

Abstract
The RNA-binding protein TDP-43, associated to amyotrophic lateral sclerosis and frontotemporal dementia, regulates the alternative splicing of several genes, including the skipping of TNIK exon 15. TNIK, a genetic risk factor for schizophrenia and causative for intellectual disability, encodes for a Ser/Thr kinase regulating negatively F-actin dynamics.
Here we show that in the human adult nervous system TNIK exon 15 is mostly included compared to the other tissues and that, during neuronal differentiation of human induced pluripotent stem cells and of human neuroblastoma cells, TNIK exon 15 inclusion increases independently of TDP-43 protein content. By studying the possible molecular interplay of TDP-43 with brain-specific splicing factors, we found that the neuronal NOVA-1 protein competitively inhibits both TDP-43 and hnRNPA2/B1 skipping activity on TNIK by means of a RNA-dependent interaction and that this competitive mechanism is common to other TDP-43 RNA targets. We also show that the TNIK protein isoforms including/excluding exon 15 differently regulate cell spreading in non-neuronal cells and neuritogenesis in primary cortical neurons.
Our data suggest a complex regulation between the ubiquitous TDP-43 and the neuron-specific NOVA-1 splicing factors in the brain that may help better understand the pathobiology of both neurodegenerative diseases and schizophrenia.
Keywords: TNIK, RNA-binding protein, Splicing, TDP-43, NOVA-1, Schizophrenia
1. Introduction
Alternative splicing is a very active process in the mammalian brain contributing to the high complexity of the nervous system proteome [1]. During neurodevelopment, a tight spatio-temporal regulation of alternative splicing is crucial for the proper development of the different brain areas, depending on the combinatorial control of both ubiquitous and neuron-specific splicing factors [2,3]. In developing neurons, NOVA protein family members regulate the alternative splicing of genes important for axon guidance, synaptic architecture and activity [4,5]. These RNA-binding proteins (RBP) are predominantly expressed in neuronal cells, with NOVA-1 particularly enriched in the hindbrain and ventral spinal cord and NOVA-2 in the hippocampus and neocortex [6].
Among the ubiquitous splicing factors, TDP-43 is of particular interest as it is associated to the neurodegenerative diseases amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), where it forms pathological aggregates in the cytoplasm of affected neuronal cells with the concomitant reduction of its nuclear localization [7,8] and splicing activity [9]. In the nucleus TDP-43 binds to a wide array of genes identified by high throughput approaches such as CLIP and mainly regulates their splicing [10]. TDP-43 was shown to bind to TNIK intron 15 and to promote the skipping of the alternative exon 15 [11]. Upon TDP-43 knock-down in human neuroblastoma cells to mimic a TDP-43 loss-of-function mechanism in ALS and FTD, we recently showed a specific increase of TNIK exon 15 inclusion and increased levels of the corresponding TNIK protein isoforms [12].
TNIK is a genetic risk factor for schizophrenia [13–15] and also causative of non-syndromic intellectual disability because of null mutations recently identified in consanguineous families [16]. TNIK encodes for a 130 KDa serine/threonine kinase of the GCK (germinal center kinase) family and, in neuronal cells, it regulates synapse composition and activity [17–19] as well as dendritic growth and arborization [20]. In particular, TNIK kinase activity regulates cytoskeleton dynamics by promoting F-actin disassembly in a regulatory pathway involving the ubiquitin ligase Nedd4–1 and the GTP-binding protein Rap2A [20]. TNIK protein is composed of a N-terminal kinase domain and a C-terminal regulatory CNH (citron homology) domain, linked by a long intermediate region (756 amino acids) with no functional domains, but involved in protein-protein interaction [20,21]. Because of the in-frame alternative splicing of three exons (ex15, ex17, ex22), all mapping within the intermediate region, eight different TNIK transcripts can be generated, although the biological functions of such isoforms have not been investigated so far [21].
Based on our previous findings about TDP-43-dependent TNIK processing, here we characterized TNIK alternative splicing in the human adult nervous system and during neuronal differentiation in vitro and studied the possible regulatory splicing mechanisms involving also the neuron-specific NOVA-1 RBP. Since TNIK negatively regulates F-actin dynamics, we also studied the biological role of the different TNIK protein splicing isoforms on neurite outgrowth in primary cortical neurons.
2. Materials and methods
2.1. Cell cultures
Human neuroblastoma SK-N-BE cells were maintained in RPMI-1640 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich), 2 g/l glucose, 2 mM l-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin and 100 μg/ml streptomycin (all from Gibco).
Human embryonic kidney (HEK) 293T cells were cultured in DMEM medium (Thermo Fisher Scientific) supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin.
Human inducent pluripotent stem cells (iPSC) were obtained by Sendai virus (Thermo Fisher Scientific) reprogramming of fibroblasts from three healthy donors after informed consent, according to the local ethics committee. iPSCs were maintained in E8 essential medium (Thermo Fisher Scientific) in Matrigel-coated wells (Corning) and characterized for the expression of pluripotency markers (Supplementary Fig. 3).
Primary cortical neurons were isolated from E.15 wild-type mouse embryos dissociated in 0.05% trypsin-EDTA (Thermo Fisher Scientific) at 37 °C for 12 min. 400,000 cells/well were plated on glass coverslips coated with 0.125 mg/ml of poly d-Lysine (Sigma) and 5 μg/ml of laminin (Corning) in 6-well plates and grown in Neurobasal medium (Thermo Fisher Scientific) supplemented with 2% B-27 (Gibco), 2 mM GlutaMAX (Gibco), 100 U/ml penicillin and 100 μg/ml streptomycin for 4–7 days in vitro (DIV).
2.2. Neural stem cell and neuronal differentiation
SK-N-BE cells were treated with 10 μM retinoic acid (RA) for six days to induce neuronal differentiation. The medium was replaced every two days and cells were analyzed before treatment (T0) and after two (T2), four (T4) and six (T6) days. The neuronal differentiation was evaluated by immunofluorescence with the neuronal cytoskeleton marker βIII-Tubulin, SMI 312 and MAP2 to visualize the neuronal outgrowth. The absence of astroglial cells was evaluated by immunofluorescence analysis of GFAP (Supplementary Fig. 2).
To obtain neural stem cells (NSC), iPSCs were seeded at low density on Matrigel-coated wells and maintained in PSC Neural Induction medium (Thermo Fisher Scientific) for six days, replacing the medium every two days. On day 7, cells were harvested, seeded on Geltrex-coated wells (Thermo Fisher Scientific) and expanded in Expansion medium (Thermo Fisher Scientific) for two passages before being characterized (Supplementary Fig. 3).
iPSCs were differentiated into neurons (iPSC-N) as already shown [22]. Briefly, iPSCs were seeded in low-adhesion dishes and grown in suspension in order to induce embryoid bodies (EB) formation. After 17-day differentiation, EBs were dissociated and 50,000 cells/ml were plated on poly-d-Lysine/laminin-coated wells and allowed to differentiate for additional 10 days (Supplementary Fig. 3).
2.3. Plasmid constructs and cell transfection
Lipofectamine 2000 (Thermo Fisher Scientific) was used for cell transfections according to the manufacturer’s instructions. For NOVA-1 over-expression experiments, HEK293T cells were seeded in 6-well plates, transfected with 3 μg pCGN-HA-NOVA-1 construct (a kind gift of Prof. Elena Battaglioli, Università degli Studi di Milano [23]) or empty vector and after 24 h they were harvested for further analyses.
For the co-immunoprecipitation assays, HEK293T cells were transfected for 24 h with pCGN-HA-NOVA-1 and pFlag-CMV2-TDP-43 or the mutant TDP-43 constructs (ΔRRM1, ΔC-terminal, 4FL), and the pFlag-CMV2-hnRNPA2B1 (all kind gifts of Dr. Emanuele Buratti, ICGEB, Trieste [24–26]) (ratio 1:1).
For TDP-43 knock-down, HEK293T cells were transfected with 80 nM siRNA duplexes (5′GCAAAGCCAAGAUGAGCCUUU3′ and 5′ AGGCUCAUCUUGGCUUUGCUU3′, Ambion) in a double round of transfection and 24 h after gene silencing, 2 μg of pCGN-HA-NOVA-1 plasmid or empty vector were transfected. Cells were harvested for the following analysis 72 h after the first transfection.
The pCI-HA-tagged plasmids for human TNIK full-length (TNIKex15) and for a mutant TNIK form (K54R, TNIK KM) were kindly provided by Prof. Ken-ichi Kariya (University of the Ryukyus, Japan [32]). The exon 15 nucleotide-sequence was deleted in the HA-TNIK FL plasmid to obtain the HA-TNIKΔ15 construct using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instruction. The following oligos 5′-CGTGCGGAGCATGAACAGGAATATAAGCGCAAACAA-3′ and 5′-TTGTTTGCGCTTATATTCCTGTTCATGCTCCGCACG-3′ were used for mutagenesis.
For morphological cell assay, HEK293T cells were seeded on glass coverslips in 24-well plates and transiently co-transfected with 0.5 μg DNA, using pEGFP-C2 vector and the different TNIK constructs in a 1:3 ratio. After 24 h, cells were fixed for image analyses.
To analyze neuronal arborization, DIV4 primary murine cortical neurons were transiently transfected with 1.3 μg DNA using a 1:3 ratio for GFP and TNIK constructs. 72 h after transfection (DIV7), cells were fixed for subsequent analyses.
2.4. RNA isolation, RT-PCR and Real time PCR
Total RNA from different human adult tissues was purchased by Clontech and includes different individuals (Supplementary Table S1). Total RNA was isolated from cells using TriZol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. 1.5 or 4 μg of total RNA from cells or tissues, respectively, were pre-treated with 1U DNaseI (Roche) for 20 min at 37 °C and retro-transcribed using 1U SuperScript II-RT (Thermo Fisher Scientific) and 3 μM oligo-dT. RT-PCR was carried out using 300 nM specific primer pairs (see sequences and amplicon size in Supplementary Table S2) and GoTaq G2 DNA polymerase (Promega) for 26–35 cycles. GAPDH and RPL10a genes were used for sample normalization. TNIK exon 15 and STAG2 exon 30b inclusion, POLDIP3 exon 3 skipping or the relative amount of TNIK splicing isoforms were quantified by densitometric analyses of agarose gel runs using ImageJ software (NIH) and were represented as percentage of the total isoforms. Nomenclature of TNIK alternative exons is referred to FAST DB 2010_2 database (EASANA).
Real time PCR was performed for 45 cycles with SYBR Green PCR Master mix (Applied Biosystems) and 300 nM -specific primers for total TNIK (exons 30–31), TNIKex15 (exons 14–16) and RPL10a genes (primer sequences and calculated efficiency in Supplementary Table S2). Reactions were run in duplicate for each sample on ABI Prism 7900HT (Applied Biosystems) and a dissociation curve was generated at the end. Threshold cycles (Ct) for each tested gene were normalized on the housekeeping RPL10a gene Ct value (ΔCt) and every experimental sample was referred to mean ΔCt of controls (ΔΔCt). Gene expression data were calculated as fold change values (2−ΔΔCt). By applying the Pfaffl method we obtained similar results (data not shown).
2.5. Minigene splicing assay
HEK293T cells were transiently co-transfected with the TNIKex15 minigene construct (pTBminigene_TNIKex15) already described in [12] and Flag-TDP-43, Flag-hnRNPA2/B1, HA-NOVA-1 plasmids or empty vectors, alone or in combination with each other, and harvested for RNA and protein extraction 24 h after transfection. Total RNA was extracted and retro-transcribed as described above and RT-PCR reactions were carried out using specific primers (a2–3For and Bra2 Rev., Table S2) designed on pTB plasmid. GAPDH gene was used for sample normalization. Transfection efficiency was assessed by western blot analysis as described below.
2.6. Western blot analysis
Cells were homogenized in lysis buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, protease inhibitor cocktail (Roche)), sonicated and incubated 15 min on ice. Protein lysates were quantified by BCA protein assay (Thermo Fisher Scientific) and 25 μg protein samples were resolved on 10% NuPAGE Bis-Tris pre-cast polyacrylamide gels (Thermo Fisher Scientific) by SDS-PAGE and transferred to nitrocellulose membranes. Immunoblots were performed with specific primary antibodies (listed in Supplementary Table S3), diluted in 5% milk in TBS with 0.1% Tween-20 (Sigma-Aldrich). The Novex ECL kit (Thermo Fisher Scientific) was used for detection of the HRP-conjugated secondary antibodies. Densitometric analyses were performed using ImageJ software (NIH).
2.7. UV-cross-linking and immunoprecipitation (UV-CLIP)
PCR amplicons containing the genomic region of TNIK exon 15 (87 bp) with 141 bp of 5′ (TNIK A) or 126 bp of 3′ (TNIK B) flanking introns were cloned into the TOPO TA vector (Thermo Fisher Scientific) downstream of the T7 promoter. TNIK A and TNIK B plasmids were linearized with HindIII restriction enzyme and radio-labeled riboprobes were obtained by transcribing 0.5 μg linearized constructs with 20 U T7 RNA polymerase, 20 μCi α-UTP32, 0.5 mM NTPs, and 20 U RNase inhibitor. After DNaseI digestion of template DNA, the resulting P32-labeled riboprobes were purified on ProbeQuant G-50 microcolumns (GE Healthcare). UV-CLIP experiments were performed as previously described [27]. Briefly, 300,000 cpm riboprobes were incubated with 200 μg protein lysates of HEK293T cells transfected with Flag-TDP-43 and/or HA-NOVA-1 constructs in 25 μl ligation buffer (1.3 mM MgCl2, 19 mM HEPES-KOH pH 7.4, 1.5 mM ATP, 19 mM Creatine phosphate) for 10 min at 30 °C. After addition of 5 μg tRNA, samples were irradiated with UV light for 5 min and RNase A-treated (25 U) for 30 min. Immunoprecipitation was then conducted with 2 μg of the selected antibody (anti-HA and anti-FLAG, both from Sigma-Aldrich) or the irrelevant IgG antibody (Santa Cruz Biotechnology) pre-coated to protein G Dynabeads (Thermo Fisher Scientific). After overnight incubation, immunocomplexes were washed five times in PBS with 0.02% Tween-20, run on a 10% SDS-PAGE and analyzed by autoradiography.
2.8. Co-immunoprecipitation assays
HEK293T protein lysates, transfected as described above, were treated with RNAse inhibitor (10 U/100 μg protein lysate) or RNAse A (5 μg/100 μg protein lysate) for 30 min at 37 °C. Immunoprecipitation assays were performed using 2 μg of the selected antibodies pre-coated to protein G Dynabeads and incubated with 200 μg of cell lysate for 45 min at room temperature (RT). Immunocomplexes were washed four times in PBS with 0.02% Tween-20, resolved on 10% SDS-PAGE and processed for western blot as described above.
2.9. Immunofluorescence and image acquisition
Cells were fixed with 4% paraformaldehyde in PBS for 15 min at RT, permeabilized with 0.3% Triton X-100 and blocked with 10% normal goat serum (NGS, Gibco) in PBS (blocking solution) for 20 min at RT. Incubation with primary antibodies (listed in Supplementary Table S3) was performed in blocking solution for 2 h at 37 °C. The fluorescent-tagged secondary antibodies (Alexa Fluor 488 and 555; Thermo Fisher Scientific) were used for detection. Nuclei were visualized by DAPI staining (Roche). Coverslips were mounted onto glass slides using FluorSave mounting medium (Calbiochem). Using the confocal inverted microscope (Nikon Eclipse C1), images were acquired as Z-stacks (0.2 μm step size) at 60× magnification.
Primary murine cortical neurons were fixed as described above, treated with hot 10 mM citrate buffer pH 6 for 20 min and permeabilized with 0.2% Triton X-100 for 5 min at RT. Cells were blocked with 5% bovine serum albumin (Sigma-Aldrich) in PBS for 45 min at RT and hybridized with primary antibodies diluted in blocking solution overnight at 4 °C. Anti-mouse goat secondary antibody and phalloidin, conjugated with Alexa Fluor 647 and Alexa Fluor 546 (Thermo Fisher Scientific) respectively, were incubated for 1 h at RT. Coverslips were mounted onto glass slides using Prolong Gold mounting medium (Thermo Fisher Scientific) and images were acquired using an epifluorescence microscope (Nikon Ti E) equipped with a cooled CMOS camera (Andor Zyla).
2.10. Cell morphological analyses
HEK293T cells, transfected and fixed as described above, were immunostained to detect HA-TNIK protein expression and confocal images were acquired as Z-stacks (0.2 μm step size) using a 40× magnification. Morphological analyses of HA-TNIK-transfected cells were performed using GFP expression as marker of whole cell shape. Images were pre-processed to eliminate noise, GFP signal was thresholded and cellular circularity was measured using the ImageJ software. At least 75 cells/group were analyzed in three biological replicates.
Primary murine cortical neurons, transfected and fixed as described, were stained with anti-HA antibody and Phalloidin, while GFP was used to visualize the cell shape. Epifluorescence images of isolated neurons were acquired as individual focal planes using 20× magnifications and Sholl analysis was used to analyze neuronal arbors by ImageJ software. Images were pre-processed to eliminate noise and background fluorescence, were segmented by applying an appropriate threshold and the obtained binary images were skeletonized. Concentric circles were drawn at 7.5 μm intervals around a common center in the cell body and the numbers of crossing neurites were counted at each concentric circles. The neurite number at 15 μm distance was considered as representative of proximal neurites for statistical analyses. Cells bodies and growth cones of isolated cortical neurons were imaged as Z-stacks (0.2 μm step size) using a 60× magnification and the epifluorescence images were deconvolved using an adaptive blind deconvolution algorithm (Autoquant X3, Media Cybernetics) before analysis. To quantify F-actin levels in these subcellular regions, the fluorescence intensity of Phalloidin was measured. The specific signal was thresholded to eliminate the background fluorescence and the integrated densities were measured into the ROI (region of interest) of the selected regions (cell body and growth cone) and normalized on the appropriate area. Raw values were normalized to the mean of the control condition for each replicate. At least 35 cells/group were analyzed in four biological replicates.
2.11. Statistical analyses
Statistical analyses were conducted with GraphPad PRISM 5 software package. The D’Agostino-Pearson Omibus test was used to evaluate data normality and the appropriate parametric or non-parametric tests were used. One-tailed Student’s t-test and one-way ANOVA followed by appropriate post-hoc tests were applied to compare two or multiple groups, respectively. Data are presented as mean ± s.e.m. (standard error of mean) of at least three independent experiments. Significance value was defined as *p < 0.05, **p < 0.01, ***p < 0.001. Detailed statistics is reported in Supplementary Table S4.
3. Results
3.1. Alternative splicing of TNIK exon 15 is regulated during neuronal differentiation
As no data was available describing the specific tissue expression of the different TNIK splicing isoforms, we first characterized the alternative splicing patterns of TNIK exons 15, 17, and 22 located in the protein intermediate region (Fig. 1a), in the human adult nervous system (brain and spinal cord) and in other tissues by RT-PCR analysis (Fig. 1b). The inclusion of TNIK exon 15, encoding for a 29 amino acid-long sequence, occurred as the main splicing event in brain, spinal cord and skeletal muscle, while the expression of both exon 15-containing (TNIKex15) and exon 15-excluding (TNIKΔ15) isoforms was observed in testis, kidney, liver and lung (Fig. 1b–c). Moreover, in all these latter tissues, with the exception for liver, the TNIKΔ15 isoforms were more abundant than the TNIKex15 ones (Fig. 1b–c). The TNIK isoforms containing the alternative exon 17 (TNIKex17), encoding for a 55 amino acid-long region, and lacking the alternative exon 22 (TNIKΔ22), encoding for a 8 amino acid-long sequence, were the predominant isoforms expressed in all human adult tissues analyzed, with the exception of the brain where TNIK isoforms including exon 22 (TNIKex22) were more abundant (Supplementary Fig. 1). These data suggest a tightly regulated splicing of TNIK gene in the different tissues with the specific expression of TNIKex15 isoforms in the nervous system.
Fig. 1.
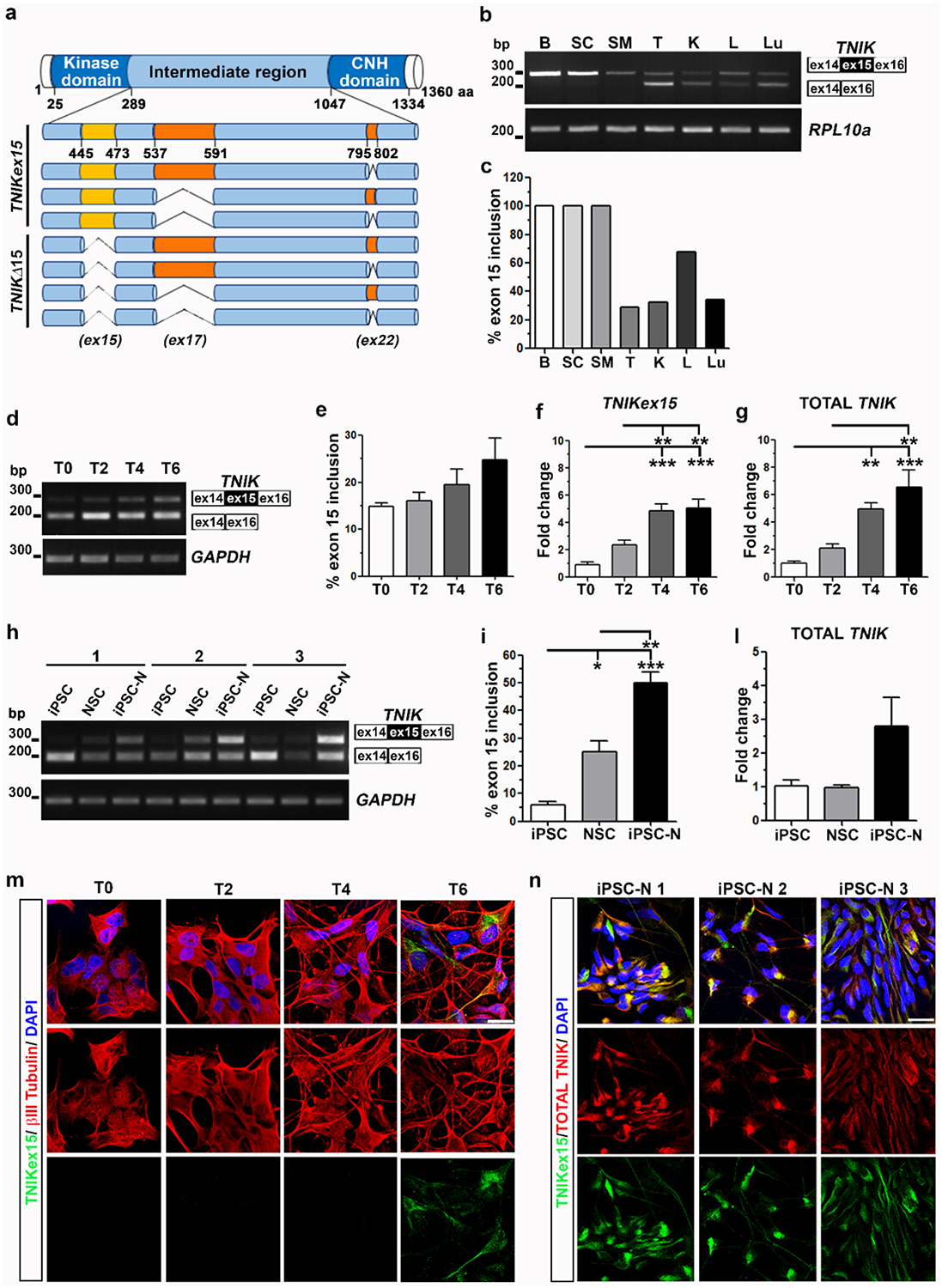
TNIK exon 15 splicing in human adult tissues and during neuronal differentiation in vitro. a) Graphic representation of TNIK protein with all splicing isoforms (CNH: Citron homology domain). b) RT-PCR assay of TNIK exon 15 splicing (Upper band: TNIKex15 isoforms; Lower band: TNIKΔ15 isoforms) in human adult tissues (B: brain; SC: spinal cord; SM: skeletal muscle; T: testis; K: kidney; L: liver; Lu: lung). RPL10a gene was used for sample normalization. c) Densitometric analysis of TNIKex15 bands shown in b). d) Representative RT-PCR assay of TNIK exon 15 splicing during SK-N-BE neuronal differentiation in a 6-day time-frame (T0, T2, T4 and T6). GAPDH was used for data normalization. e) Densitometric analysis of TNIKex15 isoforms shown in d). Real-time PCR assay of f) TNIKex15 isoforms and g) total TNIK gene expression during SK-N-BE neuronal differentiation. (e–g: mean ± s.e.m.; n ≥ 3 independent experiments; one-way ANOVA and Tukey post hoc test; **p < 0.01, ***p < 0.001). h) RT-PCR analysis of TNIK exon 15 splicing during differentiation of 3 human healthy iPSC into neural stem cells (NSC) and into iPSC-derived neurons (iPSC-N). GAPDH was used for data normalization. i) Densitometric analysis of TNIKex15 isoforms during iPSC neuronal differentiation. l) Real-time PCR assay of total TNIK during iPSC neuronal differentiation. (i–l: mean ± s.e.m., one-way ANOVA and Tukey post hoc test; n = 3 is referred to 3 different human healthy iPSC lines; *p < 0.05; **p < 0.01; ***p < 0.001). Confocal images of m) βIII-Tubulin (red) and TNIKex15 (green) immunostaining in differentiated-SK-N-BE cells and of n) TNIKex15 (green) and total TNIK (red) in differentiated iPSC-N. Nuclear staining is indicated in blue (DAPI) in all the merged images. Bar, 20 μm.
Given that TNIKΔ15 isoforms are physiologically prevalent in undifferentiated human neuroblastoma SK-N-BE cells and inducible pluripotent stem cells (iPSC) (Fig. 1d, h), we characterized changes in TNIK exon 15 alternative splicing during their differentiation into neuronal cells. Upon treatment of SK-N-BE cells with retinoic acid for 6 days that induces neurites outgrowth and branching (Supplementary Fig. 2), we observed a progressive, although not statistically significant, 1.7-fold increase of TNIK exon 15 inclusion by RT-PCR analysis (Fig. 1d–e). However, quantification by Real time PCR revealed a significant up-regulation of exon 15 inclusion (Fig. 1f) and of total TNIK mRNA content (Fig. 1g) during neuronal differentiation.
We differentiated three iPSC lines obtained from three healthy individuals into both neural stem cells (NSC) and neuronal cells (iPSC-N) (Supplementary Fig. 3). Characterization of TNIK exon 15 alternative splicing by RT-PCR under these conditions showed that exon 15 inclusion significantly increased from iPSC (5.8%) to NSC (25%) and to iPSC-N (50%) (Fig. 1h–i). An overall up-regulation of total TNIK mRNA content was observed in differentiated iPSC-N although without statistical significance (Fig. 1l).
Immunofluorescence analysis (IF) using our previously described TNIK antibody, raised specifically against the 29 amino acids encoded by exon 15 [12], showed the presence of TNIKex15 protein isoforms only in fully differentiated SK-N-BE and iPSC-N cells, when neuronal outgrowth and branching were evident (Fig. 1m–n), in line with splicing data (Fig. 1d, h). Mild TNIKex15 expression was also observed in NSC (Supplementary Fig. 4), but not in iPSC (data not shown). TNIKex15 protein isoforms localized in the cytoplasm showing a specific distribution at the perinuclear region (Fig. 1m), as already described [12], and along neurites, as confirmed also by a commercial antibody recognizing all TNIK protein isoforms (Fig. 1n).
3.2. The neuron-specific splicing factor NOVA-1 promotes TNIK exon 15 inclusion
As TDP-43 acts by promoting TNIK exon 15 skipping, we hypothesized that TDP-43 protein levels would decrease during neuronal differentiation to account for the observed increase in TNIK exon 15 inclusion. However, we found that TDP-43 protein levels were unchanged at all stages of cell differentiation both in neuroblastoma cells (Fig. 2a–b) and iPSC-N (Fig. 2d–e). Moreover, IF analysis showed that TDP-43 mainly localized in the nucleus under all experimental conditions, thereby excluding a sub-cellular TDP-43 redistribution in the cytoplasm that could lead to a partial loss of its splicing function (data not shown).
Fig. 2.
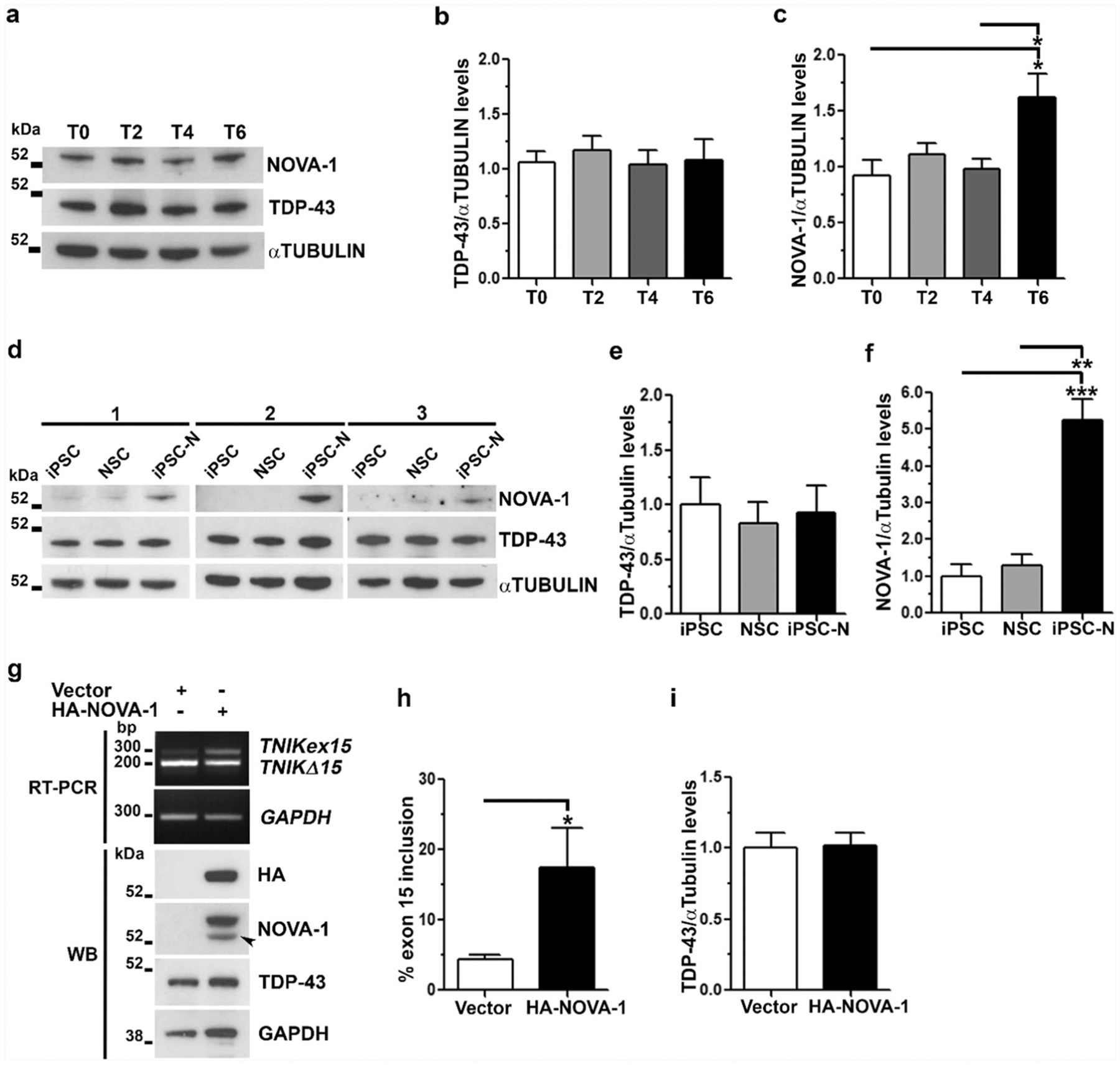
Analysis of NOVA-1 and TDP-43 proteins during neuronal differentiation in vitro. Representative WB images and densitometric analyses of TDP-43 and NOVA-1 protein levels a–c) during SK-N-BE neuronal differentiation (mean ± s.e.m.; n = 4 independent experiments; one-way ANOVA and Tukey post hoc test; *p < 0.05) and d–f) in iPSC, NSC and iPSC-N (mean ± s.e.m.; n = 3 different human healthy iPSC samples; one-way ANOVA and Tukey post hoc test; **p < 0.01, ***p < 0.001). g) RT-PCR of TNIK exon 15 splicing upon HA-NOVA-1 over-expression in HEK293T cells (Upper panels). Representative WB images to assess HA-NOVA-1 transfection efficiency and endogenous NOVA-1 and TDP-43 content (Lower panels). Arrowhead, Recombinant untagged NOVA1 protein due to the presence of the original ATG downstream the HA-tag. GAPDH was used for data normalization both in RT-PCR and WB assays. Densitometric analyses of h) TNIKex15 splicing isoforms and i) TDP-43 protein levels shown in g). (h–i: mean ± s.e.m.; n = 3 independent experiments; one-tailed Unpaired t-test; *p < 0.05).
Based on the hypothesis that other RBPs may regulate TNIK splicing in neuronal cells, we investigated NOVA-1 as a possible candidate because of its specific expression in neurons [4–6]. In contrast to TDP-43, NOVA-1 protein content significantly increased (1.6 fold) in differentiated neuroblastoma cells (Fig. 2a–c) and it was highly expressed only in iPSC-N (Fig. 2d–f). To assess if NOVA-1 regulates TNIK alternative splicing, we over-expressed NOVA-1 in non-neuronal HEK293T cells where this RBP is not physiologically expressed and exon 15 is prevalently skipped (Fig. 2g). Upon NOVA-1 over-expression, TNIK exon 15 inclusion significantly increased to 17.4% if compared to mock-transfected cells (4.4%) (Fig. 2g–h), thus supporting NOVA-1 role in TNIK exon 15 splicing regulation. Moreover, NOVA-1 over-expression in HEK293T cells did not change TDP-43 protein levels (Fig. 2i), thereby excluding that the increase of exon 15 inclusion was caused by decreased TDP-43 protein levels.
3.3. TDP-43 and NOVA-1 competitively regulate TNIK exon 15 splicing
To further investigate NOVA-1 role in regulating TNIK exon 15 splicing and its interplay with TDP-43, we performed a minigene splicing assay that we previously used to demonstrate TDP-43-mediated TNIK splicing [12] (Supplementary Fig. 5a). In our assays we also included hnRNPA2/B1, a ubiquitous RBP that is known to interact and to co-operate with TDP-43 in regulating its splicing activity [28]. The minigene construct and the three splicing factors were over-expressed in HEK293T cells and TNIK-minigene exon 15 inclusion/skipping was evaluated by RT-PCR assay (Fig. 3a). When TDP-43 was over-expressed, TNIK exon 15 inclusion significantly decreased to 53.5% compared to mock-transfected cells (88.2%) (Fig. 3b). Similar results were obtained upon transfection of the hnRNPA2/B1 construct showing a reduction to 68.8% of exon 15 inclusion, but not upon NOVA-1 over-expression (92.2%) (Fig. 3a–b). In competition experiments, by co-expression of NOVA-1 with TDP-43 or hnRNPA2/B1, we observed that NOVA-1 completely abolished both TDP-43 and hnRNPA2/B1 skipping activity on exon 15 (90.8% and 92.8% exon inclusion, respectively) (Fig. 3b). The co-expression of TDP-43 and hnRNPA2/B1 synergistically decreased exon 15 inclusion to 32.2%, while the presence of NOVA-1 was able to completely inhibit TDP-43/hnRNPA2/B1 splicing activity by restoring TNIK exon 15 inclusion level (93.5%) similar to control cells (Fig. 3a–b). Overall, these data suggest a competitive action between NOVA-1 and TDP-43/hnRNPA2/B1 in regulating splicing of TNIK alternative exon 15.
Fig. 3.
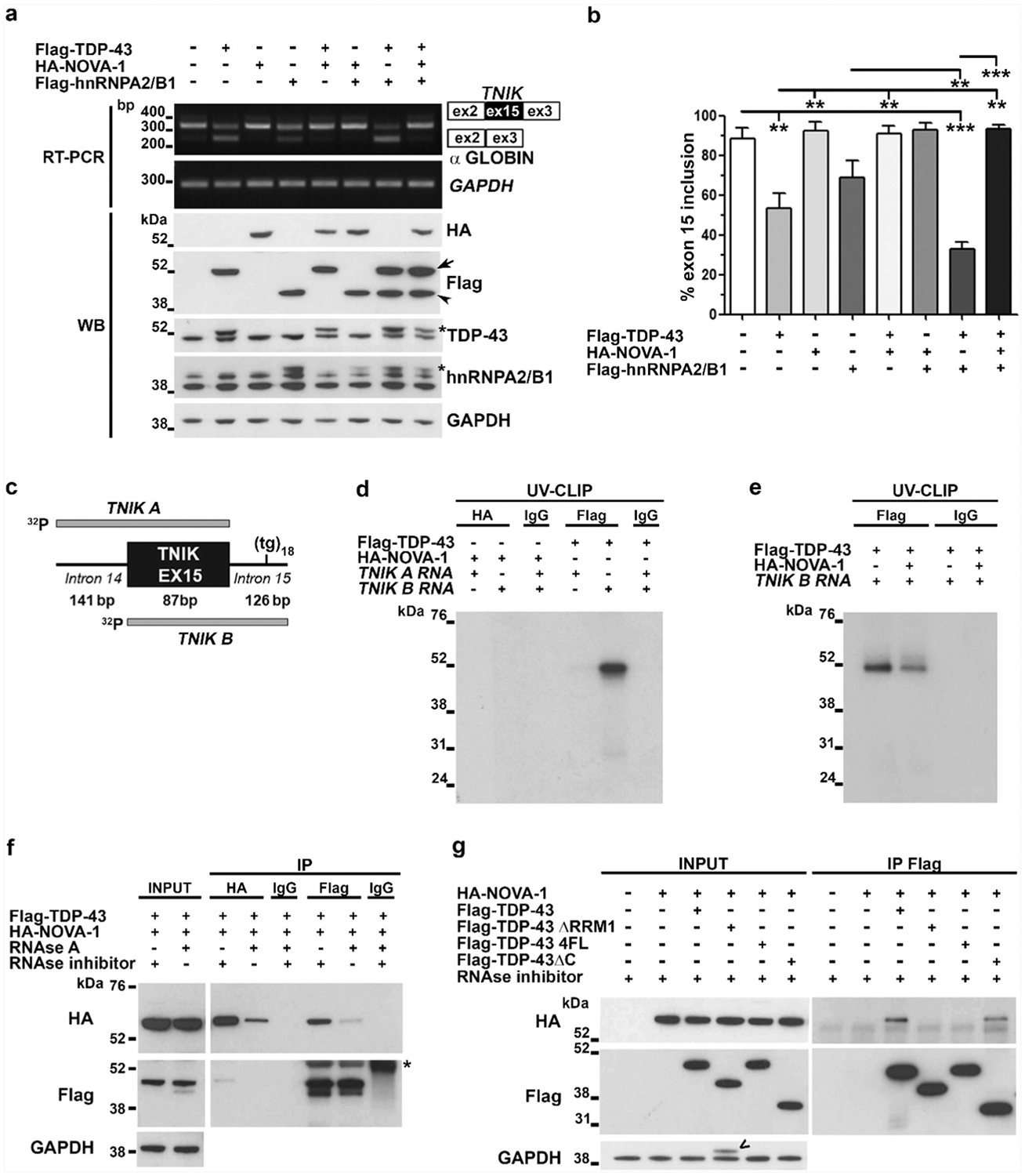
NOVA-1 and TDP-43 interplay in regulating TNIK exon 15 alternative splicing. a) Representative RT-PCR (Upper panels) and WB (Lower panels) images of the minigene splicing assay in HEK293T cells co-transfected with pTBminigene_TNIKex15 and Flag-TDP-43, Flag-hnRNPA2/B1 or HA-NOVA-1 as indicated. GAPDH was used for data normalization in both RT-PCR and WB assays. Arrow, Flag-TDP-43; arrowhead, Flag-hnRNPA2B1; asterisk, recombinant Flag-tagged TDP-43 or hnRNPA2/B1 proteins. b) Densitometric and statistical analyses of TNIK exon 15 inclusion data from the minigene assay (mean ± s.e.m.; one-way ANOVA and Tukey post hoc test; n = 3 independent experiments; **p < 0.01; ***p < 0.001). c) Schematic representation of 32P-labeled TNIK A and TNIK B riboprobes used for UV-CLIP assay. d) SDS-PAGE of UV-CLIP experiment using anti-HA or anti-Flag antibody and radiolabeled TNIK A or TNIK B riboprobes. The irrelevant IgG antibody was used as a negative control. e) Competitive UV-CLIP assay using TNIK B riboprobe and the anti-Flag or anti-IgG antibodies. f) Representative WB image of co-immunoprecipitation (co-IP) assay with HEK293T cell lysates transfected with HA-NOVA-1 and Flag-TDP43 constructs, treated with RNAse A or RNAse inhibitor and immunoprecipitated with the indicated antibodies (n = 3 independent experiments); asterisk indicates IgG heavy chain. g) Representative WB image of co-IP using anti-Flag antibody on HEK293T lysates transfected with HA-NOVA-1 and the Flag-tagged TDP-43 full length, ΔRRM1, 4FL and ΔC plasmids as indicated(n = 3 independent experiments). <, residual signal from the previous anti-Flag hybridization.
To further characterize the interplay of TDP-43 and NOVA-1 in oppositely regulating TNIK splicing, we tested their binding to TNIK pre-mRNA by UV-crosslinking immunoprecipitation (UV-CLIP) assay. Protein lysates from NOVA-1- or TDP-43-transfected HEK293T cells and two radiolabelled riboprobes, TNIK A and TNIK B, encompassing exon 15 and, respectively, the upstream and downstream TNIK intronic regions included in the TNIK minigene construct (Fig. 3c), were used in the assay. We previously showed that TNIK intron 15 contains the TDP-43 consensus binding site with an uninterrupted UG18 motif and that TDP-43 binds the TNIK pre-mRNA by RNA immunoprecipitation assay [12]. By UV-CLIP assay we confirmed that TDP-43 binds the UG18 motif present in the TNIK probe B, but we failed to detect NOVA-1 binding to either probes (Fig. 3d), although the recombinant NOVA-1 protein was successfully recovered in the IP assay (Supplementary Fig. 5b). However, when NOVA1 was over-expressed together with TDP-43 in a competitive binding assay, association of TDP-43 to TNIK RNA probe B was reduced (Fig. 3e and Supplementary Fig. 5c). The in silico analysis with the RBPmap software (http://rbpmap.technion.ac.il [29]) predicted no consensus binding motifs (YCAY clusters) for NOVA-1 within the selected TNIK sequences A and B (data not shown), supporting our negative UV-CLIP data. We therefore tested whether NOVA-1 acts indirectly on TNIK pre-mRNA splicing by binding to TDP-43 protein. By conducting co-IP experiments, NOVA-1 and TDP-43 were shown to interact only if RNA was preserved, while their interaction was abolished after RNAse A treatment (Fig. 3f).
Furthermore, co-IP assays using two TDP-43 deletion constructs, TDP-43 ΔC and TDP-43 ΔRRM1 (missing the protein-protein interaction C-terminal domain or the entire RNA-binding domain RRM1, respectively), and the RNA-binding incompetent TDP-43 4FL construct (four specific phenylalanines mutated to lysines [24]) (Supplementary Fig. 6a) confirmed NOVA-1 binding to TDP-43 only if TDP-43 ability to recognize RNA was preserved. Indeed, NOVA-1 failed to bind both TDP-43 ΔRRM1 and TDP-43 4FL mutant proteins, while its binding to TDP-43 ΔC was unaffected (Fig. 3g). As our TNIK minigene splicing assay showed that NOVA-1 counteracted also hnRNPA2/B1 exon skipping activity on TNIK exon 15 (Fig. 3a–b), we tested if NOVA-1 interacts also with this RBP and we found that, similarly to TDP-43, NOVA-1 binds hnRNPA2/B1 in a RNA-dependent manner (Supplementary Fig. 6b). IP assays using anti-tag (HA) antibody, although successful in recovering the recombinant HA-NOVA-1, failed to isolate NOVA-1-containing protein complexes including TDP-43 and hnRNPA2B1 (Fig. 3f; Supplementary Fig. 6b), probably because the N-terminal HA-tag may be masked in these complexes. Indeed, when we used a C-terminal anti-NOVA1 antibody, we were able to recover the NOVA1-TDP-43 complexes in a RNA-dependent manner (Supplementary Fig. 6c).
3.4. TDP-43-mediated splicing of POLDIP3 and STAG2 genes is also inhibited by NOVA1
We evaluated whether the competitive mechanism between TDP-43 and NOVA1 could occur also on the endogenous TNIK pre-mRNA, independently on the selected intronic sequences used in the minigene and UV-CLIP assays. Upon over-expression of TDP-43 and NOVA1 alone or in combination in HEK293 cells, we observed that NOVA1 was able to promote TNIK exon 15 inclusion and that TDP-43 was not able to counteract NOVA1 splicing activity (Fig. 4a, b and Supplementary Fig. 7).
Fig. 4.
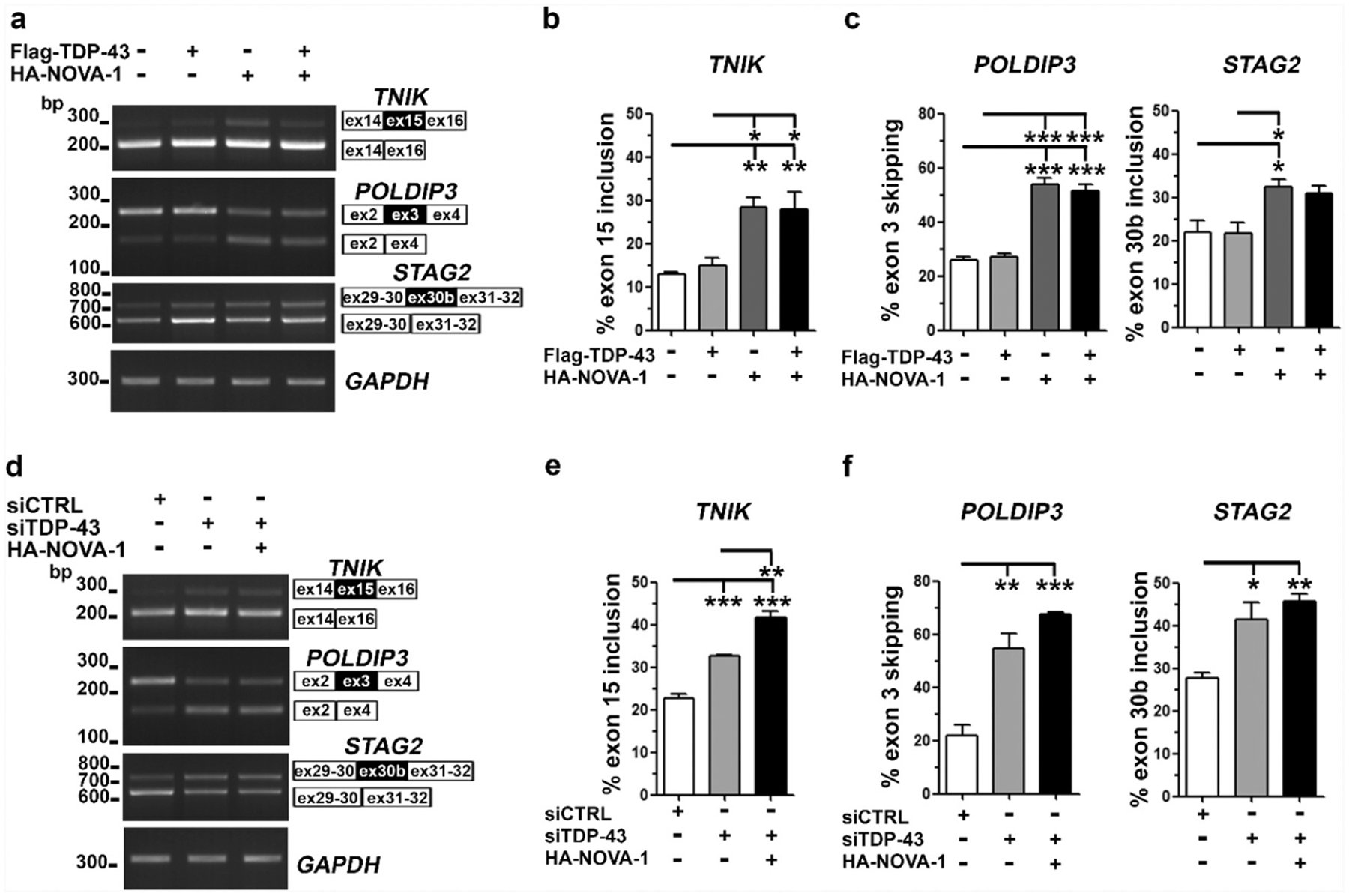
NOVA-1 competitively regulates with TDP-43 its RNA targets TNIK, POLDIP3 and STAG2 a) Representative RT-PCR of endogenous TNIK, POLDIP3 and STAG2 splicing in HEK293T cells transfected with Flag-TDP-43 and HA-NOVA-1 as indicated. GAPDH was used for data normalization. b–c) Densitometric and statistical analyses of TNIK exon 15 inclusion, POLDIP3 exon 3 skipping and STAG2 exon 30b inclusion data (mean ± s.e.m.; one-way ANOVA and Tukey post hoc test; n ≥ 3 independent experiments; *p < 0,05; **p < 0,01; ***p < 0,001). d) RT-PCR analysis of endogenous TNIK, POLDIP3 and STAG2 splicing in HEK293T upon TDP-43 knock-down and HA-NOVA-1 over-expression as indicated. GAPDH was used for data normalization. e–f) Densitometric and statistical analyses of TNIK, POLDIP3 and STAG2 alternative splicing data (mean ± s.e.m.; one-way ANOVA and Tukey post hoc test; n = 3 independent experiments; *p < 0,05; **p < 0,01; ***p < 0,001).
To test if NOVA1, by interacting directly with TDP-43 protein (Fig. 3f, g), can regulate the alternative splicing of TDP-43 RNA targets other than TNIK, we extended our splicing assays on POLDIP3 and STAG2 genes (Fig. 4a). Upon NOVA-1 over-expression, we found that the skipping of POLDIP3 exon 3 and the inclusion of STAG2 exon 30b (Fig. 4c) were significantly increased compared to mock-transfected cells, in contrast to the reported TDP-43 splicing activity on these targets [30,31]. When TDP-43 was co-expressed with NOVA1, the splicing pattern observed upon NOVA-1 over-expression was maintained, suggesting that NOVA1 inhibits TDP-43 splicing activity also on these targets (Fig. 4a, c).
Based on the hypothesis that NOVA1, by binding to TDP-43, inhibits its splicing activity, mimicking a loss of TDP-43 function, we knocked-down TDP-43 in HEK293T cells (Supplementary Fig. 7) and confirmed previous splicing data on all the three considered RNA targets (Fig. 4d) [12,30,31].TDP-43 knock-down, in fact, promoted the inclusion of TNIK exon 15 and STAG2 exon 30b and the skipping of POLDIP3 exon 3, as expected (Fig. 4e, f), and similarly to NOVA-1 over-expression (Fig. 4a–c). Moreover, upon TDP-43 gene-silencing, the over-expression of NOVA1 further increased the splicing events associated to a loss of TDP-43 function (Fig. 4d–f), suggesting that NOVA1 is able to further exert its inhibitory activity also on the residual TDP-43 protein (Supplementary Fig. 7).
3.5. TNIKex15 protein isoforms inhibit cell spreading and neurite outgrowth
TNIK over-expression has been previously described to inhibit cell spreading and affect cell morphology by promoting filamentous (F)-actin disassembly [21,32].
To evaluate if the short 29-aminoacid region encoded by the alternative exon 15 is important to regulate cytoskeleton organization, we over-expressed the human full-length TNIK (TNIKex15), the exon 15-deleted TNIK (TNIKΔ15) and the mutant TNIK (TNIK KM) constructs and analyzed their effect on HEK293T cell morphology. TNIK KM has a kinase-defective activity and was used as a negative control [21]. Our analysis showed that the over-expression of TNIKex15 significantly increased cell circularity compared to mock- and TNIK KM-transfected cells (Fig. 5a–b), as previously shown [21]. On the other hand, TNIKΔ15-expressing cells showed an intermediate phenotype between TNIKex15 and TNIK KM (Fig. 5b).
Fig. 5.
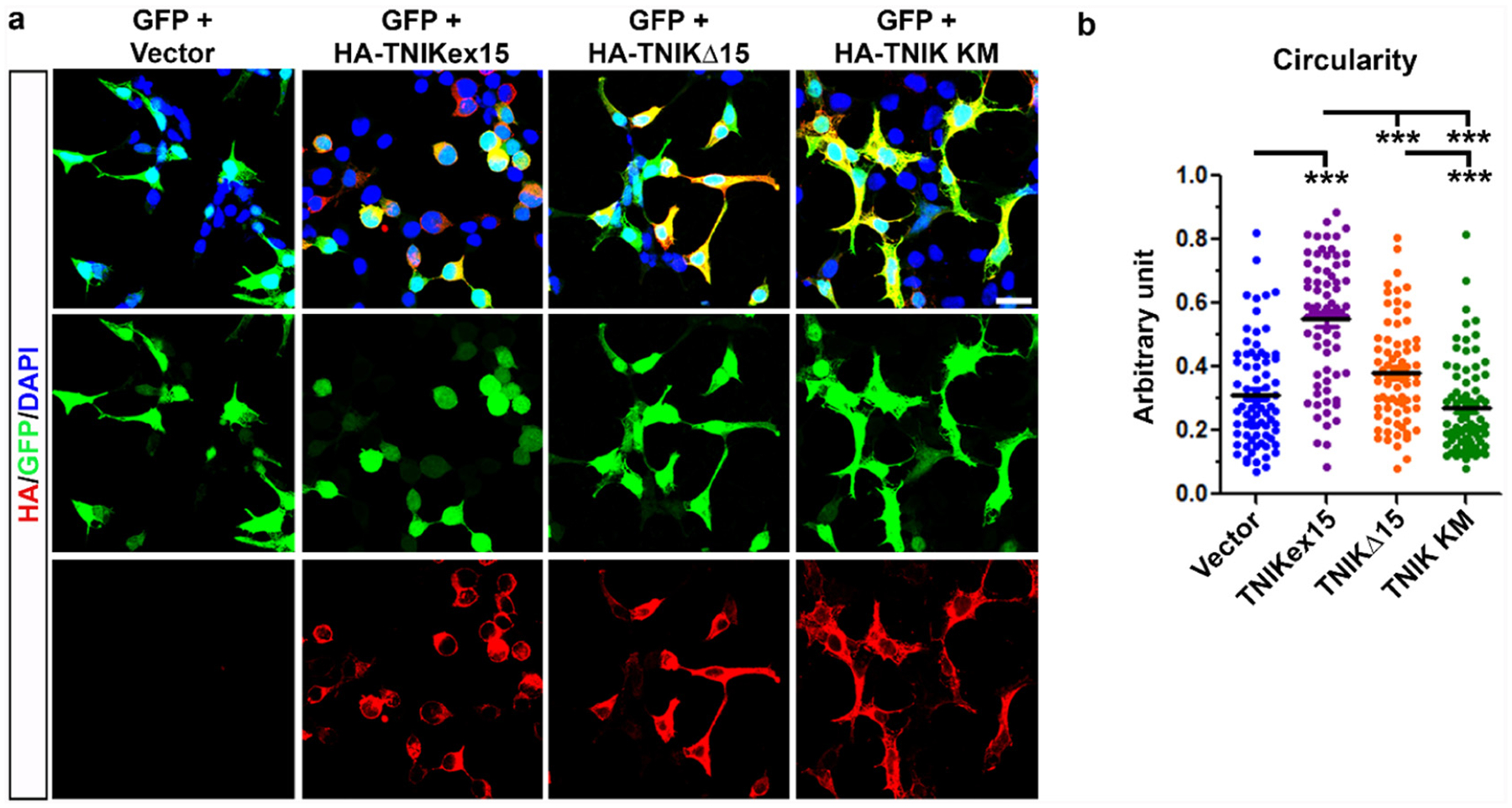
Effect of TNIK isoforms on cell spreading in HEK293T cells. a) Representative confocal images of HEK293T cells transfected with human TNIK constructs (HA-TNIKex15, HA-TNIKΔ15 and HA-TNIK KM) (red) or empty vector. GFP was co-transfected to visualize cell morphology and the nuclear staining (DAPI) is showed in blue in the merged images. Bar, 20 μM. b) Cell circularity analysis performed by ImageJ NIH software (mean ± s.e.m.; n = 3 independent experiments; 75 cells per condition were analyzed; one-way ANOVA Kruskal-Wallis, Dunn’s post hoc test; ***p < 0.001).
As our results showed a specific expression of TNIKex15 transcripts in the human nervous system and of TNIKex15 protein isoforms during neuronal differentiation in vitro (Fig. 1), we hypothesized that the amino acid region encoded by this alternative exon may be important for TNIK activity in neurons. As TNIKex15 over-expression was already described to negatively affect neurite development in primary rat neurons [20], we also investigated the effect of human TNIKΔ15 overexpression in primary murine cortical neurons (Fig. 6a). By Sholl analysis, we observed a decreased number of crossing neurites in TNIKex15-expressing neurons compared to mock-transfected neurons at each considered interval (Fig. 6b). TNIKΔ15-transfected cortical neurons showed a behavior similar to TNIK KM- and mock-transfected control cells (Fig. 6a–b). When we specifically considered the proximal neurites at 15 μm from the cell body, a significant decrease in crossing neurites number was observed in TNIKex15-transfected cells (8,4 ± 0,4) compared to both control (10,2 ± 0,4) and TNIK KM-expressing (10,7 ± 0,4) neurons (Fig. 6c). TNIKΔ15-tranfected neurons showed an intermediate phenotype (9,9 ± 0,4) with no significant differences with either control, TNIK KM and TNIKex15 cells (Fig. 6c).
Fig. 6.
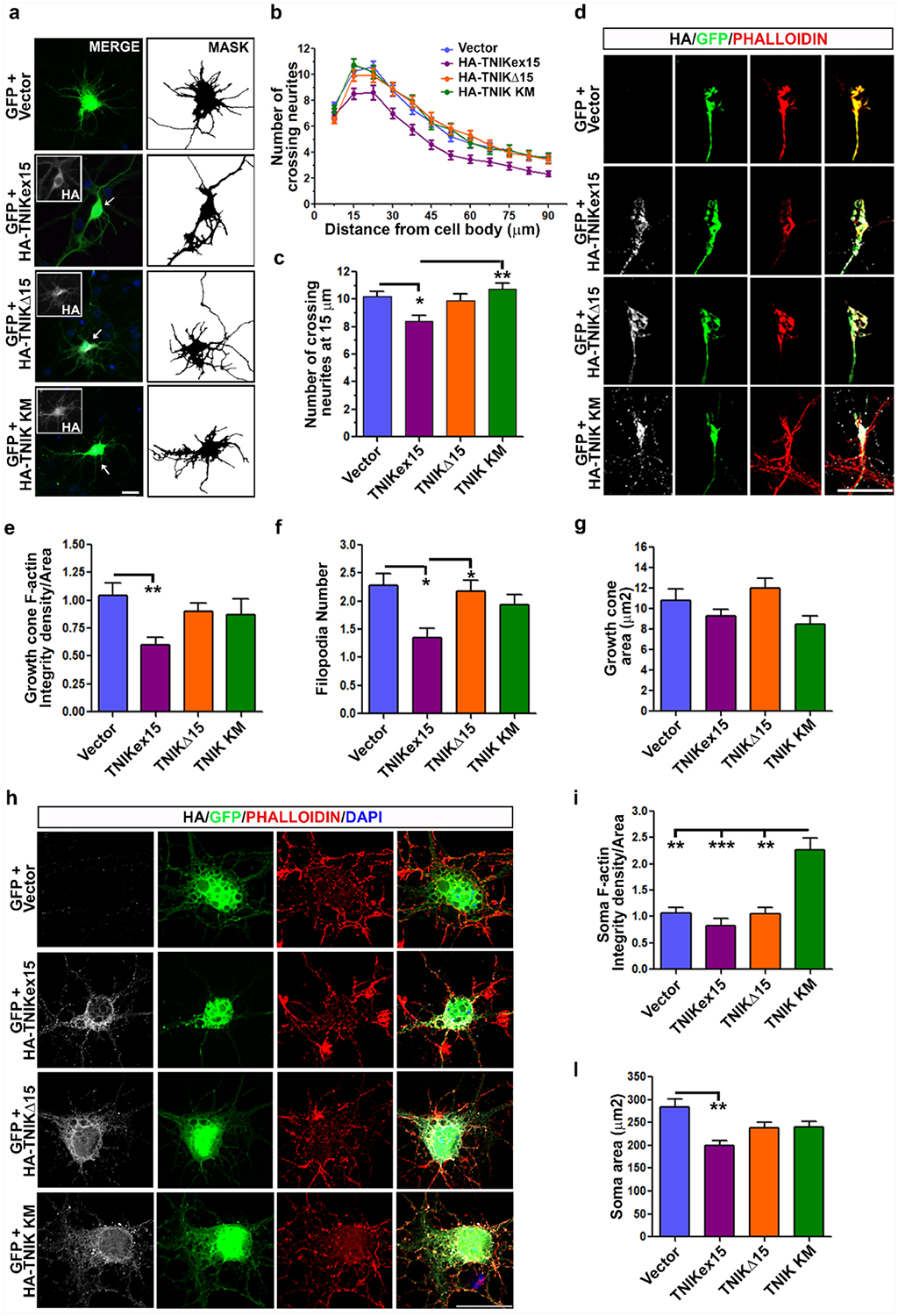
Effect of TNIK protein isoforms on neurite development and F-actin levels in primary murine cortical neurons. a) Representative IF images of DIV7 cortical neurons transfected with human TNIK constructs (HA-TNIKex15, HA-TNIKΔ15 and HA-TNIK KM) (inset, grey) or empty vector. GFP construct was co-transfected to visualize cell morphology and is showed in the merged images. Nuclei are stained with DAPI (blue). The binary images (MASK) of the GFP signal is presented to better visualize neurites. Bar, 20 μM. b) Sholl analysis of mock- (n = 72), TNIKex15- (n = 59), TNIKΔ15- (n = 72), and TNIK KM-transfected neurons (n = 59) (mean ± s.e.m.; n is referred to the number of cells analyzed for each condition from 4 independent experiments). c) Analysis of crossing neurites number at 15 μm distance from cell body in all considered conditions (mean ± s.e.m.; n = 4 independent experiments; one-way ANOVA Kruskal-Wallis, Dunn’s post hoc test; *p < 0.05, **p < 0.01). d) Representative IF images of growth cones of cortical neurons transfected as indicated. Phalloidin (red) was used to stain and quantify filamentous actin (F-actin). Bar, 10 μm. Quantification of e) F-actin levels and f) filopodia number and g) area at growth cones of TNIKex15- (n = 45), TNIKΔ15- (n = 65), TNIK KM-expressing neurons (n = 38) or control cells (n = 65) (mean ± s.e.m.; n is the cell number analyzed for each condition from 4 independent experiments; one-way ANOVA Kruskal-Wallis, Dunn’s post hoc test; *p < 0.05; **p < 0.01). h) Representative IF images of somas of cortical neurons transfected as indicated and stained for phalloidin. Bar, 20 μm. Quantification of soma F-actin levels i) and area l) in TNIKex15- (n = 35), TNIKΔ15- (n = 46), TNIK KM- (n = 42) and empty vector-transfected (n = 46) neurons (mean ± s.e.m.; n is the cell number analyzed for each condition from 4 independent experiments; one-way ANOVA Kruskal-Wallis, Dunn’s post hoc test; **p < 0.01; ***p < 0.001). HA (grey) antibody was used to visualize transfected cells and nuclear staining (DAPI, blue) is shown in the merged images.
As neurite development is tightly dependent on F-actin dynamics and TNIK kinase activity negatively affects actin polymerization [21,32], we quantified F-actin levels at the growth cone and in the soma of transfected cortical neurons. TNIKex15 over-expression significantly reduced F-actin levels in growth cones compared to control cells (Fig. 6d–e), in association with a significantly lower number of filopodia (Fig. 6f). In contrast, upon TNIKΔ15 or TNIK KM over-expression, F-actin levels and filopodia number were unchanged compared to control cells (Fig. 6d–f). The size of the growth cone was similar in all analyzed conditions (Fig. 6g). When we analyzed the soma of transfected neurons, only TNIK KM-expressing neurons showed a significant increase of F-actin levels, although their soma area showed similar values to control and TNIKΔ15-transfected cells (Fig. 6h–l). TNIKex15-positive cells had a significant smaller soma area, but no changes in soma F-actin levels (Fig. 6i–l), indicating an altered cell morphology.
No differences were observed in the sub-cellular distribution of the transfected TNIK constructs in primary cortical neurons, as all the recombinant TNIK proteins localized in the cytoplasm and were particularly enriched in the perinuclear region (Fig. 6a, h) and along neurites (Fig. 6a, d), resembling the distribution of endogenous TNIK protein in human differentiated iPSC-N (Fig. 1n).
4. Discussion
In this study we showed that during neuronal differentiation the alternative splicing of TNIK gene is oppositely regulated by the ubiquitous TDP-43 RBP, which promotes exon 15 skipping, and by the neuron-specific NOVA-1 splicing factor, which instead favours its inclusion. High-throughput studies have widely defined TDP-43-regulated transcriptome in different experimental models in the attempt to predict the TDP-43-dependent loss-of-function changes in ALS/FTD diseases [10]. However, further functional work is required to assess the biological effects of the identified gene expression/splicing changes, particularly in a neuronal context.
The Ser/Thr kinase TNIK is highly expressed in the brain, playing important roles in regulating dendrite organization, synaptic structure and functionality [17–20]. We previously showed that TDP-43 knock-down in human neuroblastoma cells is associated to a reduced skipping activity on TNIK alternative exon 15 with a consequent up-regulation of exon 15 inclusion and also of the corresponding TNIKex15 protein isoforms [12]. TNIK gene has three alternative exons but no gene expression and functional data of these different isoforms are available in the literature [21]. We found that TNIK exon 15 inclusion is the main splicing event observed in the brain and spinal cord, in contrast to the other two alternative exons 17 and 22, suggesting a specific biological role of TNIKex15 isoforms in the nervous system. By studying two neural differentiation paradigms in vitro, we found that TNIK exon 15 inclusion increases during neuronal differentiation with no change in TDP-43 protein content or sub-cellular localization, suggesting that other RBPs regulate this splicing event in a neuron-specific manner. Thus, we focused on NOVA-1 as this is one of the main splicing factors specifically expressed in neurons [33]. We found that, in non-neuronal cells, NOVA-1 ectopic over-expression promoted TNIK exon 15 inclusion with no changes in TDP-43 protein level, thereby excluding an indirect effect of NOVA-1 via the regulation of TDP-43 expression. Moreover, in differentiated neuroblastoma cells and iPSC-derived neurons, NOVA-1 protein levels correlated positively with the increase of TNIKex15 transcripts and protein isoforms.
NOVA-1 has previously been described to regulate the alternative splicing of specific genes, such as Dopamine D2 receptor [34] and NMDA receptor 1 [2], through competitive mechanisms with other ubiquitous RBPs. Similarly, TDP-43 synergistically cooperates or counteracts the activity of other ubiquitous splicing factors in processing Apo-II, Sortilin and SMN2 pre-mRNAs [35–37]. In particular, the splicing activity of TDP-43 on TNIK transcript has been recently demonstrated to be further modulated by hnRNPR, which similarly favours exon 15 skipping, and by hnRNPQ and DAZAP1, which instead promote exon 15 inclusion [38]. Our findings show that NOVA-1 is able to counteract TDP-43 skipping activity on TNIK exon 15, describing for the first time the interplay of TDP-43 with a neuron-specific splicing factor. NOVA-1 was also able to inhibit the splicing activity of hnRNPA2/B1, a well-known TDP-43 interactor, and to completely abolish the synergistic effect of hnRNPA2/B1 and TDP-43 skipping activities on TNIK pre-mRNA, proposing a model for the neuron-specific splicing pattern of TNIK exon15.
Based on the hypothesis of a competitive mechanism between TDP-43 and NOVA-1, unexpectedly we were not able to detect NOVA-1 direct binding to TNIK exon 15 and its flanking introns, while we proved TDP-43 direct binding to the consensus UG18 motif within intron 15 that was reduced when NOVA-1 was over-expressed. We instead showed that TDP-43 interacts with NOVA-1 in a RNA-dependent manner, as already described for the interaction with other splicing factors [39,40], without requiring its C-terminal domain, usually important for protein-protein interactions, as in the case of hnRNPA2/B1 [28]. We demonstrated a similar RNA-dependent binding of NOVA-1 also to hnRNPA2/B1, suggesting that NOVA-1 might promote TNIK exon 15 inclusion by interacting and competing with both TDP-43 and hnRNPA2/B1 RBPs. Based on our experimental results, we therefore propose a working model whereby, in a neuronal environment, the specific expression of NOVA-1 splicing factor may drive the formation of ribonucleoprotein complex(es) that, by sequestering the ubiquitous TDP-43 and hnRNPA2/B1 RBPs, promote TNIK exon 15 inclusion. Moreover, our results showed that also other TDP-43 RNA targets, such as POLDIP3 and STAG2, are competitively regulated by NOVA1 in a similar manner to TNIK, suggesting that the competitive mechanism between NOVA-1 and TDP-43 depends on their protein-protein interaction rather than on the cis-acting regulatory elements in the target pre-mRNAs However, further studies are needed to better define the molecular mechanisms, including the involvement of other splicing factors and of possible TDP-43 post-translational modifications [41], that may modulate TDP-43 activity in the brain and during neuronal development.
TNIK exon 15 encodes for a 29 amino acidic sequence mapping within the long intermediate protein region which was shown to mediate the interaction of TNIK with Traf2, NCK and Nedd4–1 proteins [20,21]. Using our specific antibody recognizing this short amino acid region [12], we demonstrated the expression of TNIKex15 protein isoforms specifically in differentiated neuronal cells in line with splicing data, suggesting a functional relevance of this protein region in mature neurons. Previous studies already reported that the TNIK full-length protein is necessary for dendrite organization in primary rodent neurons and both TNIK silencing or over-expression were described to affect negatively dendrite development [20]. By studying over-expression models, we showed that in primary murine cortical neurons TNIKΔ15 did not affect neurite outgrowth in contrast to the full-length protein, suggesting that the 29 amino acid region encoded by exon 15 is necessary for TNIK modulatory function on F-actin levels and filopodia number at growth cones. As the over-expression of TNIKΔ15 and of the mutant kinase TNIK KM had similar effects on neurite arborisation, we speculate that TNIK exon 15-encoded sequence might positively modulate TNIK kinase activity, thereby promoting F-actin depolymerization through the Rap2A pathway [20,32]. This in turn is important for both neurite development in neuronal cells and cell spreading in non-neuronal cells. However, given the multiple roles of TNIK in neuronal metabolism and the array of its known protein interactors, it would be interesting to study if the TNIKΔ15 isoform acts differently from the full-length protein also in other biological pathways. Interestingly, exon 15 amino acidic sequence is highly conserved phylogenetically and we validated the annotated TNIK splicing isoforms containing/excluding exon 15 in Mus musculus and Danio rerio (data not shown), suggesting specific biological functions of these TNIK protein isoforms also in these animal models that would warrant further investigation.
Genetic data support TNIK gene as a risk factor for schizophrenia, although the associated SNPs have not been functionally investigated yet [13–15]. As they map in the intergenic region upstream TNIK gene and in introns 2 and 3 [13–15], they are not likely to regulate exon 15 splicing. However, splicing dysregulation is a common pathogenic mechanism in psychiatric diseases and both RNA-seq and recent transcriptome-wide association studies in schizophrenia post-mortem brains have identified several splicing alterations in genes involved in brain development [42–45]. Although TNIK splicing changes have not been identified by these highthroughput studies, a gene-targeted approach would warrant further investigation based also on the evidence that TNIK gene expression is up-regulated in schizophrenia brains [46]. Moreover, since Tnik knock-out mice show cognitive dysfunctions similar to schizophrenia patients [19], a tight control of TNIK gene expression, and likely of its transcript isoforms, is supposed to be fundamental for normal brain functions.
5. Conclusions
In this study we describe for the first time the interplay between TDP-43 and the neuron specific splicing factor NOVA-1, highlighting the importance to consider the environmental context when studying the pathomechanisms of ubiquitous RBPs associated to neurodegenerative disorders, as in the case of TDP-43. Our results also show that a fine control of TNIK alternative splicing is important for neuronal cell differentiation since the unbalanced synthesis of functionally different protein isoforms may contribute to dysregulated neuronal activities and likely account for part of the cognitive deficits observed in schizophrenia patients. Therefore, the dysregulation of the finely tuned network of ubiquitous and neuron-specific RBPs, cooperating or competing for target RNA splicing, may play a role in many complex neurological diseases, including schizophrenia and ALS/FTD.
Supplementary Material
Acknowledgements
We thank Dr. Emanuele Buratti for critical discussion of data, Dr. Francesca Sassone for her help with iPSC cultures and Dr. Anthony Giampetruzzi for primary cortical neuron cultures.
Funding: Financial support was received by Italian Ministry of Health for Scientific Research in IRCCS (23C623). VG was supported by a fellowship of the Doctorate School of Molecular Medicine, Università degli Studi di Milano.
Abbreviations:
- RBP
RNA-binding proteins
- ALS
amyotrophic lateral sclerosis
- FTD
frontotemporal dementia
- GCK
germinal center kinase
- CNH
citron homology
- iPSC
induced pluripotent stem cell
- IPSC-N
iPSC-derived neurons
- NSC
neural stem cells
- EB
embryoid bodies
- UV-CLIP
UV Cross-linking and immunoprecipitation
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Transparency document
The Transparency document associated with this article can be found, in online version.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbagrm.2019.194413.
References
- [1].Zheng S, Black DL, Alternative pre-mRNA splicing in neurons: growing up and extending its reach, Trends Genet. 29 (8) (2013) 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ule J, Darnell RB, RNA binding proteins and the regulation of neuronal synaptic plasticity, Curr. Opin. Neurobiol 16 (1) (2006) 102–110. [DOI] [PubMed] [Google Scholar]
- [3].Weyn-vanhentenryck SM, Feng H, Ustianenko D, Duf R, Yan Q, Jacko M, Martinez JC, Goodwin M, Zhang X, Hengst U, Lomvardas S, Swanson MS, Zhang C, Precise temporal regulation of alternative splicing during neural development, Nat. Commun 9 (1) (2018) 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ule J, Jensen KB, Ruggiu M, Mele A, Darnell RB, CLIP identifies Nova-regulated RNA networks in the brain, Science vol. 302, (80-.) (2003) 1212–1215 no. 5648. [DOI] [PubMed] [Google Scholar]
- [5].Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, Zeeberg BR, Kane D, Weinstein JN, Blume J, Darnell RB, Nova regulates brain-specific splicing to shape the synapse, Nat. Genet 37 (8) (2005) 844–852. [DOI] [PubMed] [Google Scholar]
- [6].Yang YYL, Yin GL, Darnell RB, The neuronal RNA-binding protein Nova-2 is implicated as the autoantigen targeted in POMA patients with dementia, Proc. Natl. Acad. Sci 95 (22) (1998) 13254–13259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY, Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis, Science vol. 314, (80-.) (2006) 130–133 no. 5796. [DOI] [PubMed] [Google Scholar]
- [8].Arai T, Hasegawa M, Akiyama H, Ikeda K, Hashizume Y, Oda T, TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis, Biochem. Biophys. Res. Commun 351 (2006) 602–611. [DOI] [PubMed] [Google Scholar]
- [9].Ratti A, Buratti E, Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins, J. Neurochem 138 (2016) 95–111. [DOI] [PubMed] [Google Scholar]
- [10].Buratti E, Romano M, Baralle FE, TDP-43 high throughput screening analyses in neurodegeneration: advantages and pitfalls, Mol. Cell. Neurosci 56 (2013) 465–474. [DOI] [PubMed] [Google Scholar]
- [11].Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J, Hortobágyi T, Nishimura AL, Župunski V, Patani R, Chandran S, Rot G, Zupan B, Shaw CE, Ule J, Characterizing the RNA targets and position- dependent splicing regulation by TDP-43, Nat. Publ. Gr 14 (4) (2011) 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Colombrita C, Onesto E, Buratti E, de la Grange P, Gumina V, Baralle FE, Silani V, Ratti A, From transcriptomic to protein level changes in TDP-43 and FUS loss-of-function cell models, Biochim. Biophys. Acta Gene Regul. Mech 1849 (12) (2015) 1398–1410. [DOI] [PubMed] [Google Scholar]
- [13].Potkin SG, Turner JA, Guffanti G, Lakatos A, Fallon JH, Nguyen DD, Mathalon D, Ford J, Lauriello J, Macciardi F, A genome-wide association study of schizophrenia using brain activation as a quantitative phenotype, Schizophr. Bull 35 (1) (2009) 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Péer I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Crowe RR, Oksenberg JR, Mirel DB, Kendler KS, Freedman R, Gejman PV, Common variants on chromosome 6p22.1 are associated with schizophrenia, Nature 460 (7256) (2009) 753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ayalew M, Levey DF, Jain N, Changala B, Patel SD, Winiger E, Breier A, Shekhar A, Amdur R, Koller D, Nurnberger JI, Corvin A, Geyer M, Tsuang MT, Salomon D, Schork NJ, Fanous AH, Donovan MCO, Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction, Mol. Psychiatry 17 (9) (2012) 887–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Anazi S, Shamseldin HE, Alnaqeb D, Abouelhoda M, A null mutation in TNIK defines a novel locus for intellectual disability, Hum. Genet 135 (7) (2016) 773–778. [DOI] [PubMed] [Google Scholar]
- [17].Hussain NK, Hsin H, Huganir RL, Sheng M, MINK and TNIK differentially act on Rap2-mediated signal transduction to regulate neuronal structure and AMPA receptor function, J. Neurosci 30 (44) (2010) 14786–14794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang Q, Charych EI, Pulito VL, Lee JB, Graziane NM, Crozier RA, Revilla-Sanchez R, Kelly MP, Dunlop AJ, Murdoch H, Taylor N, Xie Y, Pausch M, Hayashi-Takagi A, Ishizuka K, Seshadri S, Bates B, Kariya K, Sawa A, Weinberg RJ, Moss SJ, Houslay MD, Yan Z, Brandon NJ, The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function, Mol. Psychiatry 16 (10) (2011) 1006–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Coba MP, Komiyama NH, Nithianantharajah J, Kopanitsa MV, Indersmitten T, Skene NG, Tuck EJ, Fricker DG, Elsegood KA, Stanford LE, Afinowi NO, Saksida LM, Bussey TJ, O’Dell TJ, Grant SGN, TNIK is required for post-synaptic and nuclear signaling pathways and cognitive function, J. Neurosci 32 (40) (2012) 13987–13999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kawabe H, Neeb A, Dimova K, Young SM, Takeda M, Katsurabayashi S, Mitkovski M, Malakhova OA, Zhang D, Umikawa M, Kariya K, Goebbels S, Nave K, Rosenmund C, Jahn O, Rhee J, Brose N, Regulation of Rap2A by the ubiquitin ligase Nedd4–1 controls neurite development, Neuron 65 (3) (2010) 358–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fu CA, Shen M, Huang BCB, Lasaga J, Payan DG, Luo Y, TNIK, a novel member of the germinal center kinase family that activates the c-Jun N-terminal kinase pathway and regulates the cytoskeleton, J. Biol. Chem 274 (43) (1999) 30729–30737. [DOI] [PubMed] [Google Scholar]
- [22].Bossolasco P, Sassone F, Gumina V, Peverelli S, Garzo M, and Silani V, “Motor neuron differentiation of iPSCs obtained from peripheral blood of a mutant TARDBP ALS patient,” Stem Cell Res, vol. 30, no. May, pp. 61–68, 2018. [DOI] [PubMed] [Google Scholar]
- [23].Rusconi F, Paganini L, Braida D, Ponzoni L, Toffolo E, Maroli A, Landsberger N, Bedogni F, Turco E, Pattini L, Altruda F, De Biasi S, Sala M, Battaglioli E, LSD1 neurospecific alternative splicing controls neuronal excitability in mouse models of epilepsy, Cereb. Cortex 1 (September) (2015) 2729–2740. [DOI] [PubMed] [Google Scholar]
- [24].Buratti E, Baralle FE, Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9, J. Biol. Chem 276 (39) (2001) 36337–36343. [DOI] [PubMed] [Google Scholar]
- [25].Ayala YM, Zago P, Ambrogio AD, Xu Y, Petrucelli L, Buratti E, Baralle FE, Structural determinants of the cellular localization and shuttling of TDP-43, Pp. (2008) 3778–3785. [DOI] [PubMed] [Google Scholar]
- [26].Ambrogio AD, Buratti E, Stuani C, Guarnaccia C, Romano M, Ayala YM, Baralle FE, Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo, Nucleic Acids Res. 37 (12) (2009) 4116–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ratti A, Falini C, Cova L, Fantozzi R, Calzarossa C, Zennaro E, Pascale A, Quattrone A, Silani V, A role for the ELAV RNA-binding proteins in neural stem cells: stabilization of Msi1 mRNA, J. Cell Sci 119 (7) (2006) 1442–1452. [DOI] [PubMed] [Google Scholar]
- [28].Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE, TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail an important region for the inhibition of cystic fibrosis transmembrane, J. Biol. Chem 280 (45) (2005) 37572–37584. [DOI] [PubMed] [Google Scholar]
- [29].Paz I, Kosti I, Ares M, Cline M, Mandel-Gutfreund Y, RBPmap: a web server for mapping binding sites of RNA-binding proteins, Nucleic Acids Res. 42 (W1) (2014) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fiesel FC, Weber SS, Supper J, Zell A, Kahle PJ, TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR, Nucleic Acids Res. 40 (6) (2012) 2668–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].De Conti L, Akinyi MV, Mendoza-maldonado R, Romano M, Baralle M, Buratti E, TDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathways, Nucleic Acids Res. 43 (18) (2015) 8990–9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Taira K, Umikawa M, Takei K, Myagmar B, Shinzato M, Machida N, Uezato H, Nonaka S, Kariya K, The Traf2- and Nck-interacting kinase as a putative effector of Rap2 to regulate actin cytoskeleton, J. Biol. Chem 279 (47) (2004) 49488–49496. [DOI] [PubMed] [Google Scholar]
- [33].Jensen KB, Dredge BK, Stefani G, Zhong R, Buckanovich RJ, Okano HJ, Yang YYL, Darnell RB, Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability, Neuron 25 (2) (2000) 359–371. [DOI] [PubMed] [Google Scholar]
- [34].Park E, Iaccarino C, Lee J, Kwon I, Baik SM, Kim M, Seong JY, Son GH, Borrelli E, Kim K, Regulatory roles of heterogeneous nuclear ribonucleoprotein M and Nova-1 protein in alternative splicing of dopamine D2 receptor pre-mRNA, J. Biol. Chem 286 (28) (2011) 25301–25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bose JK, Wang IF, Hung L, Tarn WY, Shen CKJ, TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing, J. Biol. Chem 283 (43) (2008) 28852–28859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE, Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene, Nucleic Acids Res. 33 (18) (2005) 6000–6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mohagheghi F, Prudencio M, Stuani C, Cook C, Jansen-west K, Dickson DW, Petrucelli L, Buratti E, TDP-43 functions within a network of hnRNP proteins to inhibit the production of a truncated human SORT1 receptor, Hum. Mol. Genet 25 (3) (2016) 534–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Appocher C, Mohagheghi F, Cappelli S, Stuani C, Romano M, Feiguin F, Buratti E, Major hnRNP proteins act as general TDP-43 functional modifiers both in Drosophila and human neuronal cells, Nucleic Acids Res. 45 (13) (2017) 8026–8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Freibaum BD, Chitta RK, High AA, Taylor JP, Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery research articles, J. Proteome Res 9 (2010) 1104–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ling S, Albuquerque CP, Seok J, Lagier-tourenne C, Tokunaga S, ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS, Pnas 107 (30) (2010) 13318–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Buratti E, TDP-43 post-translational modifications in health and disease, Expert Opin. Ther. Targets 22 (3) (2018) 279–293. [DOI] [PubMed] [Google Scholar]
- [42].Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, Mccrossin T, Cairns M, Weickert CS, Increased inflammatory markers identified in the dorsolateral pre-frontal cortex of individuals with schizophrenia, Mol. Psychiatry 18 (2) (2013) 206–214. [DOI] [PubMed] [Google Scholar]
- [43].Wu JQ, Wang X, Beveridge NJ, Tooney PA, Scott RJ, Carr VJ, Cairns MJ, Transcriptome sequencing revealed significant alteration of cortical promoter usage and splicing in schizophrenia, PLoS One 7 (4) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, Shieh AW, Haney J, Parhami S, Belmont J, Kim M, Losada PM, Khan Z, Mleczko J, Xia Y, Dai R, Wang D, Yang YT, Xu M, Fish K, Hof PR, Warrell J, Fitzgerald D, White K, Jaffe AE, Consortium P, Peters MA, Gerstein M, Liu C, Iakoucheva LM, Pinto D, and Geschwind DH, Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder, Science (80-.)., vol. 362, no. 6420, p. eaat8127, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gusev A, Mancuso N, Won H, Kousi M, Finucane HK, Reshef Y, Song L, Safi A, McCarroll S, Neale BM, Ophoff RA, O’Donovan MC, Crawford GE, Geschwind DH, Katsanis N, Sullivan PF, Pasaniuc B, Price AL, Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights, Nat. Genet 50 (4) (2018) 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Glatt SJ, Everall IP, Kremen WS, Corbeil J, Sa R, Khanlou N, Han M, Liew C, Tsuang MT, Comparative gene expression analysis of blood and brain provides concurrent validation of SELENBP1 up-regulation in schizophrenia, Proc. Natl. Acad. Sci. U. S. A 102 (43) (2005) 15533–15538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.