

Abstract
Vimentin intermediate filaments are a significant component of the cytoskeleton in cells of mesenchymal origin. In vivo, filaments assemble and disassemble and thus participate in the dynamic processes of the cell. Post‐translational modifications (PTMs) such as protein phosphorylation regulate the multiphasic association of vimentin from soluble complexes to insoluble filaments and the reverse processes. The thiol side chain of the single vimentin cysteine at position 328 (Cys328) is a direct target of oxidative modifications inside cells. Here, we used atomic force microscopy, electron microscopy and a novel hydrogen–deuterium exchange mass spectrometry (HDex‐MS) procedure to investigate the structural consequences of S‐nitrosylation and S‐glutathionylation of Cys328 for in vitro oligomerisation of human vimentin. Neither modification affects the lateral association of tetramers to unit‐length filaments (ULF). However, S‐glutathionylation of Cys328 blocks the longitudinal assembly of ULF into extended filaments. S‐nitrosylation of Cys328 does not hinder but slows down the elongation. Likewise, S‐glutathionylation of preformed vimentin filaments causes their extensive fragmentation to smaller oligomeric species. Chemical reduction of the S‐glutathionylated Cys328 thiols induces reassembly of the small fragments into extended filaments. In conclusion, our in vitro results suggest S‐glutathionylation as a candidate PTM for an efficient molecular switch in the dynamic rearrangements of vimentin intermediate filaments, observed in vivo, in response to changes in cellular redox status. Finally, we demonstrate that HDex‐MS is a powerful method for probing the kinetics of vimentin filament formation and filament disassembly induced by PTMs.
Keywords: assembly, cysteine, hydrogen–deuterium exchange mass spectrometry, S‐glutathionylation, S‐nitrosylation, vimentin
Vimentin intermediate filaments are a major component of the cytoskeleton in cells of mesenchymal origin. Oxidative modification of vimentin Cys328 is known to affect the assembly of the filament. Using atomic force microscopy, electron microscopy and a novel hydrogen–deuterium exchange mass spectrometry, the authors investigated the structural impact of S‐nitrosylation and S‐glutathionylation on human vimentin oligomerisation in vitro. They show that S‐glutathionylation does not affect the lateral association of tetramers to unit‐length filaments, but blocks the longitudinal assembly of vimentin into long filaments and causes extensive fragmentation of preformed vimentin filaments. Thus, this modification may modulate the dynamic rearrangements of vimentin intermediate filaments in vivo.
Abbreviations
- AFM
atomic force microscopy
- EM
electron microscopy
- ESI‐MS
electrospray ionisation mass spectrometry
- HDex‐MS
hydrogen–deuterium exchange mass spectrometry
- PTM
post‐translational modification
- ULF
unit‐length filaments
- TES
N‐[tris(hydroxymethyl)methyl]‐2‐aminoethanesulfonic acid, 2‐[(2‐hydroxy‐1, 1‐bis(hydroxymethyl)ethyl)amino]ethanesulfonic acid
Introduction
Intermediate filament (IF) proteins form an extended cytoplasmic filament system in all metazoan cells [1]. Vimentin, a prominent member of the large IF protein family, is expressed primarily in cells of mesenchymal origin such as fibroblasts, lens fibre cells, endothelial cells and lymphocytes [2]. In muscle, vimentin is co‐expressed and co‐assembles during embryogenesis with desmin, a protein with high amino acid sequence identity with vimentin but unique functional properties [3, 4, 5]. Vimentin is important in cell migration [6], fibroblast proliferation [7], lymphocyte extravasation [8] and the epithelial–mesenchymal transition [9]. Of note, the primary amino acid sequence of vimentin is evolutionarily highly conserved, from shark to humans, with the remarkable conservation of a single cysteine at the identical corresponding position in a conserved structural fold [4].
The IF proteins harbour a conserved tripartite structural organisation with a central α‐helical ‘rod’ of about 300 amino acids, divided into segments (1A, 1B, 2A, 2B). These segments are interconnected by linkers L1 and L12 and flanked by flexible amino‐ and carboxyl‐terminal domains (Fig. 1A). Via their rod domains, IF proteins form parallel coiled coils. Two such dimers associate into a half‐staggered, anti‐parallel complex, referred to as a tetramer (Fig. 1A). Tetramers are soluble in low‐ionic‐strength buffer, and their assembly can be initiated by raising the ionic strength through addition of monovalent ions. The first phase of assembly is characterised by lateral in‐register association of 8–10 tetramers to a unit‐length filament (ULF) (Fig. 1B). This reaction is rapid and under standard conditions completes within 1 s [10]. In the second phase, ULFs longitudinally anneal with each other and with already formed short IFs (Fig. 1C). The third phase of assembly is initiated after several minutes of incubation when filaments stochastically start to radially compact and reduce their diameter from around 17 to 11 nm [11]. As a result of the assembly process, the number of free ends decreases, but micrometre‐length filaments further elongate by end‐on annealing [12]. These mature IFs are remarkably stable in vitro with scarcely any loss of tetramers and no apparent disassembly over time [13].
Fig. 1.
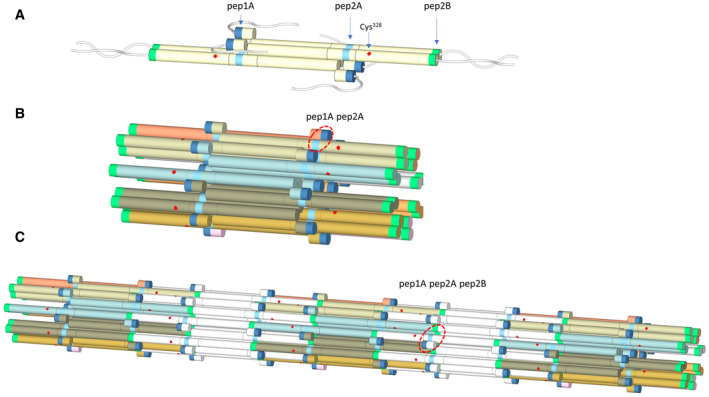
Model of vimentin tetramer (A), ULF (B) and filament (C) based on the previous hydrogen–deuterium exchange study [27]. Peptides diagnostic for assembly are marked as pep1A (dark blue), pep2A (cyan) and pep2B (green) segments. Cysteine residue at position 328 is indicated by a red star. Flexible linkers L1 and L12 are not shown for simplicity. In a tetramer, coil1A segments are schematically separated to mark their role in the inter‐tetrameric interactions as discussed in Ref. [27]. Pep1A, flexible in the tetramer, becomes stabilised in ULF upon interaction with pep2A (red circle in B). Additional protection in pep1A and pep2A is observed from the longitudinal assembly in which pep2B forms contact sites (red circle in C).
In contrast, in vivo, the IF system is highly dynamic in terms of filament movement through the cytoplasm and disassembly/assembly cycles. The apparent contradiction between in vitro stability and in vivo dynamics of vimentin filaments is easily explained by the fact that multiple post‐translational modifications (PTMs) induce intermediate filament lability [14].
Phosphorylation on serine, threonine and tyrosine residues is the best characterised way for disassembling IF proteins [15]. Other PTMs may affect IF substructure dynamics [16, 17, 18, 19, 20]. The single highly reactive cysteine 328 plays a unique role in the regulation of vimentin properties. Cys328 is a sensitive sensor of intracellular redox status and oxidative stress. Its side‐chain thiol groups are the direct targets of endogenous or exogenous oxidative modifiers.
The single highly reactive cysteine 328 plays a unique role in the regulation of vimentin properties. Cys328 is a sensitive sensor of intracellular redox status and oxidative stress. Its side‐chain thiol groups are the direct targets of endogenous or exogenous oxidative modifiers. In cells treated with electrophilic lipids or with diamide, existing vimentin filaments are disrupted but only for wild‐type vimentin and not its Cys328 mutants. In vitro, preincubation of vimentin with electrophilic lipids leads to Cys328 lipoxidation. Treatment with diamide causes the formation of vimentin disulfide. Such modifications impede NaCl‐induced filament formation in different ways either by delaying the elongation process, forming irregular filaments, aggregates, or by significantly increasing the filament diameter. If vimentin was treated with the redox modifiers after NaCl‐induced polymerisation, the chemical modifications were not hindered, but the bundling and elongation impairment was weak [20, 21].
S‐nitrosylation and S‐glutathionylation are other oxidative modifications of vimentin detected in vivo. The exact role of such modification has not been reported [22, 23, 24]. S‐nitrosylation and S‐glutathionylation are strictly regulated, reversible PTMs of proteins. Much like phosphorylation, they function as binary signalling ‘switches’ and regulate protein function, interactions and localisation [25, 26].
No methods yet exist to directly trace the consequences of a PTM of a single protein inside cells. For this reason, to follow the effect of S‐nitrosylation and S‐glutathionylation of Cys328 on the vimentin properties and its filament assembly and disassembly in vitro, we study homogeneously modified recombinant human vimentin and salt‐jump‐induced in vitro assembly conditions. In addition to using atomic force microscopy (AFM) and electron microscopy (EM) data as traditional ways to study these processes, we present a newly established method based on monitoring the hydrogen–deuterium exchange by mass spectroscopy (HDex‐MS). The HDex‐MS approach relies on our previous results showing that analysis of the deuteration of only three peptides in the vimentin sequence – pep1A, pep2A, and pep2B (Fig. 1) – distinguishes the oligomeric state of vimentin among tetramer, ULF and filament [27]. Therefore, the hydrogen–deuterium exchange level in these peptides can be used to follow the structural transitions accompanying filament formation.
Here, we show that modification of the only cysteine residue of vimentin by S‐nitrosylation and S‐glutathionylation, respectively, has distinct effects on filament assembly kinetics and stability. Whereas S‐nitrosylation only slows down the monovalent salt‐jump‐induced filament assembly, S‐glutathionylation has a striking dual effect: it obstructs the filament assembly from modified soluble oligomers and, remarkably, severs mature filaments if subjected to the derivatisation. Our data strongly support the hypothesis also propounded by others that reversible oxidative modifications of vimentin are an essential element of the cellular redox code, and the cell may use them to fine‐tune the dynamics of intermediate filaments resulting from changes in the redox environment. The presented results provide additional information that Cys328 modifications influence only the longitudinal and not the lateral assembly step.
Results
Initial characterisation of glutathionylated and nitrosylated vimentins: The purity of proteins
To address whether the in vitro assembly and disassembly properties of vimentin change because of chemical modification of the single cysteine 328 by nitrosylation and glutathionylation, we prepared homogeneous samples of the modified recombinant vimentin using chemical derivatisation with GSNO and GSSG, respectively. We used mass spectrometry (MS) and SDS/PAGE to verify the completeness of the glutathionylation and nitrosylation reactions and the integrity of the final protein products. In the nonreducing SDS/PAGE gel (Fig. 2A), both nitrosylated (Vim‐SNO) and glutathionylated (Vim‐SSG) vimentin showed similar mobility as the wild‐type protein (Vim‐WT). Often, a small band which could correspond to a dimeric vimentin species was observed. The band did not always disappear under reducing conditions. As MS spectra did not show any signs of a covalent vimentin dimer presence, we interpret this band as some not fully denatured, very stable noncovalent dimer of vimentin known to form even in 6 m urea. As expected, the addition of the ‐NO or ‐SG group to a single cysteine thiol increased the mass of vimentin by 29 and 305 Da, respectively (Fig. 2B). We also carefully examined the MS spectra to look for other vimentin modifications, but the only additional, low‐intensity signals observed corresponded to minute amounts of potassium adduct (+39 Da).
Fig. 2.
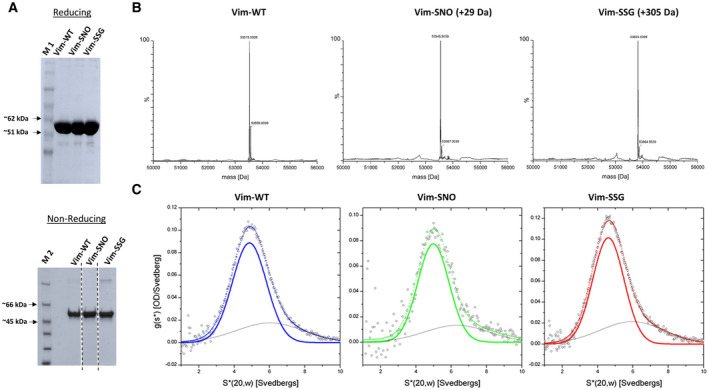
Characterisation of WT vimentin and its Cys328‐modified variants. (A) SDS/PAGE analysis of vimentin and its variants (Vim‐WT, Vim‐SNO, Vim‐SSG) at reducing and nonreducing conditions. In the bottom panel in A, the gel lines were spliced from the same gel. (B) ESI‐MS data deconvoluted with MaxEnt 1 of Vim‐WT, Vim‐SNO and Vim‐SSG. (C) Sedimentation velocity ultracentrifugation analysis of Vim‐WT, Vim‐SNO and Vim‐SSG. In panel C, data were fitted (dotted line) using two Gaussians shown as continuous coloured line for the major species. M 1, protein marker (Biomol PINK Prestained Protein Ladder); M 2, protein marker (Thermo Scientific Unstained Protein Molecular Weight Marker, Thermo Fisher Scientific, Waltham, MA, USA). The results are representative of three independent experiments.
Oligomerisation status of vimentin variants under different conditions
We used the homogenous proteins to compare the oligomeric state of wild‐type vimentin to the Cys328‐modified vimentin variants under low salt conditions (5 mm TES, pH 7.5). Analytical ultracentrifugation confirmed the formation of stable tetrameric complexes for all of them. The s‐values were 4.9 S for Vim‐WT, 5.0 S for Vim‐SNO, and 4.7 S for Vim‐SSG, in line with previously published values for recombinant vimentin. Thus, neither nitrosylation nor glutathionylation affected the sedimentation behaviour of vimentin at low ionic strength (Fig. 2C).
In vitro, the assembly of vimentin tetramers into full‐length filaments can be initiated by the addition of monovalent salts. To investigate whether the modifications of vimentin at Cys328 influenced such assembly, we performed experiments with the three protein variants by adding potassium chloride to tetramer solutions in 5 mm TES buffer, pH 7.5 (final KCl concentration 0.1 m). After 2 or 16 h at room temperature (RT), the samples were precipitated by centrifugation at 22 000 g. SDS/PAGE was used to analyse the pellet (P), and supernatant (S) fractions for the salt‐treated samples in comparison with the same samples not initially challenged with potassium chloride (Fig. 3A, left two lanes in each panel).
Fig. 3.
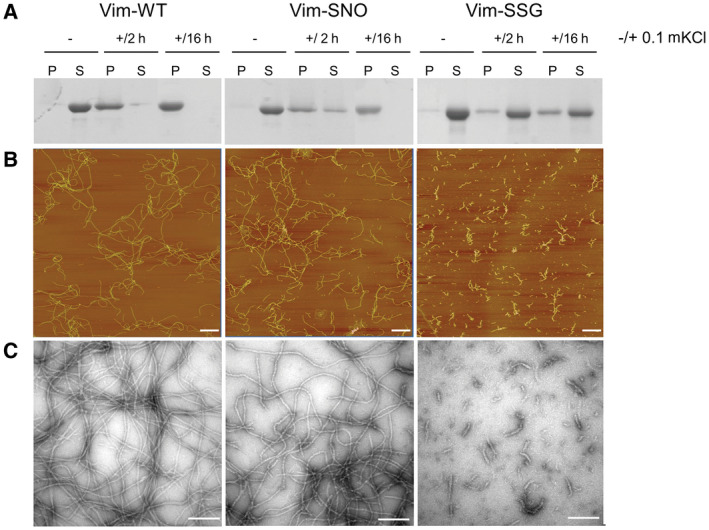
The influence of Cys328 nitrosylation and glutathionylation on vimentin filament assembly. (A) SDS/PAGE analysis of soluble (S) and insoluble (P) fractions obtained after centrifugation for Vim‐WT, Vim‐SNO and Vim‐SSG before salt addition (–), and after incubation with 0.1 m KCl for 2 h (+/2 h) as well as 16 h (+/16 h). (B) AFM and EM images (C) of Vim‐WT, Vim‐SNO and Vim‐SSG assembled for 2 h in the presence of 0.1 m KCl. Bars: (B) 1 μm; (C) 100 nm. The results are representative of the experiments performed in triplicate.
Presence of both Cys328 modifications impaired formation of insoluble vimentin oligomers in comparison with Vim‐WT. After 2 h in 0.1 m KCl, some part of Vim‐SNO was still present in the soluble fraction, and only after 16 h, total amount of the protein was in the pellet (middle panel). For Vim‐SSG, the assembly into insoluble filaments almost did not occur. Even after 16 h of incubation under assembly conditions, only a minor amount of Vim‐SSG was found in the pelleted fraction, showing just a slight increase from 2 to 16 h (right panel). In contrast, Vim‐WT fully sedimented already at 2 h with only traces of the protein left in the supernatant (left panel).
Visualisation of in vitro assembly products using atomic force microscopy and electron microscopy
To assess in more detail what are the products of in vitro vimentin assembly after Cys328 nitrosylation and glutathionylation, we visualised the appropriate pure protein samples after 2 h under assembly conditions using AFM and EM. While extended filaments readily formed for Vim‐WT (Fig. 3B,C, left panels), Vim‐SSG built only small structures (Fig. 3B,C, right panels). The obtained pictures further supported the idea that glutathionylation almost blocks the elongation step of the vimentin filament assembly. Unexpectedly, though Vim‐SNO oligomers formed less pellet when precipitated by centrifugation, AFM and EM images showed the formation of filaments with similar lengths to Vim‐WT but somewhat more tortuose (Fig. 3B,C, middle panels).
Filament‐length measurement of Vim‐WT for model‐derived assembly kinetics
To gain insight if the filament elongation process under the conditions used subsequently for HDex‐MS experiments agrees with the previously described kinetic model of vimentin assembly [28], we measured the mean length of the filaments of WT vimentin at 2 mg·mL−1 concentration at 22 °C in 5 mm TES pH 7.5, 0.1 m KCl (Fig. 4). We visualised vimentin structures by AFM at a shorter assembly time (5 min), because at the high protein concentration used here, at longer times filaments grew so long that they entangled when deposited onto the support and we could not accurately determine their length. Therefore, we employed a previously developed calculation of filament‐length growth over time. In particular, we predicted the time‐dependent filament growth by relying on the length distribution measured at a single time point for a distinct assembly condition. This approach yields an elongation constant for predicting filament length over any desired time period at the indicated temperature, buffer condition and protein concentration [28]. Figure 4A depicts a representative AFM image for the above conditions and shows a wide spectrum of short to long filaments with a mean length corresponding to 4 ULFs incorporated per filament, on average (i.e., ~ 190 nm). As filaments elongate by longitudinal annealing with each other, they reach an average length of 20 ULF by 2000 s (Fig. 4B). This elongation naturally corresponds to a decay in the number of filament ends (Fig. 4C), gradually lowering the probability of further end‐to‐end filament annealing.
Fig. 4.
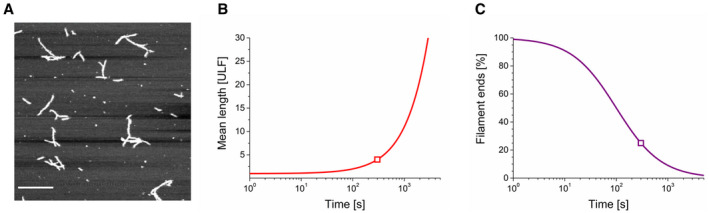
Filament‐length measurement of Vim‐WT and model‐derived assembly kinetics. (A) Vimentin (2 mg·mL−1) was assembled at 22 °C for 5 min, and filaments were visualised by AFM. Filaments exhibited a mean length of ~ 190 nm, which corresponds to 4.0 ULF integrated on average into filaments at this time point. (B) Calculation of the time‐dependent filament‐length generation over time (red line) with the help of the value determined at 5 min (red square) and the elongation constant described previously [28]. (C) Decay in the number of free filament ends (purple line) due to end‐to‐end joining of filaments during the assembly process. The values were calculated using the number of filaments determined in (B). The purple square indicates the time point of the filament‐length measurement and is equivalent to the red square in (B). Bar in (A): 1 μm. The results are representative of the experiments performed in duplicate.
HDex‐MS to study the kinetics of assembly of vimentin to filaments
The elongation process is mediated by a 3‐nm overlap reaction between the end of coil 2 of one filament and the start of coil 1 of a second filament. Thus, the tight interaction of these two ~ 20 amino acid segments reduces their domain mobility [1], an event that has indeed been quantitatively measured by HDex‐MS for vimentin assembly [27]. Here, we explored if a broader time period could be accurately investigated using HDex‐MS. In particular, we sought to track the effect of Cys328 PTMs located close to the assembly‐responsible domains on domain mobility and filament assembly kinetics.
To obtain insight into the mechanism by which the glutathionylation of a single cysteine residue affects the assembly process, we applied HDex‐MS. In this experiment, we monitored the time course of structural changes after the initiation of tetrameric vimentin assembly into higher‐order oligomeric states by rapid addition of KCl to a final concentration of 0.1 m. At various times after the salt jump, an aliquot of reaction mixture was withdrawn, and hydrogen–deuterium exchange was initiated by the addition of a tenfold volume of buffered D2O (Fig. 5). The H‐D exchange was terminated after 10 s by lowering the temperature and the pH with 2 m glycine, pH 2.5 of the samples. Such acidification sufficiently blocks the further exchange of hydrogen to deuterium. The aliquots were taken from the samples at several time points between 10 s and 16 h after the salt jump. Additionally, an aliquot was taken immediately after initiating the assembly (first row in Fig. 5) and assigned to t = 1 s.
Fig. 5.
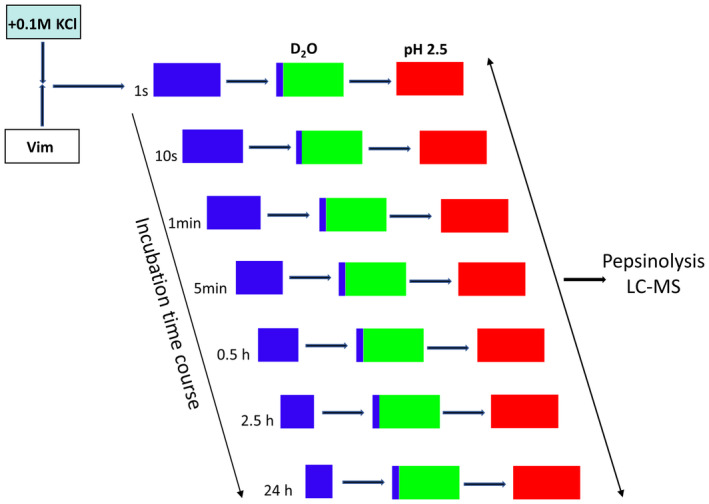
Schedule for the HDex‐MS experiment used to monitor domain dynamics during vimentin assembly. Blue squares mark neutral pH under H2O buffer conditions, green squares mark neutral pH under D2O buffer conditions, and red squares mark acidic pH conditions.
We have focused on monitoring the deuteration level of only three indicative peptides in the vimentin sequence (pep1A, pep2A, pep2B), as indicated in Fig. 1A. This selection was based on our earlier observation that exchange in these three regions is starkly different among tetramers, ULFs and filaments [27]. Therefore, the exchange level in these peptides can be used to follow the structural transitions accompanying filament formation. We observed that pep1A, which spans amino acids 108–117 (QELNDRFANY) in coil 1A, changes its status from unprotected to partly protected during ULF formation. Subsequently, the same segment becomes even more strongly protected upon elongation to filaments. Changes in pep1A are accompanied by increased protection of the peptide pep2A, which spans coil 2A segment 280–289 (AAKNLQEAEE) upon formation of ULF. Moreover, elongation also involves changes in the pep2B peptide, which spans the C‐terminal region of coil 2B from 400–408 amino acids (YRKLLEGEE).
Changes in deuteration of the three peptides after the salt jump are shown in Fig. 6A,B for Vim‐WT and Vim‐SSG, respectively. Exchange levels measured before the salt jump are indicated by a green horizontal line and correspond to the exchange value characteristic for tetrameric state. In the case of Vim‐WT (panel A), the exchange level in pep1A and pep2A decreases rapidly, and at the first time point of 10 s reaches 47% and 69%, instead of the 75% and 84%, respectively, observed in tetramers. Such values have been described before for ULFs or short filaments containing a few ULFs, in which these two regions become partly protected after tetramers associate laterally. Much smaller changes at the shortest times were observed for pep2B, in which the exchange is mainly sensitive to the ULF‐to‐filament transition step. At times longer than 10 s, in all three peptides, we observed the second stage of structural transformation, leading to much higher protection levels, that is higher structural stability in all three regions. These changes in protection follow the well‐known kinetics of the elongation step in which the end‐to‐end annealing of ULFs leads to decay of the free ULF ends and formation of filaments (Fig. 6A, red line showing decrease of filament ends; see Fig. 4).
Fig. 6.
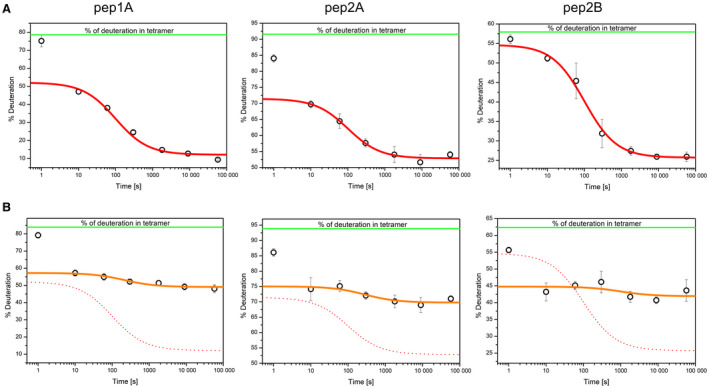
The course of HDex levels in three vimentin peptides that are diagnostic for filament assembly. HDex levels (% of deuteration) after 10 s of exchange in D2O of the pep1A peptide (left panel), pep2A (middle panel) and pep2B (right panel) in Vim‐WT (A) and Vim‐SSG (B), at 1 s, 10 s, 1 min, 5 min, 30 min, 2.5 h and 16 h after salt jump. In (A), the red lines represent the decay of the number of filament ends over time for Vim‐WT, as calculated using the elongation constant (see Fig. 4, right panel). The 1‐s time point was excluded from the fit (see Materials and methods). In (B), the orange lines depict the corresponding calculations for Vim‐SSG; the red dotted lines represent the data obtained for Vim‐WT as shown in (A). The error bars represent standard deviations of three independent experiments.
For Vim‐SSG (Fig. 6B), modification of Cys328 only weakly affected the changes in deuteration level observed at the shortest times in pep1A and pep2A. This result indicates that ULF formation seems to be undisturbed by this modification. However, a significant difference for Vim‐SSG was observed at later assembly times. We detected a strong stabilisation (30% less deuteration) in all three peptides for Vim‐WT, whereas for Vim‐SSG, we detected only a small increase in protection (2–4%). Such small changes in per cent of deuteration indicate that proper longitudinal contacts do not form in contrast to the contact regions for lateral assembly. Of interest, in comparison with Vim‐WT, in Vim‐SSG, the exchange in pep2B became significantly more decreased at 10 s of assembly. This result indicated that the glutathionylation of Cys328 induces premature stabilisation in this region already at the step when tetramers associate into ULFs, a possible reason for the inability to form the proper longitudinal contacts expected at the elongation step. Red lines in panel A and orange lines in panel B show that the HDex data obtained in this work mainly lie on the curves calculated depending on the elongation kinetic model of vimentin [28]. Thus, the direct quantitation of the change in domain mobility of the elongation‐mediating segments in coil 1A and coil 2B provides further strong support for the elongation model of vimentin, originally deduced from time‐lapse filament‐length measurements using EM [29].
The dependence of the in vitro assembly efficiency on the degree of vimentin glutathionylation
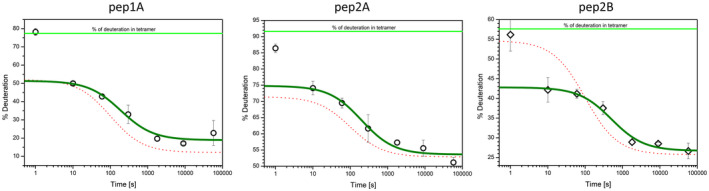
To assess how the degree of vimentin glutathionylation influences its assembly, we initially combined Vim‐SSG and Vim‐WT tetramers in a 1 : 1 molar ratio and treated the mixture with standard assembly conditions. Figure 7A shows a time dependence of HDex exchange levels for pep1A, pep2A and pep2B in such solution. The patterns of exchange suggest that if 50% of vimentin is glutathionylated, the ULFs form with almost the same efficiency as for pure Vim‐WT, but filaments elongate much slower. To find the relation between the percentage of vimentin glutathionylation and effectiveness of the ULF formation and elongation, we mixed individually prepared Vim‐SSG and Vim‐WT tetramers in several proportions and measured HDex protection levels after a single time point of 2.5 h after the salt jump. For such a time point, we saw significant effects in earlier experiments. Figure 7B presents data obtained in the titration experiment for all three marker peptides (black labels). For every Vim‐WT/Vim‐SSG mixture, red labels show calculated weighted sums of HDex exchange values measured separately for pure Vim‐WT and pure Vim‐SSG. Our data indicate that modification of only a fraction of cysteines by glutathionylation can significantly alter elongation rates of vimentin filaments. The exchange levels for each peptide increases with increasing amount of glutathionylated form. While for pep2A (Fig. 7B, middle panel) the HD exchange increases only slightly, for pep1A and pep2B the changes (Fig. 7B, left and right panels) are significant and indicate inhibition of filament longitudinal assembly. These changes are proportional to Vim‐SSG content but get even more efficient if there is more than 40% of Vim‐SSG.
Fig. 7.
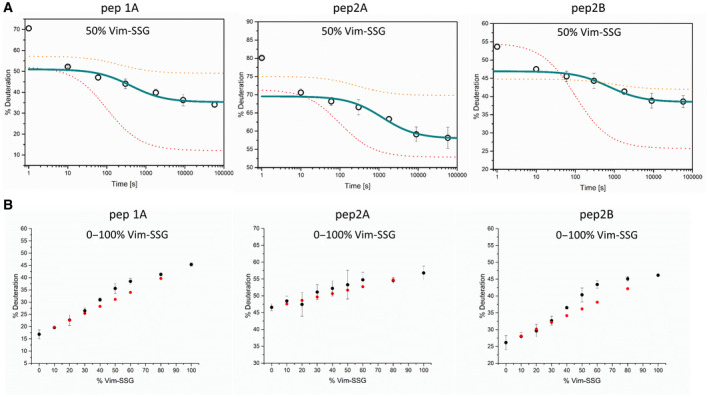
The impact of a different fraction of glutathionylated vimentin on vimentin assembly. (A) The course of HDex levels in three vimentin peptides in filaments assembled from a equimolar mixture of Vim‐WT and glutathionylated vimentin. HD exchange levels (% of deuteration) after 10 s of incubation in D2O of the peptides pep1A (left panel), pep2A (middle panel) and pep2B (right panel) in a 1 : 1 mixture of Vim‐WT and Vim‐SSG at 1 s, 10 s, 1 min, 5 min, 30 min, 2.5 h and 16 h after the salt jump. Marine lines represent the decay in the number of filament ends over time for the 1 : 1 mixture of Vim‐WT and Vim‐SSG calculated from the HDex data using the elongation constant described in [28]. The 1‐s time point was excluded from the calculation (see Materials and methods). The corresponding curves for the measurements of the individual components Vim‐WT (red dotted line) and Vim‐SSG (orange dotted line) are taken from Fig. 6A,B. (B) Dependence of HDex levels (% of deuteration after 10 s exchange) on increasing amount of glutathionylated vimentin after 2.5 h of standard assembly (black circles). Red circles represent calculated weighted sums of HD exchange level measured for pure Vim‐WT and Vim‐SSG proteins under the same conditions. The error bars represent standard deviations of three independent experiments.
The effects of glutathionylation/reduction on mature vimentin filaments
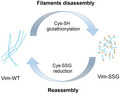
Post‐translational modification reactions such as protein kinase phosphorylation of extended filaments can disassemble them [30], so we investigated whether existing Vim‐WT filaments reorganise as a consequence of subsequent cysteine glutathionylation. For this purpose, we induced S‐glutathionylation of mature filaments with 5 mm oxidised glutathione (GSSG) and monitored the changes by AFM. The extended, several micrometre‐long filaments of Vim‐WT (Fig. 8A) were broken into short species clearly longer than a typical ULFs length (ca. 65 nm) with a maximum length of ca. 700 nm (Fig. 8B). To investigate the reversibility of this process, we treated the Vim‐SSG initially incubated in a standard assembly buffer with an efficient thiol reducing agent (TCEP). After Cys328 was reduced, incubation for 2 h was enough for vimentin to reorganise back into the longer IFs with a length up to even 3.5 µm (Fig. 8C).
Fig. 8.
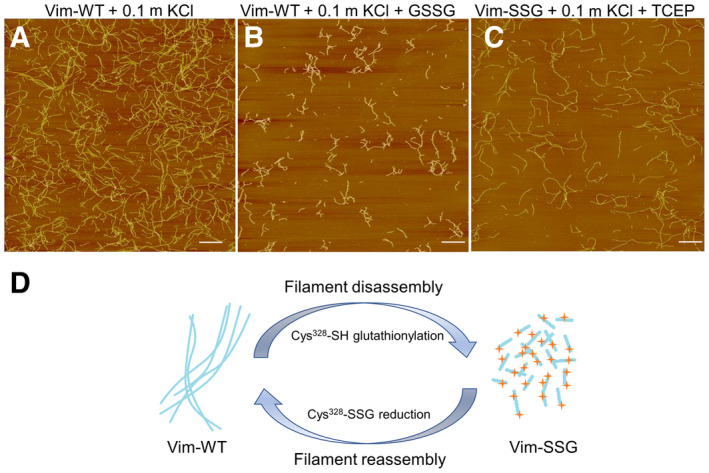
Remodelling of vimentin filaments by glutathionylation/reduction of Cys328. AFM images of Vim‐WT (2 mg·mL−1) assembled for 2 h under standard conditions (A) and treated subsequently with GSSG, leading to severed filaments (B). (C) AFM images of Vim‐SSG assembled under standard conditions after reduction of cysteines with TCEP, which restores unmodified vimentin. Scale bar: 1 µm. Panels (A), (B) and (C) are representative images that were consistently obtained from the experiments carried out in triplicate. (D) Schematic summary of the effect of glutathionylation/reduction on vimentin filaments.
We also used HDex‐MS for the three marker peptides to follow structural changes after addition of oxidised glutathione to filaments. When GSSG was added to Vim‐WT filaments, the deuteration levels increased in all three peptides during the incubation (Fig. 9, open navy circles). The deuteration of pep1A increased from roughly 10%, characteristic for filaments, to almost 50%. Pep2A and pep2B exhibited smaller changes, from 45% to 60% and from 22% to 48%, respectively (Fig. 9, middle and right panels). The observed changes in deuteration strictly followed the time course of the glutathionylation of vimentin at Cys328, which we monitored independently by measuring masses of peptides containing Cys328 (Fig. 9, left panel, orange trace). Thus, when GSSG is added after filament formation, the segments in pep1A, pep2A and pep2B become heavily deuterated, indicating that they are no longer protected, with time reaching values characteristic for ULFs or filaments consisting of a few ULFs. Because changes in HDex almost instantaneously follow the glutathionylation of cysteine, the filament fragmentation appears to occur even before a larger fraction of cysteines is glutathionylated.
Fig. 9.
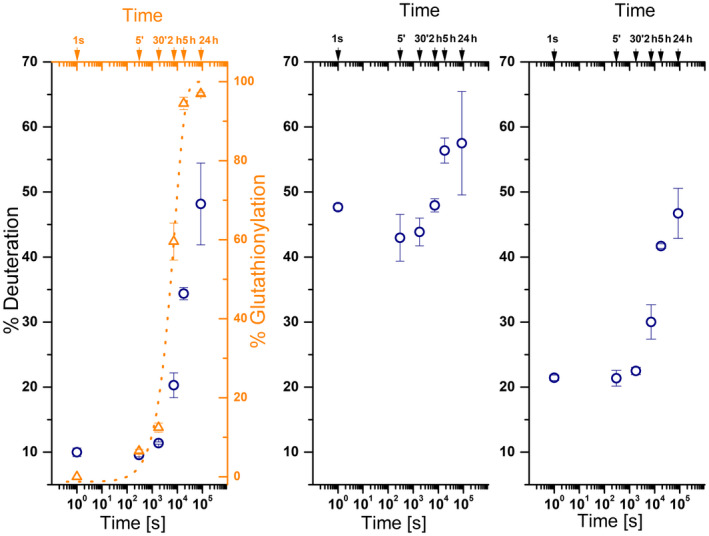
HDex‐MS study on vimentin disassembly. HD exchange levels (% deuteration – left vertical axes and navy circles) in the three marker vimentin segments as screened with pep1A (left panel), pep2A (middle panel) and pep2B (right panel), observed at different times (1 s, 5 min, 30 min, 2 h, 5 h, 24 h) after addition of oxidised glutathione to Vim‐WT filaments. The left panel (right vertical axis and open triangles, orange) shows the percentage of glutathionylated protein. Best fit to a single exponential transition between unmodified cysteine and modified cysteine is shown by an orange dotted line. The error bars represent standard deviations of three independent experiments.
HDex‐MS‐based kinetics used to study the effect of Cys328‐nitrosylation on vimentin assembly
To investigate at the molecular level whether the nitrosylation causes significant alterations with respect to domain dynamics, we subjected the protein to a detailed HDex‐MS analysis, focusing again on the three indicative peptides pep1A, pep2A and pep2B (Fig. 10).
Fig. 10.
The influence of Cys328‐nitrosylation on vimentin filament assembly. HD exchange levels (% deuteration) in the three marker vimentin segments pep1A (left panel), pep2A (middle panel) and pep2B (right panel), observed at different times after initiation of assembly of Vim‐SNO (black circles). Olive lines depict the decay of filament ends over time for Vim‐SNO as obtained from calculations using the elongation constant. Red dotted lines represent corresponding curves for Vim‐WT (Fig. 6A). Error bars represent the standard deviations of three independent experiments.
We observed that in Vim‐SNO, pep1A becomes slightly less protected than in Vim‐WT throughout the assembly process. It stayed more exchanged (19.7% versus 12.2%) even in filaments formed after a 16‐h incubation under standard assembly conditions (Fig. 10, left graph). Pep2A is less protected in Vim‐SNO during most of the incubation time, but after 16 h, the protection is nearly the same (Fig. 10, middle graph). Pep2B, like in Vim‐SSG, is more protected at 10 s of assembly (see deuteration: 42.4% in Vim‐SNO, as compared to 51.1% in Vim‐WT; Fig. 10, right graph), but at the final filamentous state, the protection is almost the same as for Vim‐WT. Over‐stabilisation of pep2B, similarly observed at earlier times in Vim‐SSG, is also present in Vim‐SNO, but in that case leads only to slower elongation. Thus, the effect of nitrosylation strongly contrasts with that of glutathionylation, and it would be interesting to explore the interplay of the two modifications in the cell.
Discussion
In this work, we demonstrate in an in vitro model system the consequences of S‐glutathionylation and S‐nitrosylation of the conserved, single cysteine 328 to the filament assembly of vimentin. In particular, for homogenous recombinant proteins, Vim‐WT, Vim‐SNO and Vim‐SSG, we compare the kinetics of salt jump–induced oligomerisation. We characterise the steps of both polymerisation and depolymerisation through measuring the HDex level at three protein regions (pep1A, pep2A and pep2B), which we previously described as critical for organisation of filament structure [27]. Our data show specific sensitivity of the oligomerisation kinetics to the modification status of vimentin's single cysteine thiol: (a) All three vimentin proteoforms studied here form stable tetrameric species under low salt conditions; (b) neither S‐nitrosylation nor S‐glutathionylation of Cys328 in tetramers alters the kinetics of lateral association of tetramers into ULFs; and (c) the presence of either thiol modification affects the kinetics of the longitudinal assembly of ULFs into long filaments. However, whereas Cys328‐nitrosylation only slows down the elongation step and the final assembly products are similar to wild‐type‐like filaments, the S‐glutathionylated vimentin cannot form the long filaments and exists only as soluble, short oligomers.
The precise molecular basis of the differential response of vimentin to various Cys328 thiol modifications is not yet well described. The oxidative PTMs diversely alter molecular mass, chemical reactivity, and charge and may impose different steric hindrance effects in the modified site. They also may induce distinct conformational and allosteric effects. Figure 11 represents a simplified scheme of vimentin oligomerisation which takes into account the possible steric effect of a bulky S‐glutathione group, where S‐glutathionylation does not disturb the pep1A/pep2A interaction that is characteristic for ULF formation but sterically interferes with the pep1A/pep2B interaction observed in filaments, thus impeding ULF elongation.
Fig. 11.
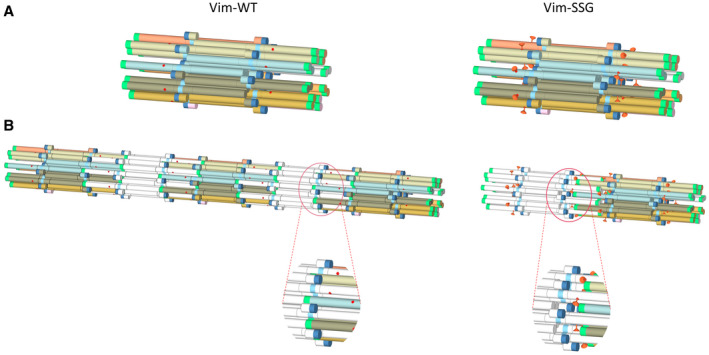
Proposed model of the steric effect of S‐glutathionylation (marked as orange pin, right panel) on the vimentin filamentation process in comparison with Vim‐WT (left panel). Contact sites between pep1A (dark blue) and pep2A (cyan) are not disturbed in Vim‐SSG when ULF is formed (A), but S‐glutathionylation severely blocks contacts between peptides pep1A and pep2B (green) during longitudinal assembly into filaments (B).
Cys328 of vimentin is a unique redox sensor. Preferential oxidation of vimentin, compared to other cytoskeletal proteins, was already reported in 1989 in cultured rheumatoid synoviocytes under mild oxidative stress. That study was the first to suggest that Cys328 oxidation may have a role in vimentin cytoskeleton rearrangement [31, 32]. Further research has shown that Cys328 fully exposes the thiol side chain to the solvent in the tetrameric structure and it does not form disulfide bonds between the cysteine thiols belonging to the same tetramer [33]. However, the Cys328 thiol is particularly reactive towards low‐molecular‐weight compounds, such as aldehyde 4‐hydroxynonenal [20], the antitumor and anti‐inflammatory prostaglandin PGA1 [34], the oxidative adduct malondialdehyde accumulated on the surface of senescence‐associated cells [35].
Perez‐Sala et al. recently demonstrated that some modifications at the Cys328 site affect vimentin assembly/disassembly in cells. In vivo effects of lipoxidation observed by these authors ranged from mild to drastic perturbations of filament assembly, depending on the nature of the thiol modifier. The same modifications altered filament morphology in vitro. They changed the length of the vimentin filaments obtained by in vitro assembly, but in contrast to what we have observed for vimentin S‐glutathionylation, did not lead to disassembly of preformed filaments [21]. Recently, it has been shown that covalent products of Cys328 thiol with exogenous compounds: ajoene from garlic, and withaferin A (WFA) from ayurvedic medicine drug, which both exert anti‐metastatic activity in cancer cells, disrupt vimentin network in cells [36, 37, 38].
S‐glutathionylation and S‐nitrosylation studied in this work are endogenous PTMs that have now been detected in vivo for multiple proteins thanks to advances in redox proteomics methods [39]. In particular, Vim‐SSG has been identified inside cells in oxidatively stressed human T lymphocytes, human red blood cell membrane‐bound proteins, and proteins secreted in glutathionylated form by macrophages stimulated with lipopolysaccharide (LPS) [23, 24, 40]. Treatment of living cells with diamide which induces vimentin glutathionylation [24] has led to disassembly of filaments and the appearance of vimentin dots [20].
Data presented in this work offer the first suggestion that S‐glutathionylation of Cys328 has the potential to serve as a reversible, redox‐regulated switch for synthesis‐independent turnover of vimentin from small soluble oligomers to the insoluble filaments and vice versa. Based on our in vitro studies, we envision that spatiotemporally selective S‐glutathionylation of vimentin could offer a way to modulate the severing of mature filaments into small soluble entities and their re‐annealing into new long filaments. These processes account for vimentin's role in cell migration, the epithelial–mesenchymal transition and its interaction with other cytoskeletal proteins, including actin and microtubules [41].
S‐nitrosylation of vimentin, other intermediate filament proteins (desmin, GFAP, NF‐H and α‐internexin), and more than 50 different cytoskeletal proteins have been detected in several physiologic and pathologic states (for a recent review, see Ref. [42]). Our results show that S‐nitrosylation does not have a significant effect on the kinetics of vimentin polymerisation. However, even the small inhibition of filament elongation observed in our studies cannot be disregarded because it may play a more subtle role in the regulation of vimentin assembly/disassembly. To date, S‐nitrosylation has been observed in more than 3000 proteins. It affects their function in different ways. A subset of cellular proteins is constitutively S‐nitrosylated and may serve as storage molecules for the quite reactive nitrogen oxide (NO) produced throughout the body [43]. Some protein SNOs are NO donors. By transnitrosylation, they transmit the NO group to other protein thiols that are distant from the local nitrosylation site inside or outside the cell [44]. Because oxidative modifications are mutually exclusive on a single cysteine thiol, the SNO group might serve to protect the thiol from derivatisation by other oxidative molecules. In contrast, unstable protein SNOs serve as active intermediates, enhancing the formation of further redox modifications, especially mixed disulfides such as the S‐glutathionylated proteins [45]. Which of the roles is played by the SNO in vimentin remains to be determined.
Previous results have indicated that cells simultaneously use S‐nitrosylation and S‐glutathionylation, together with other endogenous derivatives of a single regulatory cysteine, to process different redox‐related signals into distinct biological properties [46, 47]. Our data show substantially different consequences of S‐nitrosylation and S‐glutathionylation on vimentin assembly, especially the step of elongation of ULFs into long filaments. It would be interesting to determine if similar mechanism governs the role of other oxidative PTMs, for example the vimentin modifications formed with compounds used in natural medicine such as withaferin A and ajoene [37, 48]. Recently, there are several attempts to develop a redox proteomics methods which would analyse and quantify multiple redox PTMs of a single cysteine thiol in a protein [49]. Determining the relative levels of different thiol modifications of vimentin in living cells and finding out how they affect multiple oligomerisation equilibria states and the multiple biological functions could be of great importance in designing pharmacological approaches for treating vimentin assembly‐related disease [50]. The effect of S‐glutathionylation at a single position of the vimentin sequence also draws attention to the fact that even little modifications of a single amino acid may critically change the oligomerisation process. Thus, experimental approaches which use biotinylation or fluorescent labelling of the cysteine moiety, or analyses requiring substitutions of cysteine and its placement at other positions in the vimentin sequence, should be treated with much caution. We strongly suggest using the HDex‐MS method described in our work to prove that the modification, mutation and deletion of vimentin do not affect oligomerisation kinetics and thermodynamics.
To study the role of oxidative PTMs we have worked out a new, label‐free procedure to monitor in time the two significant steps of oligomerisation of vimentin: the lateral association of tetramers into ULFs, and ULF elongation into filaments. The new and unique feature of the procedure is that HDex‐MS measures the submicroscopic structural status at three small protein regions, which as we described in previous work are indicative of the structural status of an oligomer: tetramer, ULF, or filament [27]. This is in contrast to current methods that mostly rely on tracing oligomer sizes and shapes and thus are more sample concentration‐dependent. These methods also have limitations in characterising the oligomerisation at different time windows.
Some studies used subsecond times of the process, as in the case of SAXS measurements, combined with a fast mixing device when the rapid formation of ULFs followed the mixing of vimentin with high ionic strength buffer. But subsequent steps could not be tracked using this setup [51]. Lopez et al. used time‐resolved static light scattering and dynamic light scattering that characterise the hydrodynamic and gyration radii of molecules in solution quite well, along with the opportunity to model the mass distributions of oligomer families. These authors could follow the second step of the longitudinal growth of ULFs into filaments [52]. Time‐lapse EM and scanning force microscopy were used to examine mean filament length on timescales of seconds to minutes [53]. Viscosimetry enabled tracking of filament formation over minutes [54, 55], whereas near‐UV CD allowed tracking both steps over time, stressing the role of aromatic residues and the distinct radial compaction step [11]. Winheim et al. [12] observed the assembly of long filaments (2–3 days of incubation) from end‐to‐end annealing of shorter filaments. Recently, a stopped‐flow method enabled us to study the formation of ULFs on a time scale from milliseconds and determine the rate constant for the lateral assembly of ULFs, which occurs in a time frame below 1 s [10].
All kinetic parameters of vimentin assembly presented above depend on protein concentration, temperature and even mixing conditions. The experimental setup used in this study was established by us previously [27]. The temperature of the assembly reaction was lowered to 22 °C to slow down the kinetics of the process. To obtain optimal signal‐to‐noise ratios for MS spectra of most peptic peptides, the concentration of vimentin was set to 2 mg·mL−1 which is higher than physiological levels [56].
Under such conditions, we could observe the expected two main steps of the assembly and we could compare the main kinetic steps between WT and S‐modified variants of vimentin. Moreover, our data fit well to the kinetic models derived from experiments performed under standard assembly conditions as described in [28]. HDex‐MS provides also an opportunity to quantify the dynamics of depolymerisation of vimentin filaments and follow the degree of depolymerisation, as shown by disassembly of vimentin filaments in the presence of oxidised glutathione into shorter segments containing few ULFs.
In conclusion, we propose the HDex kinetics as a general method to compare numerous variants with mutations or PTMs, under various conditions and over a considerable time scale of seconds to days. The developed experimental setup allows to accurately determine and compare the influence of different factors on the kinetic parameters of the crucial steps in hierarchical oligomerisation of vimentin. We believe that this approach could be more generally applied to other filament‐forming IF proteins, which are challenging to study using classical methods.
Materials and methods
The reagents used during experimental procedures were purchased from Sigma Aldrich (St. Louis, MO, USA), unless stated otherwise. The experiments presented in this work were performed on different protein preparations.
Vimentin expression and purification
The expression of vimentin was performed in Escherichia coli TG1 cells, and purification was carried out following the method of Nagai and Thorgerson [57], as previously described [58].
Two millilitre of a preculture in TB medium (Terrific Broth; Formedium, Hunstanton, UK) was added to the Fernbach flask with 400 mL TB supplemented with ampicillin at 50 µg·mL−1 concentration. The expression of vimentin was performed at 37 °C for 16 h. The culture was centrifuged at 5000 g for 10 min at 4 °C, the supernatant was discarded and bacterial pellet was used for inclusion body purification. Subsequent stages of pellet treatment, before dissolving it in urea were carried out on the ice.
First, the pellet was resuspended in 8 mL of a Lysis Buffer containing 50 mm Tris/HCl pH 8.0, 25% sucrose, 1 mm EDTA and homogenised ten times in a precooled Dounce Homogenizer. The suspension was supplemented with 20 mg of lysozyme and incubated for 30 min, then 100 µL of 1 m MgCl2, 15 µL of RNAse A (6.25 mg·mL−1; Roche Diagnostic GmbH, Mannheim, Germany), 10 µL of DNAse I (Roche Diagnostic GmbH), 100 µL of a freshly prepared saturated PMSF (100 mg in 1.5 mL of ethanol) and 200 µL of Igepal CA‐630 were added and homogenised 10 times. The suspension was incubated for 10 min and then 20 mL of a Detergent Buffer (20 mm Tris/HCl pH 7.5, 200 mm NaCl, 1% sodium deoxycholate, 1% Igepal CA‐630, 2 mm EDTA) supplemented with 200 µL of 1 m DTT (Carl Roth GmbH, Karlsruhe, Germany), 300 µL of saturated PMSF and 100 µL of 50 mm Pefablock SC (SERVA Electrophoresis GmbH, Heidelberg, Germany) was added, homogenised and incubated for additional 10 min. The suspension was transferred to Nalgene tubes and centrifuged for 20 min at 7000 g. The supernatant was discarded and the pellet was resuspended in 20 mL of a Washing Buffer (10 mm Tris/HCl pH 8.0, 0.5% Triton X‐100) supplemented with 20 µL of 1 m DTT, 50 µL 50 mm Pefablock SC and 100 µL of saturated PMSF. The suspension was homogenised eight times, incubated 10 min and centrifuged again for 10 min at 7000 g. Again, supernatant was discarded, and the pellet was resuspended in 20 mL of a Washing Buffer containing 1.5 m KCl and additives (20 µL of 1 m DTT, 50 µL 50 mm Pefablock SC and 100 µL of saturated PMSF), homogenised eight times and incubated for 30 min, then centrifuged for 10 min at 7000 g. The pellet was washed with 20 mL of a Washing Buffer with 20 µL of 1 m DTT, 50 µL 50 mm Pefablock SC and 100 µL of saturated PMSF, homogenised, incubated for 10 min and centrifuged as before. Pellet was resuspended in 20 mL of a buffer containing 10 mm Tris/HCl pH 8.0, 0.1 mm EDTA, 20 µL of 1 m DTT, 50 µL 50 mm Pefablock SC and 100 µL of saturated PMSF. Again, after homogenisation, incubation and centrifugation the pellet was ready to be dissolved in 16 mL of a Urea Buffer that contain 9.5 m urea, 10 mm Tris/HCl pH 7.5, 5 mm EDTA supplemented with 160 µL of saturated PMSF and 160 µL of 1 m DTT. The solution was homogenised and finally centrifuged using a Beckman ultracentrifuge (Beckman Coulter Optima L‐90K, Brea, CA, USA, rotor 45 Ti) at 20 °C and 142 500 g. for 60 min. This time, the pellet was discarded and the supernatant containing vimentin was transferred to a Falcon tube. Methylamine hydrochloride (MAC) solution was added to a final concentration of 10 mm. The protein was frozen and kept at −20 °C for the further purification procedure.
Next day after thawing, the vimentin sample was diluted twice in a degassed Urea buffer II containing 8 m urea, 5 mm Tris/HCl, 1 mm EDTA, 0.1 mm EGTA pH 7.5 and 1 mm DTT. The protein sample was subjected to anion exchange chromatography with DEAE Sepharose Fast Flow (GE Healthcare Life Sciences, Chicago, IL, USA). About 25 mL of DEAE beads suspension was mixed with 25 mL of Urea Buffer II and degassed for 15 min. The beads were loaded onto a glass Econo‐column® from BIORAD (Hercules, CA, USA) and then were equilibrated with 50 mL of Urea Buffer II. Diluted vimentin sample was loaded onto the column and left for binding with gravity flow. The beads were washed with 50 mL of Urea buffer II. The protein was eluted with a salt gradient (0–0.3 m KCl in Urea Buffer II) using gradient mixer device with 50 mL of solution per one chamber. Fractions containing eluted protein were detected by Bradford reagent and then tested on a 12% SDS/PAGE and those with high protein concentration and reasonable purity were pooled and stored at −20 °C with 10 mm MAC. Next day after thawing, the protein sample was diluted three times with Urea Buffer II and applied for a cation exchange chromatography with CM Sepharose Fast Flow (GE Healthcare Life Sciences) with the same experimental setup like for DEAE Sepharose. The purity of eluted fractions was monitored on a 12% SDS/PAGE. Pure vimentin samples were pooled and the protein concentration was measured with Bradford assay with BSA as a standard. Vimentin sample with 10 mm MAC was aliquoted and kept at −80 °C before refolding procedures.
Vimentin refolding
The vimentin assembly into tetramers was carried out as described previously, with the exception that 5 mm TES, pH 7.5, was used as buffer instead of Tris/HCl or NaPi used in the previous studies [27, 59].
After thawing, 1 mL of a vimentin sample was applied onto a 2 mL Slide‐A‐Lyzer dialysis device (10 kDa MWCO; Thermo Scientific). The protein sample was first equilibrated in a buffer containing 8 m urea and 5 mm TES pH 7.5 and then subjected to step‐wise dialysis against 5 mm TES pH 7.5 containing 6 m urea, then 4 m urea, followed by 2 m urea with 1 mm DTT (each dialysis step performed for 20 min at RT) and 5 mm TES pH 7.5 with 1 mm DTT for 16 h at 4 °C. Finally, the dialysis was performed against 600 mL of 5 mm TES pH 7.5 for 2 h at RT. The protein was collected from the dialysis membrane, centrifuged at 22 000 g. for 10 min and applied for concentration in Millipore Centrifugal Filter Unit (10 kDa MWCO, 6 mL; Merck Millipore, Burlington, MA, USA). Protein stock was kept at 2 mg·mL−1 concentration at 4 °C.
Preparation of Cys328‐nitrosylated and Cys328‐glutathionylated forms of vimentin
To obtain Cys328‐nitrosylated vimentin (Vim‐SNO), reduced vimentin Vim‐WT (0.5 mg·mL−1) was incubated with 10 mm S‐nitroso‐glutathione (GSNO), pH 7.5, in the dark for 2 h at room temperature (RT). GSNO was prepared immediately before the S‐nitrosylation reaction by the incubation of 200 mm reduced glutathione (GSH) with 220 mm NaNO2 for 10 min at RT. S‐glutathionylated vimentin (Vim‐SSG) was obtained by incubating Vim‐WT (0.5 mg·mL−1) with 5 mm oxidised glutathione (GSSG) in 5 mm TES, pH 8.5, for 16 h. After the given time, both reactions were stopped by dilution of the samples with 5 mm TES, pH 7.5, and the remaining excess of glutathione derivatives was removed by buffer exchange in a centrifugal filter unit (10 kDa MWCO, 6 mL; Merck Millipore). The expected molecular mass after modification was confirmed by ESI‐MS (electrospray ionisation mass spectrometry). The stability was monitored by 12% SDS/PAGE. The final concentration of all protein samples was maintained at 2 mg·mL−1.
Filament assembly conditions
The assembly reaction of Vim‐WT, Vim‐SNO and Vim‐SSG in 5 mm TES, pH 7.5, was initiated by the addition of one tenth volume of 1 m KCl solution in 5 mm TES, pH 7.5. Incubation was for the specified time points at 22 °C, unless stated otherwise.
Analytical ultracentrifugation
Sedimentation velocity experiments were performed in a Beckman Coulter ProteomeLab XL‐I analytical ultracentrifuge (Indianapolis, IN, USA), equipped with AN 60Ti 4‐hole rotor and 12‐mm path length, double‐sector charcoal‐Epon cells, loaded with 400 μL of samples (0.5 mg·mL−1) and 410 μL of buffer (5 mm TES, pH 7.5). The experiments were carried out at 20 °C and 22 000 g. using the continuous scan mode and radial spacing of 0.003 cm. Scans were collected in 4‐min intervals at 280 nm. The fitting of absorbance versus cell radius data was performed using dcdt+ software, version 2.4.3 [60]. Biophysical parameters of the buffer, density and viscosity at 20 °C were measured using a Anton Paar DMA 5000 density metre (Anton Paar GmbH, Graz, Austria) and Lovis 2000 ME viscometer (Anton Paar GmbH). Protein partial specific volumes (V‐bars) were estimated using sednterp software (version 1.09, Spin Analytical, Durham, NH, USA).
Atomic force microscopy
All AFM experiments were performed in air as described previously [61]. For this purpose, 10 µL of the assembled vimentin samples (2 mg·mL−1) was diluted to 0.1 mg·mL−1 with assembly buffer (5 mm TES, pH 7.5, 0.1 m KCl), fixed by adding an equal sample volume of 0.2% glutaraldehyde in assembly buffer. The samples were applied to a freshly cleaved sheet of mica and allowed to adsorb for 10 s, and the specimen was washed with 0.5 mL of a fixing buffer to remove unbound species. After 45 s, the specimen was washed with distilled water and dried under a steady stream of nitrogen. HQ‐AU‐150 (Asylum Research, Oxford, UK) tips were used in AFM experiments. All images were captured in a tapping mode using an AFM Nanoscope V (Digital Instruments, Veeco, Santa Barbara, CA, USA) with software 7.30.
Electron microscopy
Transmission electron micrographs of unmodified and modified vimentin species assembled at 0.2 mg·mL−1 concentration for 2 h at 22 °C were collected after fixation with glutaraldehyde and negative staining with uranyl acetate, as described previously [59].
Hydrogen–deuterium exchange mass spectrometry
Hydrogen–deuterium exchange mass spectrometry experiments for Vim‐WT, Vim‐SNO and Vim‐SSG were performed as described previously [27]. Initially, the sequence coverage of vimentin by LC‐MS (liquid chromatography mass spectrometry) detected peptides was determined in H2O under the subsequently used HDex‐MS experimental conditions. Briefly, 5 µL of the protein stock (2 mg·mL−1) was diluted 10‐fold by the addition of 45 µL of 5 mm TES, pH 7.5, prepared in H2O. The sample was then acidified with 10 µL of 2 m glycine‐HCl, pH 2.5, and flash‐frozen before further analysis. The samples were hand‐thawed immediately before injection into the SYNAPT G2 High Definition Mass Spectrometry (Waters, Milford, MA, USA). First, samples were loaded onto an immobilised pepsin column (Poroszyme Immobilized Pepsin; ABI, Carlsbad, CA, USA) with 0.07% formic acid in water as a mobile phase (flow rate 200 µL·min−1). Peptides generated by the pepsin cleavage were captured on a 2.1 × 5 mm C18 trap column (ACQUITY BEH C18 VanGuard pre‐column, 1.7 μm resin; Waters) and resolved with an ACQUITY UPLC‐BEH C18 reversed‐phase column (1.0 × 50 mm, 1.7 μm; Waters) using a gradient of 6–40% acetonitrile in 0.1% (v/v) formic acid at a flow rate of 55 μL·min−1. All fluidics, valves and columns were kept at 0.5 °C, except for the pepsin column, which was kept at 20 °C inside the temperature‐controlled digestion compartment.
HDex‐MS experiments were performed for the unmodified and modified vimentin species in the absence or presence of 0.1 m KCl. All samples were subjected to the same experimental conditions as used for the sequencing step of nondeuterated samples described above, but H2O was replaced with D2O (99.9%; Cambridge Isotope Laboratories, Tewksbury, MA, USA).
Two control HDex‐MS experiments were carried out that took into account the minimum and maximum exchange of a given peptide. Briefly, to obtain a minimum exchange, D2O reaction buffer was added to the quenching buffer, cooled on ice before the addition of protein stock, and flash‐frozen before pepsin digestion and LC‐MS analysis. For the maximum exchange, 5 µL of protein stock was mixed with 45 µL of D2O reaction buffer, incubated overnight, and then mixed with quenching buffer and analysed as described above.
HDex‐MS data analysis
Deuterium uptake for each peptide resulting from the exchange was calculated using dynamx™ hdx data analysis Software 3.0 (Waters) based on the peptic peptide list obtained from the proteinlynx global server™ software (Waters, Milford, MA, USA) with the following acceptance criteria: minimum intensity threshold: 3000; and minimum products per amino acids: 0.3. Isotopic envelopes of peptides after exchange were analysed and corrected using dynamx 3.0. Final data were exported to Excel (Microsoft Office, Redmond, WA, USA). The percentage of sample deuteration was calculated with a formula that takes into consideration the minimum (M ex0) and maximum exchange (M ex100) of a given peptide:
Error bars for the percentage of deuteration D (%) were calculated and represented as standard deviations of three independent experiments. Final graphs were plotted using originpro 8.0 (OriginLab, Northampton, MA, USA) software.
Kinetics of filament assembly/disassembly monitored by HDex‐MS
Vimentin and its modified versions (2 mg·mL−1) were incubated with 0.1 m KCl at 22 °C. After specific time points (1 s, 10 s, 1 min, 5 min, 30 min, 2.5 h and 16 h), 5 µL of each sample was subjected to exchange in 5 mm TES, pH 7.5, buffer with 0.1 m KCl prepared in D2O for 10 s, quenched, flash‐frozen and then analysed by HDex‐MS as described above.
To interpret the changes in deuteration levels in three vimentin marker regions, the elongation kinetic model obtained before was used [28]. Only the 10 s and later time points were included in fitting procedures because they encompass the elongation time frame. Data collected for control just after mixing with D2O (marked 1 s) reflected low protection of marker regions in tetramers present in the sample during mixing, as expected, because the starting sample contained only tetramers. Therefore, this time point was not included in the fitting dataset.
To study vimentin depolymerisation by HDex‐MS, GSSG, pH 7.5, was added to each filament sample (final concentration, 5 mm) to induce glutathionylation of Cys328 of vimentin. After specific time points (5 min, 30 min, 2 h, 5 h, 24 h), 5 µL of each sample was diluted with D2O reaction buffer, left for 10 s, and quenched with low pH buffer (2 m glycine pH 2.5) on ice. After being flash‐frozen and then thawed, the HDex pattern of each sample was analysed as described above. The vimentin glutathionylation level at each time point was calculated from the MS spectrum of the vimentin fragment 299–408 that contains the modified cysteine 328 residue. The concentration ratio of unmodified and glutathionylated vimentin peptide was calculated based on the fraction of the MS peak intensities of modified and unmodified fragments. The sum of peak intensities did not change significantly during the experiment, so for clarity, it was assumed that protein glutathionylation does not significantly change the ESI efficiency for Cys‐containing peptides. Kinetics of glutathionylation of vimentin was calculated assuming a single exponential transition from reduced to modified vimentin.
Remodelling the vimentin filaments by glutathionylation/reduction of Cys 328 monitored by AFM
To study vimentin filament disassembly, 5 mm GSSG (pH 7.5) was added to the vimentin filament sample (2 mg·mL−1, 0.1 m KCl, 2 h at RT) and left for specific time points. For visualising the effect of cysteine reduction, TCEP neutral solution, pH 7.0, was added to Vim‐SSG and incubated for 2 h at RT under standard assembly conditions, to a final concentration of 7 mm and left for next 2 h. The samples were fixed and subjected to imaging as described above for AFM methodology. The length of vimentin structures was measured with imagej program (Research Services Branch, National Institute of Mental Health, Bethesda, MD, USA).
Conflict of interest
The authors declare no conflict of interest.
Author contributions
MK‐D designed research, performed the experiments, analysed data and wrote the paper; NM and RHS performed experiments and analysed data; TW, MC‐C and MP performed experiments; AW‐C, HH and MD designed research and wrote the paper; and AW‐C, MK‐D, HH and MD contributed to discussion.
Acknowledgements
We thank Jacek Olędzki for LC‐MS technical help. This work was supported by National Science Centre (MAESTRO 2014/14/A/NZ1/0030 and 2016/21/B/NZ1/02788), Foundation for Polish Science co‐financed by the European Union under the European Regional Development Fund (TEAM TECH CORE FACILITY/2016‐2/2) and German Research Foundation (DFG‐HE 185/11‐1 grant).
Contributor Information
Aleksandra Wysłouch‐Cieszyńska, Email: olawyslouch@ibb.waw.pl.
Michał Dadlez, Email: michald@ibb.waw.pl.
References
- 1. Herrmann H & Aebi U (2016) Intermediate filaments: structure and assembly. Cold Spring Harb Perspect Biol 8, a018242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Omary MB (2009) Review series introduction “ IF‐pathies ”: a broad spectrum of intermediate filament – associated diseases. J Clin Invest 119, 1756–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lazarides E (1982) Intermediate filaments: a chemically heterogeneous, developmentally regulated class of proteins. Annu Rev Biochem 51, 219–250. [DOI] [PubMed] [Google Scholar]
- 4. Schaffeld M, Herrmann H, Schultess J & Markl J (2001) Vimentin and desmin of a cartilaginous fish, the shark Scyliorhinus stellaris: sequence, expression patterns and in vitro assembly. Eur J Cell Biol 702, 692–702. [DOI] [PubMed] [Google Scholar]
- 5. Weckert U, Mücke N, Wedig T, Müller SA, Aebi U & Herrmann H (2005) Characterization of the in vitro co‐assembly process of the intermediate filament proteins vimentin and desmin: mixed polymers at all stages of assembly. Eur J Cell Biol 84, 379–391. [DOI] [PubMed] [Google Scholar]
- 6. Battaglia RA, Delic S, Herrmann H & Snider NT (2018) Vimentin on the move: new developments in cell migration. F1000Res 7, 1796 (1–10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eckes B, Dogic D, Colucci‐Guyon E, Wang N, Maniotis A, Ingber D, Merckling A, Langa F, Aumailley M, Delouvée A et al (1998) Impaired mechanical stability, migration and contractile capacity in vimentin‐ deficient fibroblasts. J Cell Sci 111, 1897–1907. [DOI] [PubMed] [Google Scholar]
- 8. Nieminen M, Henttinen T, Merinen M, Ichihara FM, Eriksson JE & Jalkanen S (2006) Vimentin function in lymphocyte adhesion and transcellular migration. Nat Cell Biol 8, 156–162. [DOI] [PubMed] [Google Scholar]
- 9. Satelli A & Li S (2011) Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci 68, 3033–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mücke N, Kammerer L, Winheim S, Kirmse R, Krieger J, Mildenberger M, Lara K, Norbert M, Baßler J, Hurt E et al (2018) Assembly kinetics of vimentin tetramers to unit‐length filaments : a stopped‐flow study. Biophys J 114, 2408–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Georgakopoulou S, Möller D, Sachs N, Herrmann H & Aebi U (2009) Near‐UV circular dichroism reveals structural transitions of vimentin subunits during intermediate filament assembly. J Mol Biol 386, 544–553. [DOI] [PubMed] [Google Scholar]
- 12. Winheim S, Hieb AR, Silbermann M, Surmann E‐M, Wedig T, Herrmann H, Langowski J & Mücke N (2011) Deconstructing the late phase of vimentin assembly by total internal reflection fluorescence microscopy (TIRFM). PLoS ONE 6, e19202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nöding B, Herrmann H & Köster S (2014) Direct observation of subunit exchange along mature vimentin intermediate filaments. Biophys J 107, 2923–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Snider NT & Omary MB (2014) Post‐translational modifications of intermediate filament proteins: mechanisms and functions. Nat Rev Mol Cell Biol 15, 163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Izawa I & Inagaki M (2006) Regulatory mechanisms and functions of intermediate filaments: a study using site‐ and phosphorylation state‐specific antibodies. Cancer Sci 97, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vossenaar ER (2004) Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann Rheum Dis 63, 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang L, Zhang J, Banerjee S, Barnes L, Sajja V, Liu Y, Guo B, Du Y, Agarwal MK, Wald DN et al (2010) Sumoylation of vimentin354 is associated with PIAS3 inhibition of glioma cell migration. Oncotarget 1, 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bornheim R, Müller M, Reuter U, Herrmann H, Büssow H & Magin TM (2008) A dominant vimentin mutant upregulates Hsp70 and the activity of the ubiquitin‐proteasome system, and causes posterior cataracts in transgenic mice. J Cell Sci 121, 3737–3746. [DOI] [PubMed] [Google Scholar]
- 19. Shirahata A, Sakata M, Sakuraba K, Goto T, Mizukami H, Saito M, Ishibashi K, Kigawa G, Nemoto H, Sanada Y et al (2009) Vimentin methylation as a marker for advanced colorectal carcinoma. Anticancer Res 29, 279–281. [PubMed] [Google Scholar]
- 20. Pérez‐Sala D, Oeste CL, Martínez AE, Carrasco MJ, Garzón B & Cañada FJ (2015) Vimentin filament organization and stress sensing depend on its single cysteine residue and zinc binding. Nat Commun 6, 7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mónico A, Pajares MA & Pérez‐sala D (2019) Vimentin disruption by lipoxidation and electrophiles: role of the cysteine residue and filament dynamics. Redox Biol 23, 101098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang B, Chen SC & Wang DL (2009) Shear flow increases S‐nitrosylation of proteins in endothelial cells. Cardiovasc Res 83, 536–546. [DOI] [PubMed] [Google Scholar]
- 23. Checconi P, Salzano S, Bowler L, Mullen L, Mengozzi M, Hanschmann E‐M, Lillig CH, Sgarbanti R, Panella S, Nencioni L et al (2015) Redox proteomics of the inflammatory secretome identifies a common set of redoxins and other glutathionylated proteins released in inflammation, influenza virus infection and oxidative stress. PLoS ONE 10, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E et al (2002) Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci USA 99, 3505–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stamler JS, Lamas S & Fang FC (2001) Nitrosylation: the prototypic redox‐based signaling mechanism. Cell 106, 675–683. [DOI] [PubMed] [Google Scholar]
- 26. Xiong Y, Uys JD, Tew KD & Townsend DM (2011) S‐glutathionylation: from molecular mechanisms to health outcomes. Antioxid Redox Signal 15, 233–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Premchandar A, Mücke N, Poznański J, Wedig T, Kaus‐Drobek M, Herrmann H & Dadlez M (2016) Structural dynamics of the vimentin coiled‐coil contact regions involved in filament assembly as revealed by hydrogen‐deuterium exchange. J Biol Chem 49, 24931–24950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mücke N, Winheim S, Merlitz H, Buchholz J, Langowski J & Herrmann H (2016) In vitro assembly kinetics of cytoplasmic intermediate filaments: a correlative monte carlo simulation study. PLoS ONE 11, e0157451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Herrmann H & Aebi U (1998) Intermediate filament assembly: fibrillogenesis is driven by decisive dimer‐dimer interactions. Curr Opin Struct Biol 8, 177–185. [DOI] [PubMed] [Google Scholar]
- 30. Yasui Y, Goto H, Matsui S, Manser E, Lim L, Nagata K & Inagaki M (2001) Protein kinases required for segregation of vimentin filaments in mitotic process. Oncogene 20, 2868–2876. [DOI] [PubMed] [Google Scholar]
- 31. Rogers KR, Morris CJ & Blake DR (1991) Oxidation of thiol in the vimentin cytoskeleton. Biochem J 275, 789–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rogers KR, Morris CJ & Blake DR (1989) Cytoskeletal rearrangement by oxidative stress. Int J Tissue React 11, 309–314. [PubMed] [Google Scholar]
- 33. Rogers KR, Herrmann H & Franke WW (1996) Characterization of disulfide crosslink formation of human vimentin at the dimer, tetramer, and intermediate filament levels. J Struct Biol 117, 55–69. [DOI] [PubMed] [Google Scholar]
- 34. Gharbi S, Garzn B, Gayarre J, Timms J & Perez‐Sala D (2007) Study of protein targets for covalent modification by the antitumoral and anti‐inflammatory prostaglandin PGA1: focus on vimentin. J Mass Spectrom 42, 1474–1484. [DOI] [PubMed] [Google Scholar]
- 35. Frescas D, Roux CM, Aygun‐Sunar S, Gleiberman AS, Krasnov P, Kurnasov OV, Strom E, Virtuoso LP, Wrobel M, Osterman AL et al (2017) Senescent cells expose and secrete an oxidized form of membrane‐bound vimentin as revealed by a natural polyreactive antibody. Proc Natl Acad Sci USA 114, E1668–E1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kaschula CH, Tuveri R, Ngarande E, Dzobo K, Barnett C, Kusza DA, Graham LM, Katz AA, Rafudeen MS, Parker MI et al (2019) The garlic compound ajoene covalently binds vimentin, disrupts the vimentin network and exerts anti‐metastatic activity in cancer cells. BMC Cancer 19, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bargagna‐Mohan P, Hamza A, Kim YE, Khuan Abby Ho Y, Mor‐Vaknin N, Wendschlag N, Liu J, Evans RM, Markovitz DM, Zhan CG et al (2007) The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem Biol 14, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee J, Liu J, Feng X, Salazar Hernandez MA, Mucka P, Ibi D, Choi JW & Ozcan U (2016) Withaferin A is a leptin sensitizer with strong antidiabetic properties in mice. Nat Med 22, 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang J, Carroll KS & Liebler DC (2016) The expanding landscape of the thiol redox proteome. Mol Cell Proteomics 15, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zaccarin M, Falda M, Roveri A, Bosello‐travain V, Bordin L, Maiorino M, Ursini F & Toppo S (2014) Quantitative label‐free redox proteomics of reversible cysteine oxidation in red blood cell membranes. Free Radic Biol Med 71, 90–98. [DOI] [PubMed] [Google Scholar]
- 41. Lowery J, Kuczmarski ER, Herrmann H & Goldman RD (2015) Intermediate filaments play a pivotal role in regulating cell architecture and function. J Biol Chem 290, 17145–17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Horenberg AL, Houghton AM, Pandey S, Seshadri V & Guilford WH (2019) S‐nitrosylation of cytoskeletal proteins. Cytoskeleton 76, 243–253. [DOI] [PubMed] [Google Scholar]
- 43. Furuta S (2017) Basal S‐nitrosylation is the guardian of tissue homeostasis. Trends Cancer 3, 744–748. [DOI] [PubMed] [Google Scholar]
- 44. Seth D, Hess DT, Hausladen A, Wang L, Wang Y, Stamler JS, Seth D, Hess DT, Hausladen A, Wang L et al (2018) Article A multiplex enzymatic machinery for cellular protein article a multiplex enzymatic machinery for cellular protein S‐nitrosylation. Mol Cell 69, 451–464.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wolhuter K, Whitwell HJ, Switzer CH, Burgoyne JR, Timms JF, Wolhuter K, Whitwell HJ, Switzer CH, Burgoyne JR & Timms JF (2018) Evidence against stable protein S‐nitrosylation as a widespread mechanism of post‐translational article evidence against stable protein S‐nitrosylation as a widespread mechanism of post‐translational regulation. Mol Cell 69, 438–450.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim SO, Merchant K, Nudelman R, Beyer WF, Keng T, Deangelo J, Hausladen A, Stamler JS, Carolina N & Carolina N (2002) OxyR: a molecular code for redox‐related signaling. Cell 109, 383–396. [DOI] [PubMed] [Google Scholar]
- 47. Dutka TL, Mollica JP, Lamboley CR, Weerakkody VC, Greening DW, Posterino GS, Murphy XRM & Lamb GD (2019) S‐nitrosylation and S‐glutathionylation of Cys134 on troponin I have opposing competitive actions on Ca 2+ sensitivity in rat fast‐twitch muscle fibers. Am J Physiol Cell Physiol 312, 316–327. [DOI] [PubMed] [Google Scholar]
- 48. Ridge KM, Shumaker D, Robert A, Hookway C, Gelfand VI, Janmey PA, Lowery J, Guo M, Weitz DA, Kuczmarski E et al (2016) Methods for determining the cellular functions of vimentin intermediate filaments. Methods Enzymol 568, 389–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Duan J, Gaffrey MJ & Qian WJ (2017) Quantitative proteomic characterization of redox‐dependent post‐translational modifications on protein cysteines. Mol Biosyst 13, 816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Danielsson F, Peterson M, Caldeira Araújo H, Lautenschläger F & Gad A (2018) Vimentin diversity in health and disease. Cells 7, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brennich ME, Nolting J‐F, Dammann C, Nöding B, Bauch S, Herrmann H, Pfohl T & Köster S (2011) Dynamics of intermediate filament assembly followed in micro‐flow by small angle X‐ray scattering. Lab Chip 11, 708–716. [DOI] [PubMed] [Google Scholar]
- 52. Lopez CG, Saldanha O, Huber K & Köster S (2016) Lateral association and elongation of vimentin intermediate filament proteins: a time‐resolved light‐scattering study. Proc Natl Acad Sci USA 113, 11152–11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kirmse R, Portet S, Mücke N, Aebi U, Herrmann H & Langowski J (2007) A quantitative kinetic model for the in vitro assembly of intermediate filaments from tetrameric vimentin. J Biol Chem 282, 18563–18572. [DOI] [PubMed] [Google Scholar]
- 54. Mücke N, Wedig T, Bürer A, Marekov LN, Steinert PM, Langowski J, Aebi U & Herrmann H (2004) Molecular and biophysical characterization of assembly‐starter units of human vimentin. J Mol Biol 340, 97–114. [DOI] [PubMed] [Google Scholar]
- 55. Herrmann H, Häner M, Brettel M, Müller SA, Goldie KN, Fedtke B, Lustig A, Franke WW & Aebi U (1996) Structure and assembly properties of the intermediate filament protein vimentin: the role of its head, rod and tail domains. J Mol Biol 264, 933–953. [DOI] [PubMed] [Google Scholar]
- 56. Goldman RD, Grin B, Mendez MG & Kuczmarski ER (2008) Intermediate filaments: versatile building blocks of cell structure. Curr Opin Cell Biol 20, 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nagai K & Thorgerson HC (1987) Synthesis and sequence‐specific proteolysis of hybrid proteins produced in Escherichia coli . Methods Enzymol 153, 461–481. [DOI] [PubMed] [Google Scholar]
- 58. Herrmann H, Hofmann I & Franke W (1992) Identification of a nonapeptide motif in the vimentin head domain involved in intermediate filament assembly. J Mol Biol 223, 637–650. [DOI] [PubMed] [Google Scholar]
- 59. Herrmann H, Kreplak L & Aebi U (2004) Isolation, characterization, and in vitro assembly of intermediate filaments. Methods Cell Biol 78, 3–24. [DOI] [PubMed] [Google Scholar]
- 60. Philo JS (2011) Limiting the sedimentation coefficient for sedimentation velocity data analysis: partial boundary modeling and g (s⁄) approaches revisited. Anal Biochem 412, 189–202. [DOI] [PubMed] [Google Scholar]
- 61. Mücke N, Kreplak L, Kirmse R, Wedig T, Herrmann H, Mu N, Aebi U & Langowski J (2004) Assessing the flexibility of intermediate filaments by atomic force microscopy. J Mol Biol 335, 1241–1250. [DOI] [PubMed] [Google Scholar]