

Abstract
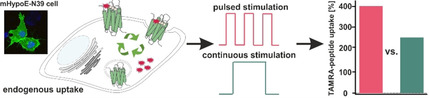
G protein‐coupled receptors (GPCRs) can be used to shuttle peptide‐drug conjugates into cells. But, for efficient therapy, a high concentration of cargo needs to be delivered. To explore this, we studied the pharmacologically interesting neuropeptide Y1 receptor (Y1R) in one recombinant and three oncogenic cell systems that endogenously express the receptor. We demonstrate that recycled receptors behave identically to newly synthesized receptors with respect to ligand binding and internalization pathways. Depending on the cell system, biosynthesis, recycling efficiency, and peptide uptake differ partially, but shuttling was efficient in all systems. However, by comparing continuous application of the ligand for four hours to four cycles of internalization and recycling in between, a significantly higher amount of peptide uptake was achieved in the pulsed application (150–250 % to 300–400 %). Accordingly, in this well‐suited drug shuttle system pulsed application is superior under all investigated conditions and should be considered for innovative, targeted drug delivery in general.
Keywords: cancer, cell lines, drug delivery, neuropeptide Y, shuttling
The sensitive equilibrium of receptor activation and desensitization is complex, and detailed knowledge is required for applications that use GPCRs as a shuttle system for intracellular drug delivery. As Y1R is overexpressed in various cancer subtypes, its shuttling capacity was quantitatively examined in diverse cell systems and by different methods to exploit its natural ligand‐induced endocytosis for targeted therapy.
Introduction
Today, cancer and obesity are among the largest public health challenges. Whereas death rates are dominated by cardiovascular diseases, for example, heart disease and stroke, frequently in consequence of overweight, oncologic diseases are listed as number 2.[ 1 , 2 , 3 , 4 ] Within the past decades, various strategies have been developed to combat these diseases effectively. However, pharmacological intervention remains challenging due to the limited selectivity of the applied drugs. Thus, innovative therapeutics that target malignant or adipose tissue selectively with minimal side effects are of great demand. The concept of selective drug targeting based on the high expression of specific cell‐surface receptors on distinct tissues has gained great attention. These receptors represent specific binding sites for their endogenous ligands as well as for engineered drug conjugates.[ 5 , 6 ] G protein‐coupled receptors (GPCRs) are the largest and most diverse class of membrane‐bound receptors conveying numerous extracellular signals into the cell. [7] Extensive investigations on GPCRs already demonstrated that the tightly regulated cyclic process comprising receptor activation and desensitization are crucial for maintaining cellular homeostasis. Thus, receptor internalization is naturally used to regulate the sensitive equilibrium by controlling the density at the cell surface and thus protect the cell from overstimulation.[ 8 , 9 ] For tissue targeting, this mechanism can be exploited and provides an elegant technique to shuttle cargos into the cell.[ 10 , 11 ]
The neuropeptide Y hormone receptor family represents an interesting group of GPCRs. It consists of four receptor subtypes – Y1R, Y2R, Y4R, Y5R, and three native ligands – neuropeptide Y (NPY), peptide YY (PYY), and pancreatic polypeptide (PP), which bind to the individual receptors with distinct preferences. [12] Whereas an overlapping expression pattern of hY1R and hY2R has been demonstrated in nephroblastomas [13] and glioblastoma, [14] highly specific hY1R expression was found in adrenal cortical tumors, Ewing sarcoma tumors and different types of breast cancer. [15] Recently, experiments by Boehme et al. and Worm, Hoppenz et al. demonstrated the delivery of toxic and non‐toxic payloads into breast cancer cells.[ 16 , 17 ] Furthermore, Wittrisch et al. reported high levels of hY1R in human adipose tissue, 3T3‐L1 preadipocytes as well as adipocytes. [18] Hence, the Y1R is of great pharmaceutical interest and displays an attractive and promising drug shuttle target for both, cancer and obesity. [15]
Requirements for an efficient targeted therapy are significant concentrations of the delivered cargo into the cell, which might be achieved by 1) high expression of the GPCR of interest at the desired cell and 2) the extent of the receptor that recycled back to the cell surface to be available for additional shuttling cycles. The recycling process enables the re‐usage of the receptor and facilitates intracellular accumulation of the desired drug. [19] Previous investigations on Y1R internalization and intracellular trafficking revealed arrestin‐dependent endocytosis, followed by endosomal sorting and recycling back to the cell membrane. These processes were found to be controlled and regulated by different motifs within the intracellular domains and the C‐terminus of the receptor.[ 20 , 21 ] However, the regulatory mechanisms are complex, and until now, there has been a lack of quantitative studies addressing the question of the capacity of GPCRs in order to be used as an efficient shuttling system. Thus, we evaluated peptide uptake within up to four cycles of receptor activation, internalization and recycling in different cellular models with endogenous and artificial receptor expression. We used a variety of assay systems and demonstrated that Y1R signaling is controlled by fast internalization and recycling. Furthermore, uptake studies with different types of recombinant and endogenous cell lines confirmed the Y1R to be a suitable drug shuttle system. The amount of internalized peptide increased in the presence of biosynthesis inhibitors up to 150–250 % after four hours of constant stimulation compared to one hour. Interestingly, much higher intracellular peptide accumulations (300–400 %) were achieved by applying a pulsed application of four cycles. These findings indicate that peptide receptors like the Y1R are indeed suitable shuttling system and pulsed delivery is preferred over continuous application.
Results
Y1R rapidly internalizes and recycles back to the membrane
The application of peptide‐drug conjugates to selectively address (over)expressed receptors is of great interest because they probably reduce side effects compared to untargeted therapy. However, using GPCRs as drug‐shuttle systems requires detailed knowledge on the internalization and trafficking of the receptors. Previous investigations already showed that the human Y1R internalizes with its ligand as ligand‐receptor complex in an arrestin 2/3 dependent mechanism after agonist stimulation. This is followed by intracellular sorting and subsequent reappearance of the receptor at the cell membrane – defined as recycling. [21] To gain further insights into the cellular transport mechanism and the kinetic profile of receptor internalization, we first examined the internalization and recycling properties at different time points after ligand addition. We used stably transfected HEK293 cells that express the Y1R C‐terminally fused to the enhanced yellow fluorescent protein (eYFP). Live‐cell fluorescent microscopy was performed to study receptor trafficking after ligand stimulation as illustrated in Figure 1A. Prior to NPY treatment, the majority of Y1Rs is localized in the plasma membrane (Figure 1B). Determination of the mean cell‐surface fluorescence (MCSF) allowed the quantification of the receptor amount and was set to 100 % (Figure 1C, w/o). Stimulation with 1 μM NPY resulted in strong internalization of the receptor, which accumulated in intracellular vesicles (34±3%, MCFS, white bar). Aspirating the stimulation solution, subsequent thorougly washing and incubation in ligand‐free medium for 60 min regained the receptor amount at the cell surface again up to 72±4 % (gray bar), which improved to 91±9 % (light gray bar) after a recovery period of 240 min. Recycling of the receptor to the surface was inhibited by applying 20 mM NH4Cl (47±4 %, dark gray bar). To further characterize the kinetic profile of the internalization and recycling of the Y1R, different time points were chosen and the amount of cell‐surface receptors was quantified. Stimulation with 1 μM NPY resulted in rapid internalization of the receptors, which reached a plateau within 20 min and an estimated t 1/2 of ∼5 min. Removing the agonist solution, subsequent washing and incubation in recovery medium revealed fast recycling of the receptors which reached a plateau after 15 min (Figure 1D).
Figure 1.
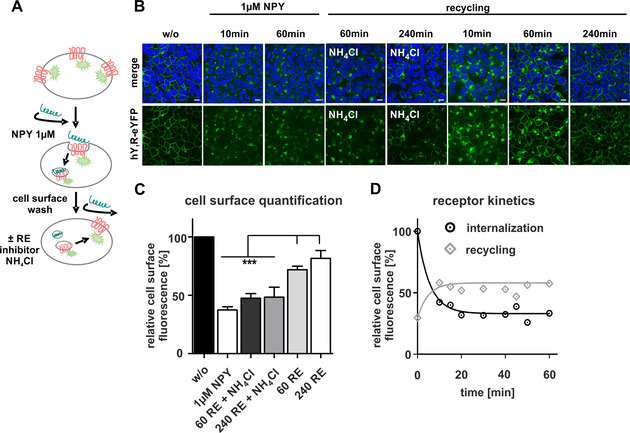
Characterization of Y1R internalization and recycling. A) Scheme of the hY1R internalization and recycling experiment. B) Live‐cell image of stably transfected HEK293‐hY1R‐eYFP cells. The cellular localization of the receptor (green) was determined by fluorescence microscopy prior to (w/o) and after stimulation with NPY. Cell nuclei were stained with Hoechst33342 (blue). C) Relative amount of cell‐surface receptors quantified by membrane fluorescence intensity using Image J. Prior to agonist stimulation, the amount of cell‐surface receptors was set to 100 % (w/o, black bar). Stimulation with 1 μM NPY led to receptor internalization and a reduction in cell‐surface fluorescence (1 μM NPY, white bar), which increased again after the recovery period of 60 or 240 min (60 RE, gray bar; 240 RE, light gray bar). Treatment with 20 mM NH4Cl significantly inhibited the reappearance of Y1R at the membrane (60 RE+NH4Cl, dark gray bar; 240 RE+NH4Cl, gray bar). D) Time‐dependent analysis of receptor internalization and receptor recycling over 60 min. Scale bars: 10 μm, experiments represent mean ±SEM values of n≥3 independent experiments (C/D), and representative data of n≥3 independent experiments (B), respectively; significance was determined by one‐way ANOVA, Tukey post test, *** p<0.0001.
Next, we investigated, whether recycled receptors are still functional and can be addressed for a second cycle of internalization. For this purpose, we examined the internalization after 60 min of recovery in ligand‐free medium. The set‐up is illustrated in Figure 2A. To quantify ligand‐induced receptor internalization, we stimulated the receptors with a fluorescently labeled NPY variant (TAMRA‐NPY) [22] and quantified the peptide uptake by measuring the TAMRA‐pixel intensity at each time point. Cells without stimulation were set to 0 %, whereas stimulation with 100 nM TAMRA‐NPY for the first time resulted in high yields of internalized peptide, which were defined as the maximum uptake per cycle of stimulation and set to 100 % (Figure 2B, C). In line with our expectation, a first stimulation with 1 μM unlabeled peptide for 60 min, subsequent washing and a direct second stimulation (no recovery phase) with 100 nM labeled NPY resulted in a reduction of peptide uptake owing to the reduced number of cell‐surface receptors after the first stimulation (0 RE, 65±4%). The amount of ligand uptake was not increased after a recovery period of 60 min or 240 min and yielded the same amount of ingested TAMRA‐NPY (60 RE, 65±4%; 240 RE, 64±10 %). By comparing the peptide uptake to control cells treated with the recycling inhibitor NH4Cl, we confirmed that the recycled receptors contribute significantly to the TAMRA‐peptide uptake as a significant reduction of internalized peptide was observed (60 RE, 65±4 %; 60 RE+NH4Cl, 31±4 %).
Figure 2.

Significantly reduced peptide uptake by inhibiting receptor recycling. A) Graphical scheme of Y1R internalization‐ and recycling experiment determining internalized peptide. B) Live‐cell image of HEK293‐hY1R‐eYFP (green) and TAMRA‐NPY (red) uptake at certain time points. Control cells (60 min) were stimulated with 100 nM TAMRA‐peptide. Stimulation with 1 μM unlabeled NPY, subsequent washing und direct second stimulation (0 RE) with 100 nM TAMRA‐NPY for 60 min or stimulation for a second time after a recovery period of 60 min ± recycling inhibitor NH4Cl (60 RE, 60 RE+NH4Cl) or 240 min ± recycling inhibitor NH4Cl (240 RE, 240 RE+NH4Cl). Cell nuclei were stained with Hoechst33342 (blue). C) The amount of ingested TAMRA‐NPY was quantified by using Image J. Control cells (60 min) were set to 100 % (white bar). Stimulation with 1 μM NPY first, washing and second stimulation with 100 nM TAMRA‐NPY after a 60 min recovery period in presence of the recycling inhibitor NH4Cl (60 RE+NH4Cl, dark gray bar; 240 RE+NH4Cl, light gray bar) revealed a significant decrease peptide internalization. Scale bars: 10 μm, experiments represent mean±SEM values of n≥3 independent experiments (C), and representative data of n≥3 independent experiments (B); significance was determined by one‐way ANOVA, Tukey post test, *** p<0.0001.
Receptor recycling contributes significantly to peptide uptake in multiple stimulation cycles
To characterize the peptide internalization in more detail, we quantified TAMRA‐NPY uptake after multiple cycles of stimulation (denoted here experiments 1 to 4) by live‐cell fluorescent microscopy, following the protocol illustrated in Figure 3A. For experiment 1, receptor‐expressing cells were incubated with a fluorescently labeled NPY variant (TAMRA‐NPY) in the presence of biosynthesis inhibitors brefeldin A (BFA) and cycloheximide (CHX). For experiments 2–4, receptor expressing cells were incubated once, twice or thrice with 1 μM NPY prior to the final incubation with fluorescently labeled NPY. Cells without stimulation were set to 0 %. Stimulation of cells for the first time with 100 nM TAMRA‐NPY resulted in high amounts of internalized peptide, which was defined as the maximum uptake and set to 100 % (Figure 3B). In line with our previous observation (Figure 2), a first stimulation with 1 μM unlabeled peptide for 60 min, a subsequent washing step, followed by a recovery period of 60 min and a second stimulation with 100 nM labeled NPY resulted in ∼25% reduced peptide uptake (74±6 %), possibly owing to the reduced number of cell‐surface receptors (cycle 2). The amount of ligand uptake was further reduced to 57±5% of the control level after three cycles of stimulation (cycle 3, gray line). Surprisingly, no further was found reduction after four cycles of stimulation (cycle 4, gray line, 71±4%). By comparing the peptide uptake to control cells treated with the recycling inhibitor NH4Cl, we confirmed that the recycled receptors significantly contribute to the TAMRA‐peptide uptake as a significant reduction of internalized peptide was observed after every cycle when the cells were treated with NH4Cl (Figure 3B, red dotted line). However, receptor biosynthesis contributes to an enhanced peptide internalization as samples without the biosynthesis inhibitors BFA/CHX exhibited higher peptide internalization values compared to samples with inhibitors (Figure 3B, black, open symbols). Under these conditions, continuous receptor expression counterbalances the partial receptor desensitization and degradation. Thus, the amount of internalized TAMRA‐peptide remains virtually constant over at least four cycles of stimulation. To calculate the total amount of internalized peptide, it can be assumed that each cycle adds up to the previous one. This results in a final peptide amount after 4 h of pulsed stimulation of ∼300 % (+BFA/CHX).
Figure 3.

Y1R serves as a suitable shuttling system in HEK293 cells. A) Schematic illustration of the protocol determining internalized peptide after different cycles of stimulation. B) To quantify the cellular peptide uptake, a fluorescently labeled TAMRA‐NPY peptide was used, and the amount of ingested TAMRA‐NPY was quantified by using ImageJ (left). Control cells were stimulated with 100 nM TAMRA‐NPY, and the internalized peptide was set to 100 % (1). To analyze the peptide uptake after certain cycles of stimulation, cells were alternately treated with NPY with or without inhibitors (−BFA,CHX black line; +BFA, CHX, gray dotted line; +BFA, CHX, NH4Cl, red dotted line) and a final TAMRA‐NPY stimulation for each cycle (2, 3, 4). Live‐cell image of internalized TAMRA‐NPY uptake (white) at distinct cycles of stimulation (right). Scale bars: 10 μm, experiments represent mean±SEM values of n≥3 independent experiments (B), and representative data of n≥3 independent experiments (B, right); significance was determined by one‐way ANOVA, Tukey post test, *** p<0.0001; * significance of +BFA/CHX over +NH4Cl/+BFA/CHX.
Recycled Y1R receptors exhibit no alterations in ligand binding affinity
To clarify whether the reduced receptor‐internalization in the second cycle is a result of reduced ligand affinity and independent of the duration of the recovery period, we performed specific [125I]‐PYY competition radioligand binding assays using stably transfected Y1R HEK293 cells. The following experiments were performed on ice, preventing receptor endocytosis and consequently internalization of the radiolabeled peptide. The specific binding of control receptors (w/o, black) was set to 100 % and represent the maximal possible binding sites (Figure 4, left). Receptors stimulated with 1 μM NPY for 60 min, followed by agonist washout and direct measuring of ligand affinity without recovery period (0 min RE, light gray) displayed a reduced Bmax (57±9%), which might be caused by a decreased amount of cell‐surface receptors in consequence of internalization after the first cycle of stimulation. Ligand binding of recycled receptors, treated with 1 μM NPY, subsequent agonist washout, and 60 min recovery period showed an increased B max (60 RE, dark gray, 76±10 %) compared to receptors without recycling period, indicating a contribution of recycled receptors. Moreover, these data fit well with our microscopy studies that revealed a reappearance to the cell membrane up to ∼70 %. To investigate the binding properties in more detail, NPY displacement experiments were performed. For clarity, the total and unspecific binding of each condition was set to 100 % and 0 % respectively (Figure 4, right). No differences in ligand affinity were observed between control receptors and receptors, which were stimulated with NPY first, independent of the recovery period (IC50[w/o]: 4.6 nM, IC50[0RE]: 8.8 nM; IC50[60RE]: 1.6 nM). These data suggest that the slight drop of receptor internalization is not caused by a loss in ligand affinity but may rather be caused by a modest reduction of receptor density, for example, degradation or the cellular status of effector proteins.
Figure 4.
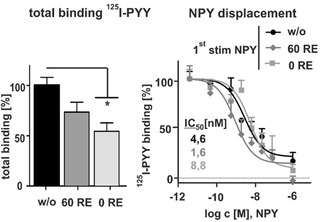
Ligand binding is not affected after recycling. A) [125I]‐PYY binding studies in stably transfected Y1R HEK293 cells. Prior to stimulation (w/o) and after incubation with 1 μM NPY, agonist washout and receptor recovery revealed no significant differences in the binding capacity of recycled receptors. Maximum binding was reduced for receptors stimulated with 1 μM NPY, agonist washout and direct second stimulation without receptor recovery (0 RE, light gray bar; n≥3). To normalize the data, total binding and unspecific binding of each condition were constrained to 100 and 0 %, respectively. Experiments represent mean±SEM values of n≥3 independent experiments. Total binding was analyzed by one‐WAY ANOVA and Dunnett's post test against unstimulated control (w/o), * p<0.03.
Recycled Y1 receptors recruit arrestin in similar measure
To clarify whether the cellular status of effector protein causes reduced Y1R internalization and peptide (cargo) uptake, we investigated arrestin recruitment using fluorescence microscopy and BRET assay experiments in transiently transfected HEK293 cells. The receptor was C‐terminally fused to eYFP and arrestin was N‐terminally tagged with mCherry or RLuc8 for live‐cell microscopy and BRET‐assay, respectively. First, we performed live‐cell microscopy to assess arrestin recruitment prior to agonist stimulation (w/o) and after treatment with 1 μM NPY at 37 °C, a following washing step and incubation in ligand‐free medium for 60 min. Prior to NPY stimulation, the receptor (green) is clearly located in the cell membrane, whereas arrestin 3 (red) is largely distributed in the cytoplasm (Figure 5A). NPY stimulation induced arrestin recruitment to the cell membrane, followed by arrestin dependent receptor endocytosis after 4 min. After 10 and 20 min, the receptor‐arrestin complex is almost completely internalized. After ligand removal and subsequent agonist washout, redistribution of both arrestin and receptor took place. While the receptor recycles back to the membrane, arrestin distributes randomly within the cell. Second stimulation experiments displayed the same recruitment and internalization properties as observed for the first stimulation. Distribution and co‐localization of arrestin was further verified using line scans. While the individual signals displayed no overlapping profile prior to NPY stimulation, NPY treatment induced signal overlapping and separation after ligand removal (Figure 5A, right). In the next step, we validated these data by specific BRET‐based arrestin‐receptor interaction assay. BRET is very sensitive and can display quantitative differences of arrestin recruitment that are not obvious from microscopy experiments. Arrestin recruitment in a concentration‐dependent setting (Figure 5B) revealed no differences regarding ligand potency for the receptors prior to (w/o) and after NPY treatment (Figure 5B, EC50[w/o]: 51 nM, EC50[60RE]: 32 nM). Similarly, the apparent rate of arrestin 3 recruitment (and thus, implicitly, the rate of receptor phosphorylation) was unchanged (Figure 5C). The amount of recruited arrestin as reflected by the netBRET signal was slightly reduced in the second cycle of stimulation, which is particularly evident in the kinetic setting. Thus, fewer receptor/arrestin complexes are formed after receptor recycling.
Figure 5.
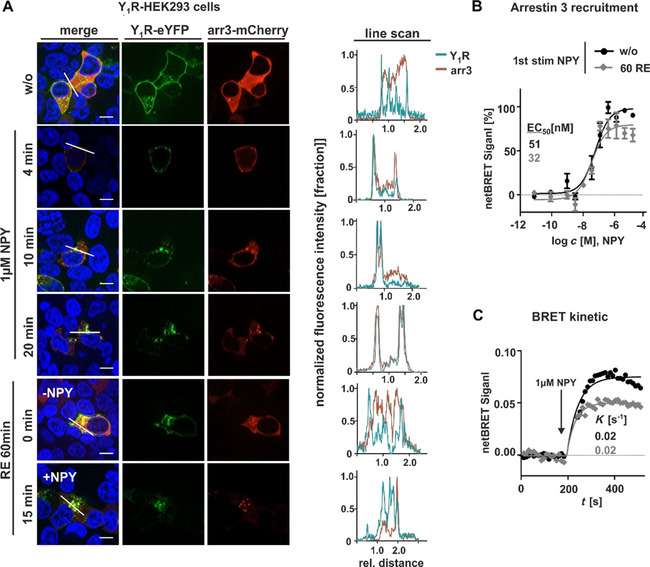
Y1Rs strongly recruit arrestin after ligand stimulation, which is not altered in second stimulation experiments. A) Live‐cell microscopy of transiently transfected HEK293 cells with Y1R carrying an eYFP on its C terminus and arrestin 3 fused to mCherry. The cellular localization of the receptor (green) and arrestin (red) was determined by fluorescence microscopy prior (w/o) and after stimulation with 1 μM NPY at 37 °C at different time points, following a washing step, incubation in ligand‐free medium for 60 min, and a second cycle experiments prior (RE 60 min, 0 min, −NPY) and after NPY treatment (RE 60 min, 15 min, NPY). Co‐localization of receptor and arrestin is indicated by yellow in the merged picture and was verified by applying line scans (right). Cell nuclei were stained with Hoechst33342 (blue). B) BRET concentration‐response curves with Y1R‐eYFP and RLuc8‐arrestin 3 prior stimulation and after stimulation with 1 μM NPY, subsequent washing and 60 min recovery period. C) Kinetic BRET studies of recycled and control Y1R were measured with receptor fused to Venus fluorophore and RLuc8‐arrestin 3 over 500 s, and the apparent first‐order rate constant was calculated. Scale bars: 10 μm, experiments represent the mean±SEM values of n≥3 independent experiments (B, C), and representative data of n≥3 independent experiments (A).
Continuous stimulation is not as efficient as a pulsed application in HEK293 cells
To study whether the cycle of Y1R internalization and recycling in complex with arrestin is still sufficient to serve as a suitable shuttling system we quantified intracellular peptide accumulation under continuous stimulation conditions. Thus, live‐cell fluorescent microscopy was performed using stably transfected Y1R‐HEK293 cells, which were stimulated with 1 μM TAMRA‐peptide over four hours. Stimulation was stopped after 1, 2, 3 or 4 hours by aspirating the ligand solution and a subsequent washing step to remove unbound TAMRA‐NPY. For quantification, cells without stimulation were set to 0 %. Stimulation of cells for one hour with 1 μM TAMRA‐NPY resulted in high yields of internalized peptide and was set to 100 %. The amount of ligand uptake was slightly enlarged after two hours of incubation, which further increased up to 155±3 % after four hours (Figure 6A). Indeed, significant peptide accumulation was achieved using stably transfected Y1R‐HEK293 cells as a recombinant model system. However, the cumulative peptide uptake was not as high as the theoretical sum of four cycles of ligand stimulation separated by recovery periods (cf. Figure 3; cumulative uptake after 4 h ∼400 % (−BFA/CHX); continuous cumulative uptake ∼150 %), thus underlining the importance of cellular recovery phases.
Figure 6.
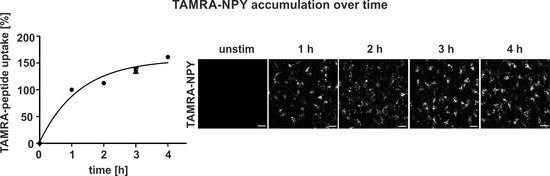
Intracellular accumulation of TAMRA‐NPY in stable Y1R‐HEK293. A) Quantification and live‐cell image of TAMRA‐NPY (white) uptake over 4 h. Stably transfected HEK293‐hY1R‐eYFP cells were stimulated with 1 μM TAMRA‐peptide in OptiMEM at 37 °C. Stimulation was stopped after 1, 2, 3 or 4 h by aspirating the ligand solution and subsequent washing. Internalized peptide was quantified using ImageJ. Unstimulated cells were set to 0 % and stimulation for 1 h was set to 100 %. TAMRA‐NPY accumulation was observed with increasing stimulation time. Scale bars: 10 μm, experiments represent mean±SEM values of n≥3 independent experiments, and representative data of n≥3 independent experiments (right).
Y1 receptor internalization and peptide accumulation in hypothalamic mHypoE‐N39 cells
To clarify whether the discrepancy between continuous and pulsed stimulation is due to transfection conditions, we investigated a more native system for receptor internalization and recycling studies, and used the murine hypothalamic cell line mHypoE‐N39. Y1R expression was verified by RT‐PCR and Ca2+‐flux assay (S1 in the Supporting Information). To clarify whether the Y1R internalizes after agonist stimulation in this native cellular system, we transiently transfected Y1R‐eYFP into mHypoE‐N39 and stimulated the cells with TAMRA‐NPY, enabling the visualization of both internalized peptide as well as the trafficking route of the receptor itself using live‐cell fluorescence microscopy. Prior to TAMRA‐NPY stimulation, the Y1R is expressed in the plasma membrane (Figure 7A, 0 min, left). Stimulation for 60 min with 100 nM TAMRA‐NPY led to a rapid co‐localization of Y1R and peptide (yellow, arrows) and high amounts of internalized receptor/ peptide complexes were observed (Figure 7A, 60 min, right, yellow dots). To investigate the internalization and arrestin distribution in more detail, fluorescence‐labeled Y1R‐eYFP and mCherry‐tagged arrestin 3 were co‐transfected into mHypoE‐N39 cells (Figure 7B). Prior to NPY treatment, receptor and arrestin were randomly distributed within the cell. Stimulation with 1 μM NPY led to a re‐organization of arrestin and co‐internalization of the protein and the Y1R in a complex (Figure 7B, 60 min, right panel, yellow dots). Receptor and arrestin redistribution and reorganization towards the initial state were detected after a recycling period in a ligand‐free medium for 60 min, according to the general protocol. Second stimulation experiments revealed nearly the same recruitment and internalization properties as seen for the first stimulation. Moreover, distribution and co‐localization of arrestin and receptor were verified using line scans. While the individual signals showed no overlapping profile prior to NPY stimulation, the lines overlapped during NPY treatment and separated again after ligand removal (Figure 7B, right). Hence, we demonstrate that Y1R internalization and arrestin recruitment are not only detected in commonly used cell lines but can be observed analogously in an endogenously expressing Y1R cell line.
Figure 7.
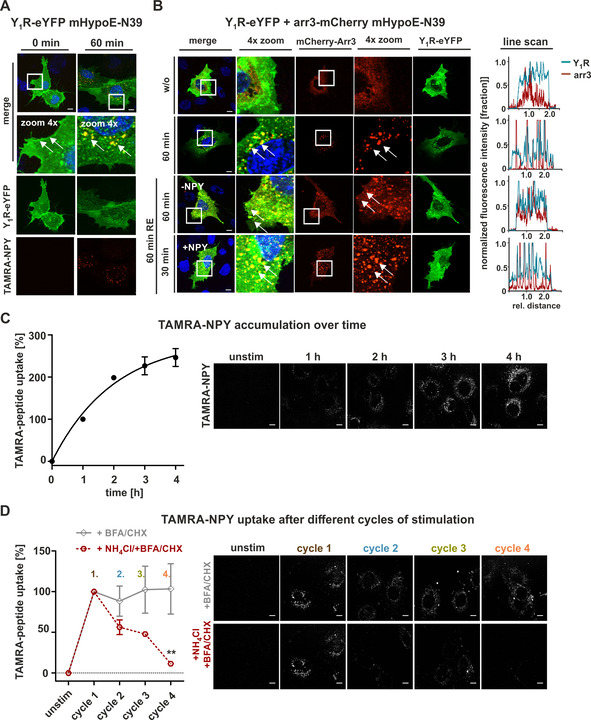
Internalization and activation properties of Y1R endogenously expressed in mHypoE‐N39 cells. A) Live‐cell microscopy of transiently transfected mHypoE‐N39 cells with hY1R fused to eYFP. Receptor (green) distribution was examined by fluorescence microscopy prior to (0 min) and after stimulation with 100 nM labeled TAMRA‐NPY (red). Co‐localization of Y1R and peptide (yellow, arrows) was observed after stimulation. B) Live‐cell microscopy of transiently transfected mHypoE‐N39 cells with Y1R fused to eYFP (green) and arrestin 3 (red) carrying mCherry. Prior to agonist stimulation, arrestin was randomly distributed within the cell (w/o). Recruitment of arrestin towards the receptors (60 min, yellow, arrows) was observed after stimulation with 1 μM NPY. No differences concerning the internalization behavior compared to the first cycle of stimulation was observed after recovery period or second‐cycle experiments prior to (RE 60 min, 0 min, ‐NPY) and after NPY treatment (RE 60 min, NPY). Co‐localization of receptor and arrestin was verified by applying line scans (right). Cell nuclei were stained with Hoechst33342 (blue). C) Quantification and live‐cell images of internalized TAMRA‐NPY (white) over 4 h. Unstimulated cells were set to 0 %, and stimulation for 1 h was set to 100 %. D) Peptide uptake after different cycles of stimulation was studied in cells treated with 1 μM NPY for 60 min first. After a recovery period, with or without recycling inhibitors (+BFA, CHX, gray line; +BFA, CHX, NH4Cl, red dotted line), a final TAMRA‐NPY stimulation was performed after each cycle (2, 3, 4). Peptide uptake in presence of BFA and CHX remains constant compared to control uptake (first cycle), which can be diminished by adding NH4Cl to the recycling period. Scale bars: 10 μm; experiments represent mean±SEM values of n≥3 independent experiments (C, D), and representative data of n≥3 independent experiments (A, B); significance was determined by one‐way ANOVA, Tukey post test, ** p<0.0014; * significance of +BFA/CHX over+NH4Cl/+BFA/CHX.
Next, we studied the behavior of untransfected mHypoE‐N39 cells. We investigated the shuttling efficiency and intracellular peptide accumulation for 4 h (Figure 7C) as well as the amount of internalized peptide after different times of NPY stimulation and subsequent recycling periods (Figure 7D) as performed in HEK293 cells (cf. Figures 6 and 3, respectively). Also in this native system, incubation of cells for one hour with 1 μM TAMRA‐NPY resulted in high yields of internalized peptide and was set to 100 %. The amount of ingested ligand was further increased over time and doubled after 2 h of continuous incubation (Figure 7C, 2 h: 198±0.5 %). However, the accumulation rate strongly decelerated over time (2 h: 198±0.5 %, 3 h: 226±21 %, 4 h: 246±21 %). Looking at separate 1 h internalization cycles with intermediate recovery periods, the TAMRA‐ligand uptake after multiple cycles of stimulation remained constant per cycle (Figure 7D, +BFA/CHX, gray line; 1st cycle 100 %, 2nd cycle 88±19 %; 3rd cycle 102±29 %; 4th cycle 103±30), thus demonstrating a functional lifecycle of the Y1R concerning internalization and resensitization in this endogenous cell system. Moreover, the application of the recycling inhibitor NH4Cl corroborated that recycled receptors are re‐integrated into the lifecycle as the amount of internalized peptide significantly decreased after every cycle of stimulation/recovery (red line) under these conditions. Taken together, arrestin recruitment and thus robust internalization of the receptor leads to great amounts of internalized peptide even after several cycles of stimulation. This suggests that cellular systems expressing the Y1R in low to moderate amounts may serve as an even more effective shuttle system, at least considering short cycles. Similar to transfected HEK293 cells, however, the amount of internalized peptide is significantly reduced under long continuous stimulation conditions compared to the theoretically summed up peptide amount achieved by short pulsed applications with separate recycling periods in between ( ∼250 to ∼395 %), even though the pulsed experiment was conducted with biosynthesis inhibitors.
Y1 receptor internalization and peptide accumulation in pathophysiologically relevant MCF7 and SK‐N‐MC cells
To confirm these findings in further endogenous, and pathophysiologically relevant, cellular systems, we repeated the peptide uptake experiments in MCF7 and SK‐N‐MC cells. SK‐N‐MC (human neuroblastoma cell line) cells express the Y1R in high amounts. [23] In contrast, MCF7 cells originate from human Caucasian breast adenocarcinoma [24] are frequently used as a model cell line for breast cancer with moderate expression levels of the Y1R. Thus, the physiological background of these cell lines as well as the individual stoichiometry of receptors/G proteins/arrestins are likely very dissimilar and will be informative about the generalizability of Y1R properties for peptide shuttling. We again assessed arrestin recruitment and TAMRA‐NPY uptake by live‐cell microscopy and quantified the images as described. Prior to NPY stimulation, the receptor (Y1R‐eYFP co‐transfected for visualization; green) is located at the cell surface, while arrestin 3 (red) is widely distributed in the cytoplasm (Figure 8A). NPY stimulation initiated arrestin recruitment to the cell membrane, followed by receptor internalization in an arrestin dependent manner after 4 min. After 10 and 20 min, the protein complex composed of receptor and arrestin was almost fully internalized. After NPY removal and subsequent agonist washout, redistribution and re‐organization of both arrestin and receptor proceeded. While the receptors recycled back to the membrane, arrestin distributed randomly within the cell again. Stimulation for a second time after a 60 min recovery period exhibited comparable recruitment and internalization properties as seen for the first stimulation and the complete overlapping profile indicated no restriction of the internalization machinery (Figure 8A, right).
Figure 8.
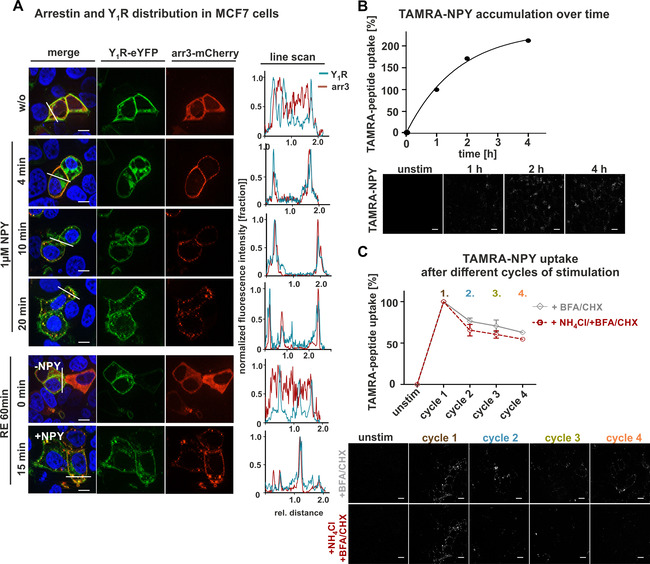
Arrestin recruitment and peptide uptake in MCF7 cells after different cycles of stimulation A) Live‐cell microscopy of transiently transfected MCF7 cells with Y1R fused to eYFP (green) and arrestin 3 (red) carrying mCherry at its C terminus. Prior to agonist stimulation, arrestin was randomly distributed within the cell (w/o), whereas the receptor was expressed at the cell surface. Stimulation with 1 μM NPY resulted in redistribution and recruitment of arrestin to the receptor even after 4 min, which becomes more obvious after 10 and 20 min. After a washing step and incubation in recycling buffer for 60 min, receptor and arrestin were separated again and either transported back to the membrane (Y1R) or redistributed randomly within the cytoplasm (RE 60 min, 0 min, −NPY). Co‐localization of receptor and arrestin is indicated by yellow in the merged picture, and was verified by applying line scans (right). Cell nuclei were stained with Hoechst33342 (blue). B) Quantification and live‐cell image of ingested TAMRA‐NPY (white) for 4 h. Cells were treated with TAMRA‐peptide and stimulation was stopped after 1, 2, 3 or 4 h by aspirating the ligand solution and subsequent washing. Internalized peptide was quantified by using Image J. Unstimulated cells were set to 0 % whereas treatment for 1 h was set to 100 %. C) Peptide uptake after different cycles of stimulation and recovery periods was studied by treatment with NPY, followed by a recovery period in ligand‐free medium supplemented either with or without recycling inhibitors (+BFA, CHX, gray line; +BFA, CHX, NH4Cl, red dotted line) and a final TAMRA‐NPY stimulation after each cycle (2, 3, 4). Peptide uptake in the presence of BFA (protein trafficking inhibitor) and CHX (a protein synthesis inhibitor) revealed a constant moderate reduction of internalized peptide compared to the first cycle. Adding NH4Cl (recycling inhibitor) to the recycling period slightly diminished the uptake of TAMRA‐NPY compared to untreated cells. Scale bars: 10 μm, experiments represent the mean±SEM values of n≥3 independent experiments (B, C), and representative data of n≥3 independent experiments.
To further characterize the shuttling capacity, we examined the cumulative peptide accumulation for 4 h continuous stimulation (Figure 8B) as well as the peptide internalization after different cycles of stimulation and recycling periods (Figure 8C), as described above without receptor transfection. In line with our observation for mHypo‐N39 cells, TAMRA‐NPY also accumulated in this tumor cell line in high amounts. The amount of cumulatively internalized peptide almost doubled after four hours of stimulation compared to 1 h. To characterize the peptide uptake in more detail, we quantified the internalization per cycle for multiple cycles of stimulation and subsequent recycling. Incubation of the Y1R with 100 nM labeled TAMRA‐NPY for the first time resulted in great amounts of internalized peptide (Figure 8C, cycle 1, brown), which was set to 100 % for quantification. In contrast to the endogenous Y1R expression in mHypoN39 cells, but similar to Y1R transfected HEK293 cells, we found a moderate reduction in the total amount of internalized peptide down to ∼60 % after four cycles of stimulation (Figure 8C, cycle 4, orange). However, also this cell system exhibited higher amounts of ingested TAMRA‐peptide by applying short and multiple stimulation cycles with recovery periods compared to continuous stimulation over 4 h (∼305 % to ∼200 %). Interestingly, the application of the recycling inhibitor NH4Cl was not as potent as observed for the other cell lines. The peptide uptake was only slightly reduced compared to control cells without recycling inhibitor (Figure 8C, red dotted line).
To further generalize these data, a second cancer cell line (SK‐N‐MC), derived from neuroblastoma, was investigated. Characterization of the arrestin recruitment prior and after agonist stimulation using line scan as well as the microscopy revealed comparable properties as seen for MCF7 cells (Figure 9A). Also in this cancer cell line, a constant reduction in the total amount of internalized peptide was observed per cycle. The sum of the peptide taken up by pulsed application was more than 300 % compared to single application. The addition of NH4Cl seems to be not as effective as seen in HEK293 and mHypoN39 cells, and the contribution of recycled receptors to peptide uptake in repeated stimulation experiments is rather small (Figure 9B).
Figure 9.
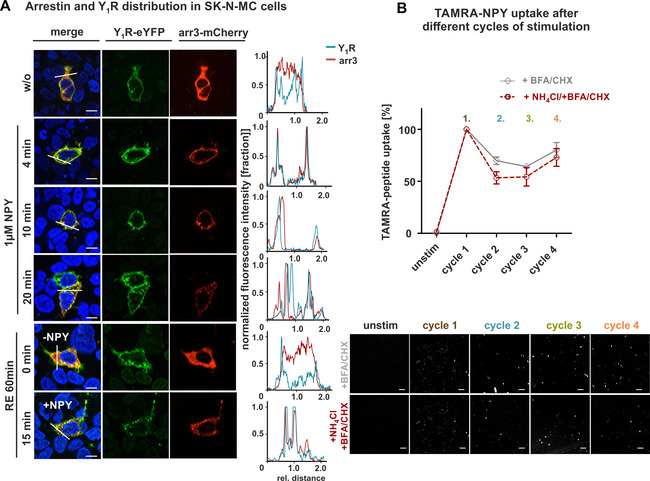
Arrestin recruitment and peptide uptake in SK‐N‐MC cells after different cycles of stimulation. A) Live‐cell microscopy of transiently transfected SK‐N‐MC cells with Y1R fused to eYFP (green) and arrestin 3 (red) carrying mCherry on its C terminus. Prior to agonist stimulation, arrestin was randomly distributed within the cytoplasm (w/o), whereas the receptor was mainly located at the cell surface. Treatment with 1 μM NPY resulted in the recruitment of arrestin to the receptor, which was observed even after 4 min and becomes more obvious after 10 and 20 min. Following acidic wash and incubation in recycling buffer for 60 min, receptor and arrestin were separated again and either transported back to the cell surface (Y1R) or redistributed within the cell (RE 60 min, 0 min, −NPY). Comparable internalization properties were observed after a second cycle of stimulation (RE 60 min, +NPY). Co‐localization of receptor and arrestin is indicated by yellow in the merged picture, and was verified by applying line scans (right). B) Peptide uptake after different circles of stimulation and recovery periods was studied by treatment with NPY, followed by a recovery period in ligand‐free medium supplemented either with or without recycling inhibitors (+BFA, CHX, gray line; +BFA, CHX, NH4Cl, red dotted line) and a final TAMRA‐NPY stimulation after each cycle (2, 3, 4). Peptide uptake in the presence of BFA (protein trafficking inhibitor) and CHX (a protein synthesis inhibitor) revealed the same pattern as seen for HEK293 cells. Constant reduction of internalized peptide compared to the first cycle was observed. Adding NH4Cl (recycling inhibitor) to the recycling period slightly diminished the uptake of TAMRA‐NPY compared to untreated cells. Scale bars: 10 μm, experiments represent the mean±SEM values of n≥3 independent experiments (B), and representative data of n≥3 independent experiments (A, B below).
Discussion
Nowadays, according to the World Health Organization, diseases like cancer and obesity are one of the largest public health challenges. Although cancer is the second leading cause of death, cardiovascular diseases such as heart disease and stroke, which are common health consequences of overweight and obesity are the leading causes of death.[ 1 , 2 , 3 , 4 ] Hence, the demand for novel therapeutics targeting specific tissues and thus minimizing side effects increased over the last years. One concept for a targeted drug delivery is based on the high expression of distinct cell‐surface receptors, which represent specific binding sites for their endogenous ligands as well as for modified peptide‐drug conjugates.[ 5 , 6 ] An elegant strategy is to exploit the natural agonist‐induced endocytosis of GPCRs, which physiologically plays a substantial role in signal termination and protecting the cell from acute or chronic overstimulation by reducing the number of receptive receptors at the cell surface. Thus, addressing certain specifically overexpressed receptors can effectively deliver modulators into desired cells. Peptide receptor radionuclide therapy (PRRT) is one strategy, which has already been successfully applied in a clinical setting. The method is based on radiopharmaceuticals composed of a peptide, a chelator and a radionuclide (e. g., lutetium‐177 or yttrium‐90) which targets peptide receptors to deliver locally limited radiotreatment. The effectiveness of current PRRT is promising but applications are still limited owing to the lack of suitable conjugates. Until now, two diverse peptide–drug conjugates (177Lu‐DOTATATE, 90Y‐DOTATOC), both addressing the somatostatin receptor are typically used to treat neuroendocrine tumors (NETs) with great potential.[ 25 , 26 ]
However, present information about the complex intracellular network as well as the sensitive balance between receptor activation and desensitization are rather limited and thus long‐term drug administration often produces adverse effects, including drug tolerance. Expanding the knowledge about the relationship between receptor endocytosis and receptor desensitization and adding quantitative measures for cargo delivery might lead to better understanding and thus to a significant improvement of therapeutic efficacy. In this context, the neuropeptide Y receptor is an excellent model system. It has received great pharmaceutical potential when the group of Reubi reported specific overexpression of the Y1R in breast cancer and derived metastases. [27] Moreover, the receptor has been targeted with toxophore‐loaded peptides [18] or boron‐containing peptides for boron‐neutron‐capture therapy, [17] which requires high amounts of boron delivered. Previous investigations have already characterized that the Y1R internalizes after ligand stimulation in an arrestin‐dependent mechanism and specific binding motifs for arrestin have been identified.[ 21 , 28 , 29 ] While significant advances have been achieved in understanding the activation and internalization of GPCRs, there were limited efforts towards quantifying resensitization in terms of repeated internalization.
According to the relative affinity of arrestin to the receptors and the different internalization and recycling properties GPCRs can be classified into two major groups. [30] Class A receptors are characterized by the dissociation of the receptor/arrestin complex prior to internalization and a fast recycling back to the plasma membrane. In contrast, the Y1R belongs to the class B receptors, that feature a persistent and strong interaction with arrestin 2 or arrestin 3 after ligand stimulation and remains bound during endocytosis. [31] Extending previous mechanistic investigations in transfected model cell lines, we confirm these characteristics now in an array of endogenously expressing Y1R cell lines. Furthermore, we report on the strong co‐internalization of the receptor‐arrestin complex in different cells.
Whereas the fast internalization of the class B receptors was verified in our experiments (Figure 1), the slow reappearance back to the membrane which is reported in literature for class B receptors [32] is in discrepancy with our findings, which are in line with the results of Wanka et al. [21] and Gicquiaux et al. [33] The kinetic profile of the Y1R by quantifying cell‐surface receptors clearly demonstrates rapid endocytosis of the receptors with a calculated t 1/2 of ∼5 min, which confirms the findings of Ouedraogo et al. [34] After removing the agonist solution, a subsequent washing step and incubation in recovery medium, fast recycling of the receptors was observed, which reached a plateau after 15 min. The fate of internalized receptors is highly regulated by distinct intracellular transport proteins. One important group of proteins controlling the recycling machinery, especially the intracellular vesicle transport are the Rab proteins, monomeric small GTPases. Recycling experiments of Ouedraogo et al. disclosed an indirect recycling pathway guided by Rab11 as well as a more rapid and direct recycling route attributing to Rab4. [34] Thus, a constant and high recycling rate is guaranteed, which is an essential requirement for a receptor to be used as a drug shuttle target. The rapid recycling in principle enables the re‐usage of the receptor and facilitate intracellular accumulation of the desired drug. [19] Indeed, great peptide accumulation was observed in our experiments (Figures 6, 7, and 8), confirming the functional shuttle system on three different cell lines with variable receptor expression. Furthermore, quantification of internalized peptide after simultaneous application of recycling inhibitor NH4Cl as well as expression and transport inhibitor CHX /BFA verified the high contribution of recycled receptors after single (Figure 2) and multiple cycles (Figures 2, 3, and 7) as the proportion of ingested NPY was dramatically reduced when recycling was blocked. Obviously, this is an endogenous function of the receptor as the closely related Y2R is not able to recover and re‐internalize. [35] Receptors mediate signals through a cyclic process of receptor activation, desensitization (termination and inactivation of downstream signals), and resensitization (reactivation for next wave of stimulation), which independently regulate GPCR function. Mohan et al. recently reviewed that receptor resensitization is not only a passive homeostasis but rather a regulated process, which significantly alters GPCR function. [36] This is supported by our observation as the amount of cumulative peptide uptake under continuous stimulation was reduced well below the sum of the individual cycles in all cell lines. Furthermore, this indicates the importance of receptor resensitization as well as successive receptor desensitization during continuous stimulation, which is seen for various GPCRs. [37] Potential peptide degradation during continuous stimulation, which might influence and sophisticate the real amount of ingested peptide can be neglected as previous investigations of Böhme et al. clearly demonstrated the stability of amide‐linked TAMRA‐peptides in cells. After 4 h, the detectable TAMRA‐fluorescence was still ∼75 %. Thus, the moderate degradation is not decisive for the discrepancy between continuous and pulsed stimulation. [38] However, also a moderate decrease in the per cycle amount of internalized peptide after four cycles of stimulation was observed for stably transfected HEK293, MCF7 and SK‐N‐MC cells when no recycling inhibitor was used (Figures 3, 8, and 9). This indicates that further cellular components may contribute to the balance of internalization and resensitization. [39] Different conditions within recombinant systems, for example, the relative stoichiometry of the receptors to other cellular downstream proteins might differ from the natural systems, which further might modulate the signaling cascade. [40] Stably transfected HEK293 cells represent an artificial and recombinant cell system with a high expression level of Y1R. Thus, the natural stoichiometry of the receptor and downstream effector proteins is manipulated genetically and shifted towards a high receptor excess. This altered stoichiometry between receptor and, for example, arrestin protein might further lead to a reduction in arrestin recruitment, which was observed particularly in the kinetic BRET experiments (Figure 5C). This might explain the decreased peptide uptake over certain cycles of stimulation by inhibiting biosynthesis and anterograde transport of proteins with CHX and BFA, respectively (Figures 3, 8, and 9). We verified that the reduced netBRET reflecting the fraction of receptor‐arrestin complexes is not due to reduced peptide affinity or potency to recruit arrestin. Rather, the reduction in receptor‐arrestin complexes must originate from altered cellular stoichiometry of donor and acceptor. The maximal netBRET is achieved only under conditions of saturating excess of binding competent receptor, that is, ligand‐bound and fully phosphorylated, over arrestin.[ 41 , 42 ] The reasons for a reduced netBRET might be an insufficient amount of GPCR kinases leading to incomplete receptor phosphorylation or receptor degradation. It may also be speculated that, in combination with or alternatively to these scenarios, the cellular arrestin expression might be upregulated (by expression or release from complexes) in response to the strong recruitment in the first phase, reducing the receptor excess and thus the fraction of receptor–arrestin complexes relative to the total amount of arrestin. Importantly, this would not go along with the same reduction in the actual number of formed receptor complexes, and hence, peptide internalization. This fits the observation that the maximal arrestin‐recruitment in the second cycle is reduced by 40 % (Figure 5C), while the peptide uptake is only reduced by 25 % (Figures 2C and 3B).
In contrast to the artificial HEK293 cells, the hypothalamic cell line (mHypoN39) represents a more endogenous system where a more natural stoichiometry between receptor and the trafficking machinery including arrestin can be assumed, and consequently the peptide uptake per cycle remain constant over different stimulation periods with intermediate recovery periods (Figure 7D). However, the equilibrium between receptor internalization and recycling is shifted under continuous stimulation conditions, and receptor recycling is not as efficient, which slows down the effective peptide uptake. Nonetheless, comparing the intracellular peptide accumulation of mHypo‐N39 cells and recombinant HEK293 cells displays differences in the peptide uptake rate as well as in the peptide amount that accumulates over time (Figures 6 and 7C). These data provide evidence that in principle a high number of effective shuttling cycles are indeed possible, also in an endogenous cell system as long as the balance between receptor and downstream effector protein is guaranteed and maintained on the cellular level and vice versa receptor overexpression displaces and unbalances the equilibrium. The hypothesis is corroborated as similar effects were also observed in the two tested cancer cell lines. MCF7 cells (originally established from metastatic lobular mammary carcinoma), as well as SK‐N‐MC cells (isolated from metastatic human neuroblastoma), reveal a relatively high number of endogenously expressed Y1 receptor,[ 23 , 24 ] and thus provide an attractive tool for investigating cancer cells for the application of targeted drug delivery systems. However, even under this natural condition, the overexpression of the Y1R is sufficient to obviously flip the balance between receptor and arrestin as the peptide uptake after every cycle of stimulation loses efficiency, likewise seen for HEK293 cells stably expressing the Y1R (Figures 8C and 9B). Despite the constant reduction of internalized peptide after certain cycles of stimulation, still a high amount of TAMRA‐NPY was taken up by the cells, which also can be seen in the accumulation experiment (Figure 8B). This confirms the Y1R receptor to be a powerful and suitable shuttling system, contrary to the recently found impaired activation and internalization of the related Y2R. [35] These findings underline the importance of comprehensive knowledge about receptor signaling and trafficking. Moreover, precise information is crucial to positively influence receptor internalization, desensitization and resensitization to exploit the maximal capacity for the development of novel drug targets. Thus, our data suggest that for a maximal cargo delivery, repetitive stimulation cycles obtained for example by a pulsed application might be beneficial to increase the amount of peptide uptake.
Conclusion
In summary, we found that the Y1 receptor undergoes robust endocytosis and recycling after NPY treatment with a rapid kinetic profile. Interestingly, in the endogenous cell systems a balanced stoichiometry between receptor and arrestin is still guaranteed, maintaining the cellular balance and vice versa the functionality of the whole internalization machinery. If this equilibrium is shifted towards a higher receptor number compared to effector proteins in artificial and recombinant cell lines (HEK293), this will result in diminished arrestin recruitment and peptide uptake over time. Thus, we postulated that in principle, a high number of effective “shuttling” cycles is technically possible and a high amount of peptide can be shuttled into the cell as long as the balance between receptor and effector expression is not changed. In very high receptor expressing cells, for example, in cancer cells, this equilibrium might collapse again and result in the reduction of effective internalization cycles. Thus, our data suggest repetitive applications to allow the cellular machinery to recover are superior for shuttling. Due to the fast receptor kinetics of internalization and recycling, one hour recycling intervals proved sufficient to increase the amount of ingested peptide, as in all tested cell lines significant higher amounts of peptide were taken up into the cells. Thus, not only the amount of receptors but also the available protein trafficking determines the success of a targeted therapy.
Experimental Section
Peptides. All peptides were synthesized by automated solid‐phase peptide synthesis using the 9‐fluorenylmethoxycarbonyl/tert‐butyl (Fmoc/tBu) strategy [43] and purified to≥95 % homogeneity by preparative HPLC. Analysis and identification were performed by MALDI‐ToF mass spectrometry (Ultraflex III MALDI ToF/ToF, Bruker, Billerica, USA) and reversed‐phase HPLC using linear gradients of solvent B (acetonitrile+0.08 % trifluoroacetic acid) in A (H2O+0.1 % trifluoroacetic acid).
Plasmids. The human Y1R within the pEYFP‐N1 expression vector (Clontech) was used for fluorescence microscopy and binding assays. [44] For fluorescence microscopy experiments, bovine arrestin 3 was C‐terminally fused to mCherry and for the BRET‐assay setup it was N‐terminally fused to Renilla luciferase 8. [45] The identity of all plasmid constructs was verified by Sanger dideoxy sequencing.
Cell culture. All cell lines were cultured in flasks to confluence prior to use in a humidified atmosphere at 37 °C and 5 % CO2. HEK293 cells (human embryo kidney, from DSMZ, ACC305) were maintained in Dulbecco's modified Eagle's medium (DMEM) with 4.5 g/L glucose and l‐glutamine and Ham's F12 (1 : 1, Lonza) supplemented with 15 % (v/v) heat‐inactivated fetal bovine serum (FBS). Stably transfected HEK293‐HA‐hY1R‐eYFP were generated by transfecting 13 μg of linearized HA‐hY1R‐eYFP‐pVitro plasmid with 20 μL Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Single clones were selected and expanded as described by Bohme et al. [20] For culturing the same conditions were used as previously described and 100 μg/mL hygromycin B gold (Invivogen) was supplied to medium to ensured stable transfection. mHypoE‐N39 (mouse hypothalamic cell line, Cedarlane Burlington, Ontario, Canada) cells were maintained in DMEM with 4.5 g/L glucose and L‐glutamine and Ham's F12 (1 : 1, Lonza) supplemented with 10 % (v/v) heat‐inactivated FBS. MCF7 (human Caucasian breast adenocarcinoma, DSMZ) cells were cultured in DMEM with 4.5 g/L glucose and L‐glutamine and Ham's F12 (1 : 1, Lonza) supplemented with 10 % (v/v) heat‐inactivated FBS. SK‐N‐MC (human neuroblastoma cell line, ATCC) were cultivated in Eagle's minimal essential medium (EMEM; Lonza) supplemented with 10 % FCS, 4 mM l‐glutamine (Lonza), 1 mM sodium pyruvate (Lonza), and 0.2× MEM nonessential amino acids (Lonza). All cell lines were routinely tested negative for mycoplasma contamination.
Live‐cell microscopy. HEK293, HEK293‐HA‐hY1R‐eYFP, mHypoE‐N39 cells, MCF7 cells and SK‐N‐MC cells were seeded (150 000/well) into sterile poly‐d‐lysine coated μ‐slide 8 wells (Ibidi) and grown in a humidified atmosphere at 37 °C and 5 % CO2. For transfection, cells were cultured up to 70–80 % confluence and subsequently transfected with 1.0 μg total DNA using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's protocol. For single transfection in empty HEK293 cells 1.0 μg of HA‐hY1R‐eYFP_N1 plasmid DNA was used. At the experimental day, cells were starved in OptiMEM reduced‐serum medium (Gibco) containing Hoechst33342 (Sigma) for 30 min at 37 °C. Internalization studies were performed by stimulating cells with 1 μM NPY or 100 nM fluorescently labeled NPY derivatives in OptiMEM reduced‐serum medium for 60 min at 37 °C. For recycling studies, cells were washed twice with acidic wash buffer (50 mM glycine, 100 mM NaCl, adjusted to pH 3.0 with glacial acetic acid) and neutralized once with Hank's balanced salt solution (HBSS; PAA), followed by a recovery period for 60 min in ligand‐free media supplemented with 100 μg/ml cycloheximide (CHX; Merck/Calbiochem); 6 μg/mL brefeldin A (BFA, Santa Cruz) and with or without 20 mM NH4Cl as expression inhibitor, trafficking inhibitor and recycling inhibitor respectively. Uptake experiments using TAMRA‐NPY were performed by stimulating cells in the first cycle with 1 μM NPY, subsequent acidic wash, followed by either a second stimulation with 100 nM TAMRA‐NPY or by incubation in ligand‐free media for 60 min (recovery period) prior to the 2nd stimulation. Experiments were performed in media supplemented with CHX and with or without 20 mM NH4Cl for 60 min. For the characterization of the receptor shuttling capacity cells were treated in the first cycle either with 100 nM TAMRA‐NPY as control or with 1 μM NPY. After 60 min of incubation, cells were washed twice with acidic wash buffer, neutralized with HBSS and prepared for 60 min recovery period. The recycling buffer was composed of either CHX and BFA or CHX, BFA and NH4Cl. After the recovery period the 2nd cycle of stimulation was initialized either with 100 nM TAMRA‐NPY for the detection of internalized peptide or 1 μM NPY. After 60 min of incubation time, cells were prepared for recovery period again following the same protocol as previously described. The same procedure was applied for the following cycles three and four. For intracellular accumulation experiments, cells were treated with 1 μM TAMRA‐NPY for 4 h. At different time points (1, 2, 3, 4 h) incubation was stopped by aspirating the stimulation solution and subsequent acidic wash. All microscopy images were obtained using the AxioObserver.Z1 microscope equipped with an ApoTome imaging system (Zeiss, Jena). Within one experimental setup, all images were taken with a fixed exposure time. For visualizing receptor internalization and arrestin recruitment simultaneously, empty HEK293, MCF7, mHypoN39 and SK‐N‐MC cells were reseeded into IBIDI‐slides and co‐transfected with 900 ng hY1R‐eYFP‐N1 plasmid and 100 ng P3‐arrestin3‐mCherry when they reach 70–80 % confluence using Lipofectamine® 2000 transfection reagent (Invitrogen) according to the manufacturer's protocol. 24 h post transfection, the cells were serum‐deprived with OptiMEM reduced‐serum medium (Gibco) containing 2.5 μg/mL Hoechst33342 (Sigma) for 30 min at 37 °C.
All microscopy pictures were processed with Axio vision software 4.8 and exported as 8 Bit grayscale TIFF files. The open‐access software ImageJ was applied for the image analysis. For the determination of MCSF (mean cell‐surface fluorescence) 10 nonadjacent cells per image were measured using the segmented line function. For the calculation of the relative TAMRA fluorescence, the raw intensity density of the microscopy image was measured. The particular background fluorescence of each image was subtracted after every evaluation. Single gray levels represented the relative fluorescence intensities and were statistically analyzed with GraphPad Prism.
BRET‐assay. HEK293 cells were grown in 25 cm2 cultivation flasks and transiently co‐transfected using Metafectene Pro transfection reagent (Biontex Laboratories GmbH) according to the manufacturer's protocol. For arrestin 3 recruitment and expression control, 7850 ng plasmid encoding for eYFP tagged receptor and 150 ng arrestin 3 fused to RLuc8 was transfected. One day post transfection, cells were seeded into poly‐D‐lysine‐coated 96‐well plates (Greiner Bio‐one). Two days post transfection, the medium was replaced with BRET buffer (HBSS buffer containing 25 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES; Merck), pH 7.3). For the BRET experiments, the Renilla luciferase substrate Coelenterazine h (Nanolights) was added to a final concentration of 4.2 μM. For kinetic BRET studies, the baseline was measured for 5 min and then 10−6 M NPY was added to the cells. The BRET signal was measured for 30 min at 37 °C with a Tecan infinite M 200 reader using filter set Blue1 (luminescence 370–480 nm) and Green1 (fluorescence 520–570 nm). For concentration‐response curves, NPY was added in a concentration ranging from 10−12 to 10−5 M, and the BRET signal was measured after 5 min of stimulation time at 37 °C. The BRET ratio was calculated as a quotient of fluorescence to luminescence values and the netBRET signal was determined by subtracting BRET signals of unstimulated cells from stimulated samples.
Specific radioligand binding assay. Stable HEK293‐HA‐Y1R‐eYFP cells were seeded into poly‐d‐lysine coated 48‐well plates (Greiner Bio‐one) and incubated overnight in a humidified atmosphere at 37 °C and 5 % CO2. On the experimental day, cells were treated either with buffer or 1 μM NPY for 60 min, subsequently washed twice with acidic wash buffer and once with HBSS, followed by a recovery period (60 min RE, 0 min RE) in ligand‐free medium containing 100 μg/mL CHX (Merck/Calbiochem®). After treatment, cells were immediately cooled down on ice, washed once with PBS and incubated with 6×10−11 M human [125I]‐PYY in binding buffer for 4 h. Binding buffer consisted of OptiMEM, 50 mM Pefabloc SC, 1 % BSA. For displacement experiments NPY were used in concentrations ranging from 10−11 to 10−6 M NPY. After incubation, cells were washed twice with ice‐cold PBS and lysed with 0.2 M NaOH. Lysates were transferred into scintillation cocktail and radioactivity was detected with a Microbeta2 counter.
Statistical analysis. Calculations of means, S.E.M., and statistical analysis were performed using PRISM 5.0 program (GraphPad Software, San Diego, USA). Significances were calculated according to one‐way ANOVA and Tukey's or paired, two‐tailed t‐test.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors gratefully thank Minh Ganther, Kristin Löbner, Janet Schwesinger, Christina Dammann and Ronny Müller for excellent assistance. This work was supported by the German Science Foundation Deutsche Forschungsgemeinschaft (German Research Foundation) through CRC 1052, project number 209933838, subproject A03 and Z05, the European Union (EU) and the Federal State of Saxony. Open access funding enabled and organized by Projekt DEAL.
I. Ziffert, A. Kaiser, P. Hoppenz, K. Mörl, A. G. Beck-Sickinger, ChemMedChem 2021, 16, 164.
References
- 1. Ferlay J., Parkin D. M., Steliarova-Foucher E., Eur. J. Cancer 2010, 46, 765–781. [DOI] [PubMed] [Google Scholar]
- 2. The Global Burden of Disease: 2004 Update (Eds.: C. Mathers, D. M. Fat, J. T. Boerma), World Health Organization, Geneva, 2008.
- 3. Must A., JAMA 1999, 282, 1523. [DOI] [PubMed] [Google Scholar]
- 4. Field A. E., Coakley E. H., Must A., Spadano J. L., Laird N., Dietz W. H., Rimm E., Colditz G. A., Arch. Intern. Med. 2001, 161, 1581. [DOI] [PubMed] [Google Scholar]
- 5. Reubi J. C., Endocrine Rev. 2003, 24, 389–427. [DOI] [PubMed] [Google Scholar]
- 6. Tesauro D., Accardo A., Aloj L., Morelli G., Aurilio M., Int. J. Nanomed. 2014, 1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rosenbaum D. M., Rasmussen S. G. F., Kobilka B. K., Nature 2009, 459, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ferguson S. S., Pharmacol. Rev. 2001, 53, 1–24. [PubMed] [Google Scholar]
- 9. Babilon S., Mörl K., Beck-Sickinger A. G., Biol. Chem. 2013, 394, DOI 101515/hsz-2013–0123. [DOI] [PubMed] [Google Scholar]
- 10. Ahrens V. M., Frank R., Boehnke S., Schütz C. L., Hampel G., Iffland D. S., Bings N. H., Hey-Hawkins E., Beck-Sickinger A. G., ChemMedChem 2015, 10, 164–172. [DOI] [PubMed] [Google Scholar]
- 11. Ahrens V. M., Kostelnik K. B., Rennert R., Böhme D., Kalkhof S., Kosel D., Weber L., von Bergen M., Beck-Sickinger A. G., J. Controlled Release 2015, 209, 170–178. [DOI] [PubMed] [Google Scholar]
- 12. Cabrele C., Beck-Sickinger A. G., J. Pept. Sci. 2000, 6, 97–122. [DOI] [PubMed] [Google Scholar]
- 13. Körner M., Waser B., Reubi J. C., Int. J. Cancer 2005, 115, 734–741. [DOI] [PubMed] [Google Scholar]
- 14. Körner M., Reubi J. C., J. Neuropathol. Exp. Neurol. 2008, 67, 741–749. [DOI] [PubMed] [Google Scholar]
- 15. Reubi J. C., Gugger M., Waser B., Schaer J. C., Cancer Res. 2001, 61, 4636–4641. [PubMed] [Google Scholar]
- 16. Worm D. J., Hoppenz P., Els-Heindl S., Kellert M., Kuhnert R., Saretz S., Köbberling J., Riedl B., Hey-Hawkins E., Beck-Sickinger A. G., J. Med. Chem. 2019, DOI 10.1021/acs.jmedchem.9b01136. [DOI] [PubMed] [Google Scholar]
- 17. Böhme D., Beck-Sickinger A. G., J. Pept. Sci. 2015, 21, 186–200. [DOI] [PubMed] [Google Scholar]
- 18. Wittrisch S., Klöting N., Mörl K., Chakaroun R., Blüher M., Beck-Sickinger A. G., Mol. Metabol. 2020, 31, 163–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Molema G., in Methods and Principles in Medicinal Chemistry (Eds.: Molema G., Meijer D. K. F.), Wiley-VCH, Weinheim, 2001, pp. 1–22. [Google Scholar]
- 20. Böhme I., Stichel J., Walther C., Mörl K., Beck-Sickinger A. G., Cell. Signalling 2008, 20, 1740–1749. [DOI] [PubMed] [Google Scholar]
- 21. Wanka L., Babilon S., Kaiser A., Mörl K., Beck-Sickinger A. G., Cell. Signalling 2018, 50, 58–71. [DOI] [PubMed] [Google Scholar]
- 22. Longmire M., Kosaka N., Ogawa M., Choyke P. L., Kobayashi H., Cancer Sci. 2009, 100, 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fabry M., Langer M., Rothen-Rutishauser B., Wunderli-Allenspach H., Höcker H., Beck-Sickinger A. G., Eur. J. Biochem. 2000, 267, 5631–5637. [DOI] [PubMed] [Google Scholar]
- 24. Wolf P., Rothermel A., Beck-Sickinger A. G., Robitzki A. A., Biosensors Bioelectronics 2008, 24, 253–259. [DOI] [PubMed] [Google Scholar]
- 25. Fröss-Baron K., Garske-Román U., Welin S., Granberg D., Eriksson B., Khan T., Sandström M., Sundin A., Neuroendocrinology 2020, DOI: 10.1159/000506746. [DOI] [PubMed] [Google Scholar]
- 26. Alsadik S., Yusuf S., AL-Nahhas A., CRP 2019, 12, 126–134. [DOI] [PubMed] [Google Scholar]
- 27. Körner M., Reubi J. C., Peptides 2007, 28, 419–425. [DOI] [PubMed] [Google Scholar]
- 28. Gimenez L. E., Babilon S., Wanka L., Beck-Sickinger A. G., Gurevich V. V., Cell. Signalling 2014, 26, 1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Walther C., Nagel S., Gimenez L. E., Mörl K., Gurevich V. V., Beck-Sickinger A. G., J. Biol. Chem. 2010, 285, 41578–41590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gainetdinov R. R., Premont R. T., Bohn L. M., Lefkowitz R. J., Caron M. G., Ann. Rev. Neurosci. 2004, 27, 107–144. [DOI] [PubMed] [Google Scholar]
- 31. Lefkowitz R. J., J. Biol. Chem. 1998, 273, 18677–18680. [DOI] [PubMed] [Google Scholar]
- 32. Cahill T. J., Thomsen A. R. B., Tarrasch J. T., Plouffe B., Nguyen A. H., Yang F., Huang L.-Y., Kahsai A. W., Bassoni D. L., Gavino B. J., Lamerdin J. E., Triest S., Shukla A. K., Berger B., Little J., Antar A., Blanc A., Qu C.-X., Chen X., Kawakami K., Inoue A., Aoki J., Steyaert J., Sun J.-P., Bouvier M., Skiniotis G., Lefkowitz R. J., Proc. Natl. Acad. Sci. USA 2017, 114, 2562–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gicquiaux H., Lecat S., Gaire M., Dieterlen A., Mély Y., Takeda K., Bucher B., Galzi J.-L., J. Biol. Chem. 2002, 277, 6645–6655. [DOI] [PubMed] [Google Scholar]
- 34. Ouedraogo M., Lecat S., Rochdi M. D., Hachet-Haas M., Matthes H., Gicquiaux H., Verrier S., Gaire M., Glasser N., Mély Y., Takeda K., Bouvier M., Galzi J.-L., Bucher B., Traffic 2008, 9, 305–324. [DOI] [PubMed] [Google Scholar]
- 35. Ziffert I., Kaiser A., Babilon S., Mörl K., Beck-Sickinger A. G., Cell Commun. Signaling 2020, 18, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mohan M. L., Vasudevan N. T., Gupta M. K., Martelli E. E., Naga Prasad S. V., Curr. Mol. Pharmacol. 2012. [PMC free article] [PubMed] [Google Scholar]
- 37. Rajagopal S., Shenoy S. K., Cell. Signalling 2018, 41, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Böhme D., Beck-Sickinger A. G., ChemMedChem 2015, 10, 804–814. [DOI] [PubMed] [Google Scholar]
- 39. Milligan G., Cell. Signalling 1996, 8, 87–95. [DOI] [PubMed] [Google Scholar]
- 40. Kenakin T., Trends Pharmacol. Sci. 1997, 18, 456–464. [DOI] [PubMed] [Google Scholar]
- 41. Borroto-Escuela D. O., Flajolet M., Agnati L. F., Greengard P., Fuxe K., Methods Cell Biol. 2013, 117, 141–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Terrillon S., Durroux T., Mouillac B., Breit A., Ayoub M. A., Taulan M., Jockers R., Barberis C., Bouvier M., Mol. Endocrinol. 2003, 17, 677–691. [DOI] [PubMed] [Google Scholar]
- 43. Hofmann S., Frank R., Hey-Hawkins E., Beck-Sickinger A. G., Schmidt P., Neuropeptides 2013, 47, 59–66. [DOI] [PubMed] [Google Scholar]
- 44. Dinger M. C., Bader J. E., Kóbor A. D., Kretzschmar A. K., Beck-Sickinger A. G., J. Biol. Chem. 2003, 278, 10562–10571. [DOI] [PubMed] [Google Scholar]
- 45. Vishnivetskiy S. A., Gimenez L. E., Francis D. J., Hanson S. M., Hubbell W. L., Klug C. S., Gurevich V. V., J. Biol. Chem. 2011, 286, 24288–24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary