

Summary
BCG vaccination can strengthen protection against pathogens through the induction of epigenetic and metabolic reprogramming of innate immune cells, a process called trained immunity. We and others recently demonstrated that mucosal or intravenous BCG better protects rhesus macaques from Mycobacterium tuberculosis infection and TB disease than standard intradermal vaccination, correlating with local adaptive immune signatures. In line with prior mouse data, here, we show in rhesus macaques that intravenous BCG enhances innate cytokine production associated with changes in H3K27 acetylation typical of trained immunity. Alternative delivery of BCG does not alter the cytokine production of unfractionated bronchial lavage cells. However, mucosal but not intradermal vaccination, either with BCG or the M. tuberculosis-derived candidate MTBVAC, enhances innate cytokine production by blood- and bone marrow-derived monocytes associated with metabolic rewiring, typical of trained immunity. These results provide support to strategies for improving TB vaccination and, more broadly, modulating innate immunity via mucosal surfaces.
Keywords: trained immunity, innate immunity, non-human primates, vaccination, BCG, MTBVAC, tuberculosis, vaccine development, immunotherapy
Graphical Abstract
Highlights
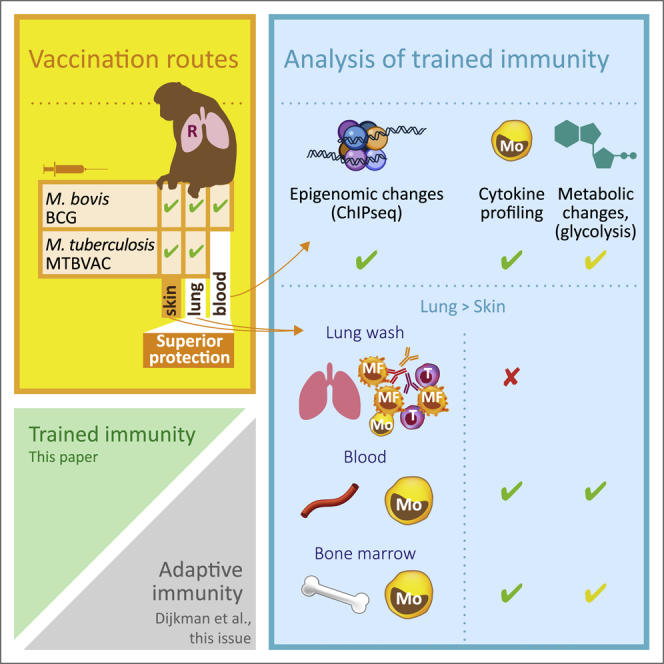
Nonhuman primates recapitulate trained immunity upon live attenuated TB vaccination
Intravenous BCG induces changes in H3K27 acetylation and enhances cytokine production
Mucosal BCG improves induction of trained immunity of monocytes over intradermal BCG
The M. tuberculosis-derived candidate vaccine MTBVAC appears equally potent as BCG
Vierboom et al. demonstrate the induction of trained immunity in blood and bone marrow monocytes after vaccination with live attenuated TB vaccines in nonhuman primates. Mucosal respiratory delivery of BCG or MTBVAC induces trained immunity more efficiently compared to standard intradermal vaccination.
Introduction
Canonical vaccinology aims at the induction of long-lived adaptive memory in antigen-specific lymphocyte populations and T and/or B cells. Innate immunity in vaccinology is primarily acknowledged in the context of vaccine formulation and the quintessential innate immune activation of professional antigen-presenting cells (APCs) that will prime and skew the adaptive response of lymphocytes.1 For years, innate immune responses and myeloid monocyte/macrophage function were considered constant and invariable over time, but it is now widely recognized that also innate immune cells can adapt and display memory-like phenotypes. This innate immune “memory” is typically referred to as trained immunity.2
Induction of trained immunity is defined by a functional adaptation of innate immunity after a primary insult resulting in a more effective response upon a “secondary” encounter with an unrelated pathogen.2 It is characterized by increased cytokine and/or chemokine production,3 metabolic rewiring,4 and epigenetic reprogramming of innate immune cells.5 Trained immunity is not associated with the characteristic immune receptor gene rearrangements that are associated with the antigen specificity of adaptive memory, but rather is driven by the epigenetic control of specific immune gene clusters resulting in the upregulation of particular pro-inflammatory cytokines.6 An epigenetic mark in particular associated with trained immunity in monocytes, and used in the present study, is the acetylation of histone 3 at the position of lysine 27 (H3K27ac).7 Recent work in mice has shown that Bacillus Calmette-Guérin (BCG)-mediated training of the myeloid monocyte/macrophage lineage is sustained by the epigenetic reprogramming of hematopoietic stem cells (HSCs), the myeloid precursors in bone marrow (BM).8
Mycobacterium bovis-derived BCG, currently the only available vaccine to fight tuberculosis (TB),9 is the prototypical biological agent for which trained immunity has been demonstrated. Trained immunity has been linked to the beneficial heterologous off-target effect of infant BCG vaccination, which from epidemiological analyses appears associated with reduced childhood mortality from causes other than TB.10, 11, 12, 13 Emerging evidence supports the hypothesis that not only adaptive memory but also the memory-like plasticity in innate immunity positively contributes to immune protection against and early clearance of M. tuberculosis (Mtb) infection.14,15 Although not established as features of innate training, a comparative analysis in macaque species suggests that markers of innate immunity and myeloid monocyte function are associated with differential TB disease susceptibility.16
Despite the widespread use of live attenuated BCG as prophylaxis against TB, it is only partially efficacious and by and large fails to protect adolescents and adults from the infectious pulmonary manifestation of Mtb infection.17 Thus, TB continues to be a major threat to global human health, and a better vaccination strategy is urgently needed to relieve the burden of TB.18,19 While intradermal injection is the standard route for BCG vaccination, historical data in rhesus macaques have shown that mucosal or intravenous administration provided superior signals of protection against experimental infection.20 We have recently demonstrated that pulmonary mucosal BCG vaccination reduced disease in a cohort of rhesus macaques in which intradermal BCG failed to show any protective effect.21 In a subsequent study, exploiting repeated limiting dose (RLD) rather than single dose Mtb challenge, we have shown that mucosal but not intradermal BCG provides signals of prevention of infection.22 Most recently, Darrah and colleagues23 have corroborated the superior protective capacity of BCG when given intravenously. Both studies did not identify an immune biomarker in the blood that correlated with the protective efficacy of either mucosal or intravenous BCG vaccination. Locally, however, antigen-specific interleukin-17A-positive (IL-17A+) polyfunctional T helper (Th) cells and IL-10 secretion in the airways did correlate with protection,22 and the article by Darrah et al.23 suggests that tissue-resident lymphocytes may be pivotal in conferring protection. While from these non-human primate (NHP) studies unconventional administration of BCG appears to be a most interesting lead for improving our current TB prophylaxis, it remains to be established, however, if and how alternative routing of BCG affects innate and trained immunity.
Various candidate TB vaccines, ranging from subunit formulations to live attenuated and recombinant mycobacteria, are at various stages of clinical testing in the current vaccine-development pipeline (www.tbvi.eu/what-we-do/pipeline-of-vaccines/). Of the live attenuated candidates, MTBVAC is unique in that it is derived from a clinical isolate of Mtb (Mt103) rather than from M. bovis (which is the parental strain of BCG and recombinant BCG candidates).24,25 MTBVAC is attenuated by deletion of the phoP and the fadD26 gene. Thereby, the expression of virulence factors is critically disrupted, while an intrinsically broader antigen repertoire in comparison to BCG is preserved.26 Aguilo et al.27 demonstrated that the expression of ESAT6 and CFP10 by MTBVAC is key to its protective capacity in a mouse models of TB. MTBVAC has shown preclinical efficacy better than BCG in animal models.24,27 MTBVAC vaccination protects rhesus macaques against aerosol challenge with Mtb and induces immune signatures analogous to those observed in clinical studies.28 It has tested safe and immunogenic in adult and infant human populations so far.9,29 Similar to BCG, MTBVAC is able to generate trained immunity through the induction of glycolysis and glutaminolysis, and the accumulation of histone methylation marks at the promoters of proinflammatory genes. Recently, Tarancón et al.30 showed that MTBVAC-induced heterologous protection against a lethal challenge with Streptococcus pneumoniae in an experimental murine model of pneumonia.
In the present study, we sought to investigate the impact of live attenuated mycobacterial vaccination via the respiratory mucosa on trained innate immunity in NHPs. To this end, we followed a 2 × 2 factorial design strategy for an immunogenicity analysis in adult rhesus macaques, vaccinating animals by intradermal injection or by endobronchial instillation either with BCG or with MTBVAC. We confirmed the distinctive adaptive response profile in the airways upon pulmonary mucosal delivery of BCG and MTBVAC, and analyzed the production of innate and adaptive cytokines after in vitro stimulation of cells obtained from lung, blood, and BM. We piloted the induction of trained immunity after intravenous BCG injection, demonstrating the increased acquisition of H3K27ac marks and resulting in prominent increased cytokine production after heterologous stimulation in monocytes isolated from blood and BM. We subsequently demonstrated that respiratory mucosal administration of live attenuated mycobacterial vaccines more efficiently induced trained immunity in blood and BM monocytes, compared to intradermal immunization, whereby MTBVAC was equally potent as BCG. Our findings underpin the innate immune stimulatory potential of the candidate TB vaccine, MTBVAC. For live attenuated mycobacterial vaccines in general, they suggest the enhancement of innate immune training via respiratory mucosal vaccine administration.
Results
In the present study, we set out to investigate whether mucosal administration of live attenuated TB vaccines in rhesus macaques could modulate innate immune responses and induce trained immunity. To this end, we selected healthy, purpose-bred adult rhesus macaques (Macaca mulatta), both males and females, and stratified them into comparable groups of 6 animals each (Table S1). By random assignment in a 2 × 2 factorial design strategy, these groups received either a standard human dose of BCG or the equivalent dose of the live attenuated candidate vaccine, MTBVAC, by intradermal injection (BCG.id; MTBVAC.id) or by endobronchial instillation into the lower right lung lobe (BCG.muc; MTBVAC.muc). For the assessment of adaptive and/or innate immunity, relevant samples were collected before and after vaccination from the airways by bronchoalveolar lavage (BAL), from the blood by venipuncture, and from the BM by needle aspiration. Before analyzing the intradermal and mucosal vaccination groups, and since it had been reported as an effective research strategy for measuring trained immunity in mice,8 a separate group of 3 rhesus macaques were vaccinated by intravenous BCG injection (BCG.iv) to pilot-trained immunity profiling of monocytes (2 weeks after vaccination). A schematic diagram of vaccination and sampling over time is displayed in Figure 1.
Figure 1.

Schematic representation of the timeline of vaccination in the different vaccination strategies
The BCG.iv group (n = 3) was analyzed at weeks −1 and 2. The groups that were immunized with BCG.id (n = 6), BCG.muc (n = 6), MTBVAC.id (n = 6), and MTBVAC.muc (n = 6) were analyzed at weeks −1 and 8. Only from mucosally vaccinated animals BAL was collected from the right lung as well as the left lung. See also Table S1.
BCG and MTBVAC are immunogenic and both display an adaptive immune profile specific to the route of vaccination
To confirm the efficiency of vaccination, we performed a specific interferon-γ (IFN-γ) enzyme-linked immunosorbent spot (ELISpot) assay after in vitro recall stimulation of peripheral blood mononuclear cells (PBMCs) with the protein-purified derivative (PPD) of Mtb (or culture medium as a negative control). For both BCG and MTBVAC and by either route of delivery, we found an increase in antigen-specific IFN-γ release after primary vaccination. In line with previous observations,21,22 the vaccine-induced IFN-γ response was higher after intradermal over mucosal BCG delivery, and a comparable response pattern was observed with MTBVAC (Figures 2A and 2B). When stimulating with a recombinant fusion protein of ESAT6 and CFP10, two antigens encoded in the region of deletion (RD)1 that is absent from M. bovis BCG but present in MTBVAC, positive IFN-γ release signals were obtained only from MTBVAC-vaccinated animals, as expected (Figure 2C).
Figure 2.

Mucosal vaccination establishes a unique local adaptive signature defined by polyfunctional Th17 cells
(A–C) Individual IFN-γ ELISpot responses after in vitro recall stimulation of PBMCs against PPD (A) before vaccination or (B) 8 weeks after vaccination or with (C) ESAT6-CFP10 8 weeks after vaccination. Horizontal lines in bars indicate group medians; n = 6 animals/group. ANOVA adjusted for multiple comparisons; Dunn’s multiple comparison test.
(D) The percentages of polyfunctional IL-17A+ PPD-specific CD4+ T cells (also producing IFN-γ, TNF-α, and IL-2) were determined by flow cytometric analysis of BAL cells before (PRE) and 8 weeks after intradermal (.id) or mucosal (.muc) vaccination. Cells were typically collected from the lower right lung lobe (R); targeted for vaccination) and for mucosally vaccinated animals also from the left lung (L) to establish dissemination of the immune response. The data are presented as medium control corrected (cc); n = 6 animals/group; Wilcoxon matched-pairs signed rank test. ULD, upper limit of detection; LLD, lower limit of detection.
See also Figure S7.
To further confirm the adaptive response profile that we registered previously after pulmonary mucosal BCG as a correlate of the prevention of Mtb infection, we assessed the cytokine production profile of CD4+ T lymphocytes from the airways by flow cytometry22 after ex vivo stimulation with PPD. As expected, we detected the most prominent levels of polyfunctional Th17 cells (producing IL-17A, IFN-γ, tumor necrosis factor α [TNF-α], and IL-2) in BALs that were collected 8 weeks after primary vaccination with BCG via the pulmonary mucosal rather than the intradermal route (Figure 2D). Mtb-derived MTBVAC was found to induce a similar response pattern and to be at least as potent as BCG. Regardless of the vaccine, significant numbers of polyfunctional Th17 cells were also recovered from the lower left lung lobe (i.e., opposite of the site targeted by endobronchial immunization), suggesting that the polyfunctional Th17 response is not contained to the site of vaccination but disseminates locally.
Establishing trained immunity in monocytes after intravenous immunization with BCG
To evaluate the induction of trained immunity after standard intradermal versus mucosal vaccination with live attenuated TB vaccines, we established the feasibility of the detection of trained immunity in the nonhuman primate model. For this, we immunized three animals with BCG intravenously, a protocol that was successfully explored in mice by Kaufmann et al.8 To minimize the interference of a developing adaptive immune response while still being able to detect robust innate immune responses, we took samples from different compartments at week 2 after vaccination. Next to the analysis of BAL cells from the lung, we performed a more selective analysis of myeloid CD14+ monocytes purified from fresh PBMCs (PBMC.mo) and BM (BM.mo), the prototypic cells in which the trained immunity phenotype has been described. CD14+ monocyte populations were obtained by positive selection (using magnetic bead-cell sorting) and typically reached a purity of ±90% (Figure S1). Both unfractionated BAL cells and enriched monocytes from blood and BM were subsequently analyzed for signals of trained immunity after in vitro restimulation with Mtb-derived whole-cell lysate (WCL) or the heterologous Escherichia coli derived Toll-like receptor (TLR)-4 agonist lipopolysaccharide (LPS). After 24 h, supernatants were harvested, frozen, and analyzed later for cytokine production, indicative of trained innate immunity, including TNF-α, IL-6, and IL-1β.
After intravenous BCG vaccination, we found marginal suppression of cytokine secretion, if any effect at all, after comparing Mtb or LPS stimulation of BAL (Figures 3A and 3B). Stimulation of PBMC.mo and BM.mo with Mtb revealed markedly increased levels of TNF-α 2 weeks after BCG.iv (Figure 3A), but no apparent modulation of IL-6 and IL-1β secretion levels. However, heterologous stimulation with LPS demonstrated prominent increased production of all three cytokines by both monocytes from the blood and BM (with the exception of a single animal with high baseline levels in BM.mo; Figure 3B, PBMC.mo and BM.mo). Metabolic rewiring, specifically a switch from oxidative phosphorylation to glycolysis under aerobic conditions,4 has been described as an initiating event underlying trained immunity. Therefore, we measured lactate, a metabolite of glycolysis, levels in the supernatants of stimulated blood- and BM-derived monocytes as a surrogate marker of metabolic rewiring, and thus trained immunity. Lactate production was observed for 2 of 3 LPS-stimulated BM.mo, but undetectable for PBMC.mo (Figure 3C).
Figure 3.

Trained immunity after intravenous BCG vaccination
(A and B) Freshly isolated BAL cells (from the right lung) and monocytes from blood (PBMC.mo) and bone marrow (BM.mo) were stimulated for 24 h with (A) Mtb whole-cell lysate (25 μg/mL) or (B) LPS (0.1 μg/mL) before and 2 weeks after intravenous vaccination with BCG. The data are presented as medium control corrected.
(C) Lactate production as an indicator of metabolic rewiring was measured in 24-h supernatant from LPS-stimulated monocytes purified from blood (PBMC.mo) or BM (BM.mo). See also Figure S1.
(D) Heatmap of H3K27ac reads (red) over BCG-specific peaks. The intensity over the center of the peak ± 12 kb is depicted.
(E) The top GO pathways associated with the nearest genes to dynamic H3K27ac, with adjusted p values.
(F) H3K27ac dynamics at interferon regulatory factor 3 (IRF3) and syndecan 2 (SDC2) locus for PBMC.mo pre- and post-vaccination. The complete list of genes and values can be found at GEO: GSE159046.
Ultimately, we determined whether BCG.iv vaccination resulted in epigenetic reprogramming, another hallmark of trained innate immunity, in blood-derived monocytes (PBMC.mo). Therefore, we examined the dynamics of histone modification H3K27ac (an epigenetic mark associated with active chromatin in the context of trained immunity7) after BCG.iv by chromatin immune precipitation, followed by high-throughput sequencing (chromatin immunoprecipitation sequencing [ChIP-seq]) to assess the changes associated with BCG.iv vaccination. H3K27ac changes were detected at many loci (Figures 3D–3F). In total, we detected 792 regions increased and 646 regions decreased in H3K27ac (Figure 3D). While the regions showing increased acetylation were associated with genes related to the cell cycle, signaling, and activation of the immune response, regions decreased in acetylation were associated with genes involved in cellular differentiation (Figure 3E). Examples for both an active region, IFN regulatory factor 3 (IRF3), and suppressed region, syndecan 2 (SDC2) are given (Figure 3F). These data are in line with vaccination-induced changes in the acetylome and the cellular program related to the induction of trained immunity.
Pro-inflammatory cytokine production after stimulation of BAL cells with Mtb or LPS
We then sought to compare the induction of trained immunity after standard intradermal versus mucosal vaccination with the live attenuated TB vaccines BCG and MTBVAC. We analyzed cytokine/chemokine production in BAL cells after ex vivo stimulation with homologous Mtb WCL or heterologous LPS. Using a tailored multiplex kit for cytokine and chemokine measurement, we assessed the production of molecules that have previously been associated with lymphocyte activation and function (IFN-γ, IL-2, IL-17A), inflammation, leukocyte differentiation, migration, and/or with trained immunity (TNF-α, IL-6, IL-1β, granulocyte-macrophage-colony-stimulating factor [GM-CSF], macrophage inflammatory protein 1α [MIP-1α; CCL3], and IFN-γ-induced protein 10 [IP-10; CXCL10]).3,11,31 In line with the flow cytometric detection of polyfunctional Th17 cells, we found significantly elevated levels of IL-17A, IFN-γ, IL-2, and TNF-α secretion from Mtb WCL-stimulated BAL cells—in particular after mucosal vaccination with BCG or MTBVAC (Figure 4A). A significant increase in the secretion of IL-2 and TNF-α (but not IL-17A and IFN-γ) was also measured after intradermal vaccination. As the response levels of BCG and MTBVAC in this regard were indistinguishable, we have depicted results and performed statistical analyses on the 12 animals of both BCG and MTBVAC arms together, per route of administration. The release of pro-inflammatory IL-6, after in vitro stimulation with Mtb WCL, comparing pre- and post-vaccination time points, was rather heterogeneous. In contrast, IL-1β, GM-CSF, and MIP-1α (CCL3) secretion was, by trend or with statistical significance, decreased in mucosally vaccinated animals (Figure 4A). CXCL-10 (IP-10), however, was markedly and significantly increased upon Mtb WCL stimulation, regardless of the nature of the vaccine or the immunization route.
Figure 4.

Cytokine production by BAL cells
Freshly isolated BAL cells were stimulated (A) with Mtb whole cell lysate or (B) LPS, before (PRE) and 8 weeks after intradermal (.id) or mucosal (.muc) vaccination (with either BCG, in circles, or MTBVAC, in triangles). Cells were typically collected from the lower right lung lobe (R), the lobe targeted by vaccination, and for mucosally vaccinated animals also from the left lung (L), to establish dissemination of the immune response. The data are presented as medium control corrected (mcc); n = 12 animals/group; Wilcoxon matched-pairs signed rank test. p values are indicated at the top of each graph. See also Figure S2.
This cytokine/chemokine secretion profile of BAL cells after stimulation with a preparation as crude as the WCL of Mtb, containing both protein antigens and (non-protein) innate receptor ligands, is the net result of adaptive and innate activation of all subsets that are present in the unfractionated BAL cell population. In addition, it must be noted that the relative abundance of immune cell subsets in BAL is affected by pulmonary mucosal but not by intradermal BCG vaccination toward a relative increase in T lymphocytes over alveolar macrophage (Figure S2). To at least circumvent the adaptive component of this response profile and toward the assessment of trained immunity by heterologous innate stimulation, we also stimulated BAL cells with LPS to measure the ensuing cytokine production capacity. Upon LPS, and in contrast to Mtb WCL stimulation, IL-17A, IFN-γ, and TNF-α concentrations after vaccination were no different from what was found at baseline (Figure 4B). Most likely as a result of the bystander activation of T lymphocytes specifically recruited into the airways after mucosal vaccination, IL-2 release upon LPS stimulation was increased with BAL cells from mucosally vaccinated animals (Figure 4B), but to a lesser extent compared to Mtb WCL stimulation (Figure 4A). Also compared to Mtb WCL stimulation, the vaccination-modulated GM-CSF secretion of BAL cells was lost with LPS, and that of CXCL-10 was much lower and heterogeneous (Figure 4B). LPS-stimulated IL-6 levels were comparably heterogeneous as with Mtb WCL stimulation. IL-1β and MIP-1α, as with Mtb WCL stimulation, showed diminished secretion upon the LPS stimulation of BAL cells after vaccination, regardless of the route of vaccine delivery (Figure 4B).
This cytokine/chemokine secretion analysis of unfractionated BAL cells upon heterologous LPS (or homologous Mtb WCL) stimulation did not reveal a signal of trained immunity in the airways after (mucosal) live attenuated mycobacterial vaccination.
Induction of trained immunity in monocytes after mucosal vaccination with BCG and MTBVAC
Since BAL cells did not provide a signal indicative of trained immunity, we sought to compare the induction of trained immunity through ex vivo heterologous LPS stimulation of blood- and BM-derived monocytes, as was shown for BCG.iv between intradermal and mucosal immunized animals, with live attenuated BCG or MTBVAC. To demonstrate that the cytokine production is due to ex vivo heterologous LPS stimulation and exclude the possibility of continued innate activation by the (persisting) vaccine, unstimulated medium controls were included that showed minimal to no production of cytokines (Figure S3).
While T cell-derived cytokines IL-17A, IFN-γ, and IL-2 were not detected upon monocyte stimulation (not shown), the enhancement of LPS-stimulated secretion of TNF-α, IL-6, and IL-1β was evident in some but not all animals that were vaccinated by intradermal injection (Figure 5A). Specifically, 4 of 11 animals (2 from each of the BCG- and the MTBVAC-vaccinated groups) showed elevated TNF-α production with an effect size (independent of baseline production) of >2-fold increase after BCG.id/MTBVAC.id (Figure 5B; due to a technical error, we missed the baseline values of a single animal of the MTBVAC.id group). Similarly, 4 of 11 showed the opposite effect of decreased TNF-α levels after BCG.id/MTBVAC.id, while 3 of 11 showed no considerable modulation of LPS-stimulated TNF-α after intradermal vaccination (Figure 5B). Of note, Mtb-derived MTBVAC appeared equally as potent as BCG in the induction of trained immunity in PBMC.mo (Figure S4A). Notably, the secretion levels of pro-inflammatory IL-6 and IL-1β showed a pattern similar to that of TNF-α. The ratio of the production for the 3 signature cytokines (TNF-α, IL-6, and IL-1β) did not deviate over a period of 8 weeks in a cohort of unvaccinated animals (Figure S4C).
Figure 5.

Trained immunity after mucosal vaccination in peripheral blood monocytes
Cytokine production was measured after 24 h of stimulation with LPS (0.1 μg/mL).
(A) Paired display of cytokine levels before and 8 weeks after vaccination. The data are presented as medium control corrected (mcc; BCG in circles; MTBVAC in triangles). Statistical significance was calculated by Wilcoxon’s non-parametric paired analysis test (n = 12 animals/group; PRE versus 8 weeks post-vaccination). p values are indicated at the top of each graph.
(B) Median fold increase in cytokine production is indicated in italics; significant p < 0.05 is indicated in bold; n = 12 animals/group; Wilcoxon signed rank test.
See also Figures S1, S3, S4, S5, and S6.
In contrast to intradermal BCG/MTBVAC vaccination, the mucosal delivery of these live attenuated vaccines resulted in a robust and significant enhancement of TNF-α, IL-6, and IL-1β production after LPS stimulation in 10 of 12 animals (Figures 5A and 5B; 5 animals from the BCG and the MTBVAC vaccinated group each). The median effect size was 15.1, 6.2, and 5.9 for the fold increase in TNF-α, IL-6, and IL-1β production, respectively (Figure 5B). For GM-CSF and MIP-1α, 2 cytokines that have been associated with trained immunity, a similar pattern of enhanced secretion after prior mucosal rather than intradermal vaccination was obtained (Figures 5A and 5B). This enhancement, however, was less prominent than for TNF-α, IL-6, and IL-1β, with a median fold-increase of GM-CSF and MIP-1α after BCG.muc/MTBVAC.muc of 2.1 and 2.9, respectively (Figure 5B). CXCL-10 was not detected from these monocyte stimulation analyses (Figure 5A). Comparing BCG to MTBVAC within each route of delivery (id versus muc) strategy demonstrated that both vaccines were equally potent in mediating trained immunity (Figure S5A). We explored the mycobactericidal activity with PBMC.mo isolated from residual frozen PBMCs, but within the limits of our efforts, we did not detect mycobactericidal activity by trained monocytes (Figure S6).
Next to PBMC.mo, we addressed the LPS-stimulated cytokine/chemokine secretion potential of BM-derived CD14+ monocytes (BM.mo) before and after BCG or MTBVAC vaccination. Similar to what was observed for PBMC.mo, after intradermal BCG/MTBVAC delivery, trained immunity by enhanced TNF-α, IL-6, and IL-1β secretion was apparent in only a subset of animals, whereas in others there was no effect or an inverse effect of yielding reduced innate cytokine release after vaccination (Figures 6A and 6B). Also, in accordance with PBMC.mo profiling, after mucosal BCG/MTBVAC delivery, cytokine production and, in particular the secretion of TNF-α and IL-1β, was significantly enhanced (Figure 6A). Again, the proportion of subjects showing trained immunity signals was higher after mucosal than after intradermal vaccination, although the effect size appeared less prominent than with PBMC.mo (3.1, 4.1, and 5.0 for TNF-α, IL-6 and IL-1β, respectively), the median effect size is increased for TNF-α and IL-1β in mucosally vaccinated compared to intradermally vaccinated animals (Figure 6B). In contrast to PBMC.mo analysis, there was no trained immunity effect apparent by the secreted amounts of GM-CSF and MIP-1α from BM.mo (Figures 6A and 6B). Also, for BM.mo, we did not observe a difference between BCG and MTBVAC in the ability to induce trained immunity (Figure S4B).
Figure 6.

Trained immunity after mucosal vaccination in BM monocytes
Cytokine production was measured after 24 h of stimulation with LPS (0.1 μg/mL).
(A) Paired display of cytokine levels before and 8 weeks after vaccination. The data are presented as medium control corrected (mcc; BCG in circles; MTBVAC in triangles). Statistical significance was calculated by Wilcoxon’s non-parametric paired analysis test (n = 12 animals/group; PRE versus 8 weeks post-vaccination). p values are indicated at the top of each graph.
(B) Median fold increase in cytokine production is indicated in italics; significant p < 0.05 is indicated in bold; n = 12 animals/group; Wilcoxon signed rank test.
See also Figures S1, S4, and S6.
To investigate whether metabolic rewiring occurs after BCG or MTBVAC vaccination, we measured lactate production in the conditioned supernatants of LPS-stimulated blood- (PBMC.mo) and BM- (BM.mo) derived monocytes. When comparing the two routes of vaccination, the change in lactate production by LPS-stimulated PMBC.mo is significantly higher after mucosal than intradermal delivery (Figure 7A; p = 0.020). The increase in lactate production after mucosal vaccination is nearly significant (Figure 7C; p = 0.0522). When analyzing BM.mo, this differential effect of vaccine route on lactate production is less prominent and not significant (Figure 7B; p = 0.26). Importantly, the change in the production of hallmark cytokines of trained immunity (TNF-α, IL-6, and IL-1β) correlated well with the change in lactate production (Figure S6), especially after mucosal vaccination with BCG/MTBVAC (Figure S6B).
Figure 7.

Increased lactate production after mucosal vaccination
(A and B) Lactate production as an indicator of metabolic rewiring was measured in 24-h supernatant from LPS-stimulated monocytes purified (A) from peripheral blood, or (B) BM. Statistical significance between group median values was calculated by Mann-Whitney rank testing and indicated at the top. The box extends from the 25th to 75th percentiles while the whiskers go down to the smallest value and up to the largest.
(C) Median fold increase in lactate production is indicated in italics; significant p < 0.05 is indicated in bold; n = 12 animals/group; Wilcoxon signed rank test.
See also Figure S6.
Discussion
In the present study we investigated the induction of trained immunity in myeloid leukocytes from adult rhesus macaques by immunization with live attenuated mycobacterial vaccine via different administration routes. The results demonstrate the induction of memory-like trained immunity in immune cells from NHPs, as determined by epigenetic reprogramming and enhanced ex vivo cytokine and lactate production upon secondary heterologous stimulation. Although we could not reveal a signal of trained immunity in unfractionated BAL cells after (mucosal) live attenuated mycobacterial vaccination, we demonstrated robust innate immune training in the periphery (PBMC.mo and BM.mo) for both MTBVAC and BCG, delivered via the respiratory mucosa as compared to intradermal injection. Combined with our previous findings of mucosal BCG delivery conferring an improved reduction of disease and prevention of Mtb infection,21,22 this study provides strong rationale for the further investigation of innate immune training and its underlying mechanisms in relation to TB immunity and vaccine efficacy.
The scope of this study was limited to immunogenicity analyses only. Therefore, the formal correlation between individual signatures of trained immunity and protective efficacy of live attenuated whole-cell vaccination remains to be established. However, the enhanced secretion of the cytokines, TNF-α, IL-6, and GM-CSF, observed in the present study, is in accordance with our previous finding of a significant correlation between the individual secretion levels for these cytokines after vaccination and the outcome of experimental RLD challenge with Mtb (see the supplemental data in Dijkman et al.22).
In line with what has been reported for intravenous BCG in mice,8 we demonstrated that upon intravenous BCG vaccination in NHPs, the effective induction of trained immunity in blood-derived monocytes, associated with epigenetic reprogramming, could be observed. It is tempting to speculate that the superior protection of BCG when delivered intravenously, as demonstrated by Darrah et al.,23 may in part be the result of innate immune training, although these investigators did not find innate immune activation in PBMCs to support this speculation. Furthermore, beyond the prevention of infection signals from the aforementioned NHP studies, it is also interesting to consider trained immunity as a relevant mechanism in light of BCG revaccination’s providing a signal of early clearance by the prevention of (sustained) Mtb infection in humans.32,33
Beyond these implications from prior observations on protective TB immunity, the present study emphasizes that innate cytokine secretion by peripheral blood monocytes, upon secondary heterologous stimulation with LPS, is positively modulated by live attenuated whole-cell vaccination. Next to TNF-α, IL-6, and IL-1β as the classical signature cytokines of trained immunity, GM-CSF and MIP-1α (CCL-3) also appeared as innate immune signals in rhesus macaques linked to trained immunity by BCG or MTBVAC vaccination. Trained immunity has been functionally associated with mycobacterial growth inhibition in human monocytes.31 Within the limits of our efforts, we did not detect mycobactericidal activity by the trained monocytes. It must be noted, however, that improved cytokine responses by monocytes can have important anti-mycobacterial effects independent of monocytes themselves, such as activation of lymphocytes, lung macrophages, and natural killer (NK) cells.
The present study corroborates local pulmonary immune correlates such as the induction of enhanced levels of antigen-specific polyfunctional Th17 cells in the airways after mucosal administration of live attenuated BCG and MTBVAC. Extended analysis of adaptive immune responses after vaccination revealed that local T cells expressed high levels of mucosal homing and tissue-residency markers that were not found after Mtb infection. Moreover, local antibodies bound to Mtb, mediating enhanced pathogen uptake. These data are presented in the accompanying paper by Dijkman et al.34 In addition, the respective innate cytokine responses from peripheral blood cell populations may provide useful correlates and biomarkers of vaccine-induced protection against TB.
The ultimate longevity of trained immunity after vaccination remains to be established and is likely to be affected by environmental exposure or endogenous microbial cues, but it is clear that the phenotype of trained immunity exceeds the lifespan of the myeloid monocytes by which we determine the training effect. Several mechanisms have been proposed to explain HSC activation in the context of infection or vaccination.32 Kaufmann and colleagues8 have provided a plausible explanation for this paradox by showing that in mice (intravenous) BCG vaccination modulates gene expression and innate response profiles of HSC, the precursors of the myeloid monocyte/macrophage lineage in the BM, by epigenetic reprogramming.8 These findings were extended by Cirovic et al.,35 who showed the induction of trained HSC in human after standard intradermal vaccination with BCG. It is suggested that the trained phenotype gained by the HSC is passed along to the CD14+ monocytic lineage. At this point we can only speculate that similar mechanisms may apply to mucosal BCG vaccination. The current study does show that innate training as observed in peripheral blood monocytes is also captured in monocytes purified from BM. The enhanced lactate production suggests metabolic reprogramming of myeloid monocytes after prior vaccination with BCG or MTBVAC, and in particular after the mucosal delivery of these vaccines. This may be indicative of host response modulation associated with long-term immune training. The epigenetic reprogramming observed after intravenous BCG supports the notion that this modulation may also underlie the robust increased production of cytokines after mucosal vaccination, but that still needs to be formally proven.
This study links mucosal vaccination via the airways to the systemic induction of trained immunity in myeloid cells, but when analyzing cytokine and chemokine release upon (heterologous) innate stimulation of BAL cells, we did not observe trained immunity locally. On the contrary, LPS stimulation of the unfractionated cell population from lung washes after live attenuated mycobacterial whole-cell vaccination, and regardless of vaccination route, did reveal significant suppression of IL-1β secretion in particular, which suggests anti-inflammatory regulation. Such anti-inflammatory training or innate tolerance response, in general, may help to preserve local pulmonary homeostasis in the face of constant environmental exposure to airborne microbial agents.36,37 However, in mice, alveolar macrophages can render a memory-like phenotype with a pro-inflammatory gene signature and increased glycolytic metabolism through respiratory viral infection and T cell help.38 The functional plasticity of alveolar macrophages and newly recruited myeloid cells in the airways as well as tissue-resident macrophages with regard to trained immunity after TB vaccination and prevention of TB infection and disease will also require future investigation.
Trained immunity has been proposed to explain epidemiological observations that BCG-vaccinated individuals show a broad reduction of unrelated respiratory infectious disease, compared to unvaccinated individuals.10 While the heterologous beneficial effect of BCG is clinically relevant, the recently suggested impact of elevated innate immunity on natural resistance to and early clearance of Mtb infection provides further support for including trained immunity in TB vaccine research and development strategies.14,15,32,33,39 In this regard, we should realize that, as much as trained immunity by BCG may have an impact on subsequent Mtb encounters, trained immune status at the time of vaccination may also have an impact on the live attenuated vaccine, its persistence, and its efficacy. It is noteworthy that not all of the animals in this study displayed trained immunity upon exposure to BCG or MTBVAC. Actually, after vaccination via the standard route of intradermal injection, an equally large proportion of animals showed the opposite effect of trained immunity (i.e., innate tolerance) by displaying reduced rather than enhanced innate cytokine release to secondary LPS stimulation. Correlation studies will be needed to shed further light on this matter, but it is tempting to speculate that the status of innate responsiveness before vaccination will relate to the variable efficacy of BCG in the clinic as much as it is observed in standardized NHP studies using different cohorts of outbred rhesus macaques.21 In addition, there may be a relevant impact of exposure to non-tuberculous mycobacteria (NTM) through innate immune modulation exerting a possibly negative effect on BCG efficacy, either by masking or by blocking protection by BCG (re)vaccination.17,40, 41, 42, 43, 44
In conclusion, the induction of trained innate immunity as it occurs in humans is recapitulated in rhesus macaques by live attenuated TB vaccination using either BCG or the Mtb-derived candidate vaccine MTBVAC. Moreover, mucosal immunization in this regard is more effective and yields more trained responders than conventional intradermal delivery. Since mucosal immunization with BCG in rhesus also confers better protection against TB infection and disease, future research efforts will address whether trained innate immunity is a correlate of vaccine-induced protection that can be registered by PBMCs and/or monocyte response analysis. In a broader perspective, these findings provide support for investigating and advancing immunomodulatory treatments based on trained innate immunity against human immune disorders via mucosal surfaces.
Limitations of study
The scope of the present study was limited to immunogenicity analyses since we demonstrated earlier that mucosal vaccination with BCG results in better protection against TB than standard intradermal vaccination.22 Therefore, the formal correlation between individual immune signatures of trained immunity and protective efficacy of live attenuated whole-cell vaccination remains to be established. Although studies in mice identified alveolar macrophages to be sensitive for the induction of trained immunity after a respiratory viral infection,38 mediating protection in the early stages of Mtb infection,45 we did not detect trained immunity in unfractionated BAL after BCG vaccination independent of the route of administration. This will be the subject of future studies. In the present study, we analyzed trained immunity in isolated monocytes from blood and BM as the prototypical cell population in which trained immunity can be established and did not explore trained immunity in unfractionated blood. Therefore, heterologous stimulation with LPS of isolated monocytes did not allow us to evaluate interactions with cells from the adaptive immune arm, such as T cells. Future studies should delineate the contribution of the adaptive immunity to trained immunity in unfractionated blood. Although we confirmed epigenetic reprogramming mediated by altered histone modification after intravenous BCG vaccination, we did not formally correlate increased cytokine production profiles with altered histone modification after intradermal and mucosal vaccination with BCG and MTBVAC.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD3 – AF700 (clone SP34-2) | BD Biosciences | Cat#: 557917; RRID: AB_396938 |
| anti-CD4 – PerCP.Cy5.5 (clone L200) | BD Biosciences | Cat#: 552838; RRID: AB_394488 |
| anti-CD8α – APC-H7 (clone SK1) | BD Biosciences | Cat#: 641400; RRID: AB_164536 |
| anti-CD14 – BV421 (clone M5E2) | BD Biosciences | Cat#: 301830; RRID: AB_10959324 |
| anti-CD20 – BV421 (clone 2H7) | Biolegend | Cat#: 302330; RRID: AB_10965543 |
| anti-CD28 – ECD (clone CD28.2) | IOTest | Cat#: 6607111; RRID: AB_1575955 |
| anti-CD45RA – PE-CF594 (clone 5H9) | BD Biosciences | Cat#: 565419; RRID: AB_2739229 |
| anti-CD95 – BV605 (clone DX2) | Biolegend | Cat#: 305628; RRID: AB_2563825 |
| anti-IL-2 – AF488 (clone MQ1-17H12) | Biolegend | Cat#: 500314; RRID: AB_493368 |
| anti-IL-17A – PE-Cy7 (clone ebio64DEC17) | Biolegend | Cat#: 25-7179-42; RRID: AB_11063994 |
| anti-TNF-α – BV650 (clone Mab11) | BD Biosciences | Cat#: 502938; RRID: AB_2562741 |
| anti-IFN-γ – BV711 (clone 4S.B3) | BD Biosciences | Cat#: 502540; RRID: AB_2563506 |
| anti-CD3ε – FITC (clone SP34) | BD Biosciences | Cat#: 556611; RRID: AB_396484 |
| anti-CD14 – V450 (Tük4) | Milteny Biotec | Cat#: 130-113-152; RRID: AB_10831023 |
| anti-CD20 – BV605 (clone 2H7) | Biolegend | Cat#: 302334; RRID: AB_2563398 |
| anti-CD45 – BV786 (clone D058-1283) | BD Biosciences | Cat#: 563861; RRID: AB_2738454 |
| anti-CD3 – BV421 (clone SP34-2) | BD Biosciences | Cat#: 562877; RRID: AB_2737860 |
| anti-CD14 – BV786 (clone M5E2) | BD Biosciences | Cat#: 301840; RRID: AB_2563425 |
| anti-CD16 – PE-CY7 (clone 3G8) | BD Biosciences | Cat#: 560716; RRID: AB_1727433 |
| anti-CD20 – BV605 (clone 2H7) | Biolegend | Cat#: 302334; RRID: AB_2563398 |
| anti-CD45 – AF700 (clone D058-1283) | BD Biosciences | Cat#: 561288; RRID: AB_10613813 |
| anti-CD66 – PE (clone TET2) | Milteny Biotec | Cat#: 130-093-133; RRID: AB_871699 |
| anti-CD206 – PerCP (clone 15-2) | BioLegend | Cat#: 321122; RRID: AB_10899411 |
| Bvbuffer | BioLegend | Cat#: 563794; RRID: AB_2869750 |
| VIVID – BV421 | Thermofisher | Cat#: L34955 |
| Life/Death – eFluor 506 | eBioscience | Cat#: 65-0866-14 |
| anti-H3K27ac | Diagenode | Cat#: pab-196-050; RRID: AB_2637079 |
| Bacterial and virus strains | ||
| BCG (strain Sofia; 5 × 105 CFU) | InterVax Ltd | Cat#: not applicable |
| MTBVAC (M.tub strain; 5 × 105 CFU) | Biofabri | Cat#: not applicable |
| Mtb Erdmann | BEI Resources | Cat#: NR-50781 |
| Chemicals, peptides, and recombinant proteins | ||
| Purified Protein Derivate (PPD; M.tub) | AJ Vaccines | Cat#: 2391 |
| Purified Protein Derivate (PPD; M.bov) | Life Technologies NV | Cat#: 760060 |
| Purified Protein Derivate (PPD; M.av) | Life Technologies NV | Cat#: 760065 |
| Whole Cell Lysate (WCL; Mtb HN878) | BEI Resources | Cat#: NR-14824 |
| Lipopolysaccharide (0111:B4 strain; Ultrapure) | Invivogen | Cat#: tlrl-3pelps |
| PMA | Sigma-Aldrich | Cat#: P8139 |
| Ionomycine | Sigma-Aldrich | Cat#: I0634 |
| Roswell Park Memorial Institute (RPMI) | Life Technologies NV | Cat#: 52400041 |
| LymphoprepTM | Axis-Shield | Cat#: AXI-1114547 |
| Fetal Calve Serum (FCS) | Life Technologies NV | Cat#: 10270106 |
| Perchloric acid | Sigma-Aldrich | Cat#: 244252-1L |
| Formaldehyde (16%) | Thermo Scientific | Cat#: 28906 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#: P8465 |
| Dynabeads protein A | Invitrogen | Cat#: 10002D |
| Dynabeads protein G | Invitrogen | Cat#: 10004D |
| BSA | Sigma-Aldrich | Cat#: A7030 |
| Tagment DNA buffer | Nextera | Cat#: 20034197 |
| AMPureXP beads | Beckman Coulter | Cat#: A63880 |
| KAPA HiFi Hotstart Ready Mix | KAPA Biosystems | Cat#: KK2601 |
| Nextera DNA Library Prep Kit | Illumina | Cat#: FC-121-1031 |
| KAPA Library Preparation Kit | KAPA Biosystems | Cat#: KK8400 |
| TMB (ELISPOT substrate) | Mabtech | Cat#: 3651-10 |
| Critical commercial assays | ||
| CD14 microbeads | Milteny Biotec | Cat#: 130-091-097 |
| Custom NHP Legendplex (11-plex) | Biolegend | Cat#: 92919 |
| Monkey IFN-γ ELISPOT | U-CyTech | Cat#: CT610-10 |
| Lactate-ELISA | Promokine | Cat#: PK-CA577-K627 |
| Deposited data | ||
| ChIP sequencing data before and after BCGiv vaccination | This paper | GEO: GSE159046 |
| Experimental models: organism/strain | ||
| Rhesus monkeys (Indian); male and female; adult (> 4 years); purpose bred | BPRC breeding colony | not applicable |
| Software and algorithms | ||
| Eli.Analyze (ELISPOT; v6.1) | A.EL.VIS GmbH | not applicable |
| FACSDiva Software v 8.0.1 (BD LSRII) | BD Biosciences | SCR_001456 |
| Flowjo software v 10 | Treestar | SCR_000410 |
| LEGENDplexTM Data Analysis Software (V8.0) | Biolegend/Vigene Tech | not applicable |
| GraphPad Prism v 8.4.2 | GraphPad Software | https://www.graphpad.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Michel PM Vierboom (vierboom@bprc.nl).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw data files of the ChIPseq analysis on BCG trained monocytes begore and after vaccination have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE159046.
Experimental model and subject details
Animals and ethics
All animal experiments were in accordance with Dutch Law on animal experimentation, which is in accordance with the EU Directive 2010/63/EU on the protection of animals used for scientific purposes. The study was approved by the institutional animal welfare body (in Dutch: Instantie voor Dierwelzijn, IvD; accession number CCD009D) of BPRC. A cohort of 8 nonvaccinated control animals was used for Figure S4C. The study in which these animals were used was approved by the institutional animal welfare body (Accession number CCD009F). The BPRC is accredited by the American Association for Accreditation of Laboratory Animal Care (AAALAC) and has an approved Assurance (#A5509-1) for the care and use of animals on file at the National Institutes of Health (NIH).
The rhesus monkeys were housed in socially compatible pairs at the Biomedical Primate Research Centre (BPRC; animal biosafety level ABSL-3). The animals were offered a daily diet consisting of monkey food pellets (Hope Farms, Woerden, the Netherlands), fruit and vegetables of the season, and bread. Drinking water was available ad libitum via automatic water systems.
The study was carried out in 27 healthy purpose-bred, pedigreed, male and female rhesus monkeys (Macaca mulatta) of Indian origin (5.99 – 13,50 kg; see Table S1). All animals were screened to be negative for pre-existing immunity against mycobacterial antigens as determined by interferon gamma (IFN-γ) ELISpot assay after in vitro recall stimulation with Purified Protein Derivate (PPD) of M.tuberculosis, M.avium and M.bovis.
All animal handling and biosampling was performed under ketamine sedation (10 mg kg−1, by intramuscular injection). For bronchoalveolar lavage (BAL) and intrabronchial vaccination inoculation with BCG or MTBVAC ketamine (5 mg kg−1) was supplemented with intramuscular medetomidine (0.04 mg kg−1) and an analgesic sprayed into the larynx.
Vaccines and vaccination
BCG standard dose & MTBVAC standard dose. Animals immunized with Bacillus Calmette–Guérin (BCG) strain Sofia (InterVax Ltd.) received a single standard adult human dose of 5 × 105 CFU. Animals immunized with MTBVAC received an equivalent human dose of 5 × 105 CFU. Vaccines were delivered in a volume of 0.1 mL intradermally and 10 mL intravenously or intrabronchially. Mucosal vaccination in the lung was performed by intrabronchial delivery of the same dose in 10mL of sterile saline solution to a segmental bronchus in the lower right lung lobe using a bronchoscope. Vaccination was executed for all animals in a single session in random order within 2-3 hours from vaccine preparation
Method details
Biological sample collection and processing
Cells from the pulmonary mucosa were recovered by BAL at week −1 for all groups from the lower right lung lobe; at week 2 after intravenous BCG and week 8 after intradermal BCG from lower right lung lobe and week 8 after mucosal vaccination from both the lower right and left lung lobe. Three volumes of 20 mL of prewarmed 0.9% saline solution were consecutively instilled and recovered. BAL fluid was harvested by centrifugation of BAL samples for 10 min at 400 g after 100 μm filtration. Supernatant was subsequently decanted and stored at −80°C pending further analysis. The BAL cell pellet was taken up in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% FCS, glutaMAX, and penicillin/streptomycin (from now on referred to as R10) and used freshly in downstream assays. Heparinized blood for immune monitoring, was collected by venipuncture. Bone marrow was obtained through needle extraction of the caput humerus. PBMCs and BM cells were obtained by density gradient centrifugation with Lymphoprep lymphocyte separation medium (Axis-Shield) and resuspended in R10 for further analysis.
Mycobacterium uptake and killing assay
To assess mycobacterium uptake, PBMC.mo (30.000 cells/well) were incubated for 1h at 37°C with Mtb Erdmann at a MOI of 5. Non-phagocytosed mycobacterium was washed away and monocytes were incubated with high dose (30 μg/mL) gentamycin for 10-minutes. PBMC.mo were subsequently lysed with 0.05% SDS in water, titrated and plated on to 7H10 agar for bacterial enumeration. To asses killing, PBMC.mo were incubated with Mtb Erdmann at a MOI of 5 for 1h at 37°C and subsequently cultured with low gentamycin (5 μg/mL) for 24 hr. Bacteria were enumerated as described before. As a control, bacteria were incubated without PBMC.mo. Data are presented as % of control (Figure S5).
Monocyte isolation
CD14+ cells (monocytes) were labeled with magnetic CD14 MicroBeads. The cell suspension was loaded onto a MACS® Column which is placed in the magnetic field of a MACS Separator. The magnetically labeled CD14+ cells are retained in the column. The unlabeled cells run through, this cell fraction is depleted of CD14+ cells. After removal of the column from the magnetic field, the magnetically retained CD14+ cells can be eluted as the positively selected cell fraction. Purity was analyzed by flowcytometry (Figure S1).
IFN-γ ELISPOT
A NHP specific IFN-γ ELISpot assay was used on PBMC, according to the manufacturer protocol (U-CyTech, Utrecht), to determine the frequency of antigen specific IFN-γ producing cells. In brief, 200.000 freshly isolated PBMC were incubated in triplicate for 24 h with specified antigens or control stimuli. Subsequently, supernatant was collected and stored (−80°C), and cells were transferred to specific anti-IFN-γ coated filter plates (PVDF, Millipore) for an additional overnight (18 h) incubation. Cells were discarded and membrane bound IFN-γ was detected using biotinylated anti-IFN-γ antibody, streptavidin-horseradish peroxidase conjugate and tetramethylbenzidine (TMB) substrate (the latter from MAbTech, Stockholm). Spots were quantified using an automated reader (AELVIS, Hannover).
Flow cytometry
Antigen-specific T cell responses were determined at WK-1 (PRE) and week 8 indicated time points by flow cytometry locally using BAL cells. Cells were stimulated overnight in the presence of Golgiplug transport inhibitor. The following mAb staining T cell panel was used: Intracellular staining: CD3 AF700 (SP34-2; BD), TNF-αPE-CY7 (Mab11; BD), IL-2BV510 (MQ1-17H12; BioLegend), IFN-γAPC (4S.B3; BD); IL-17APE-Cy7 (ebio64DEC17; Thermofisher). Surface staining: CD4PerCP-Cy5.5 (L200; BD), CD8αAPC-H7 (SK1; BD), CD14BV421 (M5E2; BD), CD20BV421 (2H7; BioLegend), CD45BV786 (D058-1283; BD).
Gating strategy for T cells (Figure S7; Related to Figure 2D)
T cells were gated as Singlets/Lymphocytes/Viable/ CD14-CD20-/CD45+ /CD3+ before CD4 and CD8 gating was applied. After doublet exclusion, lymphocytes were gated based on size and granularity. Any anomaly indicative of unstable signal acquisition was excluded using the time parameter. Events from the combined time-gates were plotted against the dump channel containing the viability, CD14 and C20 markers and subsequently gated as viable, CD14- and CD20-. Cells were further selected for CD45 and CD3 positivity before CD4 and CD8 gating was applied. Boolean gating of any cytokine expression of IL-2/IFN-γ/TNFα/IL-17A.
BAL cell composition analysis
Cells were stimulated overnight with WCL. The following mAb staining panel was used to analyze BAL cell composition: CD45AF700 (D058-1283; BD); CD66PE (TET2; Miltenyi); CD206PerCP (15-2;Biolegend); CD16PE-CY7 (3G8; BD); CD3 BV421 (SP34-2;BD); CD20BV605 (2H7; Biolegend); CD14BV786 (M5E2; BD).
Gating strategy for BAL cell composition (Figure S2B)
Singlets/ Leucocytes (CD45)/Viable/Time gate: CD206+/CD14+ (Alveolar Macrophages); CD3 (T cells); CD20 (B cells); CD16+ (NK-cells) and CD66+ (Neutrophils). BAL cell composition is expressed as % from the CD45+/viable fraction.
Isolation of CD14+ monocytes
PBMC or BM cells were incubated with CD14 magnetically labeled Microbeads (Miltenyi Biotec; Netherlands). The cell suspension is loaded onto a MACS® Column which is placed in the magnetic field of a MACS Separator. The magnetically labeled CD14+ cells are retained in the column. The column is washed once. The column was removed from the magnetic field and the magnetically retained CD14+ cells were then eluted as the positively selected cell fraction. Purity of CD14+ fraction was subsequently tested by flow cytometry.
Gating strategy for CD14+ monocytes (Figure S1B)
Singlets/CD45+/ Viable/CD14+. CD14+ cells are expressed as % from the CD45+/viable fraction.
Multiplex cytokine assay
Cells from different sources (PBMC.mo, BM.mo and BAL were stimulated with WCL and titrated amount of LPS (E.coli; Ultrapure). Freshly isolated cells 25000 cells were incubated ex-vivo for 24 hr with each stimulus after which the supernatant was collected. Medium alone was taken along for control. Cytokine production was assessed with multi-analyte flow assay kit using a custom nonhuman primate Legendplex™ (Biolegend®; USA). Assays were performed according to the manufacturer’s instructions. Briefly, the supernatants of stimulated cells were incubated with beads coated with cytokine-specific antibodies. Bound cytokines were visualized using biotin-coupled detection antibodies and R-phycoerythrin-conjugated streptavidin. Beads were acquired on 3 laser, 14 color LSR-II flow cytometer (BD Biosciences). A NHP custom made 11-plex consisted of TNF-α, IL-6, IL-1β characterizing trained immunity, GM-CSF, CCL3 (MIP-1α), CXCL10 (IP-10) and cytokines associated with adaptive immunity IFN-γ, IL-2, IL-17A.
Lactate measurements
Supernatant collected from the 24 h innate stimulation was analyzed for the production of lactate as a surrogate marker for the metabolic rewiring (Warburg-effect) underlying trained immunity. Proteins in the supernatant, interfering with the lactate analysis, were denatured precipitated with percholoric acid (PCA) and removed by high speed centrifugation. The clear deproteinized supernatant was collected and neutralized with NaOH.
Samples were subsequently diluted 15-fold and analyzed in a L-Lactate Assay Kit I (Promokine; PromoCell GmbH; Germany) according to manufacturer’s instruction.
Chromatin immunoprecipitation
Cell pellets were dissolved in PBS and crosslinked in solution by using formaldehyde (1% final volume, shaking 10 min at room temperature). Glycine (0.125 M) was added to quench the reaction. Cells were then washed two times with cold PBS. Cell pellets were lysed in a volume of 110μl using lysis buffer (20 mM HEPES pH 7.6, 1% SDS, 1 × Protease Inhibitor Cocktails). Samples were sonicated using the Biorupter Pico (Diagenode) with eight cycles (30 s on/30 s off). Afterward, the samples were spun at 16,000 × g for 5 min at room temperature and the supernatant was stored at 80°C.
Chromatin from 100,000 cells was used for ChIPmentation. ChIPmentation was performed as described by Schmidl et al.,46 with several modifications. In short, the chromatin was incubated overnight at 4°C rotating in dilution buffer (1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris pH 8, 167 mM NaCl), 1 × Protease Inhibitor Cocktail47 and 1μg of antibody [H3K27ac (C15410196, Diagenode)] in a total volume of 300 μl. The next day per ChIP 10μl protein A Dynabeads and 10μl protein G Dynabeads (both Invitrogen) were added. Beads were washed before use with dilution buffer (+0.15% SDS, +0.1% BSA) and incubated with the chromatin and antibody mix for 60 min at 4°C rotating. Afterward, the beads were washed at 4°C once with ChIP wash buffer 1 (2 mM EDTA, 20 mM Tris pH 8, 1%Triton X-100, 0.1% SDS, 150 mM NaCl), twice with ChIP washbuffer 2 (2 mM EDTA, 20 mM Tris pH 8, 1% Triton X-100, 0.1%SDS, 500 mM NaCl), and twice with TE (1 mM EDTA, 10 mM TrispH 8). Beads were resuspended in 24 μl Tagment DNA buffer (Nextera) and 1μl Tn5 enzyme (produced in-house) and incubated for 10 min at 37°C with 550 rpm shaking. Afterward, the beads were washed twice with WBI (20 mM HEPES, 150 mM NaCl, 0.1% SDS, 0.1% DOC,1% Triton X-100, 1 mM EDTA, 0.5 mM EGTA) and twice with WBIV (20 mM HEPES, 1 mM EDTA, 0.5 mM EGTA) with 5 min rotating at room temperature in between. Samples were decrosslinked for 1 h at 55°C 1,000 rpm shaking followed by an overnight incubation at 65°C using elution buffer (0.5% SDS, 300 mM NaCl,5 mM EDTA, 10 mM Tris pH 8) and proteinase K. Samples were incubated one additional hour the next day with elution buffer and proteinase K for 1 h at 55°C. The samples were purified using 2 × SPRI AMPureXP beads. qPCR was used to determine the sufficient amount of PCR cycles needed to amplify the library. The libraries were amplified using the KAPA HiFi Hotstart Ready Mix (KAPA Biosystems) and Nextera Index Kit 1 (i7) and 2 (i5) primers (Illumina). Amplified libraries were purified using a 0.65 × SPRI AMPureXP beads incubation followed by a 1.8 × SPRI AMPureXP beads incubation. Library concentration was measured using the KAPA Library Quantification Kit (KAPA Biosystems); library size was determined using the BioAnalyzer High Sensitivity DNA Kit (Agilent). Sequencing was performed using an Illumina HiSeq 2000, and 50-bp paired-end reads were generated.
ChIP-seq analysis
Sequenced reads were quality and adaptor trimmed with trim galore and cutadapt (Krueger, Felix, Trim galore. “A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files 516 (2015): 517. “Picard Toolkit.” 2020. Broad Institute, GitHub; Marin, Marcel. Cutadapt removes adaptor sequences from high-throughput sequencing reads. EMBnet.journal). These reads were aligned against the UCSC Rhesus reference genome (RheMac10) with Burrows-Wheeler Aligner (BWA) program48 with default parameters. Duplicate reads were removed with picard MarkDuplicates. Peaks were called with MACS2 and default settings49 and used to detect the binding sites for ChIP-seq tracks. Tags within a given region were counted and adjusted to represent the number of tags within a 1 kb region. Subsequently the percentage of these tags as a measure of the total number of sequenced tags of the sample was calculated and displayed as heatmaps using fluff.50 To determine genomic locations of binding sites, the peak file was analyzed using a script that annotates binding sites according to all RefSeq genes.
Necropsy and post-mortem evaluation
Rhesus monkeys were scheduled by protocol for euthanasia and necropsy at 2 weeks (3 animals for BCGIV) or 8 weeks (BCGID, BCGMUC, MTBVACID, MTBVACMUC) after vaccination. At the end of the experiment blood (< 45 ml) and bone marrow was collected before euthanasia with sodium pentobarbital (> 50 mg/kg). Samples were stored at −80°C for analysis at later time point.
Quantification and statistical analysis
Data handling of measurements of cytokine production by Multiplex
The Legendplex™ is a bead-based assay for flow cytometry allowing for the measurement of multiple cytokines in the same sample. Bead count per analyte was ± 300/sample. Data obtained with a bead count arbitrarily set at < 25 was excluded. Standard curve (pg/mL) taken along on each plate: 10000, 2500, 625, 156, 39.1, 9.8, 2.4 pg/mL. PRE and WK2/WK8 samples from individual animals were measured on the same plate to avoid inter-plate variation. Samples from animals from different treatment groups were randomly distributed over plates. If lower limit of quantification (LLOQ) was < 2.4 pg/mL, the lowest detectable concentration was set at 2.4 pg/mL. If LLOQ was > 2.4 pg/mL, the lowest detectable concentration was set at LLOQ. This data procedure ensured a conservative estimation of the fold change between PRE and WK2/WK8 post vaccination.
Statistics comparing routes of vaccination, cytokine, and lactate production
Statistical parameters including number of animals, percentage of cells levels of cytokine production, fold increase of cytokine production and absolute increase in lactate production are reported in the Figures and Figure legends. Data was analyzed by ANOVA adjusted for multiple comparisons, Wilcoxon’s non-parametric paired analysis test, Wilcoxon signed rank test to where applicable. Statistical analyses were conducted using GraphPad Prism 7 software (version 7.0d; GraphPad Software, La Jolla, CA).
Statistics epigenetics
Peaks were called with MACS2 and default settings49 and used to detect the binding sites for ChIP-seq tracks. All peaks were merged using bedtools and for each sample tags per peak were counted. Differential peaks were called by comparing the two sets of samples using these criteria: the sum of counted tags within one peak using all samples should be > 200 and the fold difference should be > median plus 2xSD. The testing for gene set/pathway enrichments was done using Fisher’s exact testing.
Acknowledgments
We thank the animal and veterinary care teams as well as the pathology team and clinical lab personnel of BPRC for their outstanding contributions to this work; F. van Hassel for support with graphics; and Prof. T. Ottenhoff at Leiden University Medical Centre for providing the ESAT6-CFP10 fusion protein. The following reagent was obtained through BEI Resources, the NIAID, and the NIH: M. tuberculosis, Strain Erdman Barcode Library, NR-50781. This NHP study and the MTBVAC vaccine are linked to the TB vaccine R&D program of the TuBerculosis Vaccine Initiative (TBVI)-governed TBVAC2020 Consortium, supported by the European Commission under the Horizon 2020 programme, grant agreement no. 643381. Extended immune response analyses were in part supported as a research effort of the TRANSVAC2 Consortium, also under the Horizon 2020 programme, grant agreement no. 730964.
Author contributions
Conceptualization & Study Design: F.A.W.V., M.P.M.V., and K.D.; Assay Performance, Data Processing, & Analysis: K.D., R.A.W.V., C.C.S., S.O.H., C.B., K.G.H., M.P.M.V., M.v.d.S., L.v.E., S.J.C.F.M., and J.D.-A.; Writing – Original Draft, M.P.M.V. and F.A.W.V.; Writing – Review & Editing, K.D., J.D.-A., J.T., E.R., E.P., R.v.C., M.G.N., N.A., and C.M.; Supervision, J.H.A.M., C.H.M.K., and F.A.W.V.
Declaration of interests
E.R., E.P., N.A., and C.M. are co-inventors on a patent on a tuberculosis vaccine held by the University of Zaragoza and Biofabri. E.R. and E.P. are employees of Biofabri. J.T. is an employee of the TuBerculosis Vaccine Initiative and an advisor to Biofabri. M.G.N. holds a patent on the inhibition of trained immunity with nanobiologics, and a patent on the stimulation of trained immunity with nanobiologics. M.G.N. is also a scientific founder of Trained Therapeutix and Discovery (TTxD). The remaining authors of the Radboud UMC and the authors from the BPRC declare no competing interests.
Published: January 19, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100185.
Supplemental information
References
- 1.Kaufmann S.H.E. Immunology’s Coming of Age. Front. Immunol. 2019;10:684. doi: 10.3389/fimmu.2019.00684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Netea M.G., Joosten L.A., Latz E., Mills K.H., Natoli G., Stunnenberg H.G., O’Neill L.A., Xavier R.J. Trained immunity: A program of innate immune memory in health and disease. Science. 2016;352:aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kleinnijenhuis J., Quintin J., Preijers F., Joosten L.A., Ifrim D.C., Saeed S., Jacobs C., van Loenhout J., de Jong D., Stunnenberg H.G. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. USA. 2012;109:17537–17542. doi: 10.1073/pnas.1202870109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng S.C., Quintin J., Cramer R.A., Shepardson K.M., Saeed S., Kumar V., Giamarellos-Bourboulis E.J., Martens J.H., Rao N.A., Aghajanirefah A. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quintin J., Saeed S., Martens J.H.A., Giamarellos-Bourboulis E.J., Ifrim D.C., Logie C., Jacobs L., Jansen T., Kullberg B.J., Wijmenga C. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12:223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Netea M.G., Domínguez-Andrés J., Barreiro L.B., Chavakis T., Divangahi M., Fuchs E., Joosten L.A.B., van der Meer J.W.M., Mhlanga M.M., Mulder W.J.M. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020;20:375–388. doi: 10.1038/s41577-020-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saeed S., Quintin J., Kerstens H.H., Rao N.A., Aghajanirefah A., Matarese F., Cheng S.C., Ratter J., Berentsen K., van der Ent M.A. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufmann E., Sanz J., Dunn J.L., Khan N., Mendonça L.E., Pacis A., Tzelepis F., Pernet E., Dumaine A., Grenier J.C. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell. 2018;172:176–190.e19. doi: 10.1016/j.cell.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 9.Spertini F., Audran R., Chakour R., Karoui O., Steiner-Monard V., Thierry A.-C., Mayor C.E., Rettby N., Jaton K., Vallotton L. Safety of human immunisation with a live-attenuated Mycobacterium tuberculosis vaccine: a randomised, double-blind, controlled phase I trial. Lancet Respir. Med. 2015;3:953–962. doi: 10.1016/S2213-2600(15)00435-X. [DOI] [PubMed] [Google Scholar]
- 10.Aaby P., Roth A., Ravn H., Napirna B.M., Rodrigues A., Lisse I.M., Stensballe L., Diness B.R., Lausch K.R., Lund N. Randomized trial of BCG vaccination at birth to low-birth-weight children: beneficial nonspecific effects in the neonatal period? J. Infect. Dis. 2011;204:245–252. doi: 10.1093/infdis/jir240. [DOI] [PubMed] [Google Scholar]
- 11.Arts R.J.W., Moorlag S.J.C.F.M., Novakovic B., Li Y., Wang S.Y., Oosting M., Kumar V., Xavier R.J., Wijmenga C., Joosten L.A.B. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe. 2018;23:89–100.e5. doi: 10.1016/j.chom.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 12.Higgins J.P., Soares-Weiser K., López-López J.A., Kakourou A., Chaplin K., Christensen H., Martin N.K., Sterne J.A., Reingold A.L. Association of BCG, DTP, and measles containing vaccines with childhood mortality: systematic review. BMJ. 2016;355:i5170. doi: 10.1136/bmj.i5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thysen S.M., Benn C.S., Gomes V.F., Rudolf F., Wejse C., Roth A., Kallestrup P., Aaby P., Fisker A. Neonatal BCG vaccination and child survival in TB-exposed and TB-unexposed children: a prospective cohort study. BMJ Open. 2020;10:e035595. doi: 10.1136/bmjopen-2019-035595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simmons J.D., Stein C.M., Seshadri C., Campo M., Alter G., Fortune S., Schurr E., Wallis R.S., Churchyard G., Mayanja-Kizza H. Immunological mechanisms of human resistance to persistent Mycobacterium tuberculosis infection. Nat. Rev. Immunol. 2018;18:575–589. doi: 10.1038/s41577-018-0025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verrall A.J., Schneider M., Alisjahbana B., Apriani L., van Laarhoven A., Koeken V., van Dorp S., Diadani E., Utama F., Hannaway R.F. Early clearance of Mycobacterium tuberculosis is associated with increased innate immune responses. J. Infect. Dis. 2020;221:1342–1350. doi: 10.1093/infdis/jiz147. [DOI] [PubMed] [Google Scholar]
- 16.Dijkman K., Vervenne R.A.W., Sombroek C.C., Boot C., Hofman S.O., van Meijgaarden K.E., Ottenhoff T.H.M., Kocken C.H.M., Haanstra K.G., Vierboom M.P.M., Verreck F.A.W. Disparate Tuberculosis Disease Development in Macaque Species Is Associated With Innate Immunity. Front. Immunol. 2019;10:2479. doi: 10.3389/fimmu.2019.02479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mangtani P., Abubakar I., Ariti C., Beynon R., Pimpin L., Fine P.E., Rodrigues L.C., Smith P.G., Lipman M., Whiting P.F. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin. Infect. Dis. 2014;58:470–480. doi: 10.1093/cid/cit790. [DOI] [PubMed] [Google Scholar]
- 18.Hatherill M., White R.G., Hawn T.R. Clinical Development of New TB Vaccines: Recent Advances and Next Steps. Front. Microbiol. 2019;10:3154. doi: 10.3389/fmicb.2019.03154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voss G., Casimiro D., Neyrolles O., Williams A., Kaufmann S.H.E., McShane H., Hatherill M., Fletcher H.A. Progress and challenges in TB vaccine development. F1000Res. 2018;7:199. doi: 10.12688/f1000research.13588.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barclay W.R., Busey W.M., Dalgard D.W., Good R.C., Janicki B.W., Kasik J.E., Ribi E., Ulrich C.E., Wolinsky E. Protection of monkeys against airborne tuberculosis by aerosol vaccination with bacillus Calmette-Guerin. Am. Rev. Respir. Dis. 1973;107:351–358. doi: 10.1164/arrd.1973.107.3.351. [DOI] [PubMed] [Google Scholar]
- 21.Verreck F.A.W., Tchilian E.Z., Vervenne R.A.W., Sombroek C.C., Kondova I., Eissen O.A., Sommandas V., van der Werff N.M., Verschoor E., Braskamp G. Variable BCG efficacy in rhesus populations: Pulmonary BCG provides protection where standard intra-dermal vaccination fails. Tuberculosis (Edinb.) 2017;104:46–57. doi: 10.1016/j.tube.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Dijkman K., Sombroek C.C., Vervenne R.A.W., Hofman S.O., Boot C., Remarque E.J., Kocken C.H.M., Ottenhoff T.H.M., Kondova I., Khayum M.A. Prevention of tuberculosis infection and disease by local BCG in repeatedly exposed rhesus macaques. Nat. Med. 2019;25:255–262. doi: 10.1038/s41591-018-0319-9. [DOI] [PubMed] [Google Scholar]
- 23.Darrah P.A., Zeppa J.J., Maiello P., Hackney J.A., Wadsworth M.H., 2nd, Hughes T.K., Pokkali S., Swanson P.A., 2nd, Grant N.L., Rodgers M.A. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature. 2020;577:95–102. doi: 10.1038/s41586-019-1817-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arbues A., Aguilo J.I., Gonzalo-Asensio J., Marinova D., Uranga S., Puentes E., Fernandez C., Parra A., Cardona P.J., Vilaplana C. Construction, characterization and preclinical evaluation of MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials. Vaccine. 2013;31:4867–4873. doi: 10.1016/j.vaccine.2013.07.051. [DOI] [PubMed] [Google Scholar]
- 25.Marinova D., Gonzalo-Asensio J., Aguilo N., Martin C. MTBVAC from discovery to clinical trials in tuberculosis-endemic countries. Expert Rev. Vaccines. 2017;16:565–576. doi: 10.1080/14760584.2017.1324303. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalo-Asensio J., Marinova D., Martin C., Aguilo N. MTBVAC: Attenuating the Human Pathogen of Tuberculosis (TB) Toward a Promising Vaccine against the TB Epidemic. Front. Immunol. 2017;8:1803. doi: 10.3389/fimmu.2017.01803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aguilo N., Gonzalo-Asensio J., Alvarez-Arguedas S., Marinova D., Gomez A.B., Uranga S., Spallek R., Singh M., Audran R., Spertini F., Martin C. Reactogenicity to major tuberculosis antigens absent in BCG is linked to improved protection against Mycobacterium tuberculosis. Nat. Commun. 2017;8:16085. doi: 10.1038/ncomms16085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White A., Sibley L., Sarfas C., Morrison A., Gullick J., Gleeson F., McIntyre A., Simon C., Lindestam C., Sette A. MTBVAC vaccination protects rhesus macaques against aerosol challenge with M. tuberculosis and induces immune signatures analogous to those observed in clinical studies. NPJ Vaccines. 2020 doi: 10.1038/s41541-020-00262-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tameris M., Mearns H., Penn-Nicholson A., Gregg Y., Bilek N., Mabwe S., Geldenhuys H., Shenje J., Luabeya A.K.K., Murillo I., MTBVAC Clinical Trial Team Live-attenuated Mycobacterium tuberculosis vaccine MTBVAC versus BCG in adults and neonates: a randomised controlled, double-blind dose-escalation trial. Lancet Respir. Med. 2019;7:757–770. doi: 10.1016/S2213-2600(19)30251-6. [DOI] [PubMed] [Google Scholar]
- 30.Tarancón R., Domínguez-Andrés J., Uranga S., Ferreira A.V., Groh L.A., Domenech M., González-Camacho F., Riksen N.P., Aguilo N., Yuste J. New live attenuated tuberculosis vaccine MTBVAC induces trained immunity and confers protection against experimental lethal pneumonia. PLoS Pathog. 2020;16:e1008404. doi: 10.1371/journal.ppat.1008404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joosten S.A., van Meijgaarden K.E., Arend S.M., Prins C., Oftung F., Korsvold G.E., Kik S.V., Arts R.J., van Crevel R., Netea M.G., Ottenhoff T.H. Mycobacterial growth inhibition is associated with trained innate immunity. J. Clin. Invest. 2018;128:1837–1851. doi: 10.1172/JCI97508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khader S.A., Divangahi M., Hanekom W., Hill P.C., Maeurer M., Makar K.W., Mayer-Barber K.D., Mhlanga M.M., Nemes E., Schlesinger L.S., Bill and Melinda Gates Foundation Collaboration for TB Vaccine Discovery Innate Immunity Working Group 18 Targeting innate immunity for tuberculosis vaccination. J. Clin. Invest. 2019;129:3482–3491. doi: 10.1172/JCI128877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nemes E., Geldenhuys H., Rozot V., Rutkowski K.T., Ratangee F., Bilek N., Mabwe S., Makhethe L., Erasmus M., Toefy A., C-040-404 Study Team Prevention of M. tuberculosis Infection with H4:IC31 Vaccine or BCG Revaccination. N. Engl. J. Med. 2018;379:138–149. doi: 10.1056/NEJMoa1714021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dijkman K., Aguilo N., Boot C., Hofman S.O., Sombroek C.C., Vervenne R.A.W., Kocken C.H.M., Marinova D., Thole J., Rodríguez E. Pulmonary MTBVAC vaccination induces immune signatures previously correlated with prevention of tuberculosis infection. Cell Rep. Med. 2021;2 doi: 10.1016/j.xcrm.2020.100187. Published online January 19, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cirovic B., de Bree L.C.J., Groh L., Blok B.A., Chan J., van der Velden W.J.F.M., Bremmers M.E.J., van Crevel R., Händler K., Picelli S. BCG Vaccination in Humans Elicits Trained Immunity via the Hematopoietic Progenitor Compartment. Cell Host Microbe. 2020;28:322–334.e5. doi: 10.1016/j.chom.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Branchett W.J., Lloyd C.M. Regulatory cytokine function in the respiratory tract. Mucosal Immunol. 2019;12:589–600. doi: 10.1038/s41385-019-0158-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lloyd C.M., Marsland B.J. Lung Homeostasis: Influence of Age, Microbes, and the Immune System. Immunity. 2017;46:549–561. doi: 10.1016/j.immuni.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 38.Yao Y., Jeyanathan M., Haddadi S., Barra N.G., Vaseghi-Shanjani M., Damjanovic D., Lai R., Afkhami S., Chen Y., Dvorkin-Gheva A. Induction of Autonomous Memory Alveolar Macrophages Requires T Cell Help and Is Critical to Trained Immunity. Cell. 2018;175:1634–1650.e17. doi: 10.1016/j.cell.2018.09.042. [DOI] [PubMed] [Google Scholar]
- 39.Koeken V.A.C.M., Verrall A.J., Netea M.G., Hill P.C., van Crevel R. Trained innate immunity and resistance to Mycobacterium tuberculosis infection. Clin. Microbiol. Infect. 2019;25:1468–1472. doi: 10.1016/j.cmi.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 40.Andersen P., Doherty T.M. The success and failure of BCG - implications for a novel tuberculosis vaccine. Nat. Rev. Microbiol. 2005;3:656–662. doi: 10.1038/nrmicro1211. [DOI] [PubMed] [Google Scholar]
- 41.Brandt L., Feino Cunha J., Weinreich Olsen A., Chilima B., Hirsch P., Appelberg R., Andersen P. Failure of the Mycobacterium bovis BCG vaccine: some species of environmental mycobacteria block multiplication of BCG and induction of protective immunity to tuberculosis. Infect. Immun. 2002;70:672–678. doi: 10.1128/iai.70.2.672-678.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fine P.E. Variation in protection by BCG: implications of and for heterologous immunity. Lancet. 1995;346:1339–1345. doi: 10.1016/s0140-6736(95)92348-9. [DOI] [PubMed] [Google Scholar]
- 43.Marinova D., Gonzalo-Asensio J., Aguilo N., Martin C. Recent developments in tuberculosis vaccines. Expert Rev. Vaccines. 2013;12:1431–1448. doi: 10.1586/14760584.2013.856765. [DOI] [PubMed] [Google Scholar]
- 44.Shah J.A., Lindestam Arlehamn C.S., Horne D.J., Sette A., Hawn T.R. Nontuberculous Mycobacteria and Heterologous Immunity to Tuberculosis. J. Infect. Dis. 2019;220:1091–1098. doi: 10.1093/infdis/jiz285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Agostino M.R., Lai R., Afkhami S., Khera A., Yao Y., Vaseghi-Shanjani M., Zganiacz A., Jeyanathan M., Xing Z. Airway Macrophages Mediate Mucosal Vaccine-Induced Trained Innate Immunity against Mycobacterium tuberculosis in Early Stages of Infection. J. Immunol. 2020;205:2750–2762. doi: 10.4049/jimmunol.2000532. [DOI] [PubMed] [Google Scholar]
- 46.Schmidl C., Rendeiro A.F., Sheffield N.C., Bock C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods. 2015;12:963–965. doi: 10.1038/nmeth.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Folegatti P.M., Ewer K.J., Aley P.K., Angus B., Becker S., Belij-Rammerstorfer S., Bellamy D., Bibi S., Bittaye M., Clutterbuck E.A., Oxford COVID Vaccine Trial Group Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet. 2020;396:467–478. doi: 10.1016/S0140-6736(20)31604-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Georgiou G., van Heeringen S.J. fluff: exploratory analysis and visualization of high-throughput sequencing data. PeerJ. 2016;4:e2209. doi: 10.7717/peerj.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data files of the ChIPseq analysis on BCG trained monocytes begore and after vaccination have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE159046.