

Abstract
Objective
To predict fetal growth restriction (FGR) by whole‐genome promoter profiling of maternal plasma.
Design
Nested case–control study.
Setting
Hospital‐based.
Population or Sample
810 pregnancies: 162 FGR cases and 648 controls.
Methods
We identified gene promoters with a nucleosome footprint that differed between FGR cases and controls based on maternal plasma cell‐free DNA (cfDNA) nucleosome profiling. Optimal classifiers were developed using support vector machine (SVM) and logistic regression (LR) models.
Main outcome measures
Genes with differential coverages in promoter regions through the low‐coverage whole‐genome sequencing data analysis among FGR cases and controls. Receiver operating characteristic (ROC) analysis (area under the curve [AUC], accuracy, sensitivity and specificity) was used to evaluate the performance of classifiers.
Results
Through the low‐coverage whole‐genome sequencing data analysis of FGR cases and controls, genes with significantly differential DNA coverage at promoter regions (−1000 to +1000 bp of transcription start sites) were identified. The non‐invasive ‘FGR classifier 1’ (CFGR1) had the highest classification performance (AUC, 0.803; 95% CI 0.767–0.839; accuracy, 83.2%) was developed based on 14 genes with differential promoter coverage using a support vector machine.
Conclusions
A promising FGR prediction method was successfully developed for assessing the risk of FGR at an early gestational age based on maternal plasma cfDNA nucleosome profiling.
Tweetable abstract
A promising FGR prediction method was successfully developed, based on maternal plasma cfDNA nucleosome profiling.
Keywords: Cell‐free DNA, classifier, fetal growth restriction, low‐coverage whole‐genome sequencing, non‐invasive prediction
Tweetable abstract
A promising FGR prediction method was successfully developed, based on maternal plasma cfDNA nucleosome profiling.
Introduction
Fetal growth restriction (FGR) refers to the fetus that fails to reach its biological growth potential. The most widely used definition of FGR is an estimated fetal weight (EFW) that is less than the 10th percentile for gestational age. 1 It is responsible for 30% of all stillbirths and is associated with increased perinatal mortality and morbidity. 2 FGR is usually diagnosed by ultrasound in the third trimester. Early diagnosis might increase the likelihood of successful treatment, minimise risks of intrauterine hypoxia and preterm birth. There are maternal serum markers and Dopplers that could be used in screening or prediction for FGR during the first and the second trimester 3 ; however, they are not considered accurate in isolation and need to be combined and examined risk factors to increase their clinical utility. More reliable biomarkers for early prediction of FGR are indeed needed and would allow for early identification of fetuses that are likely to develop FGR and might help to test hypothetical interventions for primary prevention.
Since the discovery of fetal DNA present in maternal plasma cell‐free DNA (cfDNA) in 1997, the utility of cfDNA has been well‐demonstrated by the development of non‐invasive prenatal screening tests for fetal aneuploidy, non‐invasive sequencing of fetal genomes and non‐invasive diagnostics for common monogenic diseases. 4 , 5 Recently, new methods have been developed to determine the tissue origin of cfDNA through nucleosome positioning and methylation footprint analysis. 6 , 7 , 8 Snyder et al. 7 produced a map of nucleosome occupation in the whole genome via in‐depth sequencing of cfDNA isolated from circulating plasma. Based on the nucleosome signal near the transcription start sites (TSS), a large difference between genes with high and low expression levels was observed; moreover, the intensity of their footprints upstream of the TSS was strongly related to the expression of transcriptionally active genes. 6 , 7 , 8
In pregnancies, the majority of cfDNA is derived from maternal haematopoietic cells and placental trophoblasts. 9 More importantly, pregnant complications have a root cause in the placenta, and involve the maternal immune system. 10 , 11 Therefore, we hypothesised that cfDNA fragment distribution patterns might carry information regarding source tissues of origin, particularly placental trophoblasts and maternal haematopoietic cells, and that global profiling of cfDNA fragments in promoter regions can be used to identify biomarkers that can predict FGR. The aim of the study is to identify genes with differential coverages in promoter regions (defined as –1000 to +1000 bp around TSS), through the low‐coverage whole‐genome sequencing NIPT data analysis among FGR cases and controls, and to develop high‐performance FGR classifiers using machine‐learning methods.
Methods
Study rationale
Cell‐free DNA fragments are related to the DNA released by apoptotic cells after enzyme processing. 12 Currently, cfDNA has been proven to be able to reflect the expression status of its tissue of origin. 7 In actively transcribed genes, the promoter region (−1000 to +1000 bp of TSS) showed a reduction in nucleosome occupancy. By contrast, the promoter coverage of cfDNA increased, reflecting the denser nucleosome packaging of repressed genes. 13 , 14 , 15 In this study, we detected promoter profiles using low‐coverage whole‐genome sequencing data of non‐invasive prenatal test (NIPT). By comparing the difference of promoter coverage between FGR cases and controls, we determined whether genes with differential promoter coverage are able to be used as biomarkers for screening FGR. Based on these variables, we then developed high‐performance classifiers for FGR prediction using multiple machine‐learning methods.
Study design and participants
This nested case–control study investigated whether cfDNA in maternal blood can be used to identify FGR. The study was approved by the Internal Ethics Committee of Nanfang Hospital, Southern Medical University. Samples of NIPT data were collected from patients at the Nanfang Hospital of Southern Medical University (SMU), the Third Affiliated Hospital of Sun Yat‐sen University (SYSU), and Cangzhou People’s Hospital. There was no patient or public involvement in the study and no core outcome set has been used.
From three independent Chinese institutions, we collected 3600 samples of routine low‐coverage whole‐genome sequencing NIPT data of singleton pregnant women at 12+0–27+6 weeks, in addition to general clinical information. We retrospectively analysed the data, including results of ultrasound during pregnancy and final birthweight, to derive an FGR group and a control group. The following exclusion criteria were used in the patient selection: (1) gestational age at the time of blood extraction of <12 weeks or more than 28 weeks; (2) aneuploidy and fetus structural abnormality; (3) over‐ or underweight, with tobacco or alcohol exposure; (4) FGR manifestations noted when drawing blood during the previous ultrasound screening. Referring to the expert consensus16 and based on the Chinese birthweight reference, 17 FGR group classification was based on:
birthweight ≤10th percentile, plus
prenatal evidence of uterine placental insufficiency, defined as umbilical or uterine artery pulsatility index (PI) >95th centile or absent end‐diastolic flow in the umbilical artery (<32 weeks’ gestation) or cerebroplacental ratio (CPR; calculated as the fetal middle cerebral artery PI divided by the umbilical artery PI) <5th centile (≥32 weeks’ gestation) and/or
abdominal circumference (AC) <10th centile or
birthweight <3rd centile irrespective of the prenatal ultrasonic Doppler status.
Control samples were collected from women with an appropriately grown fetus (birthweight 10–90th percentile) who subsequently delivered appropriately grown fetuses at term without obstetric complications, with a gestational age at blood collection and fetal gender and parity matched to the FGR cases, without gestational hypertension (blood pressure >140/90 mm Hg), pre‐eclampsia (proteinuria or biochemical abnormalities), autoimmune disorders and gestational diabetes mellitus. After exclusion, 2965 samples were included for further selection, of which 162 were FGR. In the rest of samples, we chose controls to match FGR cases. According to the gestational week in which the maternal plasma sample was extracted, and the fetal gender, four control cases were randomly selected to match each FGR case.
Ultimately, 810 samples (162 FGR and 648 controls) were included and the samples from the same hospital were defined as one cohort. They were divided into a Nanfang Hospital training cohort (285 samples from Nanfang Hospital), an internal validation cohort (125 samples from Nanfang Hospital), an external validation cohort 1 (190 samples from the Third Affiliated Hospital of Sun Yat‐sen University) and an external validation cohort 2 (210 samples from Cangzhou People’s Hospital). In the discovery stage, we identified the differential plasma cell‐free nucleosome footprints of the FGR and control groups. We then used support vector machine (SVM) and logistic regression (LR) models to develop a classifier based on genes with differentially coverages (training stage). Receiver operating characteristic (ROC) analysis (AUC, accuracy, sensitivity and specificity) was used to evaluate the performance of each classifier during the training stage. Finally, we devised optimal classifiers and validated their performance in one internal validation cohort and two external validation cohorts (validation stage).
Cell‐free DNA isolation and whole‐genome sequencing
First, 5 ml of maternal peripheral blood was collected into EDTA‐containing blood tubes and centrifuged at 1600 × g for 10 min at 4°C. The plasma was transferred to Eppendorf tubes and centrifuged again at 16 000 × g for 10 min at 4°C to remove residual cells. The plasma aliquots were then carefully transferred to fresh Eppendorf tubes. For each sample, cfDNA was extracted from 700 microlitres of plasma using the Circulating DNA from Plasma Kit following the manufacturer’s instructions (GenMag Biotech, Beijing, China), or from 1.2 ml of plasma using the QIAamp DSP Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany), and was stored at −80°C before testing.
Sequencing libraries were prepared according to the manufacturer’s specifications. The DNA libraries were quantified with the Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA, USA). The size of the libraries was verified using the Agilent High Sensitivity DNA Kit and a 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The libraries from different samples were pooled and sequenced on the NextSeq500 instrument (Illumina, San Diego, CA, USA) or the Ion Proton sequencing platform (Thermo Fisher Scientific, Waltham, MA, USA). The DNA sequencing performed in this study had an average depth of coverage of at least 0.3×.
Low‐coverage whole‐genome sequencing data processing
Raw reads were aligned to the hg19 human reference genome using bwa‐mem, 18 and PCR duplicates were removed using the rmdup function of SAM tools (ver. 1.2). 19 Gene information was obtained from the UCSC database using RefSeq. 20 For each transcript, the region from −1000 to +1000 bp around the transcriptional start site, defined as the primary transcription start site (pTSS), was identified. Read counts for each base at the pTSS were calculated from the aligned BAM files using SAM tools. The read coverage at the pTSS was extracted from the aligned BAM files using BEDtools (ver. 2.17.0). As the number of DNA sequencing reads between different samples was different, therefore, we used a similar normalised method with the reads per kilobase per million mapped reads (RPKM) method to normalise the raw coverages of cfDNA before comparison, using the following formula:
Classifier construction procedure
To develop classifiers for predicting pregnancies with FGR, a workflow was developed that included a discovery stage, a training stage and a validation stage (Figure 1). In the discovery stage, 57 FGR cases and 57 gestational age‐matched controls were selected and the coverage at the pTSSs was compared between the two groups. P‐values were calculated using the rank sum test and adjusted according to the false discovery rate (FDR) using the Benjamini–Hochberg procedure; an FDR ≤0.1 and |log2 fold change| ≥1.5 were cut‐offs for transcripts with significantly differential coverage at the pTSSs. Hierarchical clustering was applied to the coverage data using the average‐linkage clustering algorithms in the Cluster (ver. 3.0) programme. A cluster graph was plotted using heatmap in R software (R Development Core Team, Vienna, Austria).
Figure 1.
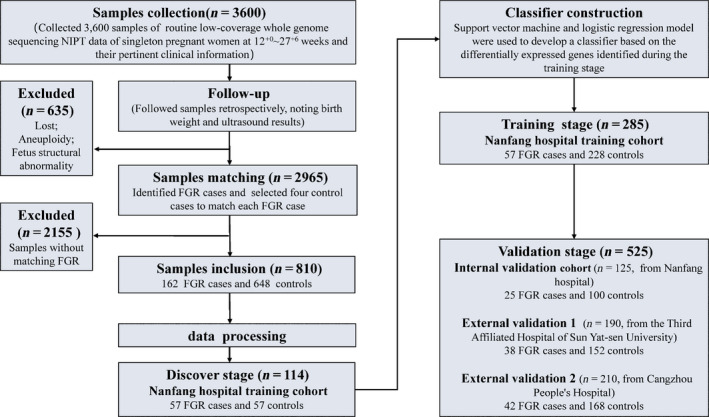
Flowchart of FGR classifier development based on low‐coverage whole‐genome sequencing. From three independent Chinese institutions, we collected 3600 samples of routine low‐coverage whole‐genome sequencing NIPT data from singleton pregnant women at 12+0–27+6 weeks, in addition to general clinical information. We used data from retrospective patient follow‐up reports, including pregnancy outcomes and final birthweight, to identify FGR cases and controls. According to gestational age at the time of maternal plasma sample extraction and fetal gender, four control cases were randomly selected to match each FGR case. Ultimately, we included 810 samples (162 FGR and 648 controls), which were divided into a Nanfang Hospital training cohort (285 samples from Nanfang Hospital), an internal validation cohort (125 samples from Nanfang Hospital), an external validation cohort 1 (190 samples from the Third Affiliated Hospital of Sun Yat‐sen University) and external validation cohort 2 (210 samples from Cangzhou People’s Hospital). In the discovery stage, gene promoters with a nucleosome footprint that differed between 57 FGR cases and 57 controls from Nanfang Hospital training cohort were identified. In the training stage, classifiers were developed via support vector machine (SVM) and logistic regression (LR) models, based on the genes with differentially coverages between the 57 FGR cases and 228 controls. In the validation stage, the optimal classifiers were further validated using the three validation cohorts.
In the training stage, an SVM and LR model were used to develop promoter profiling‐based classifiers to differentiate FGR cases from healthy controls. We used the linear kernel of SVM with the default setting. To develop classifiers, a stepwise method was used to identify promoter combinations from among genes showing differential coverage at the pTSSs. To assess the extent of over‐fitting in predictions, the robustness of the classifiers was assessed using leave‐one‐out cross‐validation (LOOCV) in the training cohorts. Briefly, each subject in the training cohort was excluded from the training model in turn, with the remaining subjects all being entered into it. The trained model was then used to predict the class (pregnancies with complications or healthy controls) of the withheld subject. This procedure continued until all subjects in the training cohort were classified. ROC analysis (AUC, accuracy, sensitivity and specificity) was used to evaluate the performance of each classifier. The classifier that achieved the highest performance, i.e. the one with the largest AUC in the training cohort, was defined as the optimal classifier.
In the validation stage, the performance of the classifier was further validated in three independent validation cohorts, including one internal cohort and two external cohorts. The function in the pROC package of R software was used to compare the performance of the optimal classifier developed based on SVM and that developed based on LR.
Results
Cohort demographic and pregnancy characteristics
To develop classifiers to predict FGR, we included 810 samples (162 FGR cases and 648 healthy pregnancies) from three independent Chinese institutions: Nanfang Hospital, the Third Affiliated Hospital of Sun Yat‐sen University and Cangzhou People’s Hospital (Figure 1). This case–control study included four cohorts: a training cohort, an internal validation cohort and two external cohorts. The demographic and pregnancy details of the cohort are shown in Tables S1 and S2.
Profiling of cfDNA from FGR and healthy pregnancies
The transcriptional activity of genes varies according to nucleosome occupancy at promoter regions, with decreased occupancy at the pTSS of the active genes. In addition, a study has found a relative cfDNA loss at transcription termination sites (TTS) with age. 21 Hence, we performed whole‐genome sequencing to profile the cfDNA from 57 FGR cases and 57 healthy pregnancies of the Nanfang Hospital training cohort. To identify nucleosome profile changes in various gene regions, we computed the average cfDNA signals across all genes relative to the TSS and TTS of the genes. We observed typical nucleosome‐depleted regions (NDRs) at the TSS, which are also commonly observed in MNase assays. In the promoter and TSS regions, we found decreased cfDNA signals in FGR cases (Figure 2).
Figure 2.
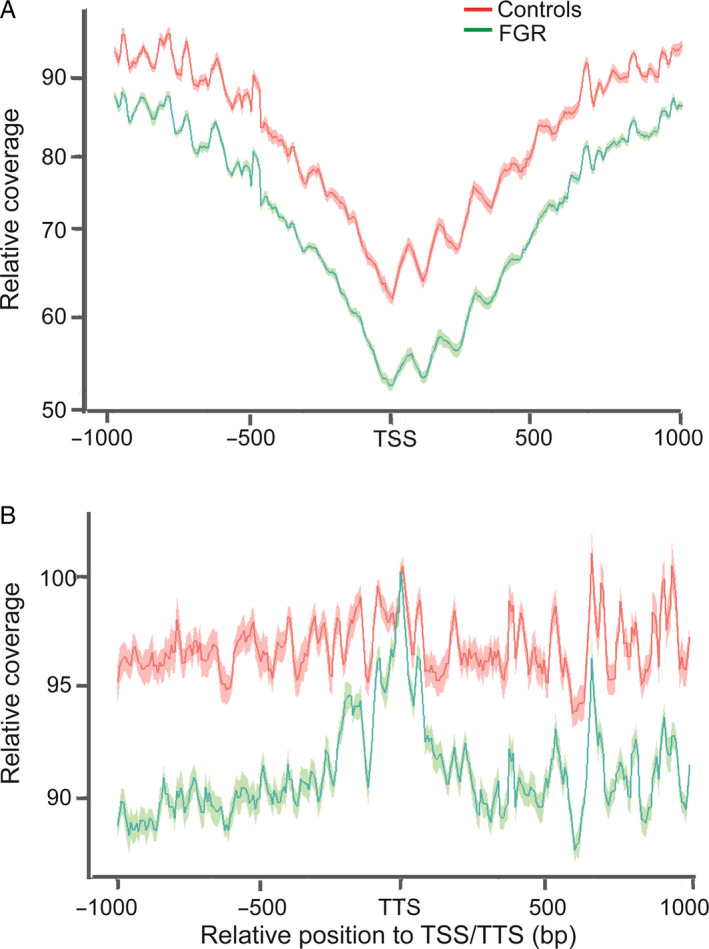
Differences in local nucleosome profiles between fetal growth restriction (FGR) and controls. Cell‐free DNA (cfDNA) signals in transcription start sites (TSS) and transcription termination sites (TTS) regions decreased in FGR cases. The red line represents mean average cfDNA signals of controls and the red shadow represents its standard error of mean. The green line represents mean average cfDNA signals of FGR cases and the green shadow represents its standard error of mean. (A) Average cfDNA signals at TSS regions. (B) Average cfDNA signals at TTS regions.
Promoter profiling of cfDNA revealed FGR‐associated patterns
In the discovery stage, 57 FGR cases and 57 gestational age‐matched controls were compared to identify genes with significantly differential coverage in order to develop classifiers for predicting FGR. In total, we found 103 gene transcripts with significantly differential coverage, including 46 transcripts with up‐regulated coverage and 57 with down‐regulated coverage (Figure 3A). Unsupervised clustering analyses based on gene coverage revealed distinct coverage patterns between FGR and their controls (Figure 3B).
Figure 3.
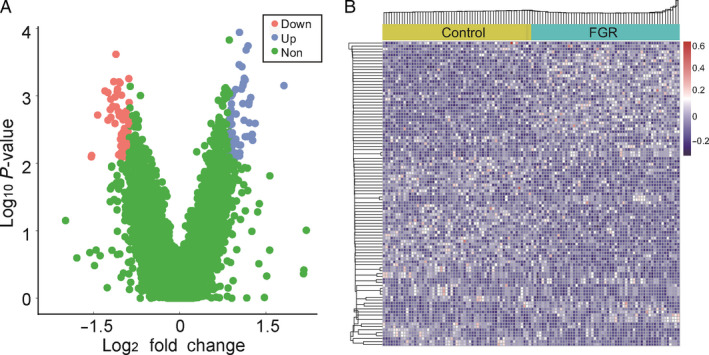
Gene transcripts with differential read coverage at transcription start sites (TSS). (A) Volcano plots of gene transcripts with differential read coverage at the TSS, as detected by whole‐genome sequencing (|Log2 fold change| ≥1.5 and FDR <0.1). Blue blots represent genes with up‐regulated promoter read depth coverage, red blots represent genes with down‐regulated promoter read depth coverage, and green blots represent genes with no significant difference. (B) Heat map of the z‐scores of promoters with differential read coverage.
Classifier development and validation for FGR prediction
In the training stage, we applied the SVM and the LR model to develop classifiers for predicting pregnancies at risk of FGR. Each gene combination created with the SVM or LR model was defined as a single classifier. We used ROC analysis to determine the sensitivity, specificity, accuracy and AUC of the FGR classifiers. Among all combinations, a 14‐gene combination (TCHH, ST5, CKB, KNOP1, NOS2, KRTAP9‐9, ARHGAP15, SLC4A10, HMGB2, SEPT11, ZKSCAN5, GPAT4, PHPT1, FANCC) achieved the highest classification performance after LOOCV (AUC, 0.800; 95% CI 0.740–0.861; accuracy, 81.8%) in the SVM model (FGR classifier 1, CFGR1), and a 12‐gene combination (TCHH, ST5, CKB, NOS2, KRTAP9‐9, ARHGAP15, SLC4A10, HMGB2, SEPT11, ZKSCAN5, GPAT4, FANCC) achieved the highest performance after LOOCV (AUC, 0.776; 95% CI 0.713–0.840; accuracy, 81.1%) in the LR model (FGR classifier 2, CFGR2) (Table 1). The probability of FGR detection by CFGR2 was calculated for each patient using a formula involving these 12 genes weighted by their regression coefficient:
Table 1.
Performance of ideal FGR classifiers
| Classifiers | SVM | LR | P‐value | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Cohort | AUC (95% CI) | Accuracy (%) | Sensitivity (%) | Specificity (%) | AUC (95% CI) | Accuracy (%) | Sensitivity (%) | Specificity (%) | |
| Training | 0.800 (0.740–0.861) | 81.8 | 77.1 | 82.9 | 0.776 (0.713–0.840) | 81.1 | 71.9 | 83.3 | 0.160 |
| Internal | 0.830 (0.743–0.917) | 84.8 | 80.0 | 86.0 | 0.790 (0.694–0.886) | 83.2 | 72.0 | 86.0 | 0.199 |
| External‐1 | 0.780 (0.700–0.859) | 83.7 | 68.4 | 87.5 | 0.737 (0.655–0.819) | 76.8 | 68.4 | 78.9 | <0.001 |
| External‐2 | 0.809 (0.739–0.880) | 83.8 | 76.2 | 85.7 | 0.729 (0.652–0.807) | 75.2 | 69.0 | 76.8 | <0.001 |
| All | 0.803 (0.767–0.839) | 83.2 | 75.3 | 85.2 | 0.757 (0.719–0.795) | 78.9 | 70.4 | 81.0 | <0.001 |
CFGR1 and CFGR2 were therefore subsequently validated using the internal validation cohort and two external validation cohorts. CFGR1 had an AUC of 0.830 (0.743–0.917), 0.780 (0.700–0.859) and 0.809 (0.739–0.880) in the internal cohort and two external validation cohorts, respectively. CFGR2 had an AUC of 0.790 (0.694–0.886), 0.737 (0.655–0.819) and 0.729 (0.652–0.807) in the internal cohort and two external validation cohorts, respectively (Figure 4, Table 1). By using the functions in the pROC package of R software to compare the performance of the two classifiers, the results show that the performance of CFGR1 is better than that of CFGR2 (P < 0.001; Table 1).
Figure 4.
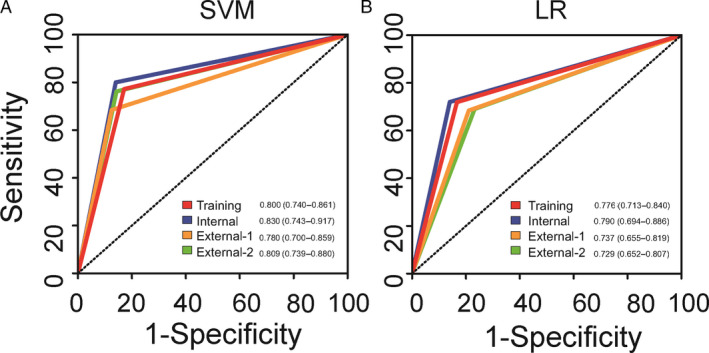
Receiver operating characteristic (ROC) curves to evaluate the performance of each classifier. (A) ROC curves for ‘FGR classifier 1’ (CFGR1) in four cohorts, (B) ROC curves for ‘FGR classifier 2’ (CFGR2) in four cohorts.
Performance of CFGR1 at different gestational weeks
The CFGR1 was selected to predict FGR in 110 samples (22 FGR) collected in the first trimester and 700 samples (140 FGR) of second trimester, respectively, to reach an AUC of 0.839 (0.744–0.933) and 0.797 (0.757–0.836). The sensitivity was 77.3 and 75.0%, and the specificity was 90.4 and 84.3%, respectively (Table S3). The CFGR1 performed similarly in the first and second trimesters, suggesting that the predictive performance of the classifier is stable and that FGR prediction can be started as early as 12 weeks of gestational age.
Discussion
Main findings
In recent years, cfDNA‐based NIPT for fetal chromosomal aneuploidies has become the first successful application of cfDNA and has quickly been transformed into clinical practice. The success of the NIPT has aroused global interest in using cfDNA in liquid biopsies for cancer. .7 found cfDNA could reflect the expression status of its tissue of origin. Chan et al. 22 found that pregnant maternal plasma cfDNA fragment end sites are specific to their placental origin. We posited that the NIPT data might provide useful information for placental‐related complications, and for the first time assessed the early prediction of FGR in this study. Specific nucleosome profiles were seen in the TSS and TTS of genes in FGR pregnancies (Figure 2), and 103 genes with significantly differential coverage at promoter regions were identified by comparing the cfDNA signals between FGR pregnancies and their matched controls. Finally, we successfully developed two high‐performance classifiers, based on SVM and LR, for predicting the risk of FGR based on maternal plasma cfDNA nucleosome profiling. For CFGR1, a 14‐gene combination (TCHH, ST5, CKB, KNOP1, NOS2, KRTAP9‐9, ARHGAP15, SLC4A10, HMGB2, SEPT11, ZKSCAN5, GPAT4, PHPT1, FANCC) achieved the highest performance and predicted FGR successfully in 83.2% of cases (AUC, 0.803, 95% CI 0.767–0.839).
Strengths and limitations
Our study sample size was large, comprising 162 cases of FGR and 648 controls. Furthermore, our prediction model is simple, can be applied during the early stages of pregnancy and only requires the NIPT data obtained during screening for chromosomal abnormalities, which is also performed in the first and second trimester; this would enable the testing of future hypothesised preventive interventions.
However, our study also had some limitations. First, it used a retrospective design, which is more prone to statistical over‐fitting and overstating of performance compared with a prospective design. In neonates considered appropriate for gestational age (AGA), there was a subgroup of normal weight neonates with features of intrauterine growth restriction due to placental dysfunction. 23 Another limitation is that the study was performed without including the above‐mentioned FGR, limiting the interpretation and universality of our results to the growth restriction of small for gestational age (SGA). Prospective studies including pregnant women with a singleton pregnancy in the first trimester are now needed to validate the prediction model further. In addition, only the TSSs of coding genes were included to construct the classifiers. Recently, by searching a panel of microarray assays, pregnancy‐associated, placenta‐specific microRNAs (miRNAs) in the plasma of pregnant women have been identified with a potential regulatory role in FGR. 24 , 25 , 26 In future studies, analysis of the TSSs of coding genes and noncoding miRNAs in combination may yield a model with an even higher positive predictive value. Furthermore, a large number of prospective studies will have to be performed before the classifiers developed herein can be used as diagnostic or screening tests in clinical trials.
Interpretation
Currently, it is difficult to block or delay the progress of FGR via management or treatment in the second and third trimesters; the course of the disease can only be observed and monitored, while also prolonging the gestation as much as possible to avoid the occurrence of severe fetal hypoxia, acidosis or even death in the uterus. FGR fetuses exhibit catch‐up growth after birth and usually reach normal size in childhood. However, there may still be a risk of long‐term neurodevelopmental abnormalities and cognitive impairment. Therefore, many researchers focus on early screening and prediction of FGR; if fetuses that will develop FGR can be identified during the first trimester (during placental formation), even though there are currently no effective treatments to control FGR, early identification will allow testing of hypothetical interventions aiming at its primary prevention.
Ultrasound and serum markers have long been the focus of research to predict FGR. Placental abnormalities that result in poor placental insufficiency are the most common pathology associated with FGR and the main cause, and an abnormal uterine arterial blood flow velocity waveform indicates insufficient recasting of the uterine spiral artery. Unfortunately, the sensitivity of uterine arterial blood flow as a predictor of FGR in the first and second trimesters of pregnancy is low. 27 , 28 Maternal characteristics, mean arterial pressure (MAP), placental growth factor (PIGF) and soluble fms‐like tyrosine kinase 1 (sFlt‐1) have also been used as predictors of FGR. 29 , 30 , 31 Compared with the detection rate of maternal characteristics (a priori risk), MAP, Doppler of the uterine arteries, PIGF, sFlt‐1 and the combination of the above markers (38, 21, 22, 62, 50, and 67%, respectively, with a false‐positive rate of 10%), the detection rate for the cfDNA‐based FGR classifiers was much higher (83.2% for CFGR1). 27 Furthermore, CFGR1 in the study had good prediction performance in the first trimester (AUC, 0.839; 95% CI 0.744–0.933; accuracy, 87.9%), although the sample size needs to be increased to improve accuracy. In strict accordance with the inclusion criteria, we included NIPT data from 12 to 28 weeks’ gestation and proved that it was feasible to predict FGR in the early stage (when NIPT can be started), which could be earlier than ultrasound screening for abnormalities. Furthermore, this classifier development strategy may be applicable to other pregnancy complications that could benefit from an early, non‐invasive screening test based on maternal plasma sampling.
Conclusions
In summary, the highest performance classifier was successfully developed for predicting the risk of FGR with low birthweight based on nucleosome profiling of maternal plasma cfDNA. This technique only requires low‐coverage DNA sequencing, similar to present NIPT procedures, without any additional tests; it may also aid in the development of predictive models for other placental‐related pregnancy complications.
Disclosure of interests
None declared. Completed disclosure of interest forms are available to view online as supporting information.
Contribution to authorship
The study was conceived and designed by FY, XY, ZZ and JZ. Patient recruitment and sample collection were undertaken by CX, QL, QT and ZL. Experiments and data collection were performed by CX, SL, KL, KW, ZT and CL. Data analyses and interpretation were performed by CX, ZG and JZ. All figures and tables were generated by CX and ZG. The manuscript was written by CX and ZG. All authors critically reviewed the manuscript and approved the final manuscript for publication.
Details of ethics approval
This study was approved by the Internal Ethics Committee of Nanfang Hospital, Southern Medical University (IRB no. NFEC‐2017‐049, approval date 28 April 2017).
Funding
This study was supported by the National Natural Science Foundation of China (81871177); Natural Science Foundation of Guangdong Province (2018A030313286); Science and Technology Program of Guangzhou (201604020104, 201803040009) and Clinical Research Program of Nanfang Hospital (2018CR039).
Acknowledgements
We sincerely thank Guangzhou Darui Biotechnology Co., Ltd. for providing sequencing platform and server. We would like to thank Dr. Liang Zhikun for providing the standard operating procedure of the sequencing. We would like to thank all doctors, midwives and sonographers who helped with patient recruitment. We are grateful to all the team members for their contributions to data collection and integrity.
Supporting information
Table S1. Clinical characteristics of the study groups.
Table S2. Relative contribution of different types of fetal growth restriction to the total samples.
Table S3. Performance of CFGR1 in the first and second trimester.
Supplementary material
Xu C, Guo Z, Zhang J, Lu Q, Tian Q, Liu S, Li K, Wang K, Tao Z, Li C, Lv Z, Zhang Z, Yang X, Yang F. Non‐invasive prediction of fetal growth restriction by whole‐genome promoter profiling of maternal plasma DNA: a nested case–control study. BJOG: Int J Obstet Gy. 2021;128:458–466.
Linked article This article is commented on by S Arora, p. 467 in this issue. To view this mini commentary visit https://doi.org/10.1111/1471-0528.16329.
Contributor Information
Z Zhang, Email: zhzhg001518@126.com.
X Yang, Email: yangxuexi2017@126.com.
F Yang, Email: fangfangy@hotmail.com.
References
- 1. ACOG Practice Bulletin No. 204: Fetal Growth Restriction. Obstet Gynecol 2019;133:e97–109. [DOI] [PubMed] [Google Scholar]
- 2. Serena C, Marchetti G, Rambaldi MP, Ottanelli S, Di Tommaso M, Avagliano L, et al. Stillbirth and fetal growth restriction. J Matern Fetal Neonatal Med 2013;26:16–20. [DOI] [PubMed] [Google Scholar]
- 3. Gaccioli F, Aye I, Sovio U, Charnock‐Jones DS, Smith GCS. Screening for fetal growth restriction using fetal biometry combined with maternal biomarkers. Am J Obstet Gynecol 2018;218(2S):S725–37. [DOI] [PubMed] [Google Scholar]
- 4. Liang D, Cram DS, Tan H, Linpeng S, Liu Y, Sun H, et al. Clinical utility of noninvasive prenatal screening for expanded chromosome disease syndromes. Genet Med 2019;21:1998–2006. [DOI] [PubMed] [Google Scholar]
- 5. Zhang J, Li J, Saucier JB, Feng Y, Jiang Y, Sinson J, et al. Non‐invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell‐free fetal DNA. Nat Med 2019;25:439–47. [DOI] [PubMed] [Google Scholar]
- 6. Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 2016;34:547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell‐free DNA comprises an in vivo nucleosome footprint that informs its tissues‐of‐origin. Cell 2016;164:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ulz P, Thallinger GG, Auer M, Graf R, Kashofer K, Jahn SW, et al. Inferring expressed genes by whole‐genome sequencing of plasma DNA. Nat Genet 2016;48:1273–8. [DOI] [PubMed] [Google Scholar]
- 9. Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM, et al. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet 1998;62:768–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huynh J, Dawson D, Roberts D, Bentley‐Lewis R. A systematic review of placental pathology in maternal diabetes mellitus. Placenta 2015;36:101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sur Chowdhury C, Hahn S, Hasler P, Hoesli I, Lapaire O, Giaglis S. Elevated levels of total cell‐free DNA in maternal serum samples arise from the generation of neutrophil extracellular traps. Fetal Diagn Ther 2016;40:263–7. [DOI] [PubMed] [Google Scholar]
- 12. Lo YM, Chan KC, Sun H, Chen EZ, Jiang P, Lun FM, et al. Maternal plasma DNA sequencing reveals the genome‐wide genetic and mutational profile of the fetus. Sci Transl Med 2010;2:61ra91. [DOI] [PubMed] [Google Scholar]
- 13. Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008;132:887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature 2011;474:516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 2015;16:178–89. [DOI] [PubMed] [Google Scholar]
- 16. Gordijn SJ, Beune IM, Thilaganathan B, Papageorghiou A, Baschat AA, Baker PN, et al. Consensus definition of fetal growth restriction: a Delphi procedure. Ultrasound Obstet Gynecol 2016;48:333–9. [DOI] [PubMed] [Google Scholar]
- 17. Dai L, Deng C, Li Y, Zhu J, Mu Y, Deng Y, et al. Birth weight reference percentiles for Chinese. PLoS ONE 2014;9:e104779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Casper J, Zweig AS, Villarreal C, Tyner C, Speir ML, Rosenbloom KR, et al. The UCSC genome browser database: 2018 update. Nucleic Acids Res 2018;46:D762–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Teo YV, Capri M, Morsiani C, Pizza G, Faria AMC, Franceschi C, et al. Cell‐free DNA as a biomarker of aging. Aging Cell 2019;18:e12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan KC, Jiang P, Sun K, Cheng YK, Tong YK, Cheng SH, et al. Second generation noninvasive fetal genome analysis reveals de novo mutations, single‐base parental inheritance, and preferred DNA ends. Proc Natl Acad Sci USA 2016;113:E8159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hendrix MLE, Bons JAP, Alers NO, Severens‐Rijvers CAH, Spaanderman MEA, Al‐Nasiry S. Maternal vascular malformation in the placenta is an indicator for fetal growth restriction irrespective of neonatal birthweight. Placenta 2019;87:8–15. [DOI] [PubMed] [Google Scholar]
- 24. Miura K, Miura S, Yamasaki K, Higashijima A, Kinoshita A, Yoshiura K, et al. Identification of pregnancy‐associated microRNAs in maternal plasma. Clin Chem 2010;56:1767–71. [DOI] [PubMed] [Google Scholar]
- 25. Higashijima A, Miura K, Mishima H, Kinoshita A, Jo O, Abe S, et al. Characterization of placenta‐specific microRNAs in fetal growth restriction pregnancy. Prenat Diagn 2013;33:214–22. [DOI] [PubMed] [Google Scholar]
- 26. Huang L, Shen Z, Xu Q, Huang X, Chen Q, Li D. Increased levels of microRNA‐424 are associated with the pathogenesis of fetal growth restriction. Placenta 2013;34:624–7. [DOI] [PubMed] [Google Scholar]
- 27. Crovetto F, Triunfo S, Crispi F, Rodriguez‐Sureda V, Dominguez C, Figueras F, et al. Differential performance of first‐trimester screening in predicting small‐for‐gestational‐age neonate or fetal growth restriction. Ultrasound Obstet Gynecol 2017;49:349–56. [DOI] [PubMed] [Google Scholar]
- 28. Miranda J, Rodriguez‐Lopez M, Triunfo S, Sairanen M, Kouru H, Parra‐Saavedra M, et al. Prediction of fetal growth restriction using estimated fetal weight vs a combined screening model in the third trimester. Ultrasound Obstet Gynecol 2017;50:603–11. [DOI] [PubMed] [Google Scholar]
- 29. Gomez‐Roig MD, Mazarico E, Sabria J, Parra J, Oton L, Vela A. Use of placental growth factor and uterine artery doppler pulsatility index in pregnancies involving intrauterine fetal growth restriction or preeclampsia to predict perinatal outcomes. Gynecol Obstet Invest 2015;80:99–105. [DOI] [PubMed] [Google Scholar]
- 30. Benton SJ, McCowan LM, Heazell AE, Grynspan D, Hutcheon JA, Senger C, et al. Placental growth factor as a marker of fetal growth restriction caused by placental dysfunction. Placenta 2016;42:1–8. [DOI] [PubMed] [Google Scholar]
- 31. Monier I, Ancel PY, Ego A, Guellec I, Jarreau PH, Kaminski M, et al. Gestational age at diagnosis of early‐onset fetal growth restriction and impact on management and survival: a population‐based cohort study. BJOG 2017;124:1899–906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical characteristics of the study groups.
Table S2. Relative contribution of different types of fetal growth restriction to the total samples.
Table S3. Performance of CFGR1 in the first and second trimester.
Supplementary material