

Abstract
Objective
To determine if PET‐based stages of regional amyloid deposition are associated with neuropathological phases of Aβ pathology.
Methods
We applied data‐driven regional frequency‐based and a‐priori striatum‐based PET staging approaches to ante‐mortem 18F‐Florbetapir‐PET scans of 30 cases from the Alzheimer’s Disease Neuroimaging Initiative autopsy cohort, and used Bayesian regression analysis to study the associations of these in vivo amyloid stages with neuropathological Thal phases of regional Aβ plaque distribution and with semi‐quantitative ratings of neocortical and striatal plaque densities.
Results
Bayesian regression revealed extreme evidence for an association of both PET‐based staging approaches with Thal phases, and these associations were about 44 times more likely for frequency‐based stages and 89 times more likely for striatum‐based stages than for global cortical 18F‐Florbetapir‐PET signal. Early (i.e., neocortical‐only) PET‐based amyloid stages also predicted the absence of striatal/diencephalic cored plaques. Receiver operating characteristics curves revealed highly accurate discrimination between low/high Thal phases and the presence/absence of regional plaques. The median areas under the curve were 0.99 for frequency‐based staging (95% credibility interval 0.97–1.00), 0.93 for striatum‐based staging (0.83–1.00), and 0.87 for global 18F‐Florbetapir‐PET signal (0.72–0.98).
Interpretation
Our data indicate that both regional frequency‐ and striatum‐based amyloid‐PET staging approaches were superior to standard global amyloid‐PET signal for differentiating between low and high degrees of regional amyloid pathology spread. Despite this, we found no evidence for the ability of either staging scheme to differentiate between low and moderate degrees of amyloid pathology which may be particularly relevant for early, preclinical stages of Alzheimer’s disease.
Introduction
Global signal increase in amyloid‐sensitive PET has become a disease‐defining biomarker in research criteria for Alzheimer’s disease (AD). 1 , 2 A range of clinico‐pathological association studies have established that global increase in amyloid‐sensitive PET signal reflects the cerebral accumulation of Aβ pathology; this was observed for all currently available amyloid PET tracers, including 11C‐PiB, 3 18F‐Florbetaben, 4 18F‐Flutemetamol, 5 and 18F‐Florbetapir. 6 Few studies went beyond global assessment of amyloid PET signal. One study assessed correlations of Thal phases 7 and CERAD neuritic plaque scores with 11C‐PIB‐PET signal in 18 bilateral cortical regions, striatum, and hippocampus. 8 However, the highest regional associations observed in that study 8 were comparable to the association with the standard global composite 11C‐PIB‐SUVR value, arguing against a marked benefit of region‐specific PET signal for measuring pathologic amyloid burden. Motivated by long‐standing neuropathological evidence for a progression sequence of amyloid pathology from neocortical to diencephalic areas, another study generated two different stages of 18F‐Flutemetamol uptake based on thresholded cortical and striatal amyloid signal in 97 cases. 9 This study found that these amyloid PET stages were associated with the regional distribution of Aβ accumulation as assessed by the neuropathological Thal phases. One shortcoming of this previous study, however, was that the cut‐offs defining the neocortical vs striatal PET amyloid stages were derived in reference to the Thal phases which likely induced some circularity in the analysis.
A similar a priori classification of amyloid PET scans into two different stages based on whether amyloid PET signal was increased in the neocortex alone or additionally affected the striatum was also proposed by a previous study using regional cut‐offs established in independent cohorts, and thus without reference to an external gold standard. 10 Alternative approaches to stage amyloid PET scans used hierarchical classification of different cortical and subcortical brain regions according to data‐driven models of regional amyloid spread based on the frequency of regional signal increases in the PET data. 11 , 12 , 13 , 14 Details of the different staging schemes can be found in Table S1. Currently, the correspondence of such PET‐defined in vivo stages of regional amyloid deposition with autopsy‐derived measures of amyloid burden, including the Thal phases, has not yet been established.
Here, we used region‐based stagings 10 , 11 of amyloid PET data not biased by reference to amyloid pathology stages from neuropathological examination to determine if amyloid PET assessments were associated with the neuropathological distribution of amyloid pathology as captured by Thal phases and CERAD grading of neuritic and diffuse plaque density. We tested this question in a patient sample from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort with available ante‐mortem 18F‐Florbetapir‐PET scans and post‐mortem neuropathological examination. Differing from the previous study, 9 the PET‐based amyloid stagings used here were unbiased with respect to the autopsy findings. The regional frequency‐based staging had been established before in a large sample of normal controls from the ADNI cohort 11 and replicated in an independent sample of subjective memory complainers from the INSIGHT‐preAD cohort. 15 The striatum‐based staging had been derived from two independent cohorts, the Harvard Aging Brain Study and ADNI. 10 In a secondary analysis, we determined whether PET‐based amyloid staging would discriminate between neocortical and striatal/diencephalic amyloid load, based on the notion that striatal and diencephalic involvement in amyloid pathology as defined by the Thal phases occurs only after neocortical involvement. 7 With this study, we tested the hypothesis that PET‐based in vivo amyloid stages would be associated with Thal phases and other neuropathological measures of progressive amyloid burden and that this association would be stronger than for standard global 18F‐Florbetapir‐PET signal. Specifically, we were interested in this approach to determine if amyloid PET stages were able to differentiate between lower phases of amyloid deposition. We chose to use a Bayesian framework to test this hypothesis because this framework allowed us to explicitly compare the likelihood of different alternative hypotheses and to directly estimate the likelihood of the presence or the absence of an effect.
Material and Methods
Data source
Data used in the preparation of this article were obtained from the ADNI database (http://adni.loni.usc.edu/). The ADNI was launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies, and non‐profit organizations, with the primary goal of testing whether neuroimaging, neuropsychologic, and other biologic measurements can be used as reliable in vivo markers of AD pathogenesis. A complete description of ADNI and up‐to‐date information is available at www.adni‐info.org.
Standard protocol approvals, registrations, and patient consents
All procedures performed in the ADNI studies involving human participants were in accordance with the ethical standards of the institutional research committees and with the 1964 Helsinki declaration and its later amendments. Written informed consent was obtained from all participants and/or authorized representatives and the study partners before any protocol‐specific procedures were carried out in the ADNI studies.
Study participants
We retrieved the last available 18F‐Florbetapir‐PET scans of 30 ADNI subjects who had come to autopsy between 2007 and 2017. Detailed inclusion criteria for the ante‐mortem diagnostic categories can be found at the ADNI web site (http://adni.loni.usc.edu/methods/). Cognitively normal (CN) subjects had MMSE scores between 24 and 30 (inclusive), a CDR = 0, were non‐depressed, non‐MCI, and non‐demented, and reported no subjective memory concerns. MCI subjects had MMSE scores between 24 and 30 (inclusive), a subjective memory concern reported by subject, informant, or clinician, objective memory loss measured by education‐adjusted scores on delayed recall, a CDR = 0.5, the absence of significant levels of impairment in other cognitive domains, essentially preserved activities of daily living, and an absence of dementia. All MCI cases were classified as amnestic MCI according to the ADNI guidelines. At inclusion into the ADNI cohort, subjects with AD dementia had initial MMSE scores between 20 and 26 (inclusive), a CDR = 0.5 or 1.0 with impaired activities of daily living and fulfilled NINCDS‐ADRDA criteria for clinically probable Alzheimer’s disease. 16
Neuropathological assessments
All neuropathological evaluations in the ADNI cohort are performed through the central laboratory of the ADNI neuropathology core (http://adni.loni.usc.edu/about/#core‐container). They assess a wide range of AD and non‐AD neuropathological lesions, including both established neuropathologic criteria as well as detailed regional assessments within 22 cortical and subcortical brain regions. 17 The neuropathological procedures follow previously established guidelines 18 that are captured in the format of the Neuropathology Data Form Version 10 of the National Alzheimer Coordinating Center (https://www.alz.washington.edu/NONMEMBER/NP/npform10.pdf).
Here, we used established rating scales for AD neuropathologic change represented by Thal amyloid phases. In addition, we used the data from the CERAD scores of neuritic plaques, and an analogous scoring of diffuse plaques. In addition, we examined semi‐quantitative neuropathological rating scores of diffuse and cored amyloid plaques in the striatum and thalamus as well as in the following neocortical regions: superior temporal gyrus, anterior cingulate gyrus, medial frontal gyrus, and inferior parietal lobule. To analyze the differential association of PET‐based amyloid stages with striatal/diencephalic and neocortical amyloid plaque scores, we used median values across striatum/thalamus (striatum/diencephalic score) and across the four neocortical regions (neocortical score), as well as binarizations thereof where a median of 0 was rated as 0 and a median above 0 was rated as 1.
Imaging data acquisition
Detailed acquisition and standardized preprocessing steps of ADNI imaging data are available at the ADNI website (https://adni.loni.usc.edu/methods/). Amyloid‐PET data were collected during a 50‐ to 70‐min interval following a 370 MBq bolus injection of 18F‐Florbetapir. To account for the multicentric acquisition of the data across different scanners and sites, all PET scans undergo standardized preprocessing steps within ADNI.
For anatomical reference and preprocessing of the PET scans, we used the corresponding structural MRI scan that was closest in time to the Florbetapir PET scan. MRI data were acquired on multiple 3T MRI scanners using scanner‐specific T1‐weighted sagittal 3D MPRAGE sequences. Similar to the PET data, MRI scans undergo standardized preprocessing steps aimed at increasing data uniformity across the multicenter scanner platforms (see https://adni.loni.usc.edu/methods/ for detailed information on multicentric MRI acquisition and preprocessing in ADNI).
Imaging data preprocessing
Images were preprocessed using Statistical Parametric Mapping software version 12 (SPM12) (The Wellcome Trust Centre for Neuroimaging, Institute of Neurology, University College London) implemented in Matlab 2019. MRI images were segmented into different tissue types and spatially normalized to a customized aging/AD‐specific reference template 19 using the CAT12 toolbox. The preprocessing pipeline for the amyloid PET images followed the routine as previously described. 11 First, each subject’s averaged PET frames were co‐registered to their corresponding T1‐weighted MRI scan. Then, partial volume effects (PVE) were corrected in native space using the three‐compartmental voxel‐based post‐reconstruction method as described by Müller‐Gӓrtner and colleagues. 20 , 21 The corrected PET images were spatially normalized to an aging/AD‐specific reference template using the deformation parameters derived from the normalization of their corresponding MRI.
Regional PVE corrected 18F‐Florbetapir‐PET mean uptake values were estimated for 52 brain regions defined by the Harvard–Oxford structural atlas, 22 including both cortical and subcortical regions (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/Atlases). Standard uptake value ratios (SUVR) were computed for the 52 brain regions by dividing the mean uptake values by the mean uptake value of the whole cerebellum as estimated in non‐PVE‐corrected PET data. 11 , 21 , 23 , 24 Similarly, global cortical SUVR values were obtained as a ratio of mean cortical uptake values extracted from PVE‐corrected images using a composite cortical mask including frontal, parietal, and temporal areas to the mean uptake of the whole cerebellum.
We based the cut‐off used for determining regional amyloid positivity on a cut‐off value of SUVR = 1.135, 11 which lies in between the two most widely used global signal cut‐offs for non‐PVE‐corrected 18F‐Florbetapir‐PET SUVRs, that is, SUVR = 1.10 6 , 25 , 26 and SUVR = 1.17. 27 , 28 This threshold was converted to the PVE‐corrected PET data used for the regional staging approach using linear regression between PVE‐corrected and non‐corrected global SUVR values, which resulted in a value of SUVR = 1.04.
PET data analysis
Frequency‐based staging of regional amyloid deposition, henceforth called frequency staging, followed the previously developed four‐stage model of amyloid pathology progression derived from 18F‐Florbetapir‐PET data of cognitively normal older individuals enrolled in the ADNI study. 11 This four‐stage model was estimated by counting the frequency of amyloid positivity across the 52 brain regions defined in the Harvard–Oxford structural atlas and then merging the regions into four broader anatomical divisions based on equal proportions of the observed range of involvement frequencies. The four anatomical divisions defining the staging scheme are illustrated in Figure 1. According to this staging approach, 11 an anatomical division was considered positive for amyloid pathology if at least 50% of the regions included in this division exceeded the cut‐off value in the respective participant. Subsequently, participants were classified as stage I if only the first division was considered positive. Then, the successive stages II‐IV were defined by the additional involvement of their corresponding divisions II, III, and IV, respectively. Participants who exhibited amyloid positivity in any division without concurrent amyloid positivity in the preceding divisions were classified as non‐stageable (mismatch).
Figure 1.
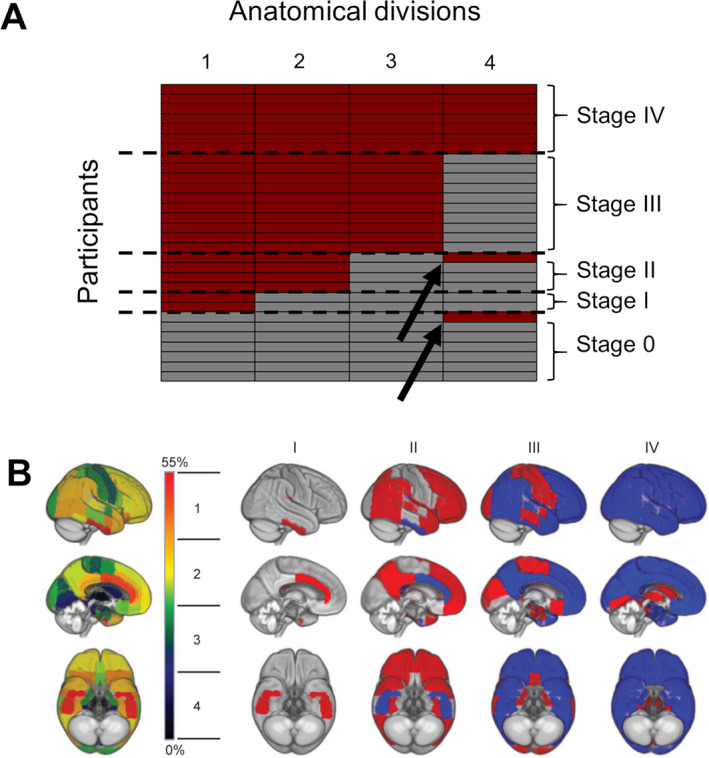
PET‐based frequency amyloid stages across cases. (A) The matrix demonstrates participants (in rows) and their amyloid positivity with respect to each of the four anatomical divisions (in columns) depicted by red. The two non‐stageable cases are indicated by arrows. (B) Brain renderings on the left illustrate the frequency of regional amyloid positivity across individuals on a color scale from black/blue (lowest) to yellow/red (highest). The 52 brain regions are merged into four larger anatomic divisions based on equal partitions of the frequency range (1–4). In the resulting four‐stage model of regional amyloid progression (I–IV), incremental stages are defined by the involvement of higher numbered anatomic divisions (in red), in addition to the affected areas of the previous stage (blue). Part B is republished with permission of Wolters Kluwer Health, Inc. from Grothe, M. J., Barthel, H., Sepulcre, J., Dyrba, M., Sabri, O., & Teipel, S. J. (2017). In vivo staging of regional amyloid deposition. Neurology, 89(20), 2031–2038, permission conveyed through Copyright Clearance Center, Inc.
For comparison, we also used the a priori two‐stage approach based on neocortical versus striatal involvement proposed by 10 using the above‐defined regional cut‐off for cortical composite score and striatum positivity, respectively. This staging approach henceforth is called striatal staging.
Statistics
We used Bayes factor (BF) hypothesis testing to compare one or more alternative hypotheses against the null hypothesis (i.e., the assumption that there is no association between amyloid measures and neuropathological markers, H0). This approach allowed us to reject the null hypotheses and accept the best possible hypotheses of our data, a desirable advantage over classical null hypothesis significance testing with P‐values which only allows the rejection or acceptance of the null hypotheses. 29 , 30 We used Jeffreys’ Amazing Statistics Program (JASP Version 0.11.1), available at jasp‐stats.org, to calculate models. JASP provides an easy to use interface to R libraries for Bayesian statistics. We report the Bayes Factor (BF10) quantifying evidence against the null hypotheses, as well as the BFM indicating the informativeness of our data given the prior (P(M)) and posterior distributions (P(M|data)). In addition, we report estimates of regression coefficients with corresponding credibility intervals (also called the highest density interval), providing the probability that the population parameters lie between the particular upper and lower bounds. This is different from the confidence interval from classical null hypothesis statistical testing which indicates the probability that the parameter will fall between the lower and upper bound when samples are repeatedly drawn from the underlying population. 31 Table 1 provides a short description of the Bayesian parameters that are reported here. To address potential issues with non‐normally distributed residuals in the multiple regressions, we applied Markov–Monte Carlo chain sampling to each analysis 1000 times. JASP was set to report the null model first, and then compare all other models against the null model. We used the JASP default JZS prior. Within this framework, we conducted two sets of multiple linear regression models.
Table 1.
Summary of the reported Bayesian Statistics
| Abbreviation | Full Name | Interpretation | Ref. |
|---|---|---|---|
| BF | Bayes Factor | Quantifies evidence | 30, 32 |
| BF10 | BF in favor of the best model | Our data are BF10 times more likely under the best H1 compared to H0 | 33 |
| BFM | Degree to which the data have changed the prior model odds | The larger, the more informative our data have been | 30 |
| P(M) | Prior (distribution) | Assumed distribution prior to data analysis | 30, 34 |
| P(M|data) | Posterior (distribution) | Distribution after the prior has been updated with our data | 30, 34 |
| 95%‐CI | Credibility interval | Parameter lies within the lower and upper bounds with 95% probability. |
First, we determined the associations between PET‐derived measures of amyloid burden (PET‐based frequency and striatal staging as well as global PVC SUVR values) with neuropathologic measures of amyloid burden (Thal phase, CERAD scores), taking age, sex, and distance between PET scanning and death into account. For a post hoc analysis, arising from an inspection of the data, we binarized Thal phases using a split at ≥4 as outcome and determined the areas under the receiver operating characteristics curves (AUC) for amyloid stages and global PVC SUVR values, respectively. AUC values were calculated using Bayesian parametric estimation as adapted for small sample sizes using the function “auc.para.bayes” in the R package “auRoc.” 35
Second, we determined the associations of PET‐based amyloid stages and global PVC SUVR values with median cored and diffuse plaque scores in striatal/diencephalic and neocortical regions, respectively, using Bayesian estimation of Kendall’s Tau correlation coefficient in JASP. AUC for the striatal and neocortical cored and diffuse plaque scores as binary outcomes (absent/present) were also determined. 35
We applied the following evidence categories: a BF10 above 3 provides “substantial evidence,” a BF10 above 10 provides “strong evidence,” a BF10 above 30 provides “very strong evidence,” and a BF10 above 100 provides “extreme evidence” against the null model. BF10 < 1/3 indicates support for the null model. 34
Results
Demographics and PET staging characteristics
Detailed demographics of the sample of 30 cases with available ante‐mortem 18F‐Florbetapir‐PET scans are given in Table 2. Of the 30 available amyloid PET scans, two could not be classified according to the previously established frequency staging scheme (Fig. 1). None of the 30 cases were in striatal amyloid stage zero, nine were in striatal amyloid stage one, that is, neocortical but no striatal amyloid uptake, and 21 were in stage two, that is, neocortical and striatal amyloid uptake. The distribution of Thal phases across the frequency stages, striatal stages, and global PVC SUVR values is shown in Figure 2.
Table 2.
Sample characteristics at time of last 18F‐Florbetapir‐PET scan.
| Clinical diagnosis 1 | Sex | Age at death [years] | Interval PET to death [years] | Global PVC SUVR | Frequency stages | Striatum‐based stages | Thal Phase | Neuritic Plaque score | Diffuse Plaque score |
|---|---|---|---|---|---|---|---|---|---|
| CN1 | f | 83 | 5.00 | 2.19 | 3 | 1 | 4 | 2 | 3 |
| CN2 | m | 89 | 1.20 | 1.59 | n.s. | 2 | 0 | 0 | 0 |
| CN3 | f | 84 | 0.50 | 1.43 | 1 | 1 | 0 | 0 | 0 |
| CN4 | m | 70 | 3.50 | 2.00 | 0 | 1 | 1 | 0 | 1 |
| CN5 | f | 84 | 2.20 | 1.15 | 0 | 1 | 3 | 0 | 2 |
| MCI1 | m | 91 | 2.40 | 1.60 | 2 | 1 | 4 | 0 | 3 |
| MCI2 | m | 80 | 2.00 | 2.46 | 4 | 2 | 4 | 1 | 3 |
| MCI3 | m | 86 | 0.90 | 2.59 | 3 | 2 | 5 | 3 | 3 |
| ADD1 | m | 81 | 1.90 | 1.48 | 0 | 1 | 1 | 1 | 1 |
| ADD2 | m | 77 | 3.80 | 1.18 | 0 | 1 | 1 | 0 | 1 |
| ADD3 | f | 77 | 0.30 | 2.25 | 3 | 2 | 4 | 3 | 3 |
| ADD4 | m | 91 | 2.10 | 1.29 | 0 | 1 | 1 | 0 | 1 |
| ADD5 | m | 93 | 4.30 | 1.59 | 0 | 1 | 1 | 0 | 1 |
| ADD6 | m | 93 | 1.60 | 4.73 | 4 | 2 | 4 | 1 | 3 |
| ADD7 | m | 76 | 1.50 | 1.87 | 2 | 2 | 5 | 3 | 3 |
| ADD8 | m | 89 | 2.00 | 2.18 | 2 | 2 | 4 | 2 | 3 |
| ADD9 | m | 91 | 4.00 | 2.36 | 3 | 2 | 4 | 3 | 3 |
| ADD10 | f | 80 | 0.60 | 1.29 | 1 | 2 | 5 | 3 | 3 |
| ADD11 | m | 59 | 3.50 | 3.46 | 3 | 2 | 5 | 3 | 3 |
| ADD12 | m | 88 | 1.10 | 1.92 | n.s. | 2 | 4 | 3 | 3 |
| ADD13 | m | 78 | 2.00 | 4.24 | 4 | 2 | 5 | 3 | 3 |
| ADD14 | f | 95 | 3.70 | 3.77 | 4 | 2 | 4 | 3 | 3 |
| ADD15 | m | 83 | 1.80 | 2.69 | 3 | 2 | 4 | 3 | 3 |
| ADD16 | m | 77 | 1.70 | 2.16 | 3 | 2 | 4 | 3 | 3 |
| ADD17 | m | 84 | 1.50 | 3.09 | 3 | 2 | 5 | 3 | 3 |
| ADD18 | f | 76 | 1.80 | 2.29 | 4 | 2 | 4 | 3 | 3 |
| ADD19 | f | 84 | 2.60 | 2.60 | 4 | 2 | 4 | 3 | 3 |
| ADD20 | m | 81 | 1.70 | 2.46 | 3 | 2 | 5 | 0 | 3 |
| ADD21 | m | 86 | 2.50 | 3.20 | 4 | 2 | 5 | 3 | 3 |
| ADD22 | m | 78 | 5.00 | 3.39 | 3 | 2 | 4 | 3 | 3 |
CN, Control; MCI, mild cognitive impairment; ADD, Alzheimer’s disease dementia; f/m, female/male; n.s., non stageable.
Last clinical diagnosis before death
Figure 2.
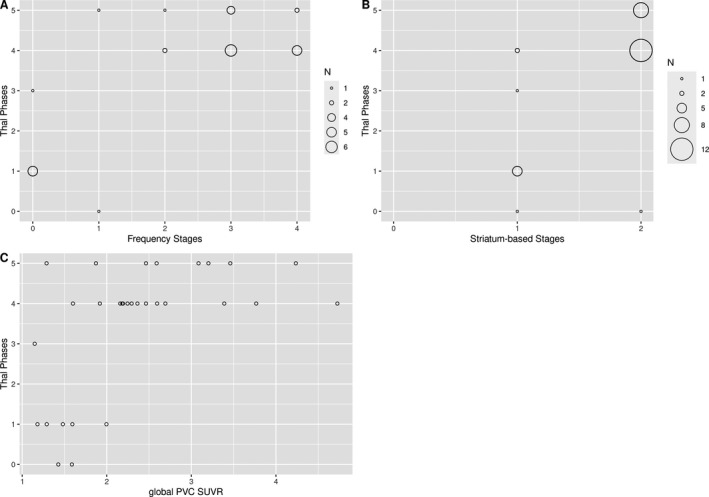
Distribution of Thal phases across amyloid stages and global PVC SUVR values. Thal phases plotted against PET‐based frequency (A) and striatal (B) amyloid stages and global PVC SUVR values (C), with the size of the circles corresponding to the number of underlying cases.
Thal phases versus PET‐based amyloid stagings and global SUVR
We found extreme evidence for an association of frequency and striatal amyloid stages with Thal phases, and strong evidence for global PVC SUVR (Table 3). Effects were similar for the outcome of CERAD neuritic plaques and diffuse plaques scores, with extreme evidence for an association with PET‐based amyloid stages and strong evidence for global PVC SUVR values (data not shown). The effect on Thal phases was about 44 times more likely for cortical frequency stages (BF10 = 729/16.6) and about 89 times more likely for striatal stages (BF10 = 4385/16.6) than for the model with global PVC SUVR (Table 3). The model with striatal stages was about six times more likely than the model with cortical frequency stages (BF10 = 4385/729).
Table 3.
Thal phases versus amyloid stagings and global SUVR.
| Models | P(M) | P(M|data) | BFM | BF10 | R 2 |
|---|---|---|---|---|---|
| Null model (incl. age, sex, PET to death) | 0.250 | 2.3e‐4 | 6.9e‐4 | 1.000 | 0.058 |
| Frequency stages | 0.083 | 0.056 | 0.647 | 729.117 | 0.585 |
| Striatum‐based stages | 0.083 | 0.334 | 5.520 | 4384.512 | 0.655 |
| Global PVC SUVR | 0.083 | 0.001 | 0.014 | 16.590 | 0.373 |
The visual inspection of the distribution of Thal phases across the stages and global PVC SUVR values (Fig. 2) suggested that the associations with PET‐based amyloid stages and global PVC SUVR values reflected the differences between Thal phases 0 to 3 versus 4 to 5 rather than a homogeneous internal differentiation across all Thal phases. This was further supported by a post hoc analysis arising from this observation: Median AUC for high versus low Thal phases predicted by PET‐based frequency stages was 0.99 (95‐CI: 0.97–1.00), 0.93 (95‐CI: 0.83–1.00) for striatal stages, and 0.87 (95‐CI: 0.72–0.98) for global PVC SUVR values.
Neocortical and striatal/diencephalic plaque scores versus PET‐based amyloid stagings and global SUVR
Table 4 shows the associations between median regional plaque scores and PET‐based amyloid stagings and global SUVR. As expected, all associations were positive. Visual inspection of the in vivo amyloid stages by striatal/diencephalic and neocortical plaque scores revealed that a PET‐based frequency stage below 3 (indicating neocortical‐only involvement, see Fig. 1) predicted the absence of striatal/diencephalic cored plaques (Fig. 3) and low likelihood of striatal/diencephalic diffuse plaques (Fig. S1). For neocortical plaques, the association appeared less pronounced, because diffuse plaques were also present at PET‐based frequency stage 0, although there were no neocortical cored plaques (Fig. S2), and in general the likelihood of neocortical plaques was lower with lower PET‐based frequency stages (Fig. S3). Similarly, striatal cored plaques were almost excluded and diffuse plaques were less likely at PET‐based striatal stage 1 (indicating only neocortical amyloid PET signal, Fig. 3, and Fig. S1).
Table 4.
Associations between PET‐based amyloid stagings and neocortical and striatal amyloid plaque scores.
| Kendall’s tau | 95‐CI: lower – upper | ||||
|---|---|---|---|---|---|
| Frequency stages | vs. | Striatum/dienceph. median CP | 0.410 | 0.126 | 0.607 |
| Frequency stages | vs. | Striatum/dienceph. median DP | 0.569 | 0.269 | 0.739 |
| Frequency stages | vs. | Neocortex median CP | 0.631 | 0.323 | 0.788 |
| Frequency stages | vs. | Neocortex median DP | 0.669 | 0.357 | 0.818 |
| Striatum‐based stages | vs. | Striatum/dienceph. median CP | 0.309 | 0.046 | 0.511 |
| Striatum‐based stages | vs. | Striatum/dienceph. median DP | 0.611 | 0.317 | 0.772 |
| Striatum‐based stages | vs. | Neocortex median CP | 0.553 | 0.267 | 0.723 |
| Striatum‐based stages | vs. | Neocortex median DP | 0.673 | 0.371 | 0.818 |
| Global SUVR | vs. | Striatum/dienceph. median CP | 0.366 | 0.098 | 0.563 |
| Global SUVR | vs. | Striatum/dienceph. median DP | 0.551 | 0.265 | 0.721 |
| Global SUVR | vs. | Neocortex median CP | 0.563 | 0.275 | 0.731 |
| Global SUVR | vs. | Neocortex median DP | 0.569 | 0.281 | 0.737 |
CP, cored plaque score; DP, diffuse plaque score.
Figure 3.
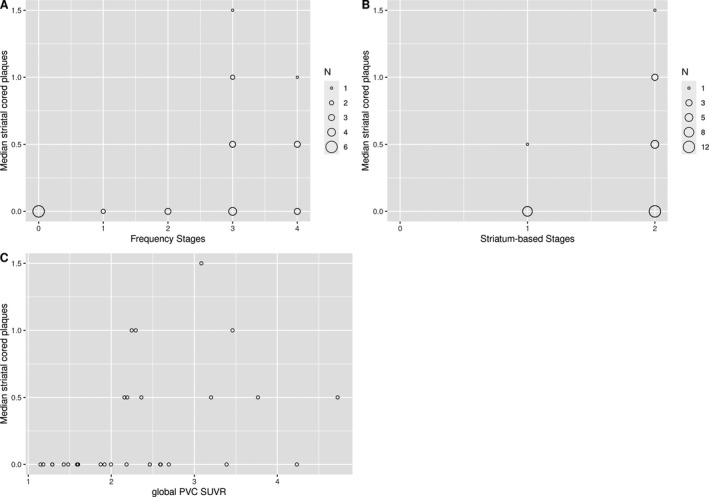
Distribution of striatal cored plaques across amyloid stages and global PVC SUVR values. Median striatal cored plaques plotted against PET‐based frequency (A) and striatal (B) amyloid stages and global PVC SUVR values (C), with the size of the circles corresponding to the number of underlying cases.
To formally test these impressions, we determined AUC under the ROC curves for PET‐based stagings and global PVC SUVR values predicting the binary outcome of the absence or presence of striatal/diencephalic and neocortical cored and dense plaques, respectively. Median AUC values were on average higher for PET‐based frequency stages than for striatal stages and global PVC SUVR values, but 95% credibility intervals overlapped with each other (Fig. 4).
Figure 4.
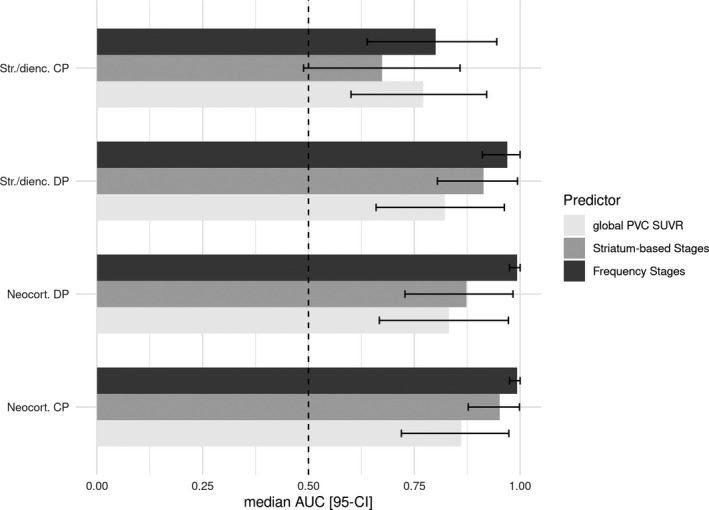
Areas under receiver operating characteristic curves and 95% credibility intervals. Median and 95% credibility interval (95‐CI) for areas under the receiver operating characteristic curves (AUC) for PET‐based frequency and striatal amyloid stages, and global PVC SUVR values predicting the binary outcomes of the presence or absence of striatal/diencephalic (Str/dienc.) and neocortical (neocort.) cored plaques (CP) and diffuse plaques (DP), respectively. The vertical red line indicates an AUC of 0.5, representing random guessing accuracy.
Discussion
Consistent with our expectation, we found that PET‐based amyloid stages, derived from either regional frequency‐based or a priori striatum‐based staging schemes, were associated with neuropathologic Thal phases of regional Aβ distribution as well as with CERAD ratings of neuritic and diffuse plaque densities. This association was approximately 44 times more likely for frequency stages and 89 times more likely for striatal stages than the association of neuropathologically‐assessed amyloid burden with global PVC 18F‐Florbetapir‐PET signal. In addition, we found that lower PET‐based amyloid stages (indicating neocortical‐only involvement) were associated with lower numbers of striatal and diencephalic cored plaques, although the credibility intervals overlapped with those from global PVC SUVR values in these analyses. Interestingly, the PET‐based frequency staging was not inferior to the striatal staging for discriminating between the absence and presence of striatal/diencephalic cored and diffuse amyloid plaques.
The finding of a much stronger association of PET‐based amyloid stages than global SUVR values with the distribution of Aβ pathology as captured by the Thal phases, as well as CERAD ratings of neuritic and diffuse plaque density, agrees with one previous study. 9 However, different to this previous study which determined neocortical vs striatal amyloid PET stages based on correspondence with Thal phases, we used unbiased approaches where the frequency staging was derived from a data‐driven analysis of independent amyloid PET data from ADNI controls and the INSIGHT‐preAD cohort, 11 , 15 and the striatal staging had previously been defined in two independent cohorts as well. 10 Despite the strong evidence for a superior association of PET‐based amyloid stages with neuropathologic Thal phases, the visual inspection of the data distribution, and our post hoc analysis of AUC values arising from it, indicated that this effect was mainly driven by a high degree of discrimination between lower Thal phases (3 and smaller) versus higher Thal phases (phases 4 and 5). Due to relatively high global cortical uptake in all cases, PET‐based striatal stages could only take one of two values (neocortical with and without additional striatal involvement) so that a differentiation among lower level Thal phases was not to be expected. Also, one has to note that numbers of early Thal phases were very low in this clinical autopsy cohort, thus preventing conclusive analyses on the association between amyloid PET signal and early Thal phases in this dataset. Our findings support a previous study of 35 cases where a global cut‐off for 11C‐PIB‐PET positivity was associated with the binary distinction between low to moderate (0–2) versus high (3–5) Thal phases. 36 In addition, one previous clinico‐pathological comparison study in a subsample of the data from 9 likewise reported that global levels of 18F‐Flutemetamol were able to discriminate between low to moderate versus high Thal phases in 68 cases. 37 This previous and our current findings also agree with our earlier observation that higher versus lower PET‐based frequency stages were able to discriminate between clinically stable versus clinically progressive cognitively normal individuals and MCI patients, and that the prediction accuracy was higher for the PET‐based amyloid stages than for the global amyloid PET signal. 38 At the same time, in our current study we could not identify a clear advantage of one PET‐based staging scheme over the other. The fit of regression parameters for the association with Thal phases was six times more likely for the striatal than for the frequency staging. However, the estimates of accuracy for predicting low versus high Thal phases were numerically higher for frequency than for striatal staging, possibly also related to the small range of values for the striatal staging.
Consistent with their association with the Thal phases, PET‐based frequency stages 2 and smaller (indicating selective neocortical amyloid signal, see Fig. 1) were associated with the absence of striatal and diencephalic cored plaques. According to the Thal staging such plaques are only present at Thal phases 3 and higher, 7 representing a more advanced stage of cerebral amyloidosis that also associates with neocortical tau pathology and clinical dementia. 39 Increased levels of striatal and diencephalic amyloid binding in PET have been found associated with a higher likelihood of clinical dementia 40 , 41 , 42 , 43 and striatal signal in amyloid PET imaging using 18F‐Flutemetamol was found to be associated with post‐mortem levels of striatal amyloid plaques. 44 These observations served as the basis for an in vivo staging of amyloid PET data based on the absence or presence of striatal amyloid binding. 10 Remarkably, the accuracy of PET‐based frequency stages to predict the absence or presence of striatal and diencephalic cored and diffuse plaques was not only numerically higher than the accuracy of global PVC SUVR values, but also than the accuracy of striatum‐based stages. This indicates that a staging mainly driven by involvement differences across distinct cortical regions was not inferior to a staging explicitly taking striatal involvement into account to detect striatal and diencephalic Aβ pathology. This is consistent with a previous study which investigated this association from an opposite viewpoint: in ten patients undergoing frontal cortex biopsy sampling during intracranial surgery, the presence of frontal lobe amyloid plaques was associated not only with increased 11C‐PIB‐PET signal in frontal, parietal, and lateral temporal cortices, but also in the striatum. 45 Our results also concur with a study of 73 cases with global amyloid positivity in 11C‐PIB‐PET that showed a high correlation between striatal and cortical 11C‐PIB binding particularly in orbitofrontal regions, 46 consistent with the transition from frequency stage 2 to 3 (see Fig. 1).
In conclusion, our data indicate that amyloid stages derived from PET data, either taking a binary or more detailed sequence of neocortical to striatal involvement into account, were more strongly associated with neuropathological phases of amyloid deposition than more standard global amyloid PET signal. However, neither metric could accurately discern specific neuropathologically defined phases. This is in contrast to one previous study 9 that was, however, based on a biased estimate of PET amyloid stages using knowledge from the Thal phases. As a maximum claim, PET‐based in vivo amyloid stages appeared to allow more accurate discrimination between high versus low‐to‐moderate stages of amyloid neuropathology compared with standard global levels of amyloid PET signal.
A caveat to our work is that the number of cases in the current analysis was small, particularly for lower Thal phases, and that our AUC analysis was post hoc after seeing the data. Related to this, here we made use of retrospectively available neuropathological scoring data. Although the ADNI data are very well‐curated, errors in the data cannot completely be excluded. As on example, case ADD20 (Table 2) showed Thal phase 5 with frequent diffuse amyloid plaques, but lack of neuritic plaques. Albeit theoretically possible, such a constellation may appear unlikely so that some transmission error of the data from the neuropathological examination to its entry into the ADNI data repository may seem possible. To check the relevance of the effect, here we conducted a sensitivity analysis, leaving the case ADD20 out of the analyses. The results remained essentially unchanged. Still, given the small number of cases and the retrospective design of the analysis, a final conclusion would require replication of our findings in a larger cohort. As another limitation, one should note that our findings relate only to 18F‐Florbetapir‐PET and other tracers with other binding properties may yield different results. Taking further confounds into account, such as comorbid TDP‐43 or Lewy body pathology, would have strengthened our results. However, the small number of cases prohibited further stratification of the sample. Of note, we combined quantitative analysis of PET data with semiquantitative ratings of neuropathologic data. Using data at different scales such as continuous SUVR values versus ordinal frequency‐based or striatal stages may influence the fit of the model as reflected in the Bayes factors. However, one would not anticipate that the scale of the predictors always has the same effect on Bayes factor, that is, an ordinal scaled predictor is not expected to necessarily lead to a more plausible model than a continuous scaled predictor. In the current dataset, there was no case with a global SUVR value below the commonly used cutoff, rendering a binarization of the continuous SUVR values unfeasible. Future studies should go beyond semiquantitative and regionally limited pathology frequency scores, such as available in the ADNI neuropathology core, and strive to use unbiased quantitative approaches for both PET and neuropathology. We are aware that Bayesian analysis is still not widely used in clinico‐pathological research. Still, we think that the beauty of Bayesian analysis is its ability to directly compare likelihood of data under different models and to directly quantify the degree of evidence for the presence or absence of an effect. This is particularly relevant for the analysis of small samples where the interpretation of a non‐significant finding in a classical null hypothesis significance testing framework is almost impossible due to a high degree of type 2 error.
In summary, we used neuropathologically unbiased approaches to demonstrate that in vivo stages of regional amyloid deposition in PET were associated with Thal phases of the regional distribution of amyloid pathology, as well as with neuropathologic ratings of neuritic and diffuse plaque density, and the presence or absence of striatal/diencephalic cored and diffuse amyloid plaques. We found strong evidence that the association with neuropathologic assessments was stronger for PET‐based in vivo amyloid stages, both based on an a priori model of neocortical versus striatal amyloid PET signal as well as a cortically more detailed regional frequency model, than for global PVC SUVR values. However, despite these stronger effects our findings challenge the notion of a previous study 9 that amyloid PET stages would allow internal differentiation between lower‐level Thal phases, a finding that may have been confounded by a biased estimation of the amyloid PET stages. Thus, if replicated in larger cohorts, our data suggest that regional frequency‐ and striatum‐based amyloid PET stages are useful to differentiate between low to moderate and high levels of regional amyloid pathology as captured by the Thal phases, and that low PET‐based amyloid stages can indicate the absence of striatal and diencephalic cored plaques, which have been linked to clinico‐pathologic AD progression. We could, however, not identify a clear advantage of one staging scheme over the other.
Conflicts of Interest
SJT participated in scientific advisory boards of Roche Pharma AG and MSD, and received lecture fees from Roche and MSD. AGMT, FL, MD, and MJG declare no conflicts of interest.
Supporting information
Table S1. PET staging schemes.
Figure S1. Distribution of striatal diffuse plaques across amyloid stages and global PVC SUVR values.
Figure S2. Distribution of neocortical cored plaques across amyloid stages and global PVC SUVR values.
Figure S3. Distribution of neocortical diffuse plaques across amyloid stages and global PVC SUVR values.
Appendix S1. Acknowledgement list for ADNI contributors.
Acknowledgment
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen Idec Inc.; Bristol‐Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. MJG is supported by the "Miguel Servet" program [CP19/00031] of the Spanish Instituto de Salud Carlos III (ISCIII‐FEDER). Open access funding enabled and organized by ProjektDEAL.
Funding InformationData collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen Idec Inc.; Bristol‐Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. MJG is supported by the "Miguel Servet" program [CP19/00031] of the Spanish Instituto de Salud Carlos III (ISCIII‐FEDER). Open access funding enabled and organized by ProjektDEAL.
Funding Statement
This work was funded by Alzheimer's Disease Neuroimaging Initiative grant ; National Institutes of Health grant U01 AG024904; Department of Defense grant W81XWH‐12‐2‐0012; National Institute on Aging grant ; National Institute of Biomedical Imaging and Bioengineering grant ; Alzheimer’s Association grant ; Alzheimer’s Drug Discovery Foundation grant ; Araclon Biotech grant ; BioClinica, Inc. grant ; Biogen Idec Inc. grant ; Bristol‐Myers Squibb Company grant ; Eisai Inc. grant ; Elan Pharmaceuticals, Inc. grant ; Eli Lilly and Company grant ; EuroImmun grant ; F. Hoffmann‐La Roche Ltd grant ; Genentech, Inc. grant ; Fujirebio grant ; GE Healthcare grant ; IXICO Ltd. grant ; Janssen Alzheimer Immunotherapy Research & Development, LLC. grant ; Johnson & Johnson Pharmaceutical Research & Development LLC. grant ; Medpace, Inc.; Merck & Co., Inc. grant ; Meso Scale Diagnostics, LLC. grant ; NeuroRx Research grant ; Neurotrack Technologies grant ; Novartis Pharmaceuticals Corporation grant ; Pfizer Inc. grant ; Piramal Imaging grant ; Servier grant ; Synarc Inc. grant ; Takeda Pharmaceutical Company grant ; Canadian Institutes of Health Research grant ; Foundation for the National Institutes of Health grant ; Northern California Institute for Research and Education grant ; Alzheimer's Disease Cooperative Study grant ; Miguel Servet grant CP19/00031; Spanish Instituto de Salud Carlos III grant ; ProjektDEAL grant .
Contributor Information
Stefan J. Teipel, Email: stefan.teipel@med.uni-rostock.de.
Michel J. Grothe, Email: neurosev@gmail.com.
References
- 1. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG‐2 criteria. Lancet Neurol 2014;13:614–629. [DOI] [PubMed] [Google Scholar]
- 3. Villeneuve S, Rabinovici GD, Cohn‐Sheehy BI, et al. Existing Pittsburgh Compound‐B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain 2015;1:2020–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sabri O, Sabbagh MN, Seibyl J, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer's disease: phase 3 study. Alzheimers Dement 2015;11:964–974. [DOI] [PubMed] [Google Scholar]
- 5. Salloway S, Gamez JE, Singh U, et al. Performance of [(18)F]flutemetamol amyloid imaging against the neuritic plaque component of CERAD and the current (2012) NIA‐AA recommendations for the neuropathologic diagnosis of Alzheimer's disease. Alzheimer's Dement 2017;9:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid‐beta plaques: a prospective cohort study. Lancet Neurol 2012;11:669–678. [DOI] [PubMed] [Google Scholar]
- 7. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 8. Seo SW, Ayakta N, Grinberg LT, et al. Regional correlations between [C‐11]PIB PET and post‐mortem burden of amyloid‐beta pathology in a diverse neuropathological cohort. Neuroimage‐Clin 2017;13:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thal DR, Beach TG, Zanette M, et al. Estimation of amyloid distribution by [(18)F]flutemetamol PET predicts the neuropathological phase of amyloid beta‐protein deposition. Acta Neuropathol 2018;136:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hanseeuw BJ, Betensky RA, Mormino EC, et al. PET staging of amyloidosis using striatum. Alzheimers Dement 2018;14:1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grothe MJ, Barthel H, Sepulcre J, et al. In vivo staging of regional amyloid deposition. Neurology 2017;89:2031–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mattsson N, Palmqvist S, Stomrud E, et al. Staging beta‐amyloid pathology with amyloid positron emission tomography. JAMA Neurol 2019. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cho H, Choi JY, Hwang MS, et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol 2016;80:247–258. [DOI] [PubMed] [Google Scholar]
- 14. Collij LE, Heeman F, Salvado G, et al. Multitracer model for staging cortical amyloid deposition using PET imaging. Neurology 2020;95:e1538–e1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sakr FA, Grothe MJ, Cavedo E, et al. Applicability of in vivo staging of regional amyloid burden in a cognitively normal cohort with subjective memory complaints: the INSIGHT‐preAD study. Alzheimers Res Ther 2019;11:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of the Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 17. Franklin EE, Perrin RJ, Vincent B, et al. Brain collection, standardized neuropathologic assessment, and comorbidity in Alzheimer's Disease Neuroimaging Initiative 2 participants. Alzheimers Dement 2015;11:815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grothe M, Heinsen H, Teipel S. Longitudinal measures of cholinergic forebrain atrophy in the transition from healthy aging to Alzheimer's disease. Neurobiol Aging 2013;34:1210–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Müller‐Gärtner HW, Links JM, Prince JL, et al. Measurement of radiotracer concentration in brain gray matter using positron emission tomography: MRI‐based correction for partial volume effects. J Cerb Blood Flow Metab 1992;12:571–583. [DOI] [PubMed] [Google Scholar]
- 21. Gonzalez‐Escamilla G, Lange C, Teipel S, et al. Alzheimer's disease neuroimaging I. PETPVE12: an SPM toolbox for Partial Volume Effects correction in brain PET ‐ Application to amyloid imaging with AV45‐PET. NeuroImage 2017;15:669–677. [DOI] [PubMed] [Google Scholar]
- 22. Desikan RS, Segonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage 2006;31:968–980. [DOI] [PubMed] [Google Scholar]
- 23. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement 2015;11:1–15.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Catafau AM, Bullich S, Seibyl JP, et al. Cerebellar amyloid‐beta plaques: how frequent are they, and do they influence 18F‐florbetaben SUV ratios? J Nucl Med 2016;57:1740–1745. [DOI] [PubMed] [Google Scholar]
- 25. Joshi AD, Pontecorvo MJ, Clark CM, et al. Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer's disease and cognitively normal subjects. J Nucl Med 2012;53:378–384. [DOI] [PubMed] [Google Scholar]
- 26. Landau SM, Breault C, Joshi AD, et al. Amyloid‐beta imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med 2013;54:70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir‐PET for imaging beta‐amyloid pathology. JAMA 2011;305:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fleisher AS, Chen K, Liu X, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol 2011;68:1404–1411. [DOI] [PubMed] [Google Scholar]
- 29. Goodman S. A dirty dozen: twelve P‐value misconceptions. Semin Hematol 2008;45:135–140. [DOI] [PubMed] [Google Scholar]
- 30. Wagenmakers EJ, Marsman M, Jamil T, et al. Bayesian inference for psychology. Part I: theoretical advantages and practical ramifications. Psychon B Rev 2018;25:35–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kruschke JK. Doing Bayesian data analysis ‐ a tutorial with R, JAGS, and Stan, 2nd ed San Diego, CA: Elsevier, 2015. [Google Scholar]
- 32. Wagenmakers EJ. A practical solution to the pervasive problems of p values. Psychon Bull Rev 2007;14:779–804. [DOI] [PubMed] [Google Scholar]
- 33. Marsman M, Wagenmakers EJ. Bayesian benefits with JASP. Eur J Dev Psychol 2017;14:545–555. [Google Scholar]
- 34. Wagenmakers EJ, Love J, Marsman M, et al. Bayesian inference for psychology. Part II: example applications with JASP. Psychon B Rev 2018;25:58–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feng D, Cortese G, Baumgartner R. A comparison of confidence/credible interval methods for the area under the ROC curve for continuous diagnostic tests with small sample size. Stat Methods Med Res 2017;26:2603–2621. [DOI] [PubMed] [Google Scholar]
- 36. Murray ME, Lowe VJ, Graff‐Radford NR, et al. Clinicopathologic and 11C‐Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer's disease spectrum. Brain 2015;138(Pt 5):1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thal DR, Beach TG, Zanette M, et al. [(18)F]flutemetamol amyloid positron emission tomography in preclinical and symptomatic Alzheimer's disease: specific detection of advanced phases of amyloid‐beta pathology. Alzheimers Dement 2015;11:975–985. [DOI] [PubMed] [Google Scholar]
- 38. Teipel SJ, Dyrba M, Chiesa PA, et al. In vivo staging of regional amyloid deposition predicts functional conversion in the preclinical and prodromal phases of Alzheimer's disease. Neurobiol Aging 2020;93:98–108. [DOI] [PubMed] [Google Scholar]
- 39. Beach TG, Sue LI, Walker DG, et al. Striatal amyloid plaque density predicts Braak neurofibrillary stage and clinicopathological Alzheimer's disease: implications for amyloid imaging. J Alzheimers Dis 2012;28:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim SE, Lee B, Park S, et al. Clinical significance of focal ss‐amyloid deposition measured by (18)F‐flutemetamol PET. Alzheimers Res Ther 2020;12:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koivunen J, Karrasch M, Scheinin NM, et al. Cognitive decline and amyloid accumulation in patients with mild cognitive impairment. Dement Geriatr Cogn Disord 2012;34:31–37. [DOI] [PubMed] [Google Scholar]
- 42. Kemppainen NM, Aalto S, Wilson IA, et al. Voxel‐based analysis of PET amyloid ligand [11C]PIB uptake in Alzheimer disease. Neurology 2006;67:1575–1580. [DOI] [PubMed] [Google Scholar]
- 43. Cho SH, Shin JH, Jang H, et al. Amyloid involvement in subcortical regions predicts cognitive decline. Eur J Nucl Med Mol Imaging 2018;45:2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beach TG, Thal DR, Zanette M, et al. Detection of striatal amyloid plaques with [18F]flutemetamol: validation with postmortem histopathology. J Alzheimers Dis 2016;52:863–873. [DOI] [PubMed] [Google Scholar]
- 45. Leinonen V, Alafuzoff I, Aalto S, et al. Assessment of beta‐amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11‐labeled Pittsburgh Compound B. Arch Neurol 2008;65:1304–1309. [DOI] [PubMed] [Google Scholar]
- 46. Sauerbeck J, Ishii K, Hosokawa C, et al. The correlation between striatal and cortical binding ratio of (11)C‐PiB‐PET in amyloid‐uptake‐positive patients. Ann Nucl Med 2018;32:398–403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. PET staging schemes.
Figure S1. Distribution of striatal diffuse plaques across amyloid stages and global PVC SUVR values.
Figure S2. Distribution of neocortical cored plaques across amyloid stages and global PVC SUVR values.
Figure S3. Distribution of neocortical diffuse plaques across amyloid stages and global PVC SUVR values.
Appendix S1. Acknowledgement list for ADNI contributors.