

Abstract
Objective
To determine the clinical and molecular features in patients with amyotrophic lateral sclerosis 4 (ALS4) due to mutations in the senataxin (SETX) gene and to develop tools for evaluating SETX variants.
Methods
Our study involved 32 patients, including 31 with mutation in SETX at c.1166 T>C (p.Leu389Ser) and 1 with mutation at c.1153 G>A (p.Glu385Lys). Clinical characterization of the patients included neurological examination, blood tests, magnetic resonance imaging (MRI), and dual‐energy x‐ray absorptiometry (DEXA). Fibroblasts and motor neurons were obtained to model the disease and characterize the molecular alteration in senataxin function.
Results
We report key clinical features of ALS4. Laboratory analysis showed alteration of serum creatine kinase and creatinine in the Leu389Ser ALS4 cohort. MRI showed increased muscle fat fraction in the lower extremities, which correlates with disease duration (thigh fat fraction R 2 = 0.35, p = 0.01; lower leg fat fraction R 2 = 0.49, p < 0.01). DEXA measurements showed lower extremities are more affected than upper extremities (average fat z scores of 2.1 and 0.6, respectively). A cellular assay for SETX function confirmed that like the Leu389Ser mutation, the Glu385Lys variant leads to a decrease in R loops, likely from a gain of function.
Interpretation
We identified clinical laboratory and radiological features of ALS4, and hence they should be monitored for disease progression. The molecular characterization of R‐loop levels in patient‐derived cells provides insight into the disease pathology and assays to evaluate the pathogenicity of candidate mutations in the SETX gene. ANN NEUROL 2020;87:547–555
Amyotrophic lateral sclerosis (ALS) is a collection of neurodegenerative disorders characterized by loss of upper and lower motor neurons. A subgroup of ALS has been found to be caused by genetic mutations.1 Among the mutated genes are those that encode RNA processing proteins, including TDP‐43, FUS, hnRNPA1, and hnRNPA2B1.2, 3, 4 Identification of these mutations has improved our understanding of how RNA synthesis and post‐transcriptional processing affect motor neuron pathophysiology.
There is a range of manifestations in ALS, including age of onset and rate of progression. The heterogeneity poses diagnostic challenges and impedes patient care. As genetic testing has become more available, more diagnoses of genetic forms of the disease have been made. However, interpretation of the results is not always simple. Even when the results are clear, most of the genetic forms of ALS are rare, and the clinical course and manifestations are not well characterized. This lack of information hampers genetic counseling and assessment of disease prognosis, and it limits evaluation of outcome in clinical trials.
A genetic form of ALS that faces these challenges is ALS4, an autosomal dominant disorder caused by mutations in the senataxin (SETX) gene.5, 6, 7, 8 SETX is a large gene that encodes a 303 kD helicase, which resolves RNA–DNA hybrids. It is often difficult to determine whether a particular sequence variant is pathogenic, as affected family members may not be available for segregation analysis or the variant may be a de novo mutation. Here we examined and followed patients from 1 large ALS4 pedigree and 1 unrelated patient to characterize the clinical signs, identify biomarkers, and develop a molecular assay to assess the functional effects of sequence variants in SETX.
Patients and Methods
Patient Population
SETX mutations were confirmed in 31 patients with the mutation Leu389Ser and 1 patient with the variant Glu385Lys. All subjects received clinical evaluations at the National Institutes of Health (NIH) in Bethesda, Maryland, under institutional review board–approved protocol 00‐N‐0043 Clinical and Molecular Manifestations of Inherited Neurological Disorders. Written informed consent was received from all participants before inclusion in the study. Laboratory testing and clinical evaluations were done at the NIH clinical center. Muscle strength was evaluated using the Medical Research Council (MRC) scale.
Radiological Testing
Seventeen ALS4 patients (10 female, 7 male) underwent T1‐weighted 1.5‐T magnetic resonance imaging (MRI) of the leg at the NIH clinical center. Nine of these subjects (5 female, 4 male) had follow‐up MRI. Images for thigh muscle quantification included a 10cm region around the midpoint between the head of the femur and the cartilage meniscus of the knee. For the lower leg, the quantified images included a 6cm region around a point one‐third of the distance from the cartilage meniscus of the knee to the articular cartilage of the tibial–talus joint. A semiautomated code with Fiji software was used to quantify the area of contractile (hypointense) and noncontractile (hyperintense) tissue with the “auto local threshold” function and the Phansalkar method for each image sequence.9 Manual image segmentation of tissue area was done for bone marrow, compact bone, and subcutaneous adipose tissue in each image. Fat fraction was calculated as the ratio of noncontractile tissue to total tissue area in the thigh muscle and leg muscle compartments.
Dual‐energy x‐ray absorptiometry (DEXA) whole‐body composition measurements were obtained at NIH Clinical Center Radiology, and z scores were calculated using normative data from the National Health and Nutrition Examination Survey dataset.10
Motor Neuron Characterization
Induced pluripotent stem cells (iPSCs) were generated from dermal fibroblasts from biopsy of the ventral forearm from 2 healthy controls. Fibroblasts were cultured as previously described.11 A third control iPSC line (WTC11) was obtained from the Coriell Institute for Medical Research. Stem cells were differentiated into motor neuron–like cells by stable insertion of an inducible transcription factor cassette expressing neurogenin‐2 (NGN2), islet‐1 (ISL1), and LIM homeobox 3 (LHX3; hNIL) into the CLYBL safe harbor locus as previously described.12 Differentiation was initiated using Dulbecco modified Eagles medium (DMEM)/F12 containing N2 supplement, nonessential amino acids, L‐glutamine, 10μM ROCK inhibitor, 0.2μM compound E, and 2μg/ml doxycycline. After 2 days of differentiation, the cells were dissociated with accutase and plated on poly‐l‐ornithine–coated dishes in DMEM/F12 media containing N2 supplement, nonessential amino acids, L‐glutamine, B27 supplement, 10ng/ml brain‐derived neurotrophic factor, and 1μg/ml laminin (density of 10,000 cells per well of a 96‐well plate for TGF‐β characterization). The cells were treated with TGF‐β (20ng/ml; R&D Systems, Minneapolis, MN) beginning at 2 days of differentiation with 50% media changes at 2‐day intervals. Morphologic analysis was done on images acquired with a 20× objective and Ti‐E motorized fluorescence microscope (Nikon Instruments, Melville, NY). Nikon's NIS‐Element Advanced Research (AR) software was used to mask the cell somas and neurites. Neurite length and number of branch points were quantified from 5 wells (25 images/well) of a 96‐well plate for each sample and normalized to the soma count.
Senataxin Assays
Control fibroblasts were transfected with plasmids corresponding to wild type (WT) and SETX variants for 1 day before collection and S9.6 dot blot. Samples for immunofluorescence were fixed 2 days after transfection with 4% paraformaldehyde for 15 minutes at room temperature, and motor neurons were fixed for 10 minutes with 4% paraformaldehyde containing 4% sucrose and then quenched in 100mM glycine for 5 minutes. Fixed cells were then washed 3 times with phosphate‐buffered saline (PBS) as previously described.11 The cells were stained with the following antibodies: S9.6 antibody (gift from Stephen Leppla), 1:500 HB9 antibody (Developmental Studies Hybridoma Bank, Iowa City, IA), 1:500 SETX (Novus Biological, Littleton, CO), or 1:2,000 SMI‐32 (Abcam, Cambridge, UK). Quantification of staining by ImageJ was done by observers blinded to the genotypes of the samples.
S9.6 Dot Blot
Genomic DNA was phenol extracted, ethanol precipitated, and reconstituted in 12μl Tris‐EDTA buffer. Then 2μl DNA solution (100ng) was loaded to Hybond N+ nylon membrane (GE Healthcare Life Sciences, Issaquah, WA) presoaked with PBS and crosslinked in UV Stratalinker 2400 (Stratagene, La Jolla, CA) at the “Auto Crosslink” setting (1,200μJ × 100). The membrane was blocked in 5% milk in Tris‐buffered saline–0.05% Tween‐20 for 1 hour and incubated with 1:1,000 S9.6 antibody overnight at 4°C to detect RNA–DNA hybrids. A duplicate blot was incubated with anti–double stranded (ds) DNA antibody (Abcam) as loading control. The dsDNA, dsRNA, and RNA–DNA hybrid controls were annealed using synthetic oligos reported previously.13 Single‐stranded (ss) RNA, top strand: 5′‐UGGGGGCUCGUCCGGGAUAUGGGAACCACUGAUCCC‐3′; ssDNA, top strand: 5′‐TGGGGGCTCGTCCGGGATATGGGAACCACTGATCCC‐3′; ssDNA, bottom strand: 5′‐GGGATCAGTGGTTCCCATATCCCGGACGAGCCCCCA‐3′; and ssRNA, bottom strand: 5′‐GGGAUCAGUGGUUCCCAUAUCCCGGACGAGCCCCCA‐3′.
Messenger RNA Expression and Western Blot Analysis
RNA was extracted and analyzed as previously described.11 Briefly, 1μg total RNA was converted to complementary DNA using the High Capacity cDNA Reverse Transcriptase kit (Applied Biosystems, Foster City, CA), and BAMBI expression levels were evaluated with TaqMan probe (Applied Biosystems; Hs03044164_m1) with HPRT1 TaqMan probe (Hs02800695_m1) as a control. Western blots were done with antibodies for SETX (Bethyl laboratories, Montgomery, TX), pSMAD2, and SMAD2/3 (Cell Signaling Technology, Danvers, MA), vinculin (Abcam), and lysates prepared in radioimmunoprecipitation assay buffer (50mM Tris pH 8, 150mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate).
Results
Genetic Evaluation
Thirty of the 31 subjects were members of a large pedigree.6 All affected members were confirmed to have the Leu389Ser mutation in SETX. Another unrelated patient was evaluated for a history of difficulty with walking since age 4 years, necessitating the use of leg braces at age 8 years. She had developed hand weakness at age 10 years. Genetic testing identified a Glu385Lys (heterozygous c.1153 G>A) change close to the previously described SETX mutation. The variant was not present in the Exome Aggregation Consortium (ExAC) sequencing database and predicts substitution of an amino acid conserved across mammalian species (Fig 1A, B). We previously showed that the Leu389Ser mutation does not affect senataxin expression.11 We found similar SETX protein expression and cellular localization in Glu385Lys patient cells as in cells from 2 unrelated controls (see Fig 1C, D).
Figure 1.
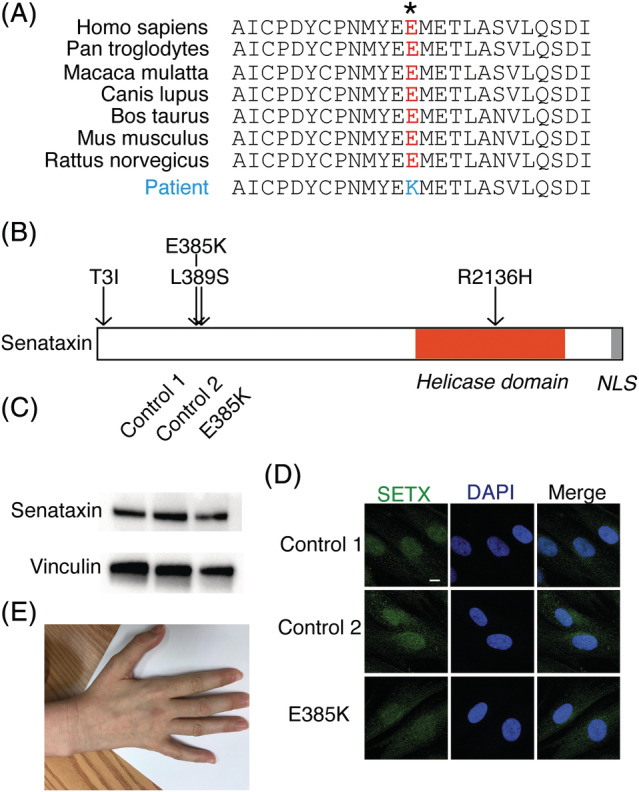
Glu385Lys mutation in senataxin (SETX). (A) The amino acid sequence at position 385 of SETX is conserved across species. (B) Map of helicase domain and nuclear localization signal (NLS) showing the location of the 3 previously known mutations causing amyotrophic lateral sclerosis 4 (Tyr3Ile, Leu389Ser, and Arg2136His) and the Glu385Lys variant. (C) Western blot of SETX from fibroblast cultures of 2 controls and the Glu385Lys patient. Vinculin was used as a loading control. (D) Immunofluorescence staining for SETX (green) in 2 controls and patient fibroblasts; 4′,6‐diamidino‐2‐phenylindole (DAPI) staining in blue. Scale bar = 5μm. (E) Atrophy in the interossei muscles of the hand and involuntary abduction of the fifth finger.
Physical Examination
The demographics of the 31 patients (14 male, 17 female) with Leu389Ser ALS4 in this study are shown in the Supplementary Table. The average age at the time of evaluation was 42 years. Sixty‐five percent of patients reported having at least 1 fall within the past year. The average age of onset of weakness was 16 years. On physical examination, a majority of patients (87%) was found to have hyperreflexia, and 7 (23%) had a Babinski reflex. Manual muscle strength testing using the MRC scale was done on all 31 patients, including 14 who had additional follow‐up visits over an average of 3 years. Muscle weakness was found in 24 patients (77%) and muscle wasting in 19 (61%). The pattern of weakness was mostly symmetric, with evidence of asymmetry (greater than 1 unit MRC difference between the left and right side) in only 3 patients. Of the 24 patients with extremity weakness, 14 had both proximal and distal involvement, and the remaining 10 had only a distal distribution. None of the patients had proximal weakness only. With increasing disease duration, we found progressive reduction in ankle dorsiflexion (R 2 = 0.31, p < 0.0001) and plantar flexion (R 2 = 0.12, p < 0.01). Seven patients had decreased sensation to at least 1 modality with a distal pattern of involvement. Three patients reported mild symptoms of dysphagia with infrequent choking, and no patients were found to have dysarthria. Only 1 patient was found to have bulbar weakness with involvement of the orbicularis oris and hypoglossal muscles. Additionally, evidence of ataxia with dysmetria or past pointing in the upper extremities was found in about half of the patients. Evidence of ocular apraxia was detected in only 2 patients.
The patient with Glu385Lys SETX variant was found to have atrophy and weakness in her distal upper and lower extremities (see Fig 1E). She was unable to extend her fingers against gravity and had no movement in her ankles or toes. Reflexes were brisk diffusely in her upper and lower extremities, and plantar reflexes were absent.
Laboratory Findings
Laboratory analysis showed a significant increase in serum creatine kinase (CK) in both males (ALS4 mean 420U/l ± 105 [standard error of the mean (SEM)], n = 11; vs control mean 177U/l ± 3, n = 1,141; p < 0.0001) and females (ALS4 mean 182U/l ± 42, n = 11; vs control mean 106U/l ± 3, n = 491; p < 0.0001). The male subjects also had a significant decrease in creatinine levels compared to healthy controls (ALS4 mean 0.56mg/dl ± 0.09, n = 8; vs control mean 1.02mg/dl ± 0.03, n = 73; p < 0.0001). There were no significant differences in the serum sex hormone levels in the male or female Leu389Ser ALS patients compared to controls. The Glu385Lys ALS4 patient had normal CK and reduced creatinine levels (0.33mg/dl).
Electrophysiology
Nerve conduction studies and electromyography in 13 patients with Leu389Ser and 1 patient with Glu385Lys showed prolonged distal motor latencies, decreased amplitudes of compound muscle action potential, and chronic neurogenic changes. A length‐dependent sensory neuropathy was detected in 1 patient.
Radiographic Findings
To determine the extent of muscle damage, we used quantitative MRI to measure fat replacement of skeletal muscle. T1‐weighted scans of the lower extremities were performed on 17 patients. The results showed a positive correlation between disease duration and muscle fat fraction in the thigh (R 2 = 0.35, p = 0.01) and lower leg (R 2 = 0.49, p < 0.01; Fig 2) consistent with muscle atrophy. We measured bone densitometry and lean body mass in 12 subjects with DEXA. Half the patients were found to have reduced overall lean body mass with a z score of <−1. In particular, the average lean body mass in the extremities was reduced with an average z score of −1.7. The lower extremities were found to be more severely involved compared to the upper extremities with increased fat replacement and average fat z scores of 2.1 and 0.6, respectively. MRI of the Glu385Lys patient showed muscle loss and diffuse fatty infiltration in muscles of the thigh and distal lower extremities (Fig 2F), and appendicular lean body mass was significantly reduced (z score = −5.7) on DEXA.
Figure 2.
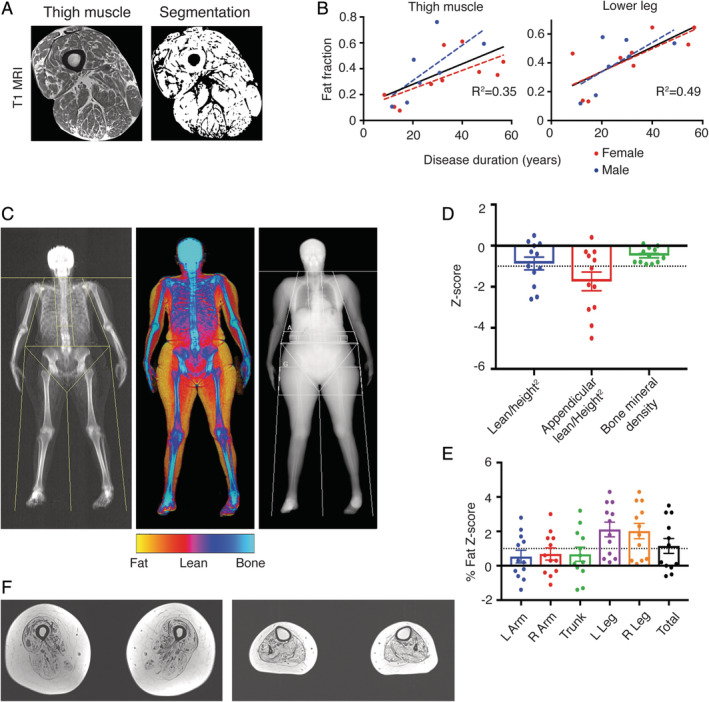
Clinical measurements in amyotrophic lateral sclerosis 4 (ALS4) patients with mutation at codon 389 (A‐E) and codon 385 (F). (A) T1 magnetic resonance image (MRI) of the thigh muscle with segmented noncontractile tissue in black. Compact bone and bone marrow removed from fat fraction calculation. (B) Disease duration is significantly correlated with MRI muscle fat fraction from the thigh (R 2 = 0.35; p = 0.01) and lower leg (R 2 = 0.49; p < 0.01). (C) Dual‐energy x‐ray absorptiometry in an ALS4 patient showing relative preservation of lean body mass (red) in the upper compared to the lower extremities. (D) Distribution of z scores shows a significant reduction in appendicular lean body mass (−1.7) and increase (E) in average leg (2.1) versus arm (0.6) fat z scores. Z scores calculated using National Health and Nutrition Examination Survey reference controls. Black dotted line indicates the z score 1 standard deviation below (D) or above (E) the mean. (F) T1 MRI of the thigh showing fatty infiltration and loss of contractile tissue in the thigh (left) and lower leg (right) in a patient with mutation at codon 385. L = left; R = right.
Other Findings
Eight patients had benign and malignant tumors, including 5 colonic polyps (1 with polyps of both the uterus and colon), 2 adenocarcinomas of the colon, and 1 of the lung. One patient with adenocarcinoma of the colon was a 35‐year‐old male, a relatively early onset given that the disease frequency is just 0.4% in men younger than 49 years.14 The benign polyps were also found in individuals younger than 49 years. In light of these findings, it is noteworthy that SETX mutations were found in 7% of colorectal tumors in the Cancer Genome Atlas,15 comparable to the rate of mutations in DNA mismatch‐repair pathway genes MLH1 (3%), MSH3 (7%), and MSH6 (8%).
Characterization of TGF‐β Biology in Fibroblasts and Stem Cell–Derived Motor Neurons
We previously found that the TGF‐β pathway is activated in Leu389Ser ALS4 patient fibroblasts with reduced expression of BAMBI, a negative regulator of the pathway.11 Here, we measured and found that the expression level of BAMBI was also reduced in the patient with Glu385Lys mutation (p < 0.01; Fig 3). To characterize the impact of TGF‐β on motor neurons, we derived motor neuron–like cells from control iPSCs. The iPSCs expressing markers of pluripotency were genotyped to confirm the stable heterozygous integration of the hNIL cassette into the CLYBL locus. Motor neuron–like identity was confirmed by the expression of the homeobox gene HB916 and the neurofilament marker SMI3217 within 1 week of differentiation.
Figure 3.
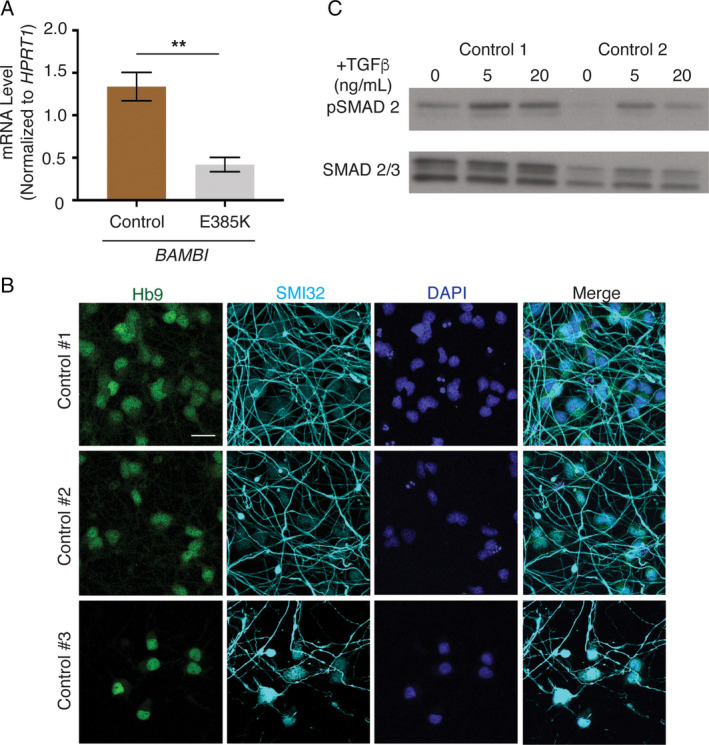
TGF‐β activity in patient fibroblasts and motor neuron characterization. (A) Reduced BAMBI transcript expression in Glu385Lys patient fibroblasts compared to 2 controls. Error bars are standard error of the mean of triplicates. **p < 0.01 (t test). (B) Immunofluorescence staining from 3 control induced pluripotent stem cell (iPSC) lines differentiated by expression of the human Neurogenin 2, Islet 1, and LIM Homeobox 3 (hNIL) transgene cassette. HB9 staining (green) in motor neuron nuclei, SMI‐32 (cyan) in motor neuron processes, and 4′,6‐diamidino‐2‐phenylindole (DAPI) staining in blue. Staining is at 5 days postdifferentiation. Scale bar = 25μm. (C) Western blot analysis of pSMAD2 and SMAD2/3 levels in iPSC motor neurons treated with 0, 5, and 20ng/ml TGF‐β.
We treated the iPSC‐derived cells with TGF‐β and confirmed activation of the pathway by an increase of SMAD2 phosphorylation. The neurons with TGF‐β treatment showed a significant reduction in neurite length and branching after 1 week of differentiation (p < 0.0001; Fig 4A–C), consistent with a toxic effect of TGF‐β activation.
Figure 4.
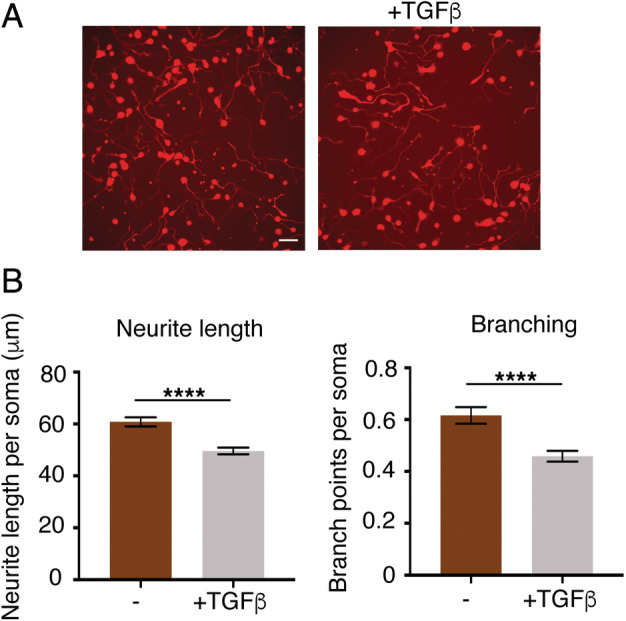
Morphological analysis of TGF‐β–treated motor neurons. (A) Fluorescence imaging of control motor neuron lines with and without TGF‐β treatment. (B) TGF‐β–treated motor neurons have reduced neurite length and branching. N = 6, 5 wells analyzed per sample, and error bars are standard error of the mean. Scale bar = 50μm. ****p < 0.0001 (t test). [Color figure can be viewed at www.annalsofneurology.org]
R‐Loop Assays
Molecular characterization of patient fibroblasts with the Leu389Ser mutation previously showed a reduction of nuclear R loops in patient fibroblasts and motor neurons.11 R loops are 3‐stranded nucleic acid structures that form when nascent RNA hybridizes with its template DNA to form an RNA–DNA hybrid and displaces the nontemplate DNA strand. Senataxin is a helicase that resolves R loops.18, 19 An antibody, S9.6, is used in many studies to characterize R loops.20 To demonstrate the specificity and sensitivity of the S9.6 antibody for R loops, we first probed synthetic hybrids of dsRNA, dsDNA, or RNA–DNA hybrid with the S9.6 antibody (Fig 5A). S9.6 signal was specific to RNA–DNA hybrid, and signal intensity was reduced with decreasing amounts of RNA–DNA hybrid. We evaluated R‐loop abundance in the Glu385Lys patient fibroblasts and found a significant decrease compared to controls by S9.6 dot blot (p < 0.05; see Fig 5B).
Figure 5.
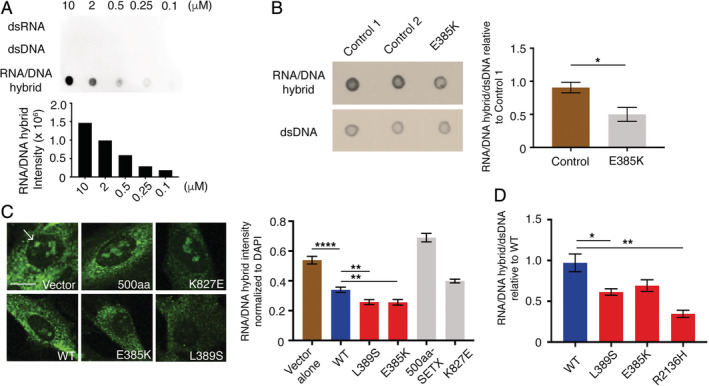
R‐loop assessments of senataxin dysfunction. (A) S9.6 antibody dot blot against a dilution series of synthetic double‐stranded (ds) RNA, dsRNA, and RNA/DNA hybrids. (B) Dot blot probed with S9.6 antibody shows fewer R loops in Glu385Lys patient fibroblasts compared to 2 age‐ and sex‐matched controls. A duplicate blot probed with anti‐dsDNA antibody was used as loading control. Error bars are standard error of the mean (SEM) of triplicates. *p < 0.05 (t test). (C) Immunofluorescence staining showing reduced nuclear S9.6 (green) in cells transfected with Leu389Ser and Glu385Lys forms of senataxin (SETX) compared to wild type (WT). Scale bar = 5μm; R loops in nuclei indicated by arrow. Ten fields per sample, 15 to 20 cells per field; error bars are SEM. ****p < 0.0001; **p < 0.01 (t test). (D) Quantification of dot blot S9.6 signal in cells transfected with WT, Leu389Ser, Glu385Lys, and Arg2136His forms of SETX. Samples normalized to dsDNA loading control. Error bars are SEM of triplicates. *p < 0.05, **p < 0.01 (t test). DAPI = 4′,6‐diamidino‐2‐phenylindole.
To confirm that the reduction in R‐loop abundance is a consequence of the SETX variant, we overexpressed the WT and mutant forms of SETX in control fibroblasts. Overexpression of the Glu385Lys and Leu389Ser mutations resulted in a significant reduction in nuclear R‐loop structure by immunofluorescence compared to transfection of WT senataxin alone (p < 0.01; see Fig 5C). Overexpression of a 500‐amino‐acid N‐terminal WT fragment of senataxin without the helicase domain or other nonpathogenic variants with high allele frequency (Lys827Glu, ExAC allele frequency 0.001) did not reduce R‐loop levels. Dot blot analysis of cells overexpressing SETX variants, including the previously reported Arg2136His ALS4 variant,5 showed a reduction in S9.6 signal intensity when compared to WT SETX (see Fig 5D).
Discussion
Defining the appropriate molecular and clinical biomarkers for disease characterization and progression are crucial steps to understanding and treating neurological disease. In this study, we identified clinical and molecular characteristics of ALS4 caused by heterozygous mutations in the SETX gene.
Relative to other forms of ALS, the onset for ALS4 is early. The average age of onset in our cohort is 17 years, which is consistent with the previous clinical characterization of patients with mutation at codon 389 of SETX.6, 21 In ALS4, males are more affected than females. Rafiq et al22 also found a significantly greater increase in male ALS patient serum CK levels compared to females. A previous study of ALS4 patients found a higher percentage of asymptomatic females (31%) compared to males (13%).6
As a motor neuron disease, ALS4 leads to progressive muscle loss. We found significantly higher serum CK and lower creatinine levels. Elevation of serum CK levels is also known to occur in patients with sporadic ALS.22 Among our ALS4 cohort, the male patients had significantly lower creatinine levels, which reflects a greater disease burden in males. Serum creatinine levels have been reported to change significantly during the progression of other motor neuron diseases.23, 24 Radiological studies showed higher thigh muscle fat fraction and lower lean body mass. ALS4 patients have fatty infiltration in the muscles of their lower extremities. Previously, we found a reduction in contractile muscle volume in a smaller cohort of ALS4 patients.11 The pattern of lean body mass on DEXA analysis supports the finding that the lower extremities are more severely affected than the upper extremities. Manual muscle testing also showed a reduction in lower leg strength with disease duration. The greater severity of lower extremity involvement in ALS4 was first described by Rabin et al.6 The increased fat replacement of muscle is likely related to a loss of lean body mass and disuse and has been found to correlate with disease status in other studies.25 Together these data suggest MRI of the lower leg for fatty infiltration to the muscles may be a useful study to follow for evidence of disease progression.
We found a similar pattern of clinical and radiological changes in a patient with mutation in SETX codon 385. Consistent with the gain of function in SETX helicase activity and reduction in R‐loop levels that had been detected in patients with mutation at Leu389Ser,11 the Glu385Lys SETX variant was also associated with a reduction in R‐loop levels. Expression levels of BAMBI, a negative regulator of TGF‐β activity, were also found to be reduced in the Glu385Lys SETX patient fibroblasts.
SETX function has been shown to modulate neuromuscular junction plasticity in drosophila through the TGF‐β pathway.26 TGF‐β signaling has been found to inhibit neurite elongation and branching in neuronal cell models27 and to promote disease progression in the SOD1 mouse model of ALS by disrupting the neuroprotective functions of activated glial and immune cells.28 Here, we show that exogenous TGF‐β treatment of control iPSC‐derived motor neurons resulted in a reduction in motor neuron neurite length and branching, indicating that activation of the TGF‐β pathway adversely affects motor neurons.
In conclusion, this study of 32 subjects with mutations in senataxin establishes the clinical and molecular features of an inherited form of upper and lower motor neuron disease. We also developed measurement of R‐loop levels and BAMBI expression as assays for interrogating the potential pathogenicity of SETX variants. Further studies with other mutations in the SETX gene causing ALS4 are needed to more thoroughly evaluate the clinical and molecular spectrum. Additional longitudinal data are also needed to determine the clinical course and molecular effects and assess heterogeneity of the disease.
Author Contributions
C.G., K.H.F., and V.G.C. contributed to the conception and design of the study. C.G., A.P., J.A., P.R., D.L., J.A.W., and A.B.S. contributed to the acquisition and analysis of data. C.G., K.H.F., and V.G.C. contributed to drafting the manuscript and figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Supplementary Table: Summary of ALS4 (L389S) patient clinical data
Acknowledgment
This study was supported by the Howard Hughes Medical Institute and Intramural Research at the National Institute of Neurological Disorders and Stroke.
We thank Dr S. Leppla for the S9.6 antibody; E. Hartnett for her help in scheduling patients during the study; the clinical staff of the outpatient NIH neurology clinic for assistance during the patients’ visits; and the NIH NHLBI iPS cell core facility for the generation of stem cell lines. We are grateful for the active participation of the patients and their families; without their involvement and encouragement, this project would not have been possible.
The copyright line for this article was changed on 15 December 2020 after original online publication.
References
- 1. Ghasemi M, Brown RH Jr. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb Perspect Med 2018;8:a024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gitcho MA, Baloh RH, Chakraverty S, et al. TDP‐43 A315T mutation in familial motor neuron disease. Ann Neurol 2008;63:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 4. Kim HJ, Kim NC, Wang YD, et al. Mutations in prion‐like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013;495:467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 2004;74:1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rabin BA, Griffin JW, Crain BJ, et al. Autosomal dominant juvenile amyotrophic lateral sclerosis. Brain 1999;122:1539–1550. [DOI] [PubMed] [Google Scholar]
- 7. Hirano M, Quinzii CM, Mitsumoto H, et al. Senataxin mutations and amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2011;12:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bassuk AG, Chen YZ, Batish SD, et al. In cis autosomal dominant mutation of senataxin associated with tremor/ataxia syndrome. Neurogenetics 2007;8:45–49. [DOI] [PubMed] [Google Scholar]
- 9. Phansalskar N, More S, Sabale A, et al. Adaptive local thresholding for detection of nuclei in diversity stained cytology images. International Conference on Communications and Signal Processing (ICCSP). Piscataway, NJ: IEEE, 2011:218–220.
- 10. Kelly TL, Wilson KE, Heymfield SB. Dual energy x‐ray absorptiometry body composition reference values from NHANES. PLoS One 2009;4:e7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grunseich C, Wang IX, Watts JA, et al. Senataxin mutation reveals how R‐loops promote transcription by blocking DNA methylation at gene promoters. Mol Cell 2018;69:426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fernandopulle MS, Prestil R, Grunseich C, et al. Transcription factor‐mediated differentiation of human iPSCs into neurons. Curr Protoc Cell Biol 2018;79:e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haruki M, Noguchi E, Kanaya S, Crouch RJ. Kinetic and stoichiometric analysis for the binding of Escherichia coli ribonuclease HI to RNA‐DNA hybrids using surface plasmon resonance. J Biol Chem 1997;272:22015–22022. [DOI] [PubMed] [Google Scholar]
- 14. American Cancer Society . Cancer facts and figures 2019. Updated 2019. Available at: http://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf. Accessed July 1, 2019.
- 15. NIH National Cancer Institute . The cancer genome atlas program. Available at: http://www.cancergenome.nih.gov. Accessed June 1, 2018.
- 16. Arber S, Han B, Mendelsohn M, et al. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron 1999;23:659–674. [DOI] [PubMed] [Google Scholar]
- 17. Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainite receptor‐mediated injury in vitro. J Neurosci 1996;16:4069–4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim HD, Choe J, Seo YS. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry 1999;38:14697–14710. [DOI] [PubMed] [Google Scholar]
- 19. Skourti‐Stathaki K, Proudfoot NJ, Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2‐dependent termination. Mol Cell 2011;42:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu Z, Zhang A, Storz G, Gottesman S, Leppla SH. An antibody‐based microarray assay for small RNA detection. Nucleic Acids Res 2006;34:e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Avemaria F, Lunetta C, Tarlarini C, et al. Mutation in the senataxin gene found in a patient affected by familial ALS with juvenile onset and slow progression. Amyotroph Lateral Scler 2011;12:228–230. [DOI] [PubMed] [Google Scholar]
- 22. Rafiq MK, Lee E, Bradburn M, et al. Creatine kinase enzyme level correlates positively with serum creatinine and lean body mass, and is a prognostic factor for survival in amyotrophic lateral sclerosis. Eur J Neurol 2016;23:1071–1078. [DOI] [PubMed] [Google Scholar]
- 23. Patin F, Corcia P, Madji Hounoum B, et al. Biological follow‐up in amyotrophic lateral sclerosis: decrease in creatinine levels and increase in ferritin levels predict poor prognosis. Eur J Neurol 2015;22:1385–1390. [DOI] [PubMed] [Google Scholar]
- 24. Hashizume A, Katsuno M, Banno H, et al. Longitudinal changes of outcome measures in spinal and bulbar muscular atrophy. Brain 2012;135:2838–2848. [DOI] [PubMed] [Google Scholar]
- 25. Dahlqvist JR, Oestergaard ST, Poulsen NS, et al. Refining the spinobulbar muscular atrophy phenotype by quantitative MRI and clinical assessments. Neurology 2019;92:e548–e559. [DOI] [PubMed] [Google Scholar]
- 26. Mushtaq Z, Choudhury SD, Gangwar SK, et al. Human senataxin modulates structural plasticity of the neuromuscular junction in drosophila through a neuronally conserved TGFβ signaling pathway. Neurodegener Dis 2016;16:324–336. [DOI] [PubMed] [Google Scholar]
- 27. Nakashima H, Tsujimura K, Irie K, et al. Canonical TGF‐β signaling negatively regulates neuronal morphogenesis through TGFIF/Smad complex‐mediated CRMP2 suppression. J Neurosci 2018;38:4791–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Endo F, Komine O, Fujimori‐Tonou N, et al. Astrocyte‐derived TGF‐β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep 2015;11:592–604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table: Summary of ALS4 (L389S) patient clinical data