

Abstract
Hereditary dentin disorders include dentinogenesis imperfecta (DGI) and dentin dysplasia (DD), which are autosomal dominant diseases characterized by altered dentin structure such as abnormality in dentin mineralization and the absence of root dentin. Shields classified DGI into three subgroups and DD into two subtypes. Although they are all hereditary dentin diseases, they do not share the same causative genes. To date, the pathogenic genes of DGI type I, which is considered a clinical manifestation of syndrome osteogenesis imperfecta, include COL1A1 and COL1A2. Mutations of the DSPP gene, which encodes the dentin sialophosphoprotein, a major non‐collagenous protein, are responsible for three isolated dentinal diseases: DGI‐II, DGI‐III, and DD‐II. However, DD‐I appears to be special in that researchers have found three pathogenicity genes―VPS4B,SSUH2, and SMOC2―in three affected families from different countries. It is believed that DD‐I is a genetically heterogeneous disease and is distinguished from other types of dentin disorders. This review summarizes the DD‐I literature in the context of clinical appearances, radiographic characteristics, and functions of its pathogenic genes and aims to serve clinicians in further understanding and diagnosing this disease.
Keywords: dentin dysplasia, pathogenic genes, SMOC2, SSUH2, VPS4B
1. INTRODUCTION
Heritable dentin defects are rare diseases affecting both deciduous and permanent teeth (Nieminen et al., 2011), which present with abnormally mineralized dentin. Based on clinical manifestations and imaging features, these diseases were classified into dentinogenesis imperfecta (three subtypes: DGI‐I, DGI‐II, and DGI‐III) and dentin dysplasia (two subtypes: DD‐I and DD‐II) (Shields, Bixler, & El‐Kafrawy, 1973). However, a large number of clinical studies have reported that teeth in DGI‐I, DGI‐II, DGI‐III, and DD‐II patients share analogous characteristics of amber translucent crowns accompanied by significant attrition, which extends even to the alveolar crest, with short and thin roots with obliterated pulp (Rafeek, Paryag, & Al‐Bayaty, 2013), while DD‐I teeth always appear normal shape and crown color, but are accompanied by sharp or absent root (Khandelwal & Likhyani, 2014). Subsequently, molecular biology and genetics studies have shown that DSPP is the pathogenic gene of DGI‐II, DGI‐III, and DD‐II, but not DD‐I. On account of the clinical manifestations and the mutant gene, investigators believe that the Shields classification is no longer applicable and have proposed a new classification: dentinogenesis imperfecta (DI, DSPP‐related disease) and radicular dentin dysplasia (Shields DD‐I) (de La Dure‐Molla, Philippe Fournier, & Berdal, 2015). DGI‐I is not involved in this new classification because it is not an independent dental disease but an oral manifestation of osteogenesis imperfecta. This new classification is consistent with the Mendelian Inheritance in Man (MIM) database, including DGI‐II (MIM 125490), DGI‐III (MIM 125500), DD‐I (MIM 125400), and DD‐II (MIM 125420). This new systematic classification system appears to be more scientific and reasonable.
Despite the fact that DD‐I is a special dentin genetic disease, there is no literature describing the disease in detail in terms of definition, classification, clinical features, etiology, diagnosis, differential diagnosis, or treatment. DD‐I is a rare disease with an autosomal dominant pattern of inheritance (Shields et al., 1973) that affects either the primary or both the primary and the secondary dentitions with an incidence of 1/100,000 (Kalk, Batenburg, & Vissink, 1998). The condition was first described as “rootless teeth” by Ballschmiede (Chamberlain & Hayward, 1983) in 1920; however, it was Rushton who named the condition “dental dysplasia” in 1939 (Malik, Gupta, Wadhwan, & Suhasini, 2015). Patients with DD‐I always present with either mobile teeth or pain associated with numerous periapical radiolucencies in non‐carious teeth (Khandelwal & Likhyani, 2014). In this review, we summarize the clinical appearance, histological performance, functions of the pathogenic genes, diagnostic criteria, differential diagnosis, and treatment of DD‐I to help clinicians gain a better understanding of this disease and to develop reasonable treatment plans.
2. CLINICAL AND HISTOLOGICAL DESCRIPTION
DD‐I teeth exhibit extreme mobility and generally premature exfoliation as a result of conical or absent roots (Ye et al., 2015), even though they are apparently normal in morphology and color. Other frequent symptoms and complaints, such as delayed dental eruption pattern, opaque incisal margins (Kalk et al., 1998; Scola & Watts, 1987), spontaneous exfoliation, and discomfort caused by severe tooth mobility, especially after meals (Ozer, Karasu, Aras, Tokman, & Ersoy, 2004), have also been reported. Based on its radiographic characteristics, DD‐I has been classified into four subtypes. Type 1a has no pulp chamber or root formation and always exhibits periapical radiolucency. Type 1b exhibits short roots a few millimeters in length, with single small crescent or herringbone shape pulp and periradicular radiolucencies. Type 1c has shortened inside roots, in which a central island of dentine is surrounded by horizontal or vertical crescent‐shape pulpal remnants and variable periapical radiolucencies. In addition to periapical radiolucencies, type 1d exhibits roots of normal length with a visible pulp chamber and large pulp stones located in the coronal portion of the canal (Carroll, Duncan, & Perkins, 1991; Shields, 1983).
Histologically, the affected teeth manifest normal coronal enamel with a thin layer of subjacent normal dentin (Sauk, Lyon, Trowbridge, & Witkop, 1972). Deeper layers of dentin exhibit an atypical tubular pattern with an amorphous, atubular area, and irregular arrangement. More centrally arranged dysplastic dentin masses may suggest multiple pulp calcifications (Rocha, Nelson‐Filho, Silva, Assed, & Queiroz, 2011; Toomarian, Mashhadiabbas, Mirkarimi, & Mehrdad, 2010). The dentinoenamel junction is either smooth or scalloped, and the scalloped structure is usually larger than the dentinoenamel junction of normal teeth. Ultrastructural study of the affected teeth reveals teardrop‐shape lacunae near the cervical enamel, fewer dental tubules, and irregular collagen fibers (Pintor et al., 2015; Ye et al., 2015).
3. IDENTIFICATION OF PATHOGENIC GENES IN DD
Investigators widely speculated about the pathogenesis of DD in earlier studies because the pathogenic gene involved in the disease was unknown. (Logan, Becks, Silverman, and Pindborg (1962) believed that variation of the dentin papillae caused abnormal tooth development, and calcification in the dentin papilla caused final occlusion of the pulpal space. Sauk et al. (1972) suggested the invagination of the root sheath occurred too soon during root development and, in a series of futile attempts to correct itself, resulted in ectopic dentin formation. Witkop (1975) proposed that internal cells of the developing dental organ would be displaced and proliferate in the dental papilla, producing ectopic dentin formation. On the other hand, Wesley, Wysoki, Mintz, and Jackson (1976) hypothesized that an ectopic interaction between odontoblasts and ameloblasts would occur, causing differentiation and/or abnormal function of the odontoblasts. Recently, three mutant genes have been detected in three affected pedigrees with different genetic modes. (Table 1, Figure 1).
Table 1.
Pathogenic genes of DD‐I
| Gene name and location | Mutant position | cDNA | Mutant type | Reference |
|---|---|---|---|---|
| SMOC2 (NM_022138, 6q27) | Intron 1 | c.84 + 1G>T | Splice mutation | Bloch‐Zupan et al. (2011) |
| VPS4B (NM_004869.3, 18q21.33) | Intron 7 | IVS7 + 46C>G | Splice mutation | Yang et al. (2016) |
| SSUH2 (NM_015931.2, 3p26.1) | Exon 2 | c.353C>A | Missense mutation | Xiong et al. (2017) |
Figure 1.
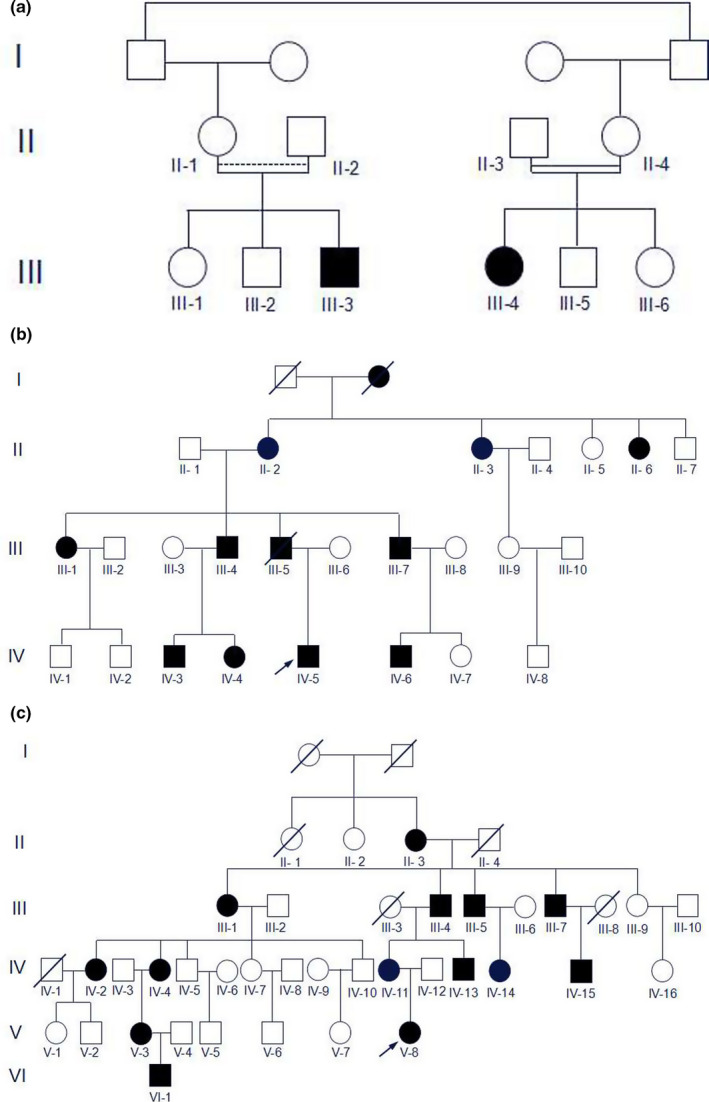
The pedigree maps of the three affected families. (a) Family with SMOC2 mutations. (b) Family with VPS4B mutations. (c) Family with SSUH2 mutations. The pedigree A is from the literature “Homozygosity Mapping and Candidate Prioritization Identify Mutations, Missed by Whole‐Exome Sequencing, in SMOC2, Causing Major Dental Developmental Defects” which is published in “The American Journal of Human Genetics” [Colour figure can be viewed at wileyonlinelibrary.com]
In 2011, Bloch‐Zupan reported a case of homozygous mutant SMOC2 in a family in which patients exhibited short roots similar to those in DD‐I teeth (Bloch‐Zupan et al., 2011). It is intriguing that the mode of inheritance in their pedigree was consistent with autosomal recessive rather than dominant. However, other scholars have also reported an autosomal recessive DD‐I family with no mutations in the SMOC2 gene (Cherkaoui Jaouad et al., 2013). The study has been stalled until our research team found two new DD‐I families. The patients of the two families manifested the typical clinical symptoms of normal or taurodontic teeth and deformed root with unexplained apices (Figure 2a–d). The results of scanning electron microscopy revealed a significantly reduced number of dentin tubules with smaller diameters (Figure 3a,b). In addition, we have found two mutant genes: VPS4B (Yang et al., 2016) in a family from northern China; and SSUH2 in a family from southern China (Xiong et al., 2017). Different mutations can result in the same disease which is highly suggestive evidence that the disease has genetic heterogeneity.
Figure 2.
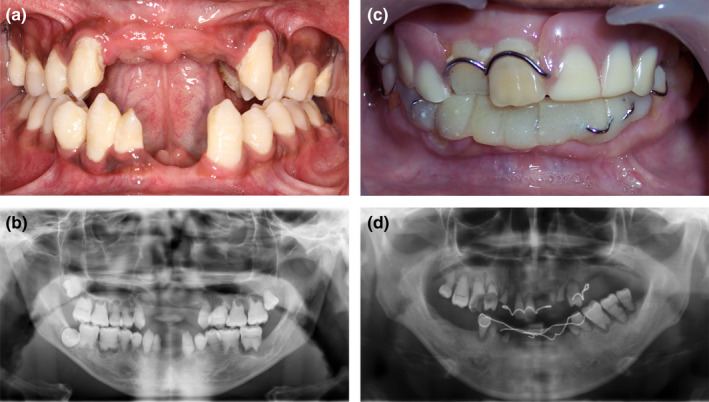
Clinical features of the patients. (a) Intraoral photo shows taurodontic teeth. (b) Panoramic radiograph revealed rootless teeth and periapical cysts. (c) A patient with normal crowns. (d) Panoramic radiograph revealed deformed root with unexplained apices
Figure 3.

SEM examination of the teeth specimens. (a) Regularly arranged dentin tubules were found in normal teeth (2000 × ). (b) Sparsely scattered dentin tubules with reduced diameter were found in DD‐I teeth (2000 × )
4. FUNCTIONAL AND PATHOGENIC MECHANISM OF THESE GENES
4.1. SMOC2
SMOC2 encompasses 14 exons encoding the secreted protein acidic and rich in cysteine (SPARC)‐related modular calcium‐binding protein‐2 (SMOC2) and has been mapped to chromosome 6q27. SMOC2, and is a member of the SPARC family, which is highly expressed during embryogenesis and wound healing (Vannahme, Gosling, Paulsson, Maurer, & Hartmann, 2003). As a matricellular protein, SMOC2 can promote matrix assembly and stimulate endothelial cell proliferation and migration as well as angiogenic activity (Bornstein & Sage, 2002). The encoded protein may serve as a target for controlling angiogenesis in tumor growth and myocardial ischemia. SMOC2 appeared to have a developmental function similar to that in the root of human teeth in a mouse model (Kim et al., 2016).
However, the specific function of SMOC2 in tooth development remains unknown. Two families with the SMOC2 mutation have been reported, and patients in both families usually exhibit extreme microdontia, oligodontia, dental shape anomalies, and short roots (Alfawaz et al., 2013). Using the well‐established morpholino knockdown technique, researchers found that the smoc2 gene affected the development of teeth by regulating the expression of related genes, including dlx2b, bmp2a, and pitx2 in zebrafish (Bloch‐Zupan et al., 2011).
4.2. VPS4B
Vacuolar protein sorting 4B (VPS4B) is encoded by the VPS4B gene, which is located on chromosome 18q21.33, and is a member of the AAA protein family (ATPases associated with diverse cellular activities). It is the homolog of the yeast VPS4 protein. VPS4B is a component of the endosomal sorting complexes required for transport (ESCRT) machinery (Gan & Gould, 2011), which has been shown to play an important role in the formation of multivesicular bodies (MVBs), virus budding (Watanabe et al., 2007), abscission of cytokinesis (Morita et al., 2010), and degradation of various membrane receptors (Jiang et al., 2015; Liu, Lv, et al., 2013). In addition, it also serves as a putative adaptor domain for the ESCRT‐III complex, which mediates the final abscission step of cytokinesis in mammals and archaea (Stuchell‐Brereton et al., 2007). The VPS4B gene is widely expressed in the human body, including pulp tissue (Bueno et al., 2011), and has been found to be a tumor suppressor gene that can regulate tumor progression. It is reported that VPS4B serves as a tumor suppressor in breast cancer via promoting the degradation of epidermal growth factor receptor (Lin et al., 2012). In addition, VPS4B can regulate the progression of non‐small‐cell lung cancer (Liu, Lv, et al., 2013), play a role in Parkinson's disease (Hasegawa et al., 2011), participate in neuronal apoptosis after cerebral ischemia in a middle cerebral artery occlusion model (Cui et al., 2014), and also facilitate intestinal epithelial cell apoptosis in Crohn's disease via the p38 MAPK signaling pathway (Zhang et al., 2015). Clearly, VPS4B is a highly multifunctional protein, although its expression and potential functions in tooth development remain unclear.
According to a study involving a family from northern China, the mutant gene was identified as VPS4B and was located on chromosome 18. Research has shown that the expression levels of VPS4B messenger RNA (mRNA) were significantly reduced in patients compared with normal controls. The overexpression of VPS4B is accompanied by a significant increase in the levels of CHMP4B, Wnt5a, and β‐catenin mRNA, while the knockdown of VPS4B inhibits the expression of CHMP4B, Wnta5a, and β‐catenin mRNA (Yang et al., 2016). Charged multivesicular body protein 4b (CHMP4B), a subunit of ESCRT‐III, in association with Wnt5a, activates the β‐catenin‐independent/Wnt pathway involved in cytokinesis (Fumoto, Kikuchi, Gon, & Kikuchi, 2012; McCullough, Fisher, Whitby, Sundquist, & Hill, 2008). The Wnt signaling pathway plays a pivotal role in the formation of root dentin and the cementum (Chen, Lan, Baek, Gao, & Jiang, 2009; Liu, Han, Wang, & Feng, 2013; Zhang et al., 2013). The VPS4B gene combined with CHMP4B can promote Wnt5a expression, thereby regulating the Wnt signaling pathway and controlling root formation (Figure 4). Experiments have shown that the knockdown of the vps4b gene in zebrafish can result in reduced tooth numbers and shorter teeth, mimicking the DD‐I phenotype. Simultaneously, the zebrafish mutant phenotype could be partially corrected by wild‐type human VPS4B mRNA; however, overexpression of wild‐type and mutant VPS4B mRNA had no significant effect on zebrafish tooth development (Yang et al., 2016). Therefore, it is speculated that the phenotype in zebrafish is caused by haploinsufficiency; however, the clinical pathogenesis in humans needs further investigation.
Figure 4.
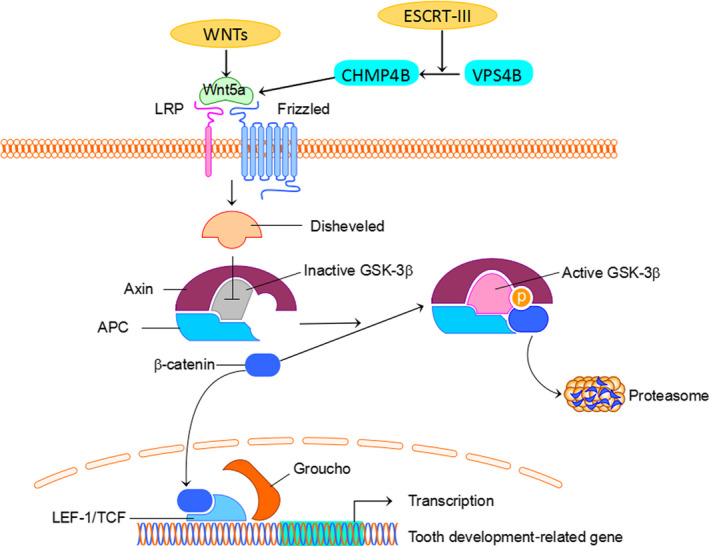
VPS4B affects tooth development‐related gene expression through Wnt/β‐catenin pathway. VPS4B regulation of CHMP4B stimulates Wnt5a, which associates with the receptor Frizzled and coreceptor LRP to trigger the Wnt signaling pathway. Wnt5a stimulates disheveled to inhibit GSK3β as well as prevent the phosphorylation of β‐catenin. Non‐phosphorylated β‐catenin induces the expression of related factors of odontoblast differentiation to further regulate root development
4.3. SSUH2
SSUH2, which is also known as SSU‐2, fls485, and C3orf32, maps to chromosome 3p26.1. It was first identified in a complementary DNA library prepared from fetal liver mRNA, which includes at least three open reading frames and is presumed to encode various translation products with different possible functions (Reinartz et al., 2010). However, no data concerning the functional relevance of SSUH2 are currently available. SSUH2 has been hypothesized to be a candidate tumor suppressor gene due to its close location to the uveal melanoma susceptibility locus UVM2 at 3p25 (Tschentscher et al., 2003). Additionally, it is also believed to be a possible chaperone with thiol‐disulfide oxidoreductase activity involved in the pathogenesis of celiac disease (Reinartz et al., 2010).
To identify essential transcription factors in dental development, specific primers with different junctions at the three mRNA spliceosomes were designed for RT‐PCR amplification. Only the transcript NM_015931.2 including a 1,803 base pair mRNA is highly expressed in human dental pulp stem cells and underjaw tissues (Xiong et al., 2017). This transcript corresponds to a putative chaperone protein (approximately 39 kDa), which is widely distributed (Reinartz et al., 2010). In addition, subcellular localization analysis shows that the SSUH2 protein is a nuclear protein and, as such, researchers hypothesize that it might be a transcription factor involved in transcriptional regulation or protein–protein interactions in tooth formation. Animal experiments have further clarified the pathogenic mechanism of this protein. In zebrafish, the incidence of agomphosis after morpholino knockdown of ssuh2 was higher than that in the control and could be partially rescued by the introduction of the wild‐type gene; meanwhile, the mutant ssuh2 gene downregulated the expression of three other reported tooth markers (dlx2b, bmp2a, and pitx2) (Xiong et al., 2017). Additionally, the expression of Bmp2 and Dlx2 was downregulated, while that of Dspp, Dmp1, Pax9, and Runx2 was dramatically upregulated in Ssuh2 +/− and Ssuh2 −/− mice (Xiong et al., 2017). These findings indicate that SSUH2 may participate in a dental development‐related signaling pathway involving several genes.
5. DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
In general, diagnosis is based on history, clinical manifestations, and radiographic features (Khandelwal & Likhyani, 2014). DD‐I teeth appear normal in morphology and color but show extreme mobility as a result of conical or absent roots. Radiographic features include sharp conical roots, pulp obliteration with crescent‐shape pulpal remnants parallel to the cementoenamel junction, or total obliteration. Numerous periapical radiolucencies in non‐carious teeth are diagnostic factor for this disorder.
DD‐I should be differentiated from DD‐II and DGI, which are characterized by amber translucent crown, and short and thin roots with obliterated pulp. In addition, some systemic disorders that exhibit features similar to dental DD‐I need to be differentiated. Disease involving skeletal anomalies, which is inherited as an autosomal dominant trait, is described by Morris and Augsburger (1977). These patients manifest not only the features of DD‐I, but also dense sclerotic bone and skeletal anomalies of the wrists and hand bones. Vitamin D‐dependent rickets type I (VDDR‐I) and vitamin D‐resistant rickets (VDRR) are characterized by a metabolic disturbance that causes defective calcification of mineralized structures. Patients with VDDR‐I exhibit not only abnormal teeth with short roots, large pulp chambers, and a widened predentin layer, but also short stature, skeletal abnormalities, genu valgum, rachitic rosary, open fontanels, pathologic fractures, muscle weakness, and convulsions (Zambrano et al., 2003). Features of VDRR include bowing of the legs, impaired growth, short stature, and dentin defects such as large pulp chambers, short roots, and abscessed non‐carious primary or permanent teeth (Souza et al., 2013). Chemotherapy or irradiation to the jaws during the period of root development leads to delayed root development and can exhibit radiographic features of DD‐I (Fayle, Duggal, & Williams, 1992). Familial tumoral calcinosis is a rare familial disorder characterized by masses of calcification in periarticular soft tissues. Teeth in patients manifest as short bulbous roots, obliterated pulp, and periapical radiolucencies in non‐carious teeth (Erbudak, Akkaya, Özbek, Hamdi Çelik, & Tatar, 2016).
6. TREATMENT AND PROGNOSIS
Treatment aims to remove infection, preserve existing teeth, enhance occlusion, and restore esthetics (Singh, Gupta, Yuwanati, & Mhaske, 2013). Due to the genetic heterogeneity of the disease, the treatment varies according to the severity of the problem and presenting complaints. Strategies should include conventional endodontic therapy, periapical curettage, and/or a preventive regimen (Khandelwal & Likhyani, 2014). However, extraction has also been suggested as a treatment option for teeth with pulp necrosis and periapical abscess (Malik et al., 2015). In the primary dentition, an overdenture can help maintain the occlusal vertical dimension. Treatment involving dental implants may be considered at approximately 18 years of age. Additionally, regular oral examination and routine conservative treatment, such as caries prevention, are essential (Ravanshad & Khayat, 2006). The outcome in patients with DD‐I always depends on his/her age and the severity of the disease. Early exfoliation of the teeth may lead to maxillomandibular atrophy in DD‐I. If the diagnosis can be made early and the treatment is appropriate, satisfactory esthetics and function can be achieved.
7. CONCLUSION
Dentinal genetic diseases have been described clinically for several years and have low incidence, similar to other hereditary pathologies. Elucidation of the genetic basis of dentin disorders should permit a better understanding of disease etiology, enabling improved classification, diagnosis, and treatment of this disease. By summarizing existing knowledge regarding the pathogenic genes of these diseases, de La Dure‐Molla proposed a new classification: DI (DSPP‐related disease) and radicular dentin dysplasia (Shield DD‐I). To date, three pathogenic genes have been found in different families, indicating that DD‐I has genetic heterogeneity. This feature can adequately explain the phenomenon that different patterns of inheritance exist in the affected families. Although DD‐I is a rare disease, clarifying its pathogenesis is necessary because dentin mineralization and root formation play important roles during tooth development. Although a series of pathogenic genes have been identified, detailed pathogenic mechanisms remain unclear. Further exploration of genetic function and signaling pathways is needed to acquire a comprehensive understanding of these diseases.
CONFLICT OF INTERESTS
None to declare.
AUTHOR CONTRIBUTIONS
Xiaocong Li composed the initial draft of the manuscript and figures. Dong Chen extended the text and edited the final manuscript. Fangli Lu and Yingying Wang edited the figures. Fu Xiong and Qiang Li reviewed and revised the manuscript to improve grammar and clarity.
ACKNOWLEDGEMENTS
This work was supported by a grant from the National Natural Science Foundation of China (Project no: 81470033) to Dong Chen and (Project no: 31371279) to Fu Xiong. The authors gratefully acknowledge Dr Jinhua Yu for his suggestions and help with structuring the article.
Chen D, Li X, Lu F, Wang Y, Xiong F, Li Q. Dentin dysplasia type I—A dental disease with genetic heterogeneity. Oral Dis. 2019;25:439–446. 10.1111/odi.12861
REFERENCES
- Alfawaz, S. , Fong, F. , Plagnol, V. , Wong, F. S. , Fearne, J. , & Kelsell, D. P. (2013). Recessive oligodontia linked to a homozygous loss‐of‐function mutation in the SMOC2 gene. Archives of Oral Biology, 58, 462–466. 10.1016/j.archoralbio.2012.12.008 [DOI] [PubMed] [Google Scholar]
- Bloch‐Zupan, A. , Jamet, X. , Etard, C. , Laugel, V. , Muller, J. , Geoffroy, V. , … Dollfus, H. (2011). Homozygosity mapping and candidate prioritization identify mutations, missed by whole‐exome sequencing, in SMOC2, causing major dental developmental defects. American Journal of Human Genetics, 89, 773–781. 10.1016/j.ajhg.2011.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein, P. , & Sage, E. H. (2002). Matricellular proteins: Extracellular modulators of cell function. Current Opinion in Cell Biology, 14, 608–616. 10.1016/S0955-0674(02)00361-7 [DOI] [PubMed] [Google Scholar]
- Bueno, D. F. , Sunaga, D. Y. , Kobayashi, G. S. , Aguena, M. , Raposo‐Amaral, C. E. , Masotti, C. , … Passos‐Bueno, M. R. (2011). Human stem cell cultures from cleft lip/palate patients show enrichment of transcripts involved in extracellular matrix modeling by comparison to controls. Stem Cell Reviews, 7, 446–457. 10.1007/s12015-010-9197-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, M. K. O. , Duncan, W. K. , & Perkins, T. M. (1991). Dentin dysplasia: Review of the literature and a proposed subclassification based on radiographic findings. Oral Surgery, Oral Medicine, and Oral Pathology, 72, 119–125. [DOI] [PubMed] [Google Scholar]
- Chamberlain, B. B. , & Hayward, J. R. (1983). Management of dentin dysplasia and facial disharmony. Special Care in Dentistry, 3, 113–116. 10.1111/j.1754-4505.1983.tb01615.x [DOI] [PubMed] [Google Scholar]
- Chen, J. , Lan, Y. , Baek, J. A. , Gao, Y. , & Jiang, R. (2009). Wnt/beta‐catenin signaling plays an essential role in activation of odontogenic mesenchyme during early tooth development. Developmental Biology, 334, 174–185. 10.1016/j.ydbio.2009.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkaoui Jaouad, I. , El Alloussi, M. , Laarabi, F. Z. , Bouhouche, A. , Ameziane, R. , & Sefiani, A. (2013). Inhabitual autosomal recessive form of dentin dysplasia type I in a large consanguineous Moroccan family. European Journal of Medical Genetics, 56, 442–444. 10.1016/j.ejmg.2013.05.003 [DOI] [PubMed] [Google Scholar]
- Cui, G. , Wang, Y. , Yu, S. , Yang, L. , Li, B. , Wang, W. , … Chen, D. (2014). The expression changes of vacuolar protein sorting 4B (VPS4B) following middle cerebral artery occlusion (MCAO) in adult rats brain hippocampus. Cellular and Molecular Neurobiology, 34, 83–94. 10.1007/s10571-013-9989-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Dure‐Molla, M. , Philippe Fournier, B. , & Berdal, A. (2015). Isolated dentinogenesis imperfecta and dentin dysplasia: Revision of the classification. European Journal of Human Genetics, 23, 445–451. 10.1038/ejhg.2014.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbudak, H. , Akkaya, N. , Özbek, M. , Hamdi Çelik, H. , & Tatar, I. (2016). Dental findings of hyperphosphatemic familial tumoral calcinosis. Oral Radiology, 33(1), 65–70. 10.1016/0003-9969(73)90075-7 [DOI] [Google Scholar]
- Fayle, S. A. , Duggal, M. S. , & Williams, S. A. (1992). Oral problems and the dentist's role in the management of paediatric oncology patients. Dental Update, 19(152–6), 158–159. [PubMed] [Google Scholar]
- Fumoto, K. , Kikuchi, K. , Gon, H. , & Kikuchi, A. (2012). Wnt5a signaling controls cytokinesis by correctly positioning ESCRT‐III at the midbody. Journal of Cell Science, 125, 4822–4832. 10.1242/jcs.108142 [DOI] [PubMed] [Google Scholar]
- Gan, X. , & Gould, S. J. (2011). Identification of an inhibitory budding signal that blocks the release of HIV particles and exosome/microvesicle proteins. Molecular Biology of the Cell, 22, 817–830. 10.1091/mbc.E10-07-0625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, T. , Konno, M. , Baba, T. , Sugeno, N. , Kikuchi, A. , Kobayashi, M. , … Takeda, A. (2011). The AAA‐ATPase VPS4 regulates extracellular secretion and lysosomal targeting of alpha‐synuclein. PLoS ONE, 6, e29460 10.1371/journal.pone.0029460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, D. , Hu, B. , Wei, L. , Xiong, Y. , Wang, G. , Ni, T. , … Lu, C. (2015). High expression of vacuolar protein sorting 4B (VPS4B) is associated with accelerated cell proliferation and poor prognosis in human hepatocellular carcinoma. Pathology, Research and Practice, 211, 240–247. 10.1016/j.prp.2014.11.013 [DOI] [PubMed] [Google Scholar]
- Kalk, W. W. , Batenburg, R. H. , & Vissink, A. (1998). Dentin dysplasia type I: Five cases within one family. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics, 86, 175–178. 10.1016/S1079-2104(98)90121-4 [DOI] [PubMed] [Google Scholar]
- Khandelwal, S. G. D. , & Likhyani, L. (2014). A case of dentin dysplasia with full mouth rehabilitation: A 3‐year longitudinal study. International Journal of Clinical Pediatric Dentistry, 7, 119–124. 10.5005/jp-journals-10005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. H. , Kim, S. , Shin, Y. , Lee, H. S. , Jeon, M. , Kim, S. O. , … Song, J. S. (2016). Comparative gene expression analysis of the coronal pulp and apical pulp complex in human immature teeth. Journal of Endodontics, 42, 752–759. 10.1016/j.joen.2016.01.024 [DOI] [PubMed] [Google Scholar]
- Lin, H. H. , Li, X. , Chen, J. L. , Sun, X. , Cooper, F. N. , Chen, Y. R. , … Ann, D. K. (2012). Identification of an AAA ATPase VPS4B‐dependent pathway that modulates epidermal growth factor receptor abundance and signaling during hypoxia. Molecular and Cellular Biology, 32, 1124–1138. 10.1128/MCB.06053-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Han, D. , Wang, L. , & Feng, H. (2013). Down‐regulation of Wnt10a affects odontogenesis and proliferation in mesenchymal cells. Biochemical and Biophysical Research Communications, 434, 717–721. 10.1016/j.bbrc.2013.03.088 [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Lv, L. , Xue, Q. , Wan, C. , Ni, T. , Chen, B. , … Mao, G. (2013). Vacuolar protein sorting 4B, an ATPase protein positively regulates the progression of NSCLC via promoting cell division. Molecular and Cellular Biochemistry, 381, 163–171. 10.1007/s11010-013-1699-2 [DOI] [PubMed] [Google Scholar]
- Logan, J. , Becks, H. , Silverman, S. Jr , & Pindborg, J. J. (1962). Dentinal dysplasia. Oral Surgery, Oral Medicine, and Oral Pathology, 15, 317–333. 10.1016/0030-4220(62)90113-5 [DOI] [PubMed] [Google Scholar]
- Malik, S. , Gupta, S. , Wadhwan, V. , & Suhasini, G. P. (2015). Dentin dysplasia type I ‐ A rare entity. Journal of Oral and Maxillofacial Pathology, 19, 110 10.4103/0973-029X.157220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough, J. , Fisher, R. D. , Whitby, F. G. , Sundquist, W. I. , & Hill, C. P. (2008). ALIX‐CHMP4 interactions in the human ESCRT pathway. Proceedings of the National Academy of Sciences of the United States of America, 105, 7687–7691. 10.1073/pnas.0801567105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita, E. , Colf, L. A. , Karren, M. A. , Sandrin, V. , Rodesch, C. K. , & Sundquist, W. I. (2010). Human ESCRT‐III and VPS4 proteins are required for centrosome and spindle maintenance. Proceedings of the National Academy of Sciences of the United States of America, 107, 12889–12894. 10.1073/pnas.1005938107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, M. E. , & Augsburger, R. H. (1977). Dentine dysplasia with sclerotic bone and skeletal anomalies inherited as an autosomal dominant trait. A new syndrome. Oral Surgery, Oral Medicine, and Oral Pathology, 43, 267–283. 10.1016/0030-4220(77)90163-3 [DOI] [PubMed] [Google Scholar]
- Nieminen, P. , Papagiannoulis‐Lascarides, L. , Waltimo‐Siren, J. , Ollila, P. , Karjalainen, S. , Arte, S. , … Alaluusua, S. (2011). Frameshift mutations in dentin phosphoprotein and dependence of dentin disease phenotype on mutation location. Journal of Bone and Mineral Research, 26, 873–880. 10.1002/jbmr.276 [DOI] [PubMed] [Google Scholar]
- Ozer, L. , Karasu, H. , Aras, K. , Tokman, B. , & Ersoy, E. (2004). Dentin dysplasia type I: Report of atypical cases in the permanent and mixed dentitions. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics, 98, 85–90. 10.1016/j.tripleo.2004.01.005 [DOI] [PubMed] [Google Scholar]
- Pintor, A. , Alexandria, A. , Marques, A. , Abrahao, A. , Guedes, F. , & Primo, L. (2015). Histological and ultrastructure analysis of dentin dysplasia type I in primary teeth: A case report. Ultrastructural Pathology, 39, 281–285. 10.3109/01913123.2014.1002960 [DOI] [PubMed] [Google Scholar]
- Rafeek, R. N. , Paryag, A. , & Al‐Bayaty, H. (2013). Management of dentinogenesis imperfecta: A review of two case reports. General Dentistry, 61, 72–76. [PubMed] [Google Scholar]
- Ravanshad, S. , & Khayat, A. (2006). Endodontic therapy on a dentition exhibiting multiple periapical radiolucencies associated with dentinal dysplasia Type 1. Australian Endodontic Journal, 32, 40–42. 10.1111/j.1747-4477.2006.00008.x [DOI] [PubMed] [Google Scholar]
- Reinartz, A. , Ehling, J. , Franz, S. , Simon, V. , Bravo, I. G. , Tessmer, C. , … Gassler, N. (2010). Small intestinal mucosa expression of putative chaperone fls485. BMC Gastroenterology, 10, 27 10.1186/1471-230X-10-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha, C. T. , Nelson‐Filho, P. , Silva, L. A. , Assed, S. , & Queiroz, A. M. (2011). Variation of dentin dysplasia type I: Report of atypical findings in the permanent dentition. Brazilian Dental Journal, 22, 74–78. 10.1590/S0103-64402011000100013 [DOI] [PubMed] [Google Scholar]
- Sauk, J. J. Jr , Lyon, H. W. , Trowbridge, H. O. , & Witkop, C. J. Jr (1972). An electron optic analysis and explanation for the etiology of dentinal dysplasia. Oral Surgery, Oral Medicine, and Oral Pathology, 33, 763–771. 10.1016/0030-4220(72)90444-6 [DOI] [PubMed] [Google Scholar]
- Scola, S. M. , & Watts, P. G. (1987). Dentinal dysplasia type I. A subclassification. British Journal of Orthodontics, 14, 175–179. 10.1179/bjo.14.3.175 [DOI] [PubMed] [Google Scholar]
- Shields, E. D. (1983). A new classification of heritable human enamel defects and a discussion of dentin defects. Birth Defects Original Article Series, 19, 107–127. [PubMed] [Google Scholar]
- Shields, E. D. , Bixler, D. , & El‐Kafrawy, A. M. (1973). A proposed classification for heritable human dentine defects with a description of a new entity. Archives of Oral Biology, 18, 543–553. [DOI] [PubMed] [Google Scholar]
- Singh, A. , Gupta, S. , Yuwanati, M. B. , & Mhaske, S. (2013). Dentin dysplasia type I. BMJ Case Reports, 2013, bcr2013009403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza, A. P. , Kobayashi, T. Y. , Lourenco Neto, N. , Silva, S. M. , Machado, M. A. , & Oliveira, T. M. (2013). Dental manifestations of patient with vitamin D‐resistant rickets. Journal of Applied Oral Science, 21, 601–606. 10.1590/1679-775720130249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuchell‐Brereton, M. D. , Skalicky, J. J. , Kieffer, C. , Karren, M. A. , Ghaffarian, S. , & Sundquist, W. I. (2007). ESCRT‐III recognition by VPS4 ATPases. Nature, 449, 740–744. 10.1038/nature06172 [DOI] [PubMed] [Google Scholar]
- Toomarian, L. , Mashhadiabbas, F. , Mirkarimi, M. , & Mehrdad, L. (2010). Dentin dysplasia type I: A case report and review of the literature. Journal of Medical Case Reports, 4, 1 10.1186/1752-1947-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschentscher, F. , Husing, J. , Holter, T. , Kruse, E. , Dresen, I. G. , Jockel, K. H. , … Zeschnigk, M. (2003). Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Research, 63, 2578–2584. [PubMed] [Google Scholar]
- Vannahme, C. , Gosling, S. , Paulsson, M. , Maurer, P. , & Hartmann, U. (2003). Characterization of SMOC‐2, a modular extracellular calcium‐binding protein. The Biochemical Journal, 373, 805–814. 10.1042/bj20030532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, T. , Sorensen, E. M. , Naito, A. , Schott, M. , Kim, S. , & Ahlquist, P. (2007). Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proceedings of the National Academy of Sciences of the United States of America, 104, 10205–10210. 10.1073/pnas.0704000104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley, R. K. , Wysoki, G. P. , Mintz, S. M. , & Jackson, J. (1976). Dentin dysplasia type I. Clinical, morphologic, and genetic studies of a case. Oral Surgery, Oral Medicine, and Oral Pathology, 41, 516–524. 10.1016/0030-4220(76)90279-6 [DOI] [PubMed] [Google Scholar]
- Witkop, C. J. Jr (1975). Hereditary defects of dentin. Dental Clinics of North America, 19, 25–45. [PubMed] [Google Scholar]
- Xiong, F. , Ji, Z. , Liu, Y. , Zhang, Y. , Hu, L. , Yang, Q. , … Xu, X. (2017). Mutation in SSUH2 causes autosomal‐dominant dentin dysplasia type I. Human Mutation, 38, 95–104. 10.1002/humu.23130 [DOI] [PubMed] [Google Scholar]
- Yang, Q. , Chen, D. , Xiong, F. , Chen, D. , Liu, C. , Liu, Y. , … Xu, X. (2016). A splicing mutation in VPS4B causes dentin dysplasia I. Journal of Medical Genetics, 53, 624–633. 10.1136/jmedgenet-2015-103619 [DOI] [PubMed] [Google Scholar]
- Ye, X. , Li, K. , Liu, L. , Yu, F. , Xiong, F. , Fan, Y. , … Chen, D. (2015). Dentin dysplasia type I‐novel findings in deciduous and permanent teeth. BMC Oral Health, 15, 163 10.1186/s12903-015-0149-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano, M. , Nikitakis, N. G. , Sanchez‐Quevedo, M. C. , Sauk, J. J. , Sedano, H. , & Rivera, H. (2003). Oral and dental manifestations of vitamin D‐dependent rickets type I: Report of a pediatric case. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics, 95, 705–709. 10.1067/moe.2003.116 [DOI] [PubMed] [Google Scholar]
- Zhang, D. , Wang, L. , Yan, L. , Miao, X. , Gong, C. , Xiao, M. , … Tang, Q. (2015). Vacuolar protein sorting 4B regulates apoptosis of intestinal epithelial cells via p38 MAPK in Crohn's disease. Experimental and Molecular Pathology, 98, 55–64. 10.1016/j.yexmp.2014.12.007 [DOI] [PubMed] [Google Scholar]
- Zhang, R. , Yang, G. , Wu, X. , Xie, J. , Yang, X. , & Li, T. (2013). Disruption of Wnt/beta‐catenin signaling in odontoblasts and cementoblasts arrests tooth root development in postnatal mouse teeth. International Journal of Biological Sciences, 9, 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]