandara D.R., Hiret S., Blumenschein G.R., Barlesi F., Delord J.-P., Madelaine J., Infante J.R., Reckamp K.L., Lara P., Audebert C. Oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with docetaxel in KRAS-mutant and wild-type (WT) advanced non-small cell lung cancer (NSCLC): a phase I/Ib trial. J. Clin. Oncol. 2013;31:8028. [Google Scholar]
- 91.Gandara D.R., Leighl N., Delord J.P., Barlesi F., Bennouna J., Zalcman G., Infante J.R., Reckamp K.L., Kelly K., Shepherd F.A. A Phase 1/1b Study Evaluating Trametinib Plus Docetaxel or Pemetrexed in Patients With Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017;12:556–566. doi: 10.1016/j.jtho.2016.11.2218. [DOI] [PubMed] [Google Scholar]
- 92.Misale S., Fatherree J.P., Cortez E., Li C., Bilton S., Timonina D., Myers D.T., Lee D., Gomez-Caraballo M., Greenberg M. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin. Cancer Res. 2019;25:796–807. doi: 10.1158/1078-0432.CCR-18-0368. [DOI] [PubMed] [Google Scholar]
- 93.Berndt N., Hamilton A.D., Sebti S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer. 2011;11:775–791. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gajewski T.F., Salama A.K.S., Niedzwiecki D., Johnson J., Linette G., Bucher C., Blaskovich M.A., Sebti S.M., Haluska F., Cancer and Leukemia Group B Phase II study of the farnesyltransferase inhibitor R115777 in advanced melanoma (CALGB 500104) J. Transl. Med. 2012;10:246. doi: 10.1186/1479-5876-10-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Adjei A.A., Mauer A., Bruzek L., Marks R.S., Hillman S., Geyer S., Hanson L.J., Wright J.J., Erlichman C., Kaufmann S.H., Vokes E.E. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2003;21:1760–1766. doi: 10.1200/JCO.2003.09.075. [DOI] [PubMed] [Google Scholar]
- 96.Heymach J.V., Johnson D.H., Khuri F.R., Safran H., Schlabach L.L., Yunus F., DeVore R.F., 3rd, De Porre P.M., Richards H.M., Jia X. Phase II study of the farnesyl transferase inhibitor R115777 in patients with sensitive relapse small-cell lung cancer. Ann. Oncol. 2004;15:1187–1193. doi: 10.1093/annonc/mdh315. [DOI] [PubMed] [Google Scholar]
- 97.Kim E.S., Kies M.S., Fossella F.V., Glisson B.S., Zaknoen S., Statkevich P., Munden R.F., Summey C., Pisters K.M.W., Papadimitrakopoulou V. Phase II study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in patients with taxane-refractory/resistant nonsmall cell lung carcinoma. Cancer. 2005;104:561–569. doi: 10.1002/cncr.21188. [DOI] [PubMed] [Google Scholar]
- 98.Simanshu D.K., Nissley D.V., McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cox A.D., Der C.J., Philips M.R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 2015;21:1819–1827. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Spencer-Smith R., Koide A., Zhou Y., Eguchi R.R., Sha F., Gajwani P., Santana D., Gupta A., Jacobs M., Herrero-Garcia E. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017;13:62–68. doi: 10.1038/nchembio.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Janes M.R., Zhang J., Li L.-S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S.J., Darjania L. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell. 2018;172:578–589.e17. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 102.Ostrem J.M., Peters U., Sos M.L., Wells J.A., Shokat K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patricelli M.P., Janes M.R., Li L.S., Hansen R., Peters U., Kessler L.V., Chen Y., Kucharski J.M., Feng J., Ely T. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 104.Canon J., Rex K., Saiki A.Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C.G., Koppada N. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 105.Fakih M., O’Neil B., Price T., Falchook G., Desai J., Kuo J., Govindan R., Rasmussen E., Morrow P., Ngang J. Phase 1 study evaluating the safety, tolerability, pharmacokinetics, and efficacy of AMG 510, a novel small molecule KRASG12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019;37:3003. [Google Scholar]
- 106.Govindan R., Fakih M., Price T., Falchook G., Desai J., Kuo J., Strickler J., Krauss J., Li B., Denlinger C. Phase 1 Study of AMG 510, a Novel Molecule Targeting KRAS G12C Mutant Solid Tumors. Ann. Oncol. 2019;30(Suppl_5):v159–v193. [Google Scholar]
- 107.Hallin J., Engstrom L.D., Hargis L., Calinisan A., Aranda R., Briere D.M., Sudhakar N., Bowcut V., Baer B.R., Ballard J.A. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020;10:54–71. doi: 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xue J.Y., Zhao Y., Aronowitz J., Mai T.T., Vides A., Qeriqi B., Kim D., Li C., de Stanchina E., Mazutis L. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. 2020;577:421–425. doi: 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lito P., Solomon M., Li L.S., Hansen R., Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351:604–608. doi: 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ryan M.B., Fece de la Cruz F., Phat S., Myers D.T., Wong E., Shahzade H.A., Hong C.B., Corcoran R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin. Cancer Res. 2020;26:1633–1643. doi: 10.1158/1078-0432.CCR-19-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mainardi S., Mulero-Sánchez A., Prahallad A., Germano G., Bosma A., Krimpenfort P., Lieftink C., Steinberg J.D., de Wit N., Gonçalves-Ribeiro S. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018;24:961–967. doi: 10.1038/s41591-018-0023-9. [DOI] [PubMed] [Google Scholar]
- 112.Chen N., Fang W., Lin Z., Peng P., Wang J., Zhan J., Hong S., Huang J., Liu L., Sheng J. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017;66:1175–1187. doi: 10.1007/s00262-017-2005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kalvala A., Wallet P., Yang L., Wang C., Li H., Nam A., Nathan A., Mambetsariev I., Poroyko V., Gao H. Phenotypic Switching of Naïve T Cells to Immune-Suppressive Treg-Like Cells by Mutant KRAS. J. Clin. Med. 2019;8:1726. doi: 10.3390/jcm8101726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Coelho M.A., de Carné Trécesson S., Rana S., Zecchin D., Moore C., Molina-Arcas M., East P., Spencer-Dene B., Nye E., Barnouin K. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity. 2017;47:1083–1099.e6. doi: 10.1016/j.immuni.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scheel A.H., Ansén S., Schultheis A.M., Scheffler M., Fischer R.N., Michels S., Hellmich M., George J., Zander T., Brockmann M. PD-L1 expression in non-small cell lung cancer: correlations with genetic alterations. OncoImmunology. 2016;5:e1131379. doi: 10.1080/2162402X.2015.1131379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dietrich M., Hunis B., Raez L. P2.07-052 Detection of KRAS Mutation in Blood Predicts Favorable Response to Immunotherapy in NSCLC. J. Thorac. Oncol. 2017;12:S2149. [Google Scholar]
- 117.D’Incecco A., Andreozzi M., Ludovini V., Rossi E., Capodanno A., Landi L., Tibaldi C., Minuti G., Salvini J., Coppi E. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br. J. Cancer. 2015;112:95–102. doi: 10.1038/bjc.2014.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu C., Zheng S., Jin R., Wang X., Wang F., Zang R., Xu H., Lu Z., Huang J., Lei Y. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020;470:95–105. doi: 10.1016/j.canlet.2019.10.027. [DOI] [PubMed] [Google Scholar]
- 119.Mok T.S.K., Wu Y.-L., Kudaba I., Kowalski D.M., Cho B.C., Turna H.Z., Castro G., Jr., Srimuninnimit V., Laktionov K.K., Bondarenko I., KEYNOTE-042 Investigators Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393:1819–1830. doi: 10.1016/S0140-6736(18)32409-7. [DOI] [PubMed] [Google Scholar]
- 120.Herbst R.S., Baas P., Kim D.-W., Felip E., Pérez-Gracia J.L., Han J.-Y., Molina J., Kim J.-H., Arvis C.D., Ahn M.-J. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–1550. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 121.Reck M., Rodríguez-Abreu D., Robinson A.G., Hui R., Csőszi T., Fülöp A., Gottfried M., Peled N., Tafreshi A., Cuffe S., KEYNOTE-024 Investigators Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 122.Rittmeyer A., Barlesi F., Waterkamp D., Park K., Ciardiello F., von Pawel J., Gadgeel S.M., Hida T., Kowalski D.M., Dols M.C., OAK Study Group Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–265. doi: 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gandhi L., Rodríguez-Abreu D., Gadgeel S., Esteban E., Felip E., De Angelis F., Domine M., Clingan P., Hochmair M.J., Powell S.F., KEYNOTE-189 Investigators Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 124.Herbst R., Lopes G., Kowalski D., Kasahara K., Wu Y., De Castro G., Jr., Cho B., Turna H., Cristescu R., Aurora-Garg D. Association of KRAS mutational status with response to pembrolizumab monotherapy given as first-line therapy for PD-L1-positive advanced non-squamous NSCLC in KEYNOTE-042. Ann. Oncol. 2019;30(Suppl 11):XI63–XI64. [Google Scholar]
- 125.Moderna . 2020. KRAS vaccine (mRNA-5671)https://investors.modernatx.com/static-files/12f15d58-0a61-4ca4-a3e1-61d06ab28e0c [Google Scholar]
- 126.Quandt J., Schlude C., Bartoschek M., Will R., Cid-Arregui A., Schölch S., Reissfelder C., Weitz J., Schneider M., Wiemann S. Long-peptide vaccination with driver gene mutations in p53 and Kras induces cancer mutation-specific effector as well as regulatory T cell responses. OncoImmunology. 2018;7:e1500671. doi: 10.1080/2162402X.2018.1500671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang Q.J., Yu Z., Griffith K., Hanada K., Restifo N.P., Yang J.C. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol. Res. 2016;4:204–214. doi: 10.1158/2326-6066.CIR-15-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tran E., Robbins P.F., Lu Y.-C., Prickett T.D., Gartner J.J., Jia L., Pasetto A., Zheng Z., Ray S., Groh E.M. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016;375:2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sim M.J.W., Lu J., Spencer M., Hopkins F., Tran E., Rosenberg S.A., Long E.O., Sun P.D. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl. Acad. Sci. USA. 2020;117:12826–12835. doi: 10.1073/pnas.1921964117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Scheffler M., Ihle M.A., Hein R., Merkelbach-Bruse S., Scheel A.H., Siemanowski J., Brägelmann J., Kron A., Abedpour N., Ueckeroth F. K-ras Mutation Subtypes in NSCLC and Associated Co-occuring Mutations in Other Oncogenic Pathways. J. Thorac. Oncol. 2019;14:606–616. doi: 10.1016/j.jtho.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 131.Izar B., Zhou H., Heist R.S., Azzoli C.G., Muzikansky A., Scribner E.E., Bernardo L.A., Dias-Santagata D., Iafrate A.J., Lanuti M. The prognostic impact of KRAS, its codon and amino acid specific mutations, on survival in resected stage I lung adenocarcinoma. J. Thorac. Oncol. 2014;9:1363–1369. doi: 10.1097/JTO.0000000000000266. [DOI] [PubMed] [Google Scholar]
- 132.Pan W., Yang Y., Zhu H., Zhang Y., Zhou R., Sun X. KRAS mutation is a weak, but valid predictor for poor prognosis and treatment outcomes in NSCLC: A meta-analysis of 41 studies. Oncotarget. 2016;7:8373–8388. doi: 10.18632/oncotarget.7080. [DOI] [PMC free article] [PubMed] [Google Scholar]