

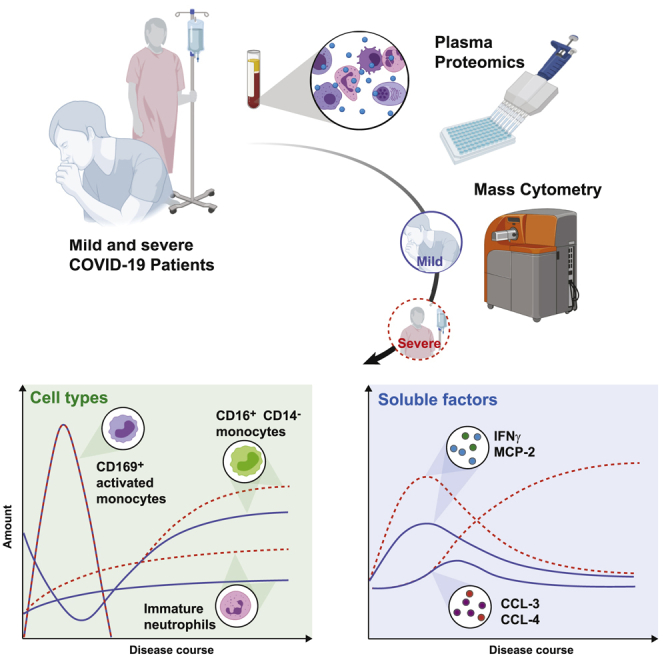
Summary
Coronavirus disease 2019 (COVID-19) manifests with a range of severities, but immune signatures of mild and severe disease are still not fully understood. Here, we use mass cytometry and targeted proteomics to profile the innate immune response of patients with mild or severe COVID-19 and of healthy individuals. Sampling at different stages allows us to reconstruct a pseudo-temporal trajectory of the innate response. A surge of CD169+ monocytes associated with an IFN-γ+MCP-2+ signature rapidly follows symptom onset. At later stages, we observe a persistent inflammatory phenotype in patients with severe disease, dominated by high CCL3 and CCL4 abundance correlating with the re-appearance of CD16+ monocytes, whereas the response of mild COVID-19 patients normalizes. Our data provide insights into the dynamic nature of inflammatory responses in COVID-19 patients and identify sustained innate immune responses as a likely mechanism in severe patients, thus supporting the investigation of targeted interventions in severe COVID-19.
Keywords: SARS-CoV-2, COVID-19, innate immune response, mass cytometry, proteomics, monocytes, inflammation, emergency granulopoiesis, sialoadhesin, CD169, CCL3, MIP1α
Graphical Abstract
Highlights
Systems analysis of the innate immune responses in COVID-19 up to 47 days after onset
Surge of CD169+ and depletion of CD16+CD14− monocytes early after symptom onset
High levels of inflammatory cytokines and immature neutrophils in severe COVID-19
Persistent inflammation in late severe cases, while normalization in mild cases
Chevrier et al. characterize the innate immune response in mild and severe COVID-19 patients and healthy individuals by mass cytometry and serum proteomics. They demonstrate profound phenotypic changes after SARS-CoV-2 infection and ongoing inflammation, with an innate signature switch in severe COVID-19 patients late in the disease course.
Introduction
Coronavirus disease 2019 (COVID-19) was first identified in December 2019 in Wuhan, China.1 The disease developed into a global pandemic, with 60 million confirmed cases and close to 1.5 million confirmed deaths as of the end of November 2020.2 The clinical presentation of COVID-19 can vary from asymptomatic cases to an acute respiratory distress syndrome (ARDS), requiring mechanical ventilation.3 Approximately 5% of those clinically diagnosed with COVID-19 develop ARDS and generally experience a sudden deterioration after ∼1week of symptom onset.4
Severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2), a positive-sense, single-stranded RNA virus, has been identified as the causative pathogen of COVID-19. This virus shows a tropism for cells expressing the angiotensin-converting enzyme 2, which serves as an entry receptor for SARS-CoV-2 into cells of the respiratory tract, kidneys, liver, heart, brain, and blood vessels.5 Upon infection of epithelial cells, pattern recognition receptors that sense viral RNA initiate interferon (IFN) production and innate immune cell recruitment, triggering an inflammatory response that has also been linked to inflammasome activation in COVID-19.6,7
Early data indicated that patients with severe disease mount a strong inflammatory response, as shown by increased levels of proinflammatory cytokines, such as tumor necrosis factor (TNF), monocyte chemoattractant protein 1 (MCP-1/CCL2), and macrophage inflammatory protein 1α (MIP-1α/CCL3).8 Other studies further revealed a distinct cytokine response with activated interleukin-1 (IL-1) and IL-6 pathways as well as chemokine-enriched signatures.9,10 Type I IFN response during SARS and SARS-CoV-2 infection has gained particular attention since a deficient or delayed type I IFN response may be associated with a severe disease course as observed in patients with inborn errors in type I IFN response or with neutralizing autoantibodies against type I IFN.11, 12, 13, 14, 15, 16
Myeloid cells have been implicated in the pathophysiology of COVID-19 by contributing to local tissue damage and as potential producers of cytokines that lead to the hyperinflammatory state seen in severe COVID-19.17, 18, 19 Studies applying high-dimensional single-cell analysis have shown distinct phenotypic changes within the monocytic compartment in patients with COVID-19.20,21 A recent single-cell RNA-sequencing study of bronchoalveolar lavage fluid showed changes in the local myeloid environment toward a proinflammatory, peripheral monocyte-derived phenotype and a depletion of alveolar macrophages in severe COVID-19 patients.22
These data suggest a distinct role of the myeloid compartment in the pathogenesis of COVID-19; however, these studies have focused mainly on the early disease course of the infection, and data on the cellular innate immune response combined with the underlying cytokine and chemokine network, at high phenotypic and temporal resolution, are still sparse. Here, we describe an in-depth, cross-sectional characterization of the myeloid compartment in 66 patients with mild to severe COVID-19 and 22 healthy controls by using 40-parameter mass cytometry and targeted proteomics up to 47 days after symptom onset. Using this systems approach, we could reconstruct phenotypic changes arising throughout the course of the disease both in mild and severe patients.
Results
Clinicopathological assessment of patients with mild versus severe COVID-19
To better understand the role of the myeloid compartment in the pathophysiology of COVID-19, we established a multicenter cohort, comprising 66 COVID-19 patients. At blood sampling, 28 patients were classified as mild COVID-19 with either mild flu-like illness or mild pneumonia, whereas 38 patients were classified as severe COVID-19 experiencing severe pneumonia or ARDS (Figure 1A; Table 1).23 The patient outcomes were prospectively recorded, and one individual died of COVID-19. Samples of 22 healthy donors were included in the study as controls. Patients suffering from severe disease were on average older than those with mild disease (Figure 1B; Table 1), which is consistent with previously published results.3 Furthermore, hypertension and heart disease were significantly associated with a severe disease course (Table 1).
Figure 1.

Experimental approach, clinical characteristics, and identification of the main immune cell types in COVID-19 patients based on mass cytometry
(A) Schematic of the study design of the cohort.
(B) Boxplots showing the age distribution, selected clinical parameters at admission, and the NK cell counts in the patient cohort split by disease severity (n = 22 healthy controls, 28 mild COVID-19 patients, and 38 severe COVID-19 patients).
(C) Correlation map of the indicated parameters and clinical features grouped by a hierarchical clustering on the COVID-19 patients. The circle color reflects the magnitude of the Pearson’s correlation coefficient (red indicates positive correlation, blue indicates negative correlation). Asterisks represent the statistically significant correlations (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(D) t-SNE plot of a random subset of 1,000 immune cells of the mass cytometry analysis from each sample (n = 78 individuals) colored by main cell types as identified based on a random forest cell classification.
(E) Heatmap of the normalized marker expression in the main cell types. Relative abundances of each cell type are plotted to the right of the heatmap.
(F) Boxplots comparing the frequencies of the indicated cell types in healthy controls and patients with mild and severe disease.
Statistical analyses were performed with a Mann-Whitney-Wilcoxon test corrected for multiple testing using the Holm method, and p values are shown if the results were significant (p < 0.05).
Table 1.
Clinical and laboratory characteristics of the healthy controls and the COVID-19 patients
| Disease severitya |
Healthy controls (n = 22) |
Mild cases (n = 28) |
Severe cases (n = 38) |
||||
|---|---|---|---|---|---|---|---|
| Disease gradeb | Mild illness | Mild pneumonia | Severe pneumonia | Mild ARDS | Moderate ARDS | Severe ARDS | |
| Grade at sampling, no. | – | 18 | 10 | 20 | 7 | 7 | 4 |
| Maximal grade, no. | – | 16 | 8 | 19 | 7 | 8 | 8 |
| Characteristics | |||||||
| Age, y, median (IQR) | 32.50 (29.25–48.0) | 50.5 (34.50–60.25)d | 67.5 (59.0–79.0)cd | ||||
| Gender, M/F | 11/11 | 12/16 | 24/14 | ||||
| Time after symptom onset, days | – | 12.86 ± 10.71 | 20.21 ± 11.96c | ||||
| Level of care at blood sampling | |||||||
| Outpatient, no. (%) | – | 14 (50) | – | ||||
| Inpatient, no. (%) | – | 14 (50) | 38 (100)c | ||||
| Ward, no. (%) | – | 14 (50) | 28 (73.7) | ||||
| Respiratory supporte required, no. (%) | – | – | 36 (94.7) | ||||
| ICU, no. (%) | – | – | 10 (26.3) | ||||
| Comorbidities | |||||||
| Hypertension, no. (%) | – | 7 (25) | 22 (57.9)c | ||||
| Diabetes, no. (%) | 1 (4.5) | 4 (14.3) | 12 (31.6) | ||||
| Heart disease, no. (%) | – | 3 (10.7) | 17 (44.7)c | ||||
| Cerebrovascular disease, no. (%) | – | 1 (3.6) | 4 (10.5) | ||||
| Lung disease, no. (%) | – | 3 (10.7) | 6 (15.8) | ||||
| Kidney disease, no. (%) | – | 7 (25) | 10 (26.3) | ||||
| Malignancy, no. (%) | – | – | 4 (10.5) | ||||
| Systemic immunosuppression, no. (%) | – | 3 (10.7) | 4 (10.5) | ||||
| Outcome | |||||||
| Released/recovered, no. (%) | – | 28 (100) | 37 (97.4) | ||||
| Death, no. (%) | – | – | 1 (2.6) | ||||
IQR, interquartile range.
COVID-19 disease severity at the time of blood sample collection. Mild illness and mild pneumonia are considered mild COVID-19 disease, and severe pneumonia and any grade of ARDS are considered severe COVID-19 disease.
COVID-19 grade according to World Health Organization guidelines, recorded at sampling and prospectively followed until recovery.23
Indicates significance (p value threshold < 0.05) compared to “mild.”
Indicates significance in comparison to “healthy”. Mann-Whitney-Wilcoxon test was used to test for differences between continuous variables adjusted for multiple testing using the Holm method. Categorical variables were compared between “mild” and “severe” using Fisher’s exact test.
Supplemental oxygen, non-invasive ventilation, or mechanical ventilation are considered respiratory support.
The laboratory findings at admission revealed a prominent inflammatory state for patients with both mild and severe disease, as evidenced by higher levels of C-reactive protein (CRP) and pathological values of lactate dehydrogenase (LDH) in mild and severe COVID-19 patients (Table S1). Complete differential blood counts showed normal white blood cell counts (WBCs), despite significant changes in the granulocyte subsets, with an increase in neutrophils in severe COVID-19 patients and a decrease in basophils and eosinophils in both mild and severe COVID-19 patients compared to healthy controls (Figure 1B; Table S1). Natural killer (NK) cell cytopenia in the CD3−CD56brightCD16dim population was also associated with a severe disease course (Figure 1B; Table S1), confirming previous publications.24, 25, 26 A correlation map that included additional clinical metadata of COVID-19 patients revealed that high CRP and neutrophil counts positively correlate with measures of disease severity, whereas lymphocytes and NK cells show an inverse relationship to these parameters (Figure 1C). Thus, we could recapitulate the strong inflammatory state, the changes in the complete blood count, and the prolonged clinical course before deterioration, which make COVID-19 a distinct disease.27
Systems-wide profiling of innate compartment in COVID-19 patients
To comprehensively characterize the innate immune response against SARS-CoV-2, we took a systems-level approach based on mass cytometry and targeted proteomics (Figure 1A). We used a 40-plex mass cytometry panel designed to identify main immune cell types and further characterize myeloid cell subsets in depth (Key Resources Table; Figures S1A–S1D). The main cell types were identified using a random forest classifier trained on manually gated cells from a representative subset of data (Figure S1E). The cell annotation was consistent with the t-distributed stochastic neighbor embedding (t-SNE) map visualization (Figures 1D and S1F) and the expression of canonical markers (Figure 1E). In line with previous reports,26 we observed a decrease in T cell frequencies in severe COVID-19 cases compared to mild cases and healthy controls, which was accompanied by an increased monocyte frequency (Figure 1F). Since peripheral blood mononuclear cells (PBMCs) were isolated following a density gradient separation, only low-density neutrophils were included in the analysis. Consistent with a previous report,28 this subset was present at very low frequency in healthy controls but increased in patients infected with SARS-CoV-2, accounting for >50% of the PBMCs in some patients (Figure 1F).
Different myeloid landscape in patients with mild and severe COVID-19
Neutrophils have been reported to play a key role in the development of severe COVID-19. In particular, a high neutrophil:lymphocyte ratio has been associated with poor clinical outcomes, and a CD16intCD44lowCD11bint low-density neutrophil population, associated with high IL-6 and TNF concentrations, was increased in severe COVID-19 patients compared to healthy controls.28 To assess the low-density neutrophil subsets in our cohort, we used t-SNE to visualize the expression of relevant markers on this cell type (Figure 2A). Although all cells were positive for the canonical neutrophil markers CD15 and CD66ace, differences in the abundance of CD11b, CD11c, and CD16 were observed. Visualizing the disease status on the t-SNE map revealed an enrichment of CD16low neutrophils in patients with severe disease (Figure 2B). To confirm this observation, we classified neutrophils into CD16hi, CD16int, and CD16low populations based on manual annotation of PhenoGraph clusters (Figures S2A and S2B). The proportion of CD16low neutrophils was significantly higher in patients with severe disease than in the other 2 groups (Figure 2C). The CD16low subset was associated with an increased proliferation rate, based on Ki-67 expression (Figures 2A and S2C). Since mature neutrophils do not proliferate in the periphery, this observation indicates the release of immature or alternatively activated neutrophils into the circulation, consistent with their CD11blowCD16low phenotype.29, 30, 31
Figure 2.

In-depth characterization of the myeloid cells in the peripheral blood of COVID-19 patients
(A) t-SNE plots of normalized expression of the indicated markers across a maximum of 1,000 neutrophils per patient (n = 22 healthy controls, 27 mild COVID-19 patients, and 29 severe COVID-19 patients).
(B) t-SNE plot generated as indicated in (A) and colored by disease severity.
(C) Left: t-SNE plot generated as indicated in (A) and colored by CD16 expression level based on manual assignment of PhenoGraph clusters. Right: boxplot showing the frequency of CD16low neutrophils in COVID-19 patients and healthy controls. Statistical analyses were performed with a Mann-Whitney-Wilcoxon test corrected for multiple testing using the Holm method, and p values are shown if the results were significant (p < 0.05).
(D) t-SNE plots of normalized expression of the indicated markers across a maximum of 1,000 monocytes per patient.
(E) t-SNE plots generated as indicated in (D) colored by disease severity (top) and by clusters identified with the PhenoGraph algorithm (bottom).
(F) Contour plots showing the distribution of a subset of 10,000 cells from the indicated clusters for a selected set of markers. For this analysis, classical monocytes M1–M7 were merged in a single cluster.
(G) Heatmap of the normalized marker expression in PhenoGraph monocyte clusters. The frequency of each cluster in patients with mild and severe disease and in healthy controls is indicated as a stacked histogram to the right of the heatmap. Cell numbers for each cluster are plotted to the right of the stacked histogram.
(H) Overlaid histograms showing arcsinh transformed counts on a linear axis for selected markers for the CD169+-activated monocyte clusters.
(I) Overlaid histograms showing arcsinh transformed counts on a linear axis for the indicated markers for the classical monocyte clusters. Values in the plot indicate median untransformed count intensities.
In (H) and (I), values on the plot indicate the median raw ion count intensity for each cluster and marker indicated.
To characterize the phenotypic diversity in the monocytic compartment across the cohort, we visualized the myeloid-related markers on the t-SNE map of the monocyte population (Figure 2D). To identify monocyte subsets in an unsupervised way, we performed automated clustering using the PhenoGraph algorithm, which led to the identification of 13 distinct cell subsets (Figure 2E). Clusters M1–M7 were characterized by a high abundance of CD14, CD36, and CCR2 and the absence of CD16, corresponding to classical monocytes. Based on CD16 expression and reduced levels of CD14, CCR2, and CD36, M8 and M9 were identified as non-classical (CD16+CD14dim) and intermediate (CD16+CD14+) monocytes, respectively.32 M10–M13 showed an activated CD169+ phenotype. While clusters M11–M13 shared similarities with classical monocytes, M10 was closely related to intermediate monocytes (Figure 2F). Strikingly, CD169+ activated monocytes (M10–M13) were found exclusively in SARS-CoV-2-infected patients (Figures 2E and 2G). The fact that CD169+ monocytes could derive both from classical and intermediate monocytes was supported by a diffusion map analysis (Figures S2D and S2E), which aligns cells along putative developmental trajectories.33 CD169 upregulation was accompanied by the induction of other activation markers, including CD64, CD68, and CD38, compared to the classical monocyte clusters M1–M3 (Figure 2G). CD141, which is associated with anticoagulant properties,34 was particularly induced on M11 (Figure 2H). Markers involved in antigen presentation and co-stimulation, including HLA-DR, CD86, TIM-3 and VISTA, progressively decreased in clusters M10–M13, suggesting these clusters were part of a phenotypic continuum (Figures 2G and 2H). Proliferation, based on Ki-67 expression, was found to be induced in these clusters, especially in M12, compared to non-classical or CD169− intermediate and classical monocytes (Figures 2G and 2H).
Among CD169− classical monocytes, clusters M1–M3 were found predominantly in healthy controls, whereas clusters M4–M7 were mostly found in SARS-CoV-2-infected patients (Figures 2E and 2G). Overall, CD169− classical monocytes found in COVID-19 patients tended to express markers associated with activation, including CD38 and CD86, compared to the classical monocytes found in healthy donors (Figure 2I). Cluster M7, which expressed intermediate levels of CD169, could constitute a stage between CD169− classical monocytes and CD169+ classical monocytes. Similar to the M11 CD169+ cluster, the M6 cluster displayed an increased expression of thrombomodulin (CD141) and CD163. M4 showed a distinct phenotype with decreased expression of CCR2, HLA-DR, CD38, CD86, and the co-inhibitory receptors TIM-3 and VISTA, consistent with chemotactic paralysis in a murine model.35 In summary, high-dimensional single-cell mass cytometry analysis allowed us to characterize the monocyte compartments of COVID-19 patients and healthy controls with unprecedented depth and to uncover profound changes upon SARS-CoV-2 infection.
Stratification of COVID-19 patients based on monocyte composition
We next assessed the distribution of the 13 identified monocyte clusters across patients. This analysis revealed 3 main groups, which were enriched for either mild COVID-19 patients, healthy controls, or severe cases of COVID-19 (Figure 3A). In the group that included more mild cases, the monocyte compartment consisted almost exclusively of CD169+ activated clusters (M10–M13) in different ratios, with only a minor fraction of cells from clusters M4–M7. The healthy controls were relatively homogeneous: ∼80% of cells consisted of classical monocytes (M1–M3) and ∼15%–20% of cells were intermediate and non-classical monocytes (M9 and M8, respectively), consistent with the literature.32 The group dominated by patients with severe COVID-19 was characterized by a high frequency of distinct classical monocyte subsets (M4–M7), with frequencies of intermediate and non-classical monocytes slightly higher than in the group dominated by healthy controls.
Figure 3.

Patient stratification based on myeloid signature
(A) Stacked histogram of the PhenoGraph monocyte clusters per patient (n = 75 individuals), ordered by cluster composition similarities. Disease severity and grade for each patient are shown on top of the stacked histogram.
(B) Boxplots of frequencies of the indicated monocyte clusters in the different disease severities. Statistical analyses were performed with a Mann-Whitney-Wilcoxon test corrected for multiple testing based on the Holm method, and p values are shown if the results were significant (p < 0.05).
(C) Top: principal-component analysis (PCA) of monocyte and neutrophil cluster frequencies and myeloid immune cell subset frequencies across the cohort. The PCA plot (top) shows the first 2 principal components separating the samples. The percentage of explained variance for each component is shown in brackets. Each dot represents a patient, colored by disease status. A concentration ellipse and the mean point are shown for each group. Bottom: biplot displaying simultaneously the observations (patients) as gray dots and the variables (cell subsets) as vectors.36 Vectors indicate the direction and strength of each cell component to the overall distribution. Variables grouping together are positively correlated.
We observed similar patterns when directly comparing differences of the monocyte cluster frequencies in healthy subjects and patients with mild and severe disease (Figure 3B). Most strikingly, the CD169+ clusters M10–M13 were completely absent in healthy controls. Conversely, the classical monocyte clusters M1–M3 were present at high abundance in healthy donors, at lower abundance in patients with mild disease, and at intermediate abundance in patients with severe disease. The HLA-DRlow CCR2low and the CD141+ monocytes (M4 and M6, respectively) were virtually absent in healthy controls and were present at higher abundance in patients with severe than in mild disease, while the phenotypically similar M5 monocytes originated mainly from a single patient with severe illness. The non-classical (M8) and intermediate monocytes (M9) were significantly reduced in patients with mild disease compared to healthy controls and were present at higher abundance in a subset of patients with severe disease than in healthy controls. To exclude a confounding effect due to age or gender differences between the severity groups, we compared the cluster frequencies in the different age and gender groups. While non-classical (M8) and intermediate (M9) monocytes increased slightly with age in both COVID-19 patients and healthy controls, which is consistent with the literature,37 we could not observe any evident age or gender association for the other clusters (Figures S3A and S3B).
To gain further insight into the relationship between the innate immune signature and disease status, we performed a principal-component analysis (PCA) of monocyte and neutrophil cluster frequencies across the cohort and found that the first two principal components enabled the stratification of subjects based on disease status (Figure 3C, top panel). A biplot graph displaying simultaneously the subjects as dots and the contribution of the different cell types on the principal components as arrows revealed a strong association between clusters M1–M3 and healthy controls (Figure 3C, bottom panel). A group dominated by mild patients was characterized by a high abundance of clusters M10–M13. A more heterogeneous set of predominantly severe COVID-19 cases was defined by a higher abundance of the clusters M4–M6, M8, and M9, as well as by CD16low low-density neutrophils. A correlative analysis performed across innate cell subsets and patients confirmed the pattern observed based on the PCA analysis (Figure S3C). Thus, despite the expected diversity across individuals, these multiparametric analyses identified innate immune signatures that stratified different groups of patients. However, this stratification could be only partly explained by disease severity, suggesting that other factors are also at play.
Changes in innate cell frequencies over the course of SARS-CoV-2 infection
Since the samples for different patients were collected at different points in their disease course, the time component is likely to have an influence on the cell cluster composition. To investigate this possibility, we plotted the different cell type frequencies as a function of time after symptom onset. The total monocyte compartment remained relatively constant over the disease course, while the low-density neutrophils tended to increase in COVID-19 patients, compared to healthy controls early after symptom onset, before their counts went back to baseline values at later stages of the disease (Figure 4A). These changes were accompanied by a decrease in CD16hi neutrophils over time in patients with severe disease, whereas CD16low neutrophils remained consistently high (Figure 4B).
Figure 4.

Myeloid cell frequencies over the course of the SARS-CoV-2 infection
(A–C) Scatterplot of indicated subset frequencies of (A) total PBMCs, (B) total neutrophils, and (C) total monocytes, relative to time after symptom onset. The dots are colored by disease grade at sampling time (n = 75 individuals). The frequencies in healthy controls are shown as a reference on the left, with a horizontal line indicating the median. The pseudo-time course was modeled using a generalized additive model for the disease severities separately (mild, blue lines; severe, red lines). p values of generalized additive models using time as a covariate were calculated for both the mild (blue) and severe (red) patient group and are shown when significant (p < 0.05).
(D) Contour plots showing expression of CD14 and CD16 on monocytes from a representative sample of a healthy donor as well as an early- and late-stage patient (left column). Gates and percentage of cells in each gate for classical, intermediate, and non-classical monocytes are indicated for each sample. Expression of CD169 in each gate for each sample is displayed as overlaid histograms on the left.
(E) Scatterplots of indicated marker median ion count (MIC) in the classical monocyte gate relative to the time after symptom onset. Dots are colored by disease grade at sampling time and healthy controls are displayed in green on the left as reference.
The monocyte clusters M1–M13 also showed distinct changes over the disease course. Classical monocytes typically found in healthy controls (M1–M3) were observed at very low frequencies in mild and severe cases sampled early during the disease course but gradually increased in patients sampled later (Figure 4C, purple frame). The frequencies of these clusters tended to remain at lower levels than in the healthy controls even as late as 47 days after symptom onset. In contrast, frequencies of disease-specific CD169− classical monocytes (M4 and M6) increased during the disease course of severe COVID-19 cases, peaking at ∼3 weeks after symptom onset (Figure 4C, purple frame). Intriguingly, mild COVID-19 cases with increased frequencies of cluster M4 corresponded to patients who required hospitalization or showed prolonged symptoms (Figure S4A).
The activated CD169+ monocyte clusters M10–M13 were present at high frequencies early during the disease course (Figure 4C, orange frame). Within the first 20 days after symptom onset, the monocyte compartment was strongly dominated by this phenotype, which decreased thereafter and was undetectable in most patients sampled >20 days after symptom onset. This pattern was similar in mild and severe COVID-19, suggesting that the time after symptom onset had a stronger influence on CD169+-activated cluster frequency than disease severity.
The non-classical (M8) and intermediate (M9) monocyte clusters were virtually absent early upon viral infection (Figure 4C, green frame). The initial reduction was followed by their recovery at later stages, showing a trend toward higher frequencies in patients with severe compared to mild disease for both clusters. This finding could be explained in part by the fact that severe COVID-19 patients tend to be older (Figure S3A).
The disappearance of non-classical monocytes and the increase of CD169+ cells among classical, intermediate, and non-classical subset at early time points was confirmed based on a manual gating approach (Figure 4D, S4B, and S4C). Using this strategy, we further investigated the evolution of marker expression in classical monocytes throughout the disease course and found that the quick induction of CD169 was accompanied by the upregulation of CD141, CD38, indoleamine 2,3-dioxygenase (IDO), and Ki-67, which is consistent with the fact that these markers are predominantly found in CD169+-activated clusters (Figure 4E). In summary, the analysis of cluster frequencies in relation to the time after symptom onset revealed distinct dynamic patterns of monocyte frequencies in patients with mild and severe disease, which were most obvious at later stages.
Temporal changes in cytokine signatures in COVID-19 patients
Monocyte development, homeostasis, and fate are strongly interlinked with the cytokine and chemokine environment. To probe potential correlates to the changes seen in the myeloid compartment during COVID-19 progression, we used the targeted proteomics Olink assay to measure 92 inflammation-associated serum proteins. The comparison of data from healthy controls and patients with severe COVID-19 showed a strong upregulation of proinflammatory cytokines and chemokines (Figure 5A). IL-6, TNF, IFN-γ, and IL-18 were significantly upregulated (false discovery rate [FDR] 1%) in patients with severe disease (Figure 5A) consistent with previous reports.9,10 The elevated concentrations of IL-18 and LDH may indicate a contribution of inflammasome activation,7,13,18 although we did not detect an increase in IL-1β (Figure S5A).
Figure 5.

Cytokine signature shift between early and late stages of disease
(A) Volcano plot of the Olink proteomics data comparing the healthy control data (n = 17) to that from patients with severe COVID-19 (n = 35). An FDR of 1% was taken as significance cutoff. The identity is given for factors reaching significance, with the color code indicated on the right.
(B) Scatterplot of serum protein expression levels relative to the time after symptom onset. Plotted is normalized protein expression (NPX) on a log2 scale. The dots are colored by disease grade at sampling time (n = 77 individuals). The expression levels of the healthy controls are shown as a reference on the left, with a horizontal line indicating the median. The pseudo-time course was modeled as described in Figure 4A, and p values are shown when significant (p < 0.05).
Several chemokines important for myeloid cell trafficking were significantly upregulated in patients with severe COVID-19. These chemokines included MCP-1, an IFN-stimulated gene (ISG), MCP-2, MCP-3, CX3CL1 (chemotactic for non-classical monocytes), CXCL1 (a potent neutrophil-recruiting chemokine), the more promiscuous CCL3 and CCL4, and macrophage-colony-stimulating factor (M-CSF), which is crucial for myeloid precursor survival and lineage commitment (Figure 5A).38,39
Distinct expression patterns emerged by plotting cytokine expression versus time after symptom onset. In comparison to healthy controls, IFN-γ and MCP-2 were present at significantly higher concentrations in patients with severe disease sampled early after symptom onset but returned to near normal values later in the disease course. Interestingly, in the mild group, concentrations of proinflammatory cytokines remained lower in patients with mild illness compared to individuals with pneumonia (Figure 5B). Consistent with ELISA measurements, M-CSF, IL-6, and TNF were present at higher concentrations in patients with severe disease compared to mild disease; these cytokines tended to be present at high concentrations throughout the disease course in patients with severe disease, whereas a decrease was observed in patients with mild symptoms as the disease progressed (Figures 5B and S5A). The chemokines CXCL1, CX3CL1, MCP-1, and MCP-3 were significantly increased compared to healthy controls in patients with severe COVID-19 independent of time after symptom onset (Figure S5B). In striking contrast, CCL3 and CCL4 were not elevated in the first days of the disease but were significantly increased at later stages in patients with severe disease (Figure 5B). These time-dependent changes were confirmed with a differential expression analysis and by binning the time after symptom onset (Figures S5C and S5D).40 In summary, these results are indicative of a more exacerbated inflammatory phenotype late during the disease course in patients with severe disease than in those with mild symptoms.
Temporal correlation of cytokine signatures and innate cell subsets
To better understand the interplay between these serum proteins and innate immune cell subsets, we performed a hierarchical clustering on a correlation map of all significantly changed (FDR 1%) serum proteins, immune cell subsets, and myeloid clusters (Figure S6A). This approach demonstrated a strong association of the activated CD169+ monocyte clusters M10–M13 with proinflammatory cytokines, including IFN-γ, MCP-2, IL-18, and IL-6, as well as an association of the disease-related CD169− classical monocyte clusters M4 and M6 with transforming growth factor-α (TGF-α), CCL3, and CCL4. To further understand these relationships in the context of the patients in our cohort, we performed PCA. A biplot graph revealed that certain combinations of cell types and soluble factors stratified patients and healthy controls (Figures 6A and 6B). One group, dominated by patients with mild symptoms and patients sampled early in the disease course, was defined by activated CD169+ monocytes (M10–M13) in combination with MCP-2 and IFN-γ. A second group of patients, enriched for more severe, late-stage cases, was defined primarily by disease-related CD169− classical monocytes (M4–M6) in combination with CCL3, CCL4, and CCL23 (Figures 6A and 6B). The healthy donors were defined by classical monocytes (clusters M1–M3) and were located close to the majority of late-stage mild patients. Intermediate (M9) and non-classical (M8) monocytes contributed to both healthy and severe groups. These data strongly suggest an innate signature shift between the early and late stages of the disease, leading to a divergence of patients with mild and severe COVID-19 over the disease course. While the former become similar to healthy controls, the latter exhibit signs of hyperinflammation.
Figure 6.

Temporal correlation of cytokine signature and innate cell subsets
(A) Biplot of the first two principal components of a PCA based on monocyte and neutrophil cluster frequencies, myeloid immune cell subset frequencies, and expression values of selected serum proteins. Dots represent the COVID-19 patients and healthy controls (n = 70 individuals), and the arrows indicate the direction and strength of each cell and soluble components to the overall distribution. Variables that group together are positively correlated. The percentage of explained variance for each component is shown in brackets.
(B) Scatterplot of the first two principal components of a PCA generated as indicated in (A), colored by the time since symptom onset. The shape of each dot corresponds to the patient groups.
(C) Scatterplots of frequencies of the indicated clusters versus NPX of selected serum proteins in individual patients. The dots indicate data for individual patients colored by disease grade. Relationship between the 2 variables is visualized with a linear regression line and quantified using a Spearman’s correlation coefficient (Rho) with the corresponding p value. Fq, frequency.
(D and E) Scatterplots of frequencies of the indicated monocyte clusters (D) or the NPX of the indicated soluble factors (E) as a function of time after symptom onset in individual patients who were sampled twice during the course of the study. Dots are colored by patient number, and lines connect paired samples. Sample indicated with an asterisk was excluded from the cohort analysis due to low cell number.
To further characterize the relationships between monocyte subsets and soluble factors identified in the PCA, we performed a direct correlation between the cluster frequencies and cytokine levels. There was a strong correlation between the activated Ki-67+ monocyte cluster M12 and M-CSF, IFN-γ, IL-6, and TNF, which was most evident in early-stage patients (Figures 6C and S6B). We also found that the non-classical monocyte M8 and the intermediate monocyte cluster M9 were strongly correlated with CCL3 and CCL4, but in this case the correlation was most prominent in late-stage patients (Figures 6C, right column, and S6C). These results suggest that the inflammatory environment found in late stages of COVID-19 is predominantly associated with the presence of the intermediate monocytes.
To determine whether the switch in signatures observed in our pseudo-temporal analysis could be observed in individual patients over time, we compared the cluster frequencies and cytokine levels in 4 patients who were sampled twice. We confirmed that the frequencies of the activated CD169+ clusters M10–M13 were reduced, and the frequencies of the non-classical (M8) and intermediate (M9) monocytes were increased over time (Figure 6D). Confirming our pseudo-longitudinal analysis, we observed decreases in IFN-γ and MCP-2 over time in these re-sampled patients, accompanied by increases in CCL3 and CCL4 (Figure 6E). These observations were also confirmed in a separate, publicly available, longitudinal dataset of 37 hospitalized COVID-19 patients (Figure S6D).24 Overall, these data suggest there is a coordinated change from a signature characterized by a high abundance of activated CD169+ monocytes, IFN-γ, and MCP-2 to a signature with increased intermediate and non-classical monocytes, CCL3 and CCL4, in the first 2 weeks after SARS-CoV-2 infection. Severe COVID-19 patients showed long-lasting high concentrations of CCL3 and CCL4, demonstrating a marked phenotypic switch.
Discussion
Early in the COVID-19 pandemic, data began to suggest that patients with severe disease show hyperinflammatory immune responses with changes in the myeloid compartment toward a proinflammatory phenotype.8,22,41 Using a systems-wide characterization of immune parameters in a multicenter cohort of COVID-19 patients and healthy controls, we identified marked changes in the innate immune signature in SARS-CoV-2-infected individuals.
Among the phenotype changes occurring early upon SARS-CoV-2 infection, we identified a decrease in non-activated classical monocytes, a depletion of non-activated intermediate and non-classical monocytes, and a surge of CD169+ activated monocytes as well as low-density neutrophils.42
CD169 is quickly induced in a type I IFN-dependent manner on the surface of monocytes upon Epstein-Barr virus (EBV) or human immunodeficiency virus (HIV) infection.43,44 CD169 has been found on circulating monocytes in mild COVID-19 patients.20,42,45 Furthermore, CD169+ monocytes have been shown to have a strong ISG signature in single-cell RNA sequencing (scRNA-seq) data, which is consistent with the induction of ISGs observed in our study, including CD64, CD38, and IDO. We find that this phenotype can also be present in severe COVID-19. Although the fate of these CD169+ CCR2+ monocytes remains elusive, the recruitment to the lung through the CCR2-CCL2 axis is a possible option, as MCP-1 (CCL2) is significantly upregulated in COVID-19 patients. SARS-CoV mouse models have shown that the recruitment of proinflammatory monocytes to the lungs, promoted by delayed type I IFN signaling, drives immunopathology, and interestingly, a scRNA-seq study in bronchoalveolar lavage fluid of SARS-CoV-2 infected individuals found an accumulation of monocyte-derived macrophages and high levels of inflammatory cytokines in severe patients.16,22 Several factors, such as a maladapted IFN response or an inadequate T cell response in severe COVID-19,46 may contribute to the dysregulated monocyte response in severely ill patients. The latter could be due to suboptimal viral control, although T cells have also been shown to control an overactive innate response in a murine coronavirus infection model.47,48
The reduction of non-classical CD16+ monocytes early in the disease course is striking and has been evaluated as a potential marker of severe COVID-19.20 Several mechanisms such as differentiation or migration, potentially dependent on CX3CL1,49 could be involved. A recent study in bronchoscopy samples from intensive care unit (ICU)-admitted COVID-19 patients found an enrichment of CD16+ monocytes in the lung.50 Based on our diffusion map analysis, CD169+ monocytes could also arise from CD16+ monocytes, similar to what was reported upon simian immunodeficiency virus (SIV) or HIV infection.51,52 Intriguingly, we found a tendency toward elevated levels of activated CD169+ HLA-DRhi intermediate monocytes in individuals with mild COVID-19, which is of interest as CD16+ monocytes are critical for viral sensing. Therefore, a strong response of this subset may be important for an effective adaptive response to be mounted.53, 54, 55
Analysis of the inflammatory cytokine and chemokine profile to SARS-CoV-2 infection identified a response dominated by TNF and IL-6, in agreement with a previous report.9 The initial inflammatory response was dominated by IFN-γ, MCP-2, M-CSF, and IL-6, which were co-regulated with the activated CD169+ monocyte subsets, most prominently with the cluster showing increased proliferation, and the low-density granulocytes. IL-6 and M-CSF contribute to increased hematopoiesis under inflammatory conditions, a process called emergency granulopoiesis,38 potentially explaining the initial increase in activated monocytes and granulocytes after SARS-CoV-2 infection. Alternatively, a small subset of monocytes has been shown to retain proliferative capacity, especially under inflammatory conditions, which may also contribute to the initial surge, as we detected increased Ki-67 positivity in these cells. However, Ki-67 positivity may also be explained by a more immature cell phenotype.56, 57, 58 Our proteomics panel did not include granulocyte-CSF (G-CSF) and GM-CSF, 2 other cytokines crucially involved in myeloid homeostasis.59 Immature granulocytes were increased in severely ill patients, and such dysregulated, emergency granulocytes may contribute to severe COVID-19 pathogenesis, as previously suggested.20,21,28
Late in the disease course, COVID-19 patients with severe disease continued to show signs of ongoing inflammation, including abnormally high levels of TNF. The cellular signature at this stage was driven by a surge of severe COVID-19-associated CD141+ CD163+ classical monocytes, markers recently associated with severe COVID-19,20,21,55 and HLA-DRlow CCR2low VISTAlow classical monocytes, reminiscent of an immune paralysis phenotype in monocytes. Such dysregulated monocytes may contribute to an insufficient antiviral immune response; however, the phenotype can also be the product of an hyperinflammatory environment.27 We find that the chemokine signature was dominated by CCL3 and CCL4, which were correlated with the reappearance of intermediate and non-classical monocytes. These CCR5 ligands drive the recruitment of a variety of immune cells such as neutrophils and monocytes, but also adaptive immune cells, and are increased early in the serum and lungs of patients with certain acute respiratory viral infections.60,61 In contrast, these chemokines quite distinctly increased late during the disease course in our cohort, and their production at this stage in severe COVID-19 seems to be a correlate of ongoing local inflammation and may contribute to tissue damage. Both chemokines have been shown to be produced on a transcriptional level by monocytes isolated from the lungs of severe COVID-19 patients.13,22
In summary, our systems-level analysis of the innate immune response to SARS-CoV-2 shows that there are profound changes in the peripheral monocyte compartment of COVID-19 patients, with a previously underappreciated temporal dynamic. Even though patients with mild and severe disease showed similar phenotypic changes early after symptom onset, we found a stronger inflammatory phenotype throughout the disease course of patients experiencing severe symptoms, and could show for the first time that severe COVID-19 patients had a distinct innate signature at later stages of the disease. These results provide evidence for a strong inflammatory response to SARS-CoV-2 infection, further supporting the investigation of targeted interventions in severe cases of COVID-19.18 The distinct temporal changes in immune signatures indicate that specific interventions may benefit from precise timing to maximize therapeutic efficacy.59,62
Limitations of study
A limitation of our study is that the cellular trajectories identified during acute infection rely on cross-sectional samples collected from patients at different times after symptom onset. We were able to analyze only 4 patients in a longitudinal manner, but confirmed our findings by analyzing a publicly available longitudinal dataset,24 which provided strong support for our pseudo-temporal analysis.
Another limitation of the study is that the different groups could not be completely age and gender matched due to COVID-19-associated clinical risk factors. However, our analyses revealed that time after symptom onset and disease severity are the key factors that explain signature shifts upon SARS-CoV-2 infection. Furthermore, mild COVID-19 patients are underrepresented at time points later than 30 days after symptom onset, which reduces the statistical power of our analyses.
STAR★methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD1c (L161), Purified | Biolegend | Cat# 331502; RRID: AB_1088995 |
| Anti-human CD3e (UCHT1), Purified | Biolegend | Cat# 317302; RRID: AB_571927 |
| Anti-human CD4 (RPA-T4), Purified | Biolegend | Cat# 300502; RRID: AB_314070 |
| Anti-human CD7 (M-T701), Purified | BD Bioscience | Cat# 555359; RRID: AB_395762 |
| Anti-human CD8 (RPA-T8), Purified | Biolegend | Cat# 301002; RRID: AB_314120 |
| Anti-human CD11b (M1/70), Purified | BioLegend | Cat# 101202; RRID: AB_312785 |
| Anti-human CD11c (Bu15), Purified | BioLegend | Cat# 337202; RRID: AB_1236381 |
| Anti-human CD14 (M5E2), Purified | Biolegend | Cat# 301802; RRID: AB_314184 |
| Anti-human CD15 (HI98), Purified | Biolegend | Cat# 301902; RRID: AB_314194 |
| Anti-human CD16 (3G8), Purified | Biolegend | Cat# 302002; RRID: AB_314202 |
| Anti-human CD20 (2H7), Purified | Biolegend | Cat# 302302; RRID: AB_314250 |
| Anti-human CD32 (Fun-2), Purified | Biolegend | Cat# 303202; RRID: AB_314334 |
| Anti-human CD36 (5-271), Purified | Biolegend | Cat# 336202; RRID: AB_1279228 |
| Anti-human CD38 (HIT2), Purified | Biolegend | Cat# 303502; RRID: AB_314354 |
| Anti-human CD39 (A1), Purified | Biolegend | Cat# 328202; RRID: AB_940438 |
| Anti-human CD40 (5c3), Purified | eBioscience | Cat# 14-0409-82; RRID: AB_467232 |
| Anti-human CD45 (HI30), Purified | Biolegend | Cat# 304002; RRID: AB_314390 |
| Anti-human CD64 (10.1), Purified | Biolegend | Cat# 305002, RRID: AB_314486 |
| Anti-human CD66ace (ASL-32), Purified | Biolegend | Cat# 342302, RRID: AB_1626265 |
| Anti-human CD68 (KP1), Purified | eBioscience | Cat# 14-0688-82, RRID: AB_11151139 |
| Anti-human CD73 (AD2), Purified | Biolegend | Cat# 344002, RRID: AB_2154067 |
| Anti-human CD86 (2331 (FUN-1)), Purified | BD Bioscience | Cat# 555655; RRID: AB_396010 |
| Anti-human CD103 (SP301), Purified | Abcam | Cat# ab245746, RRID: AB_2884021 |
| Anti-human CD123 (6H6), Purified | Biolegend | Cat# 306002; RRID: AB_314576 |
| Anti-human CD141 (M80), Purified | Biolegend | Cat# 344102; RRID: AB_2201808 |
| Anti-human CD163 (GHI/61), Purified | Biolegend | Cat# 333602; RRID: AB_1088991 |
| Anti-human CD169 (7-239), Purified | Biolegend | Cat# 346002; RRID: AB_2189031 |
| Anti-human CD192 (K036C2), Purified | Biolegend | Cat# 357202; RRID: AB_2561851 |
| Anti-human CD206 (15-2), Purified | Biolegend | Cat# 321102; RRID: AB_571923 |
| Anti-human CD274 (E1L3N), Purified | CST | Cat# 13684, RRID: AB_2687655 |
| Anti-human CD370 (EPR22324), Purified | Abcam | Cat# ab223188, RRID: AB_2884022 |
| Anti-human CXCR2 (48311), Purified | R&D Systems | Cat# MAB331-100, RRID: AB_2296102 |
| Anti-human Granzyme B (GB11), Purified | Invitrogen Antibodies | Cat# MA1-80734; RRID: AB_931084 |
| Anti-human HLA-DR (L243), Purified | Biolegend | Cat# 307602; RRID: AB_314680 |
| Anti-human Indoleamine 2,3-dioxygenase (SP260), Purified | Abcam | Cat# ab228468, RRID: AB_2884023 |
| Anti-human IL-6 (MQ-13A5), Purified | Biolegend | Cat# 501115; RRID: AB_2562841 |
| Anti-human Ki-67 (B56), Purified | BD Bioscience | Cat# 556003; RRID: AB_396287 |
| Anti-human PPARg (14G4B17), Purified | Biolegend | Cat# 683402; RRID: AB_2572020 |
| Anti-human Tim-3 (Polyclonal), Purified | R&D Systems | Cat# AF2365, RRID: AB_355235 |
| Anti-human TMEM173 (SP339), Purified | Abcam | Cat# ab238796, RRID: AB_2884024 |
| Anti-human VISTA (D1L2G), Purified | Fluidigm | Cat# 3160025D, RRID: AB_2811070 |
| Biological samples | ||
| Peripheral blood mononuclear cells (PBMCs) and Serum samples from COVID-19 patients and healthy control individuals | Hospital Uster, Hospital Limmattal, Triemli Hospital, and the University Hospital Zurich (Switzerland) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Antibody Stabilizer PBS | Candor Bioscience | Cat# 131 050 |
| Bis(2,2′-bipyridine)-4′-methyl-4-carboxybipyridine-ruthenium-N-succidimyl ester-bis(hexafluorophosphate) (96Ru, 98-102Ru, 104Ru) | Sigma Aldrich | Cat# 96631 |
| Bismuth trichloride (209Bi) | Sigma Aldrich | Cat# 450723 |
| maleimidomono-amido-DOTA (mDOTA) | Macrocyclics | Cat# B272 |
| Cell Acquisition Solution | Fluidigm | Cat # 201240 |
| Cell-ID Intercalator-Ir | Fluidigm | Cat# 201192B |
| DMSO | Sigma Aldrich | Cat# D2438 |
| EDTA | StemCell Technologies, Inc. | Cat# EDS-100G |
| EQ Four Element Calibration Beads | Fluidigm | Cat# 201078 |
| FcR Blocking Reagent, human | Miltenyi Biotec | Cat# 130-059-901 |
| Indium (113In, 115In) | Fluidigm | N/A |
| Isothiocyanobenzyl-EDTA | Dojindo Laboratories | M030-10 |
| Lanthanide (III) metal isotopes as chloride salts | Fluidigm | N/A |
| Lymphodex | Inno-Train Diagnostik GmBH | Cat# 002041500 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15710 |
| Saponin | Sigma Aldrich | Cat# S7900 |
| SepMate™ PBMC Isolation Tubes | StemCell Technologies, Inc. | Cat# 85460 |
| Yttrium (89Y) | Sigma Aldrich | N/A |
| Critical commercial assays | ||
| IL-1β/IL-1F2 Quantikine ELISA Kit | R&D Systems | Cat# DLB50 |
| IL-6 Quantikine ELISA Kit | R&D Systems | Cat# D6050 |
| IFNγ Quantikine ELISA Kit | R&D Systems | Cat# DIF50 |
| TNFα Quantikine HS ELISA Kit | R&D Systems | Cat# HSTA00D |
| Maxpar X8 Multimetal Labeling Kit | Fluidigm | Cat# 201300 |
| Olink Inflammation panel | Olink® Proteomics | N/A |
| Deposited data | ||
| Mass cytometry data | This paper | https://doi.org/10.17632/vyy8ttw7n9.1 |
| Olink Proteomics data | This paper | https://doi.org/10.17632/vyy8ttw7n9.1 |
| Software and algorithms | ||
| CATALYST 3.12 | Chevrier et al.63 | http://bioconductor.org/packages/release/bioc/html/CATALYST.html |
| Cytobank | Kotecha et al.64 | https://www.cytobank.org/ |
| Diffusion map | Angerer et al.65 | https://cran.r-project.org/web/packages/diffusionMap/index.html |
| FactoMineR | N/A | https://cran.r-project.org/web/packages/FactoMineR/FactoMineR.pdf |
| FlowJo v10.7.1 | N/A | https://www.flowjo.com/ |
| ggpubr | N/A | https://cran.r-project.org/web/packages/ggpubr/index.html |
| randomForest 4.6-14 | Liaw and Wiener66 | https://cran.r-project.org/web/packages/randomForest/index.html |
| Rphenograph_0.99.1.9003 | Levine et al.67 | https://github.com/i-cyto/Rphenograph/ |
| R 4.0.0 | R Core Team, 2016 | https://www.R-project.org |
| Rtsne 0.15 | Van Der Maaten and Hinton68 | https://github.com/jkrijthe/Rtsne |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bernd Bodenmiller (bernd.bodenmiller@uzh.ch).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The raw mass cytometry data and the Olink protein data generated during this study are available for download at Mendeley Data: https://doi.org/10.17632/vyy8ttw7n9.1.
Experimental model and subject details
Patients were recruited at the Hospital Uster, Hospital Limmattal, Triemli Hospital, and the University Hospital Zurich (Switzerland) from an outpatient as well as inpatient setting. The patients were eligible if they were symptomatic at the time of inclusion, had a newly diagnosed SARS-CoV-2 infection confirmed by quantitative reverse-transcriptase polymerase chain reaction (RT-qPCR), and were more than 18 years old. Healthy donors (n = 22) were recruited as controls. All participants, patients and healthy controls, signed a written informed consent. This non-interventional, observational study was approved by the Cantonal Ethics Committee of Zurich (BASEC #2016-01440) and performed in accordance with the Declaration of Helsinki. The sample size was based on availability of the samples. Investigators were blinded to disease severity, while performing experiments. While the analysis was cross-sectional, the patient outcomes were recorded prospectively after inclusion.
Standard clinical laboratory data (CRP, LDH, complete blood count with differential) was collected from the first day of hospitalization until the end of hospitalization. Patients were classified according to WHO guidance23 into mild cases (mild illness and mild pneumonia) and (b) severe cases (severe pneumonia and ARDS). Pneumonia was considered severe if the patient required supplemental oxygen or had an oxygen saturation < 94%, in accordance with WHO guideline.23 ARDS was classified as defined by the Berlin definition.69 36 of the severe COVID-19 patients required respiratory support at the time of sampling, such as supplemental oxygen, non-invasive ventilation or mechanical ventilation. A blood sample was collected from each patient, if possible coordinated with the usual care. For longitudinal analysis of SARS-CoV-2-specific immune responses, two subjects with mild COVID-19 and two subjects with severe COVID-19 were sampled twice during their disease course. All samples were processed in the same hospital laboratory.
At time of the manuscript, 70 COVID-19 patients and 22 healthy subjects were recruited. Four patients were excluded from analysis (two due to chronic lymphocytic leukemia and two due to unclarity whether the current disease was the primary infection). All healthy controls were tested for SARS-CoV-2 specific IgA and IgG antibodies and all were below the diagnostic reference value. We have previously described the SARS-CoV-2-specific antibody response in a subset of this cohort.70 The complete characteristics of the cohort are given in Table 1.
All patients received a standard clinical laboratory sampling and cytokines were measured. Furthermore, samples from 27 COVID-19 patients with mild, 29 with severe disease and all healthy subjects were processed for CyTOF. Samples from 26 COVID-19 patients with mild, 36 with severe disease and 17 healthy patients were evaluated with Olink proteomics. 54 patients and 17 healthy individuals were analyzed by CyTOF and Olink concomitantly. Longitudinal samples from patient CovP45 homogeneously failed the Olink incubation control, and could thus only be compared with each other, but were excluded from other analysis, together with one other sample which was not correctly processed prior to analysis. Three samples had less than 100 myeloid cells and were excluded from the monocyte analyses (samples: CovP17, CovP58, CovP74). Routine flow cytometry for NK cell quantification was performed on all samples in the accredited immunological laboratory at the University Hospital Zurich, as previously described.46 The cohort characteristics and a selection of the Olink dataset are also shown in Adamo et al.46 describing the T cell response of this cohort.
Method details
Blood collection and sample preparation for CyTOF
Venous blood samples were collected in BD vacutainer EDTA tubes, centrifuged, plasma removed, and the remaining blood diluted with an equal amount of PBS. This mixture was then layered into a SepMate tube (STEMCELL) filled with lymphodex (Inno-Train Diagnostik GmBH) solution. The tube was centrifuged, and the PBMCs were washed with PBS and re-centrifuged. Aliquots of 1x106 PBMCs were then centrifuged, resuspended in 200 μL 1.6% PFA (Electron Microscopy Sciences) diluted with RPMI 1640 medium, and fixed at room temperature for 10 min. Subsequently the reaction was stopped by adding 1 mL of cell staining medium (CSM, PBS with 0.5% bovine serum albumin and 0.02% sodium azide). The cells were centrifuged and the disrupted pellet was frozen at −80°C. The remaining PBMCs were frozen and stored in 1.5 mL 90% FBS, 10% DMSO at −80°C for at least 4 h. For long-term storage, the frozen cells were moved to liquid nitrogen. Reference cells for CyTOF analysis were derived from PBMCs from a healthy donor, which were stimulated with either 0.1 μg/mL phytohemagglutinin for 24 h or 1 μg/mL lipopolysaccharide and 1.5 μg/mL monensin for 48 h. One-third of the PBMCs were unstimulated. The three sets of PBMCs were fixed and frozen as described above.
Mass cytometry barcoding
We ensured homogeneous staining by barcoding 1 × 106 PBMCs from each patient using a 60-well barcoding scheme consisting of unique combinations of four out of eight barcoding reagents as previously described.71 Six palladium isotopes (102Pd, 104Pd, 105Pd, 106Pd, 108Pd, and 110Pd, Fluidigm) were chelated to 1-(4-isothiocyanatobenzyl)ethylenediamine-N,N,N’,N’ tetraacetic acid (Dojino). Two indium isotopes (113In and 115In, Fluidigm) were chelated to 1,4,7,10-tetraazacy-clododecane-1,4,7-tris-acetic acid 10-maleimide ethylacetamide (Dojino) following standard procedures.72 We titrated mass tag barcoding reagents to ensure equivalent staining for each reagent; final concentrations were between 50 nM and 200 nM. We used the previously described transient partial permeabilization approach to barcode the cells.73 PBMCs from all samples were randomly loaded into wells of two 96-well plates and were analyzed in two independent experiments. Three standard samples were loaded onto each plate to enable assessment of inter-run variability. Cells were washed with 0.03% saponin in PBS (PBS-S, Sigma Aldrich) and incubated for 30 min with 200 μL of mass tag barcoding reagents diluted in PBS-S. After washing three times with CSM, samples from each plate were pooled and stained with the antibody panel.
Antibodies and antibody labeling
The antibodies used in this study, including provider, clone, and metal tag, are listed in the key resources table. Antibody conjugation was performed using the MaxPAR antibody labeling kit (Fluidigm). Upon conjugation, the yield of recovered antibody was assessed on a Nanodrop (Thermo Scientific) and then supplemented with Candor Antibody Stabilizer. We performed titrations to determine optimal concentrations of all conjugated antibodies. All antibodies used in this study were managed using the cloud-based platform AirLab.74
Sample staining and data acquisition
After barcoding, pooled cells were incubated with FcR blocking reagent (Miltenyi Biotec) for 10 min at 4°C. Cells were stained with 400 μL of the antibody panel per 107 cells for 45 min at 4°C. Cells were washed three times in CSM, once in PBS, resuspended in 0.4 mL of 0.5 μM nucleic acid Ir- labeled intercalator (Fluidigm) and incubated overnight at 4°C. Samples were then prepared for CyTOF acquisition by washing the cells once in CSM, once in PBS, and once in water. Cells were then diluted to 0.5 × 106 cells/mL in Cell Acquisition Solution (Fluidigm) containing 10% EQ Four Element Calibration Beads (Fluidigm). Samples were acquired on a Helios upgraded CyTOF 2 in independent FCS files.
Cytokine ELISA
Serum was collected in BD vacutainer tubes. The samples were processed in the accredited immunological laboratory at the University Hospital Zurich. IL-1β, IL-6, IFNγ, TNFα were quantified using R&D Systems ELISA kits.
Proteomics analysis using Olink
For serum proteomics, the commercially available proximity extension assay-based technology from Olink® Proteomics was used. Heat-inactivated plasma samples were sent to the Olink analysis laboratory in Davos, Switzerland for analysis using the inflammation panel. The Olink technology has been described previously.75 Briefly, binding of paired cDNA-tagged antibodies directed against the targeted serum proteins lead to hybridization of the corresponding DNA oligonucleotides allowing subsequent extension by a DNA polymerase. The protein level is quantified using real-time PCR. Only samples that passed the quality control tests are reported. If expression was below the detection limit, the value reported is the lower limit of detection. Only proteins that were detectable in at least 50% of samples were used for subsequent analysis.
Quantification and statistical analysis
Mass cytometry data preprocessing and automated cell classification
Individual FCS files collected from each set of samples were pre-processed using a semi-automated R pipeline based on CATALYST to perform individual file concatenation, bead based normalization, compensation, debarcoding, and batch correction as previously described.76 During this process, inter-sample doublets were excluded based on the debarcoding scheme and intra-sample doublets were excluded based on DNA content. Spillover matrix for CyTOF compensation was assessed on all antibodies used in this study as previously suggested.63
Upon pre-processing, a subset of 1,000 randomly selected cells from each sample were exported as FCS files and loaded on Cytobank. Immune cell subsets were manually gated according to the scheme described in Figure S1E. FCS files corresponding to each gate were exported and used to train a random forest classifier (R package randomForest), based on 500 trees and 6 variables tried at each split, leading to an OOB estimate of error rate of 0.43%. The resulting random forest model was used to assign each cell of the dataset to the predefined cell types. Based on a 40% assignment probability cutoff and a 20% delta cutoff, 98% of the cells were retained in the analysis.
Dimensionality reduction and clustering analysis
To visualize the high-dimensional data in two dimensions, the t-SNE algorithm was applied on data from a maximum of 1,000 randomly selected cells from each sample, with a perplexity set to 80, using the implementation of t-SNE available in CATALYST.77 Channels which were not relevant for these cell subsets or which were affected by different background stainings across batches were excluded and not used for monocyte cluster characterization (CD15, CD66ace, CD3, CD45, CD8a, CD20, CXCR2, Granzyme B). Data were displayed using the ggplot2 R package or the plotting functions of CATALYST.77
Visualization of marker expression on t-SNE maps was performed upon data normalization between 0 and 1. The maximum intensity was defined as the 99th percentile.
Clustering analysis of the myeloid and neutrophil subsets was performed using the R implementation of PhenoGraph run on all samples simultaneously, with the parameter k, defining the number of nearest neighbors, set to 100.67 For the myeloid subset, clusters with less than 600 cells were excluded from the analysis.
In order to perform hierarchical clustering, pairwise distances between samples were calculated using the Spearman correlation or Euclidean distance, as indicated in the figure legend. Dendrograms were generated using Ward.2's method. Heatmaps were generated based on the pheatmap package.
To identify putative single-cell trajectories among monocyte clusters, we used the implementation of the diffusion map algorithm available in the R package scater using the default parameters and the same channels used to perform the t-SNE analysis. A maximum of 1,000 cells randomly selected from each cluster were included in the analysis.
The principal component analysis to identify the variations in the data described by the cluster frequencies or the combination of cluster frequencies and cytokine levels was performed based on the FactoMineR package. Data were visualized using the Factoextra R package.
Statistical analyses
The statistical analysis was performed using GraphPad Prism (version 8.4.3, GraphPad Software, La Jolla California USA) and R software (version 4.0.1) using the package “mgcv.” Statistical normality testing was not performed. In general, non-parametric Mann-Whitney Wilcoxon test was used to test for differences between continuous variables and p values were adjusted for multiple testing using the Holm method. The Olink data was tested using parametric tests as recommended by the company. Categorical variables were compared using Fisher’s exact test. Generalized additive models were used to evaluate relationships between time since symptom onset and different variables, with the number of knots used to represent the smooth term set at three. The correlation maps were displayed using the corrplot or pheatmap packages and the implemented hclust method.
The correlations between cellular subsets and the serum protein expressions were analyzed using non-parametric Spearman correlations. The significance threshold was set at an alpha < 0.05. For the differential expression analysis, a false-discovery rate78 of 1% was used as significance threshold, except for the late versus early comparison where 5% was used as indicated. The respective statistical tests used, as well as the exact n values, are given in each figure legend.
Acknowledgments
We thank all of the patients for their participation in this study, the teams of the Diagnostic Immunology and the Transplantation Immunology Laboratories at the University Hospital Zurich for their great effort, as well as Alessandra Guaita, Claudia Meloni, Jennifer Jörger, Ulrike Held, Sara Hasler, and the members of the Bodenmiller and Boyman laboratories for their support and helpful discussions. Furthermore, we would like to thank Lucie Rodriguez and Petter Brodin for kindly sharing data. The graphical abstract was created with BioRender.com. This work was funded by the Swiss National Science Foundation (4078P0_198431 to O.B., B.B., and J.N. and 310030-172978 to O.B.), the Pandemic Fund of the University of Zurich (to O.B.), the Innovation Fund of the University Hospital Zurich (to O.B.), Swiss Academy of Medical Sciences fellowships (323530-191220 to C.C., 323530-191230 to Y.Z., and 323530-177975 to S.A.), the Young Talents in Clinical Research Fellowship by the Swiss Academy of Medical Sciences and the Bangerter Foundation (YTCR 32/18 to M.E.R.), the Clinical Research Priority Program of the University of Zurich for the CRPP CYTIMM-Z (to O.B.), and a SNSF R’Equip grant (to B.B.).
Author contributions
S.C., Y.Z., and C.C. contributed to the study design, patient recruitment, data collection, analysis, interpretation, and writing of the manuscript. S.S., A.J., and S.C. developed the CyTOF antibody panel and performed the CyTOF experiments. S.A. contributed to the experiments, data collection, analysis, and interpretation. M.E.R., E.B., A.R., M.S.-H., L.C.H., and D.J.S. contributed to patient recruitment and clinical management. N.d.S. contributed to the writing of the manuscript. J.N., O.B., and B.B. contributed to the study conception and design, data analysis, interpretation, and writing of the manuscript. All of the authors reviewed and approved the final version of the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: December 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100166.
Contributor Information
Jakob Nilsson, Email: jakob.nilsson@uzh.ch.
Onur Boyman, Email: onur.boyman@uzh.ch.
Bernd Bodenmiller, Email: bernd.bodenmiller@uzh.ch.
Supplemental information
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., China Novel Coronavirus Investigating and Research Team A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu Z., McGoogan J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323:1239–1242. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 4.Wiersinga W.J., Rhodes A., Cheng A.C., Peacock S.J., Prescott H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA. 2020;324:782–793. doi: 10.1001/jama.2020.12839. [DOI] [PubMed] [Google Scholar]
- 5.Puelles V.G., Lütgehetmann M., Lindenmeyer M.T., Sperhake J.P., Wong M.N., Allweiss L., Chilla S., Heinemann A., Wanner N., Liu S. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020;383:590–592. doi: 10.1056/NEJMc2011400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwasaki A., Medzhitov R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yap J.K.Y., Moriyama M., Iwasaki A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J. Immunol. 2020;205:307–312. doi: 10.4049/jimmunol.2000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van de Veerdonk F.L., Janssen N.A.F., Grondman I., de Nooijer A.H., Koeken V.A.C.M., Matzaraki V., Boahen C.K., Kumar V., Kox M., Koenen H.J.P.M. A systems approach to inflammation identifies therapeutic targets in SARS-CoV-2 infection. medRxiv. 2020 doi: 10.1101/2020.05.23.20110916. [DOI] [Google Scholar]
- 10.Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park A., Iwasaki A. Type I and Type III Interferons – Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe. 2020;27:870–878. doi: 10.1016/j.chom.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee J.S., Park S., Jeong H.W., Ahn J.Y., Choi S.J., Lee H., Choi B., Nam S.K., Sa M., Kwon J.-S. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020;5:1554. doi: 10.1126/sciimmunol.abd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bastard P., Rosen L.B., Zhang Q., Michailidis E., Hoffmann H.-H., Zhang Y., Dorgham K., Philippot Q., Rosain J., Béziat V. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q., Bastard P., Liu Z., Le Pen J., Moncada-Velez M., Chen J., Ogishi M., Sabli I.K.D., Hodeib S., Korol C. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Channappanavar R., Fehr A.R., Vijay R., Mack M., Zhao J., Meyerholz D.K., Perlman S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKechnie J.L., Blish C.A. The Innate Immune System: Fighting on the Front Lines or Fanning the Flames of COVID-19? Cell Host Microbe. 2020;27:863–869. doi: 10.1016/j.chom.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merad M., Martin J.C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 2020;20:355–362. doi: 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vabret N., Britton G.J., Gruber C., Hegde S., Kim J., Kuksin M., Levantovsky R., Malle L., Moreira A., Park M.D. Immunity. 2020;52:910–941. doi: 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silvin A., Chapuis N., Dunsmore G., Goubet A.-G., Dubuisson A., Derosa L., Almire C., Hénon C., Kosmider O., Droin N. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 23.World Health Organization . 2020. Clinical management of severe acute respiratory infection (SARI) when COVID-19 disease is suspected: interim guidance.https://www.who.int/publications/i/item/clinical-management-of-covid-19 [Google Scholar]
- 24.Rodriguez L., Pekkarinen P.T., Lakshmikanth T., Tan Z., Consiglio C.R., Pou C., Chen Y., Mugabo C.H., Nguyen N.A., Nowlan K. Systems-Level Immunomonitoring from Acute to Recovery Phase of Severe COVID-19. Cell Rep. Med. 2020;1:100078. doi: 10.1016/j.xcrm.2020.100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J.J., Dong X., Cao Y.Y., Yuan Y.D., Yang Y.B., Yan Y.Q., Akdis C.A., Gao Y.D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy. 2020;75:1730–1741. doi: 10.1111/all.14238. [DOI] [PubMed] [Google Scholar]
- 26.Kuri-Cervantes L., Pampena M.B., Meng W., Rosenfeld A.M., Ittner C.A.G., Weisman A.R., Agyekum R.S., Mathew D., Baxter A.E., Vella L.A. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020;5:eabd7114. doi: 10.1126/sciimmunol.abd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giamarellos-Bourboulis E.J., Netea M.G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M.E., Katsaounou P. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe. 2020;27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrissey S.M., Geller A.E., Hu X., Tieri D., Cooke E.A., Ding C., Woeste M., Zhange H., Cavallazi R., Clifford S.P. Emergence of Low-density Inflammatory Neutrophils Correlates with Hypercoagulable State and Disease Severity in COVID-19 Patients. medRxiv. 2020 doi: 10.1101/2020.05.22.20106724. [DOI] [Google Scholar]
- 29.Scapini P., Marini O., Tecchio C., Cassatella M.A. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol. Rev. 2016;273:48–60. doi: 10.1111/imr.12448. [DOI] [PubMed] [Google Scholar]
- 30.Ng L.G., Ostuni R., Hidalgo A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019;19:255–265. doi: 10.1038/s41577-019-0141-8. [DOI] [PubMed] [Google Scholar]
- 31.Impellizzieri D., Ridder F., Raeber M.E., Egholm C., Woytschak J., Kolios A.G.A., Legler D.F., Boyman O. IL-4 receptor engagement in human neutrophils impairs their migration and extracellular trap formation. J. Allergy Clin. Immunol. 2019;144:267–279.e4. doi: 10.1016/j.jaci.2019.01.042. [DOI] [PubMed] [Google Scholar]
- 32.Thomas G.D., Hamers A.A.J., Nakao C., Marcovecchio P., Taylor A.M., McSkimming C., Nguyen A.T., McNamara C.A., Hedrick C.C. Human Blood Monocyte Subsets: A New Gating Strategy Defined Using Cell Surface Markers Identified by Mass Cytometry. Arterioscler. Thromb. Vasc. Biol. 2017;37:1548–1558. doi: 10.1161/ATVBAHA.117.309145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haghverdi L., Buettner F., Theis F.J. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics. 2015;31:2989–2998. doi: 10.1093/bioinformatics/btv325. [DOI] [PubMed] [Google Scholar]
- 34.Wang K.C., Li Y.H., Shi G.Y., Tsai H.W., Luo C.Y., Cheng M.H., Ma C.Y., Hsu Y.Y., Cheng T.L., Chang B.I. Membrane-Bound Thrombomodulin Regulates Macrophage Inflammation in Abdominal Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2015;35:2412–2422. doi: 10.1161/ATVBAHA.115.305529. [DOI] [PubMed] [Google Scholar]
- 35.Broughton T.W.K., ElTanbouly M.A., Schaafsma E., Deng J., Sarde A., Croteau W., Li J., Nowak E.C., Mabaera R., Smits N.C. Defining the Signature of VISTA on Myeloid Cell Chemokine Responsiveness. Front. Immunol. 2019;10:2641. doi: 10.3389/fimmu.2019.02641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdi H., Williams L.J. Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2010;2:433–459. [Google Scholar]
- 37.Seidler S., Zimmermann H.W., Bartneck M., Trautwein C., Tacke F. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 2010;11:30. doi: 10.1186/1471-2172-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boettcher S., Manz M.G. Regulation of Inflammation- and Infection-Driven Hematopoiesis. Trends Immunol. 2017;38:345–357. doi: 10.1016/j.it.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Ginhoux F., Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 40.Prévost J., Gasser R., Beaudoin-Bussières G., Richard J., Duerr R., Laumaea A., Anand S.P., Goyette G., Benlarbi M., Ding S. Cross-Sectional Evaluation of Humoral Responses against SARS-CoV-2 Spike. Cell Rep. Med. 2020;1:100126. doi: 10.1016/j.xcrm.2020.100126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carissimo G., Xu W., Kwok I., Abdad M.Y., Chan Y.H., Fong S.W., Puan K.J., Lee C.Y.P., Yeo N.K.W., Amrun S.N. Whole blood immunophenotyping uncovers immature neutrophil-to-VD2 T-cell ratio as an early marker for severe COVID-19. Nat. Commun. 2020;11:5243. doi: 10.1038/s41467-020-19080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rempel H., Calosing C., Sun B., Pulliam L. Sialoadhesin Expressed on IFN-Induced Monocytes Binds HIV-1 and Enhances Infectivity. PLoS One. 2008;3:e1967. doi: 10.1371/journal.pone.0001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farina A., Peruzzi G., Lacconi V., Lenna S., Quarta S., Rosato E., Vestri A.R., York M., Dreyfus D.H., Faggioni A. Epstein-Barr virus lytic infection promotes activation of Toll-like receptor 8 innate immune response in systemic sclerosis monocytes. Arthritis Res. Ther. 2017;19:39. doi: 10.1186/s13075-017-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bedin A.-S., Makinson A., Picot M.-C., Mennechet F., Malergue F., Pisoni A., Nyiramigisha E., Montagnier L., Bollore K., Debiesse S. Monocyte CD169 expression as a biomarker in the early diagnosis of COVID-19. medRxiv. 2020 doi: 10.1101/2020.06.28.20141556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adamo S., Chevrier S., Cervia C., Zurbuchen Y., Raeber M.E., Yang L., Sivapatham S., Jacobs A., Bächli E., Rudiger A. Lymphopenia-induced T cell proliferation is a hallmark of severe COVID-19. bioRxiv. 2020 doi: 10.1101/2020.08.04.236521. [DOI] [Google Scholar]
- 47.Kim K.D., Zhao J., Auh S., Yang X., Du P., Tang H., Fu Y.X. Adaptive immune cells temper initial innate responses. Nat. Med. 2007;13:1248–1252. doi: 10.1038/nm1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Channappanavar R., Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017;39:529–539. doi: 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas G., Tacke R., Hedrick C.C., Hanna R.N. Nonclassical patrolling monocyte function in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2015;35:1306–1316. doi: 10.1161/ATVBAHA.114.304650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sánchez-Cerrillo I., Landete P., Aldave B., Sánchez-Alonso S., Sánchez-Azofra A., Marcos-Jiménez A., Ávalos E., Alcaraz-Serna A., de Los Santos I., Mateu-Albero T., REINMUN-COVID and EDEPIMIC groups COVID-19 severity associates with pulmonary redistribution of CD1c+ DCs and inflammatory transitional and nonclassical monocytes. J. Clin. Invest. 2020;130:6290–6300. doi: 10.1172/JCI140335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Kuyl A.C., van den Burg R., Zorgdrager F., Groot F., Berkhout B., Cornelissen M. Sialoadhesin (CD169) Expression in CD14+ Cells Is Upregulated Early after HIV-1 Infection and Increases during Disease Progression. PLoS One. 2007;2:e257. doi: 10.1371/journal.pone.0000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim W.K., McGary C.M., Holder G.E., Filipowicz A.R., Kim M.M., Beydoun H.A., Cai Y., Liu X., Sugimoto C., Kuroda M.J. Increased Expression of CD169 on Blood Monocytes and Its Regulation by Virus and CD8 T Cells in Macaque Models of HIV Infection and AIDS. AIDS Res. Hum. Retroviruses. 2015;31:696–706. doi: 10.1089/aid.2015.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cros J., Cagnard N., Woollard K., Patey N., Zhang S.Y., Senechal B., Puel A., Biswas S.K., Moshous D., Picard C. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–386. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwissa M., Nakaya H.I., Onlamoon N., Wrammert J., Villinger F., Perng G.C., Yoksan S., Pattanapanyasat K., Chokephaibulkit K., Ahmed R., Pulendran B. Dengue virus infection induces expansion of a CD14(+)CD16(+) monocyte population that stimulates plasmablast differentiation. Cell Host Microbe. 2014;16:115–127. doi: 10.1016/j.chom.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kvedaraite E., Hertwig L., Sinha I., Ponzetta A., Myrberg I.H., Lourda M., Dzidic M., Akber M., Klingström J., Folkesson E. Perturbations in the mononuclear phagocyte landscape associated with COVID-19 disease severity. medRxiv. 2020 doi: 10.1101/2020.08.25.20181404. [DOI] [Google Scholar]
- 56.Clanchy F.I.L., Holloway A.C., Lari R., Cameron P.U., Hamilton J.A. Detection and properties of the human proliferative monocyte subpopulation. J. Leukoc. Biol. 2006;79:757–766. doi: 10.1189/jlb.0905522. [DOI] [PubMed] [Google Scholar]
- 57.Patel A.A., Zhang Y., Fullerton J.N., Boelen L., Rongvaux A., Maini A.A., Bigley V., Flavell R.A., Gilroy D.W., Asquith B. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017;214:1913–1923. doi: 10.1084/jem.20170355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geissmann F., Manz M.G., Jung S., Sieweke M.H., Merad M., Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lang F.M., Lee K.M.C., Teijaro J.R., Becher B., Hamilton J.A. GM-CSF-based treatments in COVID-19: reconciling opposing therapeutic approaches. Nat. Rev. Immunol. 2020;20:507–514. doi: 10.1038/s41577-020-0357-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Melchjorsen J., Sørensen L.N., Paludan S.R. Expression and function of chemokines during viral infections: from molecular mechanisms to in vivo function. J. Leukoc. Biol. 2003;74:331–343. doi: 10.1189/jlb.1102577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nuriev R., Johansson C. Chemokine regulation of inflammation during respiratory syncytial virus infection. F1000Res. 2019;8 doi: 10.12688/f1000research.20061.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.The RECOVERY Collaborative Group Dexamethasone in Hospitalized Patients with Covid-19 - Preliminary Report. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chevrier S., Crowell H.L., Zanotelli V.R.T., Engler S., Robinson M.D., Bodenmiller B. Compensation of Signal Spillover in Suspension and Imaging Mass Cytometry. Cell Syst. 2018;6:612–620.e5. doi: 10.1016/j.cels.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kotecha N., Krutzik P.O., Irish J.M. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010;Chapter 10:17. doi: 10.1002/0471142956.cy1017s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Angerer P., Haghverdi L., Büttner M., Theis F.J., Marr C., Buettner F. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics. 2016;32:1241–1243. doi: 10.1093/bioinformatics/btv715. [DOI] [PubMed] [Google Scholar]
- 66.Liaw A., Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 67.Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir A.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van der Maaten L., Hinton G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- 69.Ranieri V.M., Rubenfeld G.D., Thompson B.T., Ferguson N.D., Caldwell E., Fan E., Camporota L., Slutsky A.S., ARDS Definition Task Force Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 70.Cervia C., Nilsson J., Zurbuchen Y., Valaperti A., Schreiner J., Wolfensberger A., Raeber M.E., Adamo S., Weigang S., Emmenegger M. Systemic and mucosal antibody responses specific to SARS-CoV-2 during mild versus severe COVID-19. J. Allergy Clin. Immunol. 2020 doi: 10.1016/j.jaci.2020.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zunder E.R., Finck R., Behbehani G.K., Amir A.D., Krishnaswamy S., Gonzalez V.D., Lorang C.G., Bjornson Z., Spitzer M.H., Bodenmiller B. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 2015;10:316–333. doi: 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zivanovic N., Jacobs A., Bodenmiller B. A practical guide to multiplexed mass cytometry. Curr. Top. Microbiol. Immunol. 2014;377:95–109. doi: 10.1007/82_2013_335. [DOI] [PubMed] [Google Scholar]
- 73.Behbehani G.K., Thom C., Zunder E.R., Finck R., Gaudilliere B., Fragiadakis G.K., Fantl W.J., Nolan G.P. Transient partial permeabilization with saponin enables cellular barcoding prior to surface marker staining. Cytometry A. 2014;85:1011–1019. doi: 10.1002/cyto.a.22573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Catena R., Özcan A., Jacobs A., Chevrier S., Bodenmiller B. AirLab: a cloud-based platform to manage and share antibody-based single-cell research. Genome Biol. 2016;17:142. doi: 10.1186/s13059-016-1006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lundberg M., Eriksson A., Tran B., Assarsson E., Fredriksson S. Homogeneous antibody-based proximity extension assays provide sensitive and specific detection of low-abundant proteins in human blood. Nucleic Acids Res. 2011;39:e102. doi: 10.1093/nar/gkr424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crowell H.L., Jacobs A., Tumor Profiler Consortium. Bodenmiller B., Robinson M.D. An R-based reproducible and user-friendly preprocess- ing pipeline for CyTOF data. F1000Res. 2020;9:1263. doi: 10.12688/f1000research.26073.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nowicka M., Krieg C., Crowell H.L., Weber L.M., Hartmann F.J., Guglietta S., Becher B., Levesque M.P., Robinson M.D. CyTOF workflow: differential discovery in high-throughput high-dimensional cytometry datasets. F1000Res. 2017;6:748. doi: 10.12688/f1000research.11622.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Benjamini Y., Krieger A.M., Yekutieli D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika. 2006;93:491–507. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw mass cytometry data and the Olink protein data generated during this study are available for download at Mendeley Data: https://doi.org/10.17632/vyy8ttw7n9.1.