

Abstract
Background and Aims
Hepatic ischemia‐reperfusion (IR) injury is a major complication of liver transplantation, resection, and hemorrhagic shock. Hypoxia is a key pathological event associated with IR injury. MicroRNA‐210 (miR‐210) has been characterized as a micromanager of hypoxia pathway. However, its function and mechanism in hepatic IR injury is unknown.
Approach and Results
In this study, we found miR‐210 was induced in liver tissues from patients subjected to IR‐related surgeries. In a murine model of hepatic IR, the level of miR‐210 was increased in hepatocytes but not in nonparenchymal cells. miR‐210 deficiency remarkably alleviated liver injury, cell inflammatory responses, and cell death in a mouse hepatic IR model. In vitro, inhibition of miR‐210 decreased hypoxia/reoxygenation (HR)–induced cell apoptosis of primary hepatocytes and LO2 cells, whereas overexpression of miR‐210 increased cells apoptosis during HR. Mechanistically, miR‐210 directly suppressed mothers against decapentaplegic homolog 4 (SMAD4) expression under normoxia and hypoxia condition by directly binding to the 3′ UTR of SMAD4. The pro‐apoptotic effect of miR‐210 was alleviated by SMAD4, whereas short hairpin SMAD4 abrogated the anti‐apoptotic role of miR‐210 inhibition in primary hepatocytes. Further studies demonstrated that hypoxia‐induced SMAD4 transported into nucleus, in which SMAD4 directly bound to the promoter of miR‐210 and transcriptionally induced miR‐210, thus forming a negative feedback loop with miR‐210.
Conclusions
Our study implicates a crucial role of miR‐210‐SMAD4 interaction in hepatic IR‐induced cell death and provides a promising therapeutic approach for liver IR injury.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ChIP
chromatin immunoprecipitation
- DMEM
Dulbecco’s modified Eagle’s medium
- HIF1α
hypoxia inducible factor 1 alpha subunit
- HR
hypoxia/reoxygenation
- I
miR‐210 inhibitor
- IC
inhibitor control
- IgG
immunoglobulin G
- IL
interleukin
- IR
ischemia reperfusion
- KO
knockout
- LDH
lactate dehydrogenase
- M
miR‐210 mimic
- MC
mimic control
- miRNA
microRNA
- ns
not significant
- TNF α
tumor necrosis factor α
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling
- WT
wild type
Ischemia‐reperfusion (IR) injury is a common phenomenon that occurs during the clinical settings such as liver surgery, liver transplantation, and trauma.( 1 , 2 ) IR injury involves two distinct stages with different mechanisms of hepatic damage: The ischemia stage is a process in which the blood supply was restricted and then caused cellular metabolic disturbances, tissue hypoxia, glycogen consumption, and adenosine triphosphate depletion, whereas the reperfusion stage often results in not only tissue damage but also inflammatory responses.( 3 ) Complex molecular and cellular mechanisms are involved in IR injury. The present strategies used to minimize IR injury include primarily avoidance of inflow occlusion, ischemic preconditioning, and the administration of pharmacologic agents.( 4 ) However, the hepatic IR injury still remains a severe and frequent complication that may result in hepatic dysfunction and increases the chances of postoperative mortality and morbidity, complicating recovery and overall outcome.( 5 ) Thus, it is necessary to make great efforts to explore this mechanism as an effective therapeutic strategy for IR injury.
MicroRNAs (miRNAs) are small noncoding RNAs of 18‐22 nucleotides that mediate posttranscriptional regulation of gene expression through repressing the translation of proteins from mRNAs or increasing the degradation of mRNAs.( 6 , 7 , 8 ) miRNAs are involved in numerous pathophysiological processes, such as hypoxia and inflammation.( 9 ) Reducing the levels of pathogenic or aberrantly expressed miRNAs or elevating the levels of miRNAs with beneficial functions has been proven to be effective in the treatment of various diseases in mice.( 10 , 11 , 12 ) Therefore, the identification of key miRNAs in ischemia may provide a new clinical therapeutic strategy for the treatment of ischemic diseases.
miR‐210 exerts diverse effects on numerous pathological processes in various diseases, such as TH17 differentiation,( 13 ) psoriasis,( 10 ) and human cancers.( 14 , 15 ) miR‐210 can be significantly induced by hypoxia–hypoxia inducible factor 1 alpha subunit (HIF1α)‐mediated transcriptional activation.( 13 ) Under ischemic condition, the function of miR‐210 is context‐specific. In hypoxic‐ischemic brain injury, elevated miR‐210 aggravates mitochondrial dysfunction, oxidative stress, and neuronal loss.( 16 ) By contrast, in renal IR injury, miR‐210 plays a protective role by enhancement of angiogenesis through vascular endothelial growth factor (VEGF) signaling pathway.( 17 ) Moreover, miR‐210 can alleviate myocardial infarction through mediating proliferation, survival, and angiogenesis of cardiomyocytes.( 18 , 19 ) However, there are no reports about the effects of miR‐210 on liver IR.
Our present study showed that the level of miR‐210 was increased after hepatic IR injury. miR‐210 knockout (KO) mice displayed lower inflammatory responses and hepatocytes apoptosis after hepatic IR injury. miR‐210 directly suppressed the expression of SMAD4, which could transport into the nucleus and activate miR‐210 transcription under hypoxia condition. We further revealed that the protective effect of SMAD4 overexpression could be weakened by miR‐210 mimic in LO2 cells, implicating the interaction of miR‐210 and SMAD4 as an important requirement in hepatic IR injury.
Materials and Methods
Animals and Human Liver Specimens
The miR‐210 KO mice were generated by the CRISPR/Cas9‐mediated genome editing. The sgRNA sequence is GGCCCACCGCACACTGCGTTGCTC. C57BL/6 WT mice were purchased from the Animal Experiment Center of Wuhan University. All mice were housed in an environment with controlled light (12 hours light/12 hours dark), temperature (23°C ± 2°C), and humidity; food and water were available ad libitum. Only 8‐week‐old to 10‐week‐old (24‐27 g) males were used. All experimental procedures involving animals were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publications 80‐23, revised 1996) and were approved by the Animal Care and Use Committee of Wuhan Union Hospital.
The use of human liver specimens was approved by the Ethics Committee of Wuhan Union Hospital, and informed consent was obtained from all participants before the specimen harvest. All subjects in the study suffered from hepatic carcinoma or traumatic hepatic rupture, and received a hepatic portal occlusion surgery. For the patients with hepatic carcinoma, para‐carcinoma tissues were collected.
Hepatic IR Model and Hepatocytes Isolation
Nonlethal segmental (70%) liver ischemia was performed as previously described.( 20 ) Mice were sacrificed after 1 hour of ischemia and 6 hours of reperfusion. The isolation of hepatocytes from IR‐model mice livers was briefly summarized as the livers were excised, minced, and strained through a steel mesh, then purified by low‐speed centrifugation (3 times at 50g for 5 minutes each).
Histology Staining and TUNEL Staining
The hematoxylin and eosin staining was performed to detect the necrosis in the ischemic lobes according to protocols as previously described.( 21 ) Histopathology images were captured with a light microscope (Aperio versa8, Leica Germany). TUNEL assay was performed to detect hepatocyte apoptosis according to the manufacturer’s instructions (ApopTag Plus In Situ Apoptosis Fluorescein Detection Kit; EMD Millipore, Burlington, MA).
Cell Culture and HR Model
Primary hepatocytes were isolated from livers as the protocol previously reported.( 22 )
The isolated cells were cultured overnight in Dulbecco’s modified Eagle’s medium (DMEM) contained 10% fetal bovine serum (FBS). LO2 cells were grown in complete DMEM supplemented with 10% FBS. For the HR model, the medium was replaced with serum‐free DMEM medium before hypoxia, and cells were incubated for 6 hours at 1% oxygen in a hypoxia chamber (Biospherix, Lacona, NY), and then for another 1 hour under normoxic conditions (air/5% CO2).
Plasmid Constructs and Stable Transfected Cell Line
miR‐210 mimics, inhibitors, and the corresponding negative control oligos were purchased from Guangzhou RiboBio Co., Ltd. (Guangzhou, China). SMAD4 complementary DNA (cDNA) was amplified following the primers 5′‐ATGGACAATATGTCTATTAC‐3′ (forward) and 5′‐TCAGTCTAAAGGTTGTGGG‐3′ (reverse). The shRNA sequences against SMAD4 were as follows: 1, CGAGTTGTATCACCTGGAATT; 2, GTACTTCATACCATGCCGATT; 3, GCAGACAGAAACTGGATTAAA; and the scramble sequence, AATTCTCCGAACGTGTCACGT. These fragments and corresponding vector were ligated by Ligation high (Cat: LGK‐101, TOYOBO, Osaka, Japan). Cells were transfected with mimic control/mimic (50 nmol/L), inhibitor control/inhibitor (100 nmol/L) or plasmids (3 μg), using Lipofectamine2000 (Invitrogen, Carlsbad, CA) at a density of 1.2 × 106 cells. The stable transfected SMAD4 overexpression and knock‐down cell lines were produced by lentivirus; then the cell lines were screened by puromycin (2 μg/mL).
Luciferase Reporter, Flow Cytometry, Serum Chemistry, and Cytokine Assays
Luciferase assays were performed using a dual‐luciferase assay kit (Cat: E1960; Promega, Madison, WI). Apoptosis was tested using PI/annexin V‐FITC staining (Cat: 556547; BD Biosciences, San Jose, CA) according to the manufacturer’s protocol. The serum AST and ALT were measured using a spectrophotometer (ELx800; BioTek, Winooski, VT). The serum IL‐6 and TNF‐α was detected using cytometric bead array kits (Cat: 51‐9000147; BD Biosciences), and IL‐1β was detected using an enzyme‐linked immunosorbent assay kit (Cat: RK00006; ABclonal, Woburn, MA).
ChIP and Quantitative Real‐Time PCR
ChIP analysis was conducted following the manufacturer’s instructions for the SimpleChIP Plus Sonication Chromatin IP Kit (#56383; Cell Signaling Technology, Danvers, MA). The reagent includes anti‐SMAD4 antibody (#38454; Cell Signaling Technology), rabbit IgG (#2729S; Cell Signaling Technology), and ChIP‐Grade Protein G Magnetic Beads (#9006; Cell Signaling Technology). The PCR primer for ChIP assays was forward primer (5′‐GTTTAGGGCCAGGGAGCTG‐3′) and reverse primer (5′‐AGCCTTGACGGTTTGACCTTC‐3′).
Total RNA was extracted using TRIzol reagent (#9109; Takara, Kyoto, Japan). miRNAs were reverse‐transcribed into the cDNA using specific Bulge‐Loop RT primers and RT reagent kit (#K1622; Thermo Fisher Scientific, Waltham, MA). The relative miRNA levels were normalized to small nuclear RNA U6. All of the Bulge‐Loop RT primers and U6 were purchased from RiboBio Co., Ltd. (Guangzhou, China).
Antibodies and Cell Fractionation
The antibodies used in this study were as follows: SMAD4 (#38454), Bax (#5023), Bcl‐2 (#15071), caspase3 (#9665), cleved‐caspase3 (#9661), β‐actin (#3700), Lamin A/C (#4777), β‐tubulin (#2128), and HIF1α (#36169) (all from Cell Signaling Technology).
Cell fraction assay was performed according to the protocol previously reported.( 23 ) Briefly, cells were lysed in lysis buffer, followed by incubation at 4°C for 20 minutes. Thereafter, lysates were subjected to centrifugation at 1,000 rpm to separate cytoplasmic protein from nuclei.
Statistical Analysis
All of the data in this study are expressed as means ± SEM and analyzed with the statistical analysis software SPSS 15.0 (SPSS, Inc., Chicago, IL). Statistical differences were performed using one‐way analysis of variance analysis. Statistical significance was considered when P < 0.05.
Result
miR‐210 Was Induced by Ischemia and Hypoxia
To investigate whether miR‐210 was involved in hepatic IR injury, we detected the levels of miR‐210 in hepatocytes from mice following liver IR by quantitative PCR. The results showed that the expression of miR‐210‐3p was increased in hepatocytes from mice subjected to 1 hour of ischemia and 6 hours of reperfusion, compared with the sham‐operated group (Fig. 1A). On the contrary, the expression of miR‐210‐5p was decreased following IR (Supporting Fig. S1A). Moreover, miR‐210‐3p was the dominant one expressed in mice liver tissue, so we focused on the function of miR‐210‐3p in further study (Supporting Fig. S1B). Then the levels of miR‐210 in specimens from clinical patients with IR‐related surgeries were detected, and we found that miR‐210 was gradually increased along with hepatic portal occlusion time (Fig. 1B). To investigate the main cells differentially expressed miR‐210, we analyzed miR‐210 levels in primary hepatocytes and nonparenchymal cells. There was no significant change of miR‐210 in nonparenchymal cells after IR injury (Supporting Fig. S2). Next, primary hepatocytes were isolated and exposed to hypoxia for 0 hours, 3 hours, 6 hours, and 9 hours with 1% oxygen and 5% carbon dioxide. We observed that miR‐210 levels were gradually increased along with exposure time (Fig. 1C). Similarly, LO2 cell line exposed to hypoxia exhibited high miR‐210 levels in a time‐dependent manner (Fig. 1D). These results indicated that miR‐210 was induced by ischemia or hypoxia, suggesting a possible role of miR‐210 in hepatic IR.
Fig. 1.
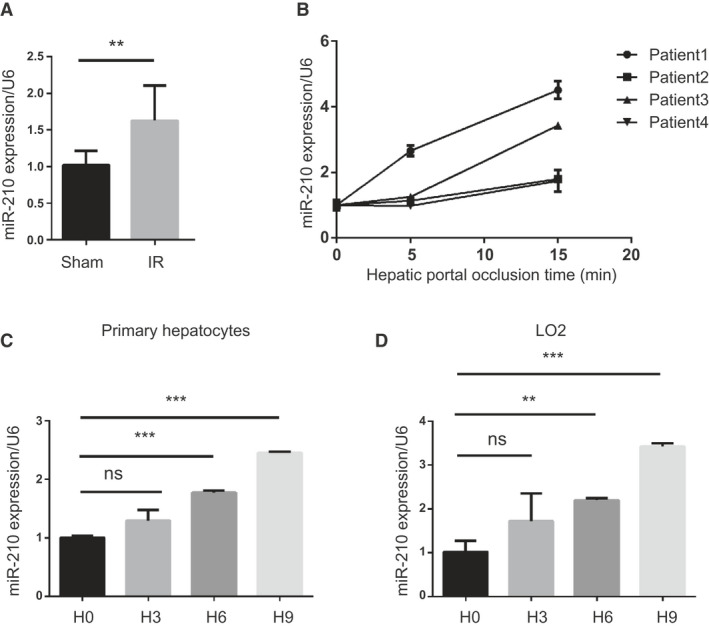
miR‐210 is increased under IR stress and hypoxia condition. (A) Quantitative PCR analysis of miR‐210‐3p in hepatocytes isolated from IR injury mice liver (mice n = 2‐8, **P < 0.01, vs. sham). (B) Levels of miR‐210 in liver samples from clinical patients with IR‐related surgeries (n = 4). (C) Quantification of miR‐210 levels in primary hepatocytes subjected to hypoxia stimulation (n = 3, ***P < 0.001, vs. control). (D) Quantification of miR‐210 levels in liver cells (LO2) subjected to hypoxia stimulation (n = 3, **P < 0.01, ***P < 0.001, vs. control). Abbreviation: ns, not significant.
miR‐210 Deficiency Protects Liver Against Ischemia Reperfusion Injury
To explore the role of miR‐210 in hepatic IR injury, miR‐210 KO mice were established using the CRISPR‐Cas9 genome editing system. The genome sequencing result showed a deletion of eight bases at the miR‐210 gene in miR‐210 KO mice. Quantitative PCR and miR‐210 in situ hybridization assay further confirmed the successful knockout of miR‐210 (Fig. 2A). Then, miR‐210 KO and wild‐type (WT) mice were subjected to liver IR, and damaged liver tissues were harvested for histopathologic analysis. IR induced obvious liver necrosis, but miR‐210 deficiency resulted in remarkably lower necrotic rate (Fig. 2B). The levels of pro‐inflammatory factors including interleukin (IL)‐1β, IL‐6, and tumor necrosis factor α (TNF‐α) were markedly reduced in liver tissues or serum of IR‐challenged mice (Fig. 2C,D). Accordingly, significantly lower levels of the serum markers alanine aminotransferase (ALT) and aspartate transaminase (AST) were noted in miR‐210 KO mice than that in WT mice after IR injury (Fig. 2E). Next, primary hepatocytes were exposed to 6 hours of hypoxia and 1 hour of reoxygenation to mimic mouse IR model, and lactate dehydrogenase (LDH) release was measured to evaluate cell damage. We observed a reduction of LDH release in miR‐210 KO hepatocytes in both hypoxic and normoxic conditions (Fig. 2F). Taken together, these data indicate that miR‐210 deletion protects liver against IR‐induced tissue damage and the inflammatory insult.
Fig. 2.
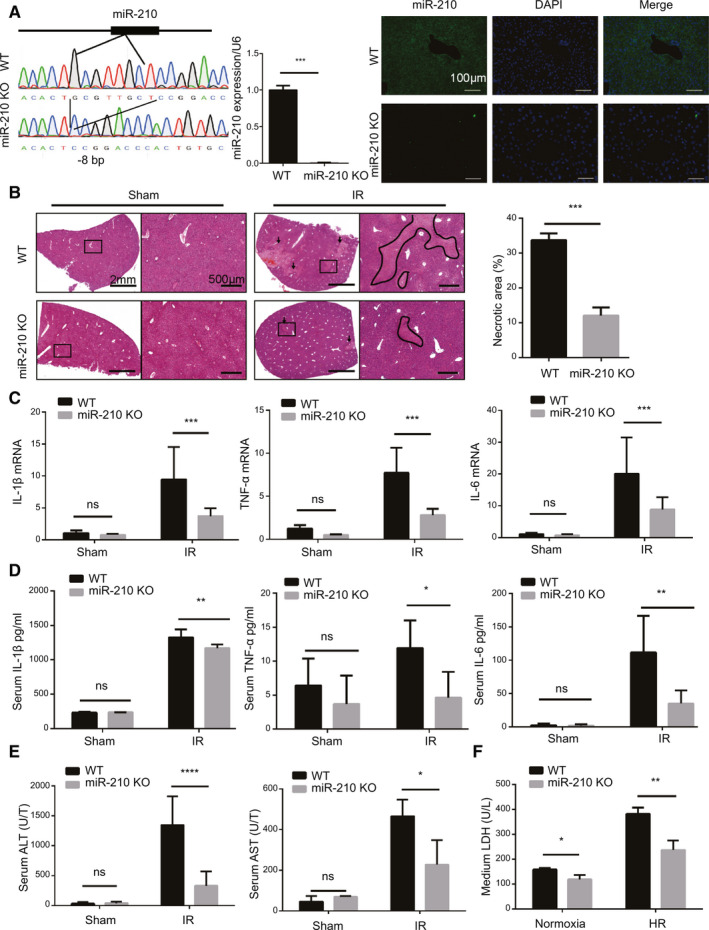
Effect of miR‐210 deficiency on IR‐induced liver damage. (A) Genome sequencing analysis (left), quantification of miR‐210 expression (middle, n = 3, ***P < 0.001) and representative images of miR‐210 in situ hybridization (right) in liver tissues of miR‐210 KO and WT mice. (B) Representative images of hematoxylin and eosin staining and quantification of necrotic areas in miR‐210 KO and WT mice after IR injury (mice n = 3 per group, ***P < 0.001). (C,D) Levels of pro‐inflammatory factors in liver tissues (C) and serum (D) from sham or IR‐operated mice (mice n = 8 per group, *P < 0.05, **P < 0.01, ***P < 0.001). (E) Quantification of serum ALT and AST levels in miR‐210 KO and WT mice after IR injury (mice n = 8 per group, *P < 0.05, ****P < 0.0001). (F) Levels of LDH in the medium from the indicated primary hepatocytes after HR stimulation (n = 3, *P < 0.05, **P < 0.01). DAPI, 4′,6‐diamidino‐2‐phenylindole.
miR‐210 Promotes Hepatocytes Apoptosis During IR Injury
The levels of cell proliferation and cell death determine the extent of liver damage after IR. To investigate whether miR‐210 is a modulator of hepatocyte activity or survival during IR, we analyzed cell proliferation and apoptosis of hepatocytes by using Ki67 and terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) staining, respectively. There was no significant difference in the number of Ki67‐positive cells between WT and miR‐210 KO mice (Fig. 3A); however, miR‐210 deficiency significantly diminished cell apoptosis in liver tissues after IR injury (Fig. 3B). Next, miR‐210 mimic/inhibitor and corresponding control oligos were used to modulate the endogenous miR‐210 level in LO2 (Fig. 3C). Protease inhibitor (PI)/annexin V– fluorescein isothiocyanate (FITC) cell apoptosis analysis demonstrated that hypoxia/reoxygenation (HR) triggered significant cell apoptosis. miR‐210 overexpression increased the apoptotic rate of LO2 cells, but inhibition of miR‐210 reduced cell death under HR stimulation (Fig. 3D). However, miR‐210‐5p had no effect on cell apoptosis of LO2 following HR (Supporting Fig. S1C). Consistently, the level of pro‐apoptotic factors cleaved‐caspase3 and Bax were increased, while anti‐apoptotic marker Bcl‐2 was reduced in primary hepatocytes under HR stimulation. miR‐210 deletion led to a decrease of cleaved‐caspase3 and Bax and an increase of Bcl‐2, whereas miR‐210 overexpression induced the levels of cleaved‐caspase3 and Bax and reduced Bcl‐2 (Fig. 3E). In LO2 cells, inhibition of miR‐210 also resulted in less apoptosis, whereas miR‐210 overexpression aggravated cell apoptosis, compared with their corresponding control groups (Fig. 3E). These results illustrate that miR‐210 mediated hepatocytes apoptosis during IR/HR stimulation.
Fig. 3.
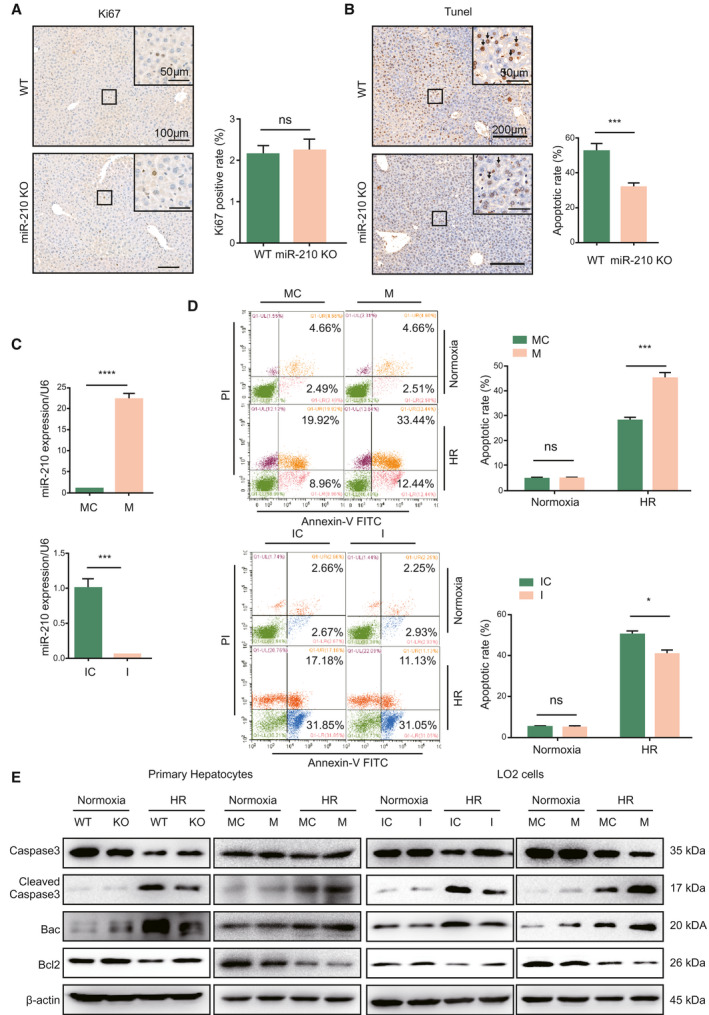
Effect of miR‐210 on cell death under IR or HR stimulation. (A) Ki67 staining and quantification of Ki67‐positive cells of miR‐210 KO and WT mice after IR injury (n = 3 per group). (B) TUNEL staining and quantification of TUNEL‐positive cells of miR‐210 KO and WT mice after IR injury (n = 3, ***P < 0.001). (C) Quantitative PCR analysis of endogenous miR‐210 levels in LO2 cells transfected with miR‐210 mimics, inhibitors, and corresponding control oligos (n = 3, ***P < 0.001, ****P < 0.0001). (D) Flow cytometry cell apoptosis analysis of LO2 cells transfected with indicated oligos under normoxia and HR conditions (n = 2, *P < 0.05, ***P < 0.001). (E) Western blot analysis of the indicated proteins in primary hepatocytes and LO2 cells under normoxia or HR condition. Abbreviations: I, miR‐210 inhibitor; IC, inhibitor control; M, miR‐210 mimic; and MC, mimic control.
miR‐210 Targets SMAD4
Next, we explored the downstream targets of miR‐210 by target prediction databases, including TargetScan (http://www.targetscan.org/vert_72/) and miRanda (http://www.microrna.org/microrna/home.do). Several candidate targets related to cell apoptosis were selected, including SMAD4, PTPN2, and IGFBP3. We found that only SMAD4 3′UTR reporter activity was suppressed by miR‐210 (Fig. 4A). The results from luciferase assay indicated that miR‐210 can repress the reporter activity of WT SMAD4 3′UTR, but not the mutant SMAD4 3′UTR, indicating a direct binding of miR‐210 to SMAD4 3′UTR (Fig. 4B). This phenomenon was also observed in primary hepatocytes (Supporting Fig. S3A,B). Biotinylated miRNA pull‐down assay further identified the interaction between SMAD4 3′UTR and miR‐210 (Fig. 4B). Further western blot analysis showed that miR‐210 overexpression could depress the expression of SMAD4, whereas miR‐210 inhibition promoted SMAD4 expression (Fig. 4C). We further examined SMAD4 protein level in response to hypoxia. Hypoxia challenge abated the expression of SMAD4 over time (Fig. 4D). Hepatocytes isolated from liver of IR injury displayed a lower level of SMAD4 than that from the sham‐operated group. In addition, the level of SMAD4 was significantly higher in miR‐210‐deficient hepatocytes than the WT group, both under sham and IR injury conditions (Fig. 4E). Accordingly, in vitro HR stimulation also decreased the protein levels of SMAD4 in primary hepatocytes and LO2 cells, as shown in Fig. 4F. Under the HR condition, deletion or inhibition of miR‐210 increased the level of SMAD4, whereas miR‐210 mimics further down‐regulated SMAD4 expression compared with their corresponding controls (Fig. 4F). Collectively, miR‐210 directly targets the 3′ UTR of SMAD4 to suppress SMAD4 expression.
Fig. 4.
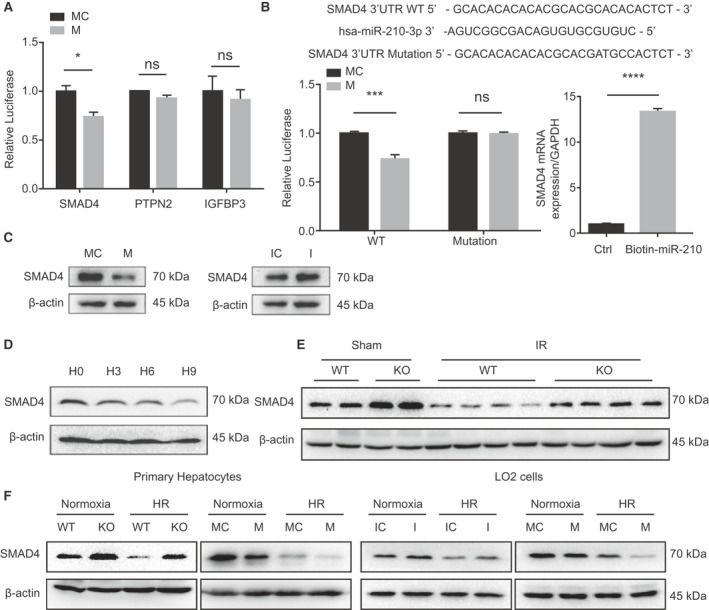
miR‐210 targets SMAD4. (A) luciferase analysis of SMAD4, PTPN2, and IGFBP3 3′UTR reporter activities (n = 3, *P < 0.05). (B) Predicted binding site of miR‐210 in the SMAD4 3′UTR and luciferase analysis of SMAD4 3′UTR reporter activities with WT or mutated binding seed regions in LO2 transfected with miR‐210 mimic or mimic control (left, n = 3, ***P < 0.001) and biotinylated miRNA pull‐down assay (right, n = 3, ****P < 0.0001). (C) Western blot analysis of SMAD4 levels in LO2 cells transfected with the indicated oligos under normoxia condition. (D) Western blot analysis of SMAD4 levels in primary hepatocytes with gradient hypoxia. (E) Western blot analysis of SMAD4 in hepatocytes isolated from WT and miR‐210 KO mice liver with indicated stimulation. (F) Western blot analysis of SMAD4 in primary hepatocytes and LO2 cells transfected with indicated oligos under normoxia or HR condition. Abbreviation: Ctrl, control.
Inhibition of SMAD4 by miR‐210 Increases Cell Death
To investigate the effect of miR‐210‐SMAD4 interaction on hepatocyte apoptosis induced by HR, we obtained stable cell line–expressed pHAGE‐SMAD4 and pLKO.1–short hairpin SMAD4 (sh‐SMAD4), which could effectively increase or decrease the protein level of endogenous SMAD4, respectively (Fig. 5A). The results showed that overexpression of SMAD4 could reduce cell apoptosis, whereas knock‐down SMAD4 increased cell apoptosis induced by HR (Fig. 5B). An In vitro co‐transfection study was performed to investigate the relationship between miR‐210 and SMAD4. The results showed that down‐regulation of SMAD4 increased cell apoptosis, and the protective function of miR‐210 inhibitor could be abrogated by sh‐SMAD4 (Fig. 5C). Overexpression of SMAD4 reduced cell apoptosis under HR condition, and the effect of SMAD4 overexpression could be partly impaired by miR‐210 mimic (Fig. 5D). Taking these results together, we could show that the effect of miR‐210 in hypoxia‐induced cell death was mediated by inhibition of SMAD4.
Fig. 5.
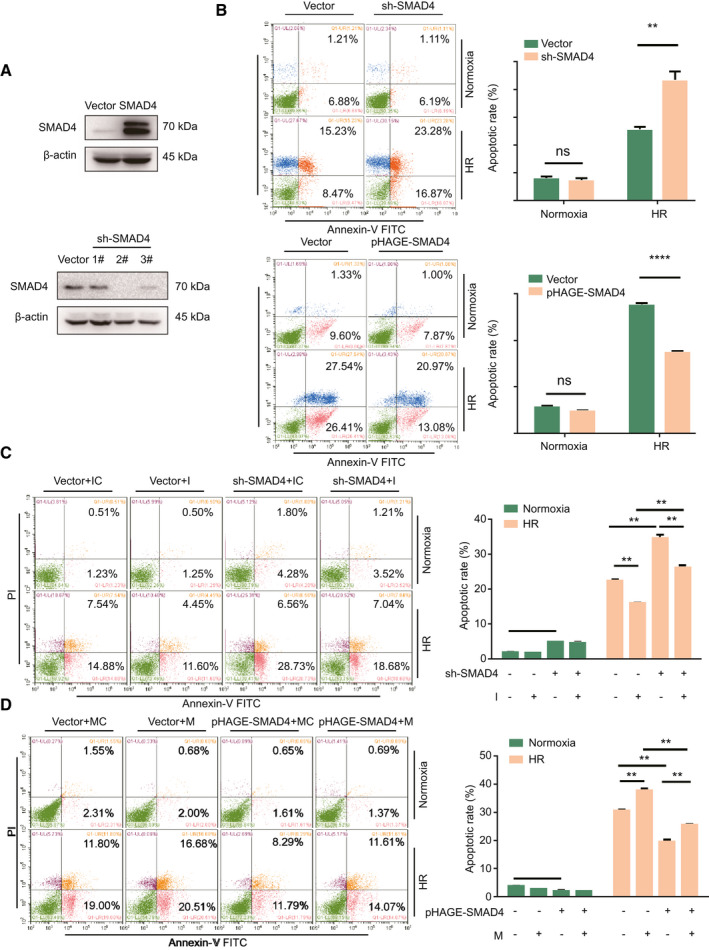
Interaction of miR‐210 and SMAD4 is important for cell apoptosis under hypoxia condition. (A) Western blot analysis of SMAD4 levels in stable transfected overexpression and knock‐down cell lines. (B) Flow cytometry analysis of stable transfected cell lines after HR treatment (n = 2, **P < 0.01, ****P < 0.0001). (C,D) Flow cytometry analysis of cell apoptosis of LO2 cells co‐transfected with the indicated oligos after HR treatment (n = 2, **P < 0.01, ***P < 0.001).
SMAD4 Transcriptionally Activates miR‐210
miR‐210 has been reported be regulated by HIF1α.( 13 ) To investigate whether there were other transcription factors to regulate miR‐210, we used HIF1α‐inhibitor LW6 to suppress endogenous HIF1α production. As Fig. 6A shows, LW6 could effectively eliminate HIF1α protein under normoxia or hypoxia condition; meanwhile, hypoxia‐induced SMAD4 reduction was weakened following LW6 treatment. We also found that the level of miR‐210 was still induced in LO2 cells treated with LW6 under hypoxia (Fig. 6A), indicating a HIF1α‐independent transcriptional regulation of miR‐210.
Fig. 6.
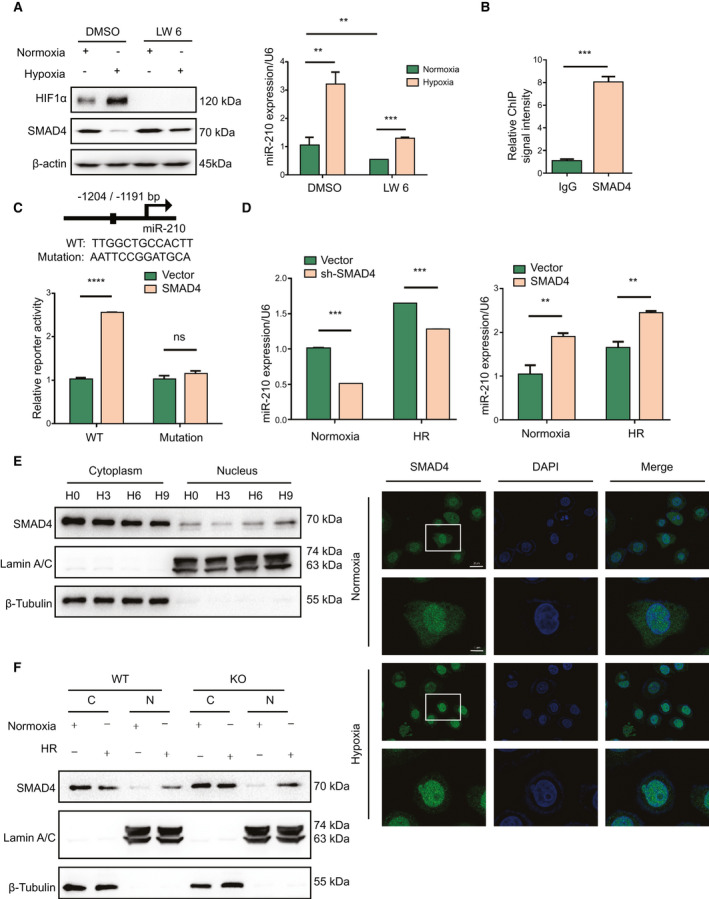
SMAD4 promotes miR‐210 expression. (A) Western blot analysis of indicated protein levels and quantitative PCR analysis of miR‐210 expression in LO2 cells treated with HIF1α inhibitor LW6 (20 μM) under normoxia or hypoxia (n = 3, **P < 0.01, ***P < 0.001). (B) ChIP analysis of the binding of SMAD4 to miR‐210 promoter (n = 3, ***P < 0.001). (C) SMAD4 binding sequence on miR‐210 promoter predicted by JASPAR database and luciferase analysis of the effect of SMAD4 on miR‐210 promoter activity in LO2 cells (n = 3, ****P < 0.0001). (D) Quantitative PCR analysis of miR‐210 expression in LO2 cells transfected with indicated plasmids under normoxia or HR stimulation (n = 3, **P < 0.01, ***P < 0.001). (E) Cell fractionation western blot analysis and immunofluorescence analysis of the cellular distribution of SMAD4 in LO2 cells following normoxia or 6 hours of hypoxia (63× lens, scale [up] = 25 μm, scale [down] = 7.5 μm). (F) Cell fractionation western blot analysis of SMAD4 in primary hepatocytes following normoxia or HR.
As SMAD4 is a key transcription factor for various miRNAs( 24 , 25 , 26 ) and participates in IR injury,( 27 , 28 ) we analyzed the predicted targets of SMAD4 through hTFtarget Bioinformatic tools (http://bioinfo.life.hust.edu.cn/guo_lab#!/). As expected, we found that miR‐210 was one of the predicted targets of SMAD4. Then we analyzed the miR‐210 gene‐promoter region by the JASPAR database (http://jaspar.genereg.net/). The results revealed a putative binding site of SMAD4 in the miR‐210 promoter region, suggesting that miR‐210 might be a potential transcriptional target of SMAD4. To identify whether SMAD4 could regulate miR‐210 transcription, chromatin immunoprecipitation (ChIP) assay was performed in LO2 cells. The results showed that the ChIP fragments of the SMAD4 group could be enriched in the predicted promoter region compared with the immunoglobulin G (IgG) group (Fig. 6B). Approximately 1‐kb sequences of the putative 5′‐promoter region of miR‐210 (WT) and the region with the mutated binding site (Mutation) were cloned into luciferase promoter reporter vectors. We found that SMAD4 induced a 2.5‐fold increase in the activity of WT miR‐210 promoter, but the promotion effect was abrogated in cells transfected with mutant miR‐210 promoter (Fig. 6C). The luciferase assay was also performed in primary hepatocytes, and the result was consistent with that from LO2 cells (Supporting Fig. S3C). Then we measured the levels of miR‐210 in SMAD4 up‐regulated or down‐regulated LO2 cells by quantitative PCR under normoxia and HR conditions. As Fig. 6D shows, overexpression of SMAD4 promoted the miR‐210 mRNA level, whereas inhibition of SMAD4 decreased the miR‐210 level both in stimulation of normoxia and HR. These data indicate that SMAD4 could bind to the promoter region of miR‐210 and activate its expression.
It may appear paradoxical that SMAD4 induced the expression of miR‐210 under hypoxia while the total protein level of SMAD4 was decreased following hypoxia treatment. It is well known that SMAD4 synergistically cooperated with other SMAD members or transcription factors, translocated into the nucleus, bound to the promoters, and mediated transcription of target genes. Therefore, we further investigated the cellular distribution of SMAD4 under hypoxia. Western blot assay indicated an increase of nucleic SMAD4 and a decrease of cytoplasmic SMAD4 along with hypoxia time (Fig. 6E). Correspondingly, we observed a translocation of SMAD4 into the nucleus by using confocal microscopy under hypoxia stimulation (Fig. 6E). The lower magnification images are shown in Supporting Fig. S4. Moreover, primary hepatocytes with miR‐210 deficiency exhibited higher cytoplasmic and nucleic SMAD4 protein levels compared with the WT group (Fig. 6F). Together, these results indicate that hypoxia‐mediated translocation of SMAD4 into the nucleus transcriptionally activated the expression of miR‐210.
Discussion
Hepatic IR is a common phenomenon that occurs in clinical settings, such as liver surgery, liver transplantation, and trauma.( 1 , 2 ) miRNAs, as negative regulators of gene expression, could participate in IR injury and be therapeutic targets in acute ischemic diseases.( 29 ) In the current study, we describe a highly significant function of miR‐210 in liver IR injury. We found an increased expression of liver miR‐210 in the mouse hepatic IR model; deletion of miR‐210 significantly reduced liver IR induced damage. We also found that hypoxia induced SMAD4 to transport into the nucleus and activated the expression of miR‐210. Meanwhile, SMAD4 was a target of miR‐210, which repressed SMAD4 expression to induce cell apoptosis during liver IR injury.
Hypoxia is a common phenomenon, not only following acute and chronic ischemia, but also during several physio‐pathological circumstances such as rapid tissue growth and organ and tumor development.( 30 ) miR‐210 is a “master” miRNA of hypoxic response, which was found up‐regulated by hypoxia in all of the cells and tissues tested to date.( 31 ) As a solid tumor biomarker,( 32 ) miR‐210 can regulate tumor cell proliferation,( 33 ) apoptosis,( 34 ) and metastasis.( 35 ) In IR injury, miR‐210 expression is activated.( 31 ) In the current study, we found that miR‐210 was induced both in human liver specimens after IR‐related surgeries and in murine primary hepatocytes from IR‐challenged murine liver. In addition, we showed that miR‐210 inhibition alleviated cell apoptosis in vivo and in vitro, which is consistent with previous reports indicating miR‐210 as an inducer of neural cell apoptosis through targeting iron‐sulfur cluster scaffold homolog in brain IR injury.( 16 , 36 ) Consistently, miR‐210 deficiency resulted in lower inflammatory responses by reducing pro‐inflammatory markers IL‐1β, TNF‐α, and IL‐6 both in liver tissues and serum, which is in contrast to the previous studies that miR‐210 exhibits anti‐inflammatory and pro‐vascularization properties by enhancing VEGF signaling in renal IR injury( 17 ) and myocardial infarction.( 18 , 19 ) Therefore, the organ‐specific or cell‐specific role of miR‐210 in IR injury may be due to the different regulation targets or signals.
The SMAD family of transcription factors includes eight known members (SMAD1–8) in mammals, which can be divided into the receptor‐regulated SMADs/R‐SMADs (Smads‐1, ‐2, ‐3, ‐5, and ‐8) and the co‐SMADs, of which SMAD4 is the only member.( 37 ) In the nucleus, SMADs are associating with additional transcription factors or cofactors to positively or negatively control the expression of various genes,( 38 ) including miRNAs.( 25 , 26 , 39 ) In the present study, SMAD4 was characterized as a transcription activator of miR‐210, apart from the well‐known HIF1α or nuclear factor kappa B.( 13 , 40 ) We further illustrated a significant increase of nucleic SMAD4 levels after hypoxia, although the total protein level of SMAD4 was reduced by hypoxia. It is worth noting that IR or HR challenges significantly decreased the level of SMAD4, which could not be fully restored by miR‐210 deletion, suggesting that hypoxia‐induced SMAD4 reduction was partially mediated by miR‐210. Further studies are needed to explore the upstream factors contributed to SMAD4 reduction during IR injury and the downstream genes of SMAD4 to mediate the anti‐apoptotic effect of SMAD4.
In conclusion, as shown in Fig. 7, the present study provides evidence that miR‐210 deficient mice are resistant to liver IR injury, and the expression of mir‐210 under hypoxia was regulated by SMAD4, apart from the well‐known HIF1α. Moreover, miR‐210 directly suppresses SMAD4 expression. This negative‐feedback loop is crucial for cell apoptosis under IR or HR conditions. Our study suggests inhibition of miR‐210 in vivo as an effective therapeutic strategy for hepatic IR injury.
Fig. 7.
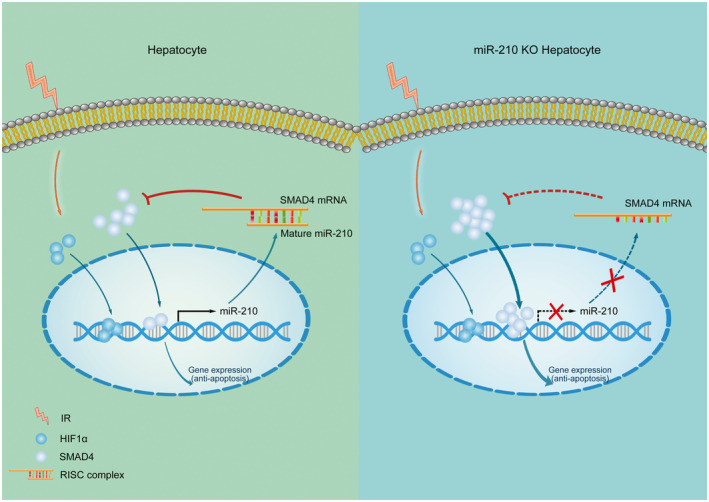
Schematic model of miR‐210‐SMAD4 interaction during hepatic IR injury. Hypoxia or ischemia induced the transportation of HIF1α and SMAD4 into nucleus, which triggered the transcription of miR‐210 by directly binding to its promoter region. Up‐regulated miR‐210 matured in cytoplasm, incorporated into RNA‐induced silencing complex to pair to SMAD4 3′UTR, and subsequently suppressed the expression of SMAD4. The negative feedback of SMAD4 and miR‐210 suppressed the levels of SMAD4, which further impaired SMAD4‐mediated anti‐apoptosis and induced cell death and liver damage.
Author Contributions
W‐M.P., H.W., X.‐F.Z., and G.‐L.W performed and analyzed the experiments. H.W., J.‐X.Z., W.‐M.P., and X.‐F.Z. performed the statistical analyses. X‐D.Z. and R‐L.D. provided plasmids and reagents. H.Z., Y.‐J.L., and P.X. helped to construct the plasmids and stable cell lines. Y‐W.Z. and K‐P.H. performed the histopathologic analysis. H.W., J‐X.Z., and H.H. designed the overall research and directed the work. W‐M.P., X‐F.Z., and J‐X.Z. wrote the manuscript. All authors discussed the results, and reviewed and approved the final manuscript. W‐M.P. and H.W. contributed equally to this work as first authors, respectively.
Supporting information
Fig S1‐S4
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81670575, 81070355, 81570570, 81700558, 81801923, 81470902, and 81770650), Program of HUST Academic Frontier Youth Team (2018QYTD02), Pre‐research Fund for Free Innovation of Union Hospital, and Huazhong University of Science and Technology (02.03.2017‐59, 02.03.2017‐312, and 02.03.2018‐126).
Potential conflict of interest: Nothing to report.
Contributor Information
Xiao‐Dong Zhang, Email: zhangxd@whu.edu.cn, Email: zhangjinxiang@hust.edu.cn.
Jin‐Xiang Zhang, Email: zhangxd@whu.edu.cn, Email: zhangjinxiang@hust.edu.cn.
References
Author names in bold designate shared co‐first authorship.
- 1. Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec‐Weglinski JW. Ischaemia‐reperfusion injury in liver transplantation—from bench to bedside. Nat Rev Gastroenterol Hepatol 2013;10:79‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shaked A, Ghobrial RM, Merion RM, Shearon TH, Emond JC, Fair JH, et al. Incidence and severity of acute cellular rejection in recipients undergoing adult living donor or deceased donor liver transplantation. Am J Transplant 2009;9:301‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abu‐Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver ischemia/reperfusion injury: processes in inflammatory networks—a review. Liver Transpl 2010;16:1016‐1032. [DOI] [PubMed] [Google Scholar]
- 4. Cannistra M, Ruggiero M, Zullo A, Gallelli G, Serafini S, Maria M, et al. Hepatic ischemia reperfusion injury: a systematic review of literature and the role of current drugs and biomarkers. Int J Surg 2016;33(Suppl. 1):S57‐S70. [DOI] [PubMed] [Google Scholar]
- 5. Li J, Li RJ, Lv GY, Liu HQ. The mechanisms and strategies to protect from hepatic ischemia‐reperfusion injury. Eur Rev Med Pharmacol Sci 2015;19:2036‐2047. [PubMed] [Google Scholar]
- 6. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pratt AJ, MacRae IJ. The RNA‐induced silencing complex: a versatile gene‐silencing machine. J Biol Chem 2009;284:17897‐17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Valencia‐Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 2006;20:515‐524. [DOI] [PubMed] [Google Scholar]
- 9. Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell 2010;40:205‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu R, Zeng J, Yuan J, Deng X, Huang Y, Chen L, et al. MicroRNA‐210 overexpression promotes psoriasis‐like inflammation by inducing Th1 and Th17 cell differentiation. J Clin Invest 2018;128:2551‐2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caballero‐Garrido E, Pena‐Philippides JC, Lordkipanidze T, Bragin D, Yang Y, Erhardt EB, et al. In vivo inhibition of miR‐155 promotes recovery after experimental mouse stroke. J Neurosci 2015;35:12446‐12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ishii H, Vodnala SK, Achyut BR, So JY, Hollander MC, Greten TF, et al. miR‐130a and miR‐145 reprogram Gr‐1(+)CD11b(+) myeloid cells and inhibit tumor metastasis through improved host immunity. Nat Commun 2018;9:2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang H, Flach H, Onizawa M, Wei L, McManus MT, Weiss A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia‐regulated microRNA miR‐210. Nat Immunol 2014;15:393‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ren D, Yang Q, Dai Y, Guo W, Du H, Song L, et al. Oncogenic miR‐210‐3p promotes prostate cancer cell EMT and bone metastasis via NF‐kappaB signaling pathway. Mol Cancer 2017;16:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kai AK, Chan LK, Lo RC, Lee JM, Wong CC, Wong JC, et al. Down‐regulation of TIMP2 by HIF‐1alpha/miR‐210/HIF‐3alpha regulatory feedback circuit enhances cancer metastasis in hepatocellular carcinoma. Hepatology 2016;64:473‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma Q, Dasgupta C, Li Y, Huang L, Zhang L. MicroRNA‐210 downregulates ISCU and induces mitochondrial dysfunction and neuronal death in neonatal hypoxic‐ischemic brain injury. Mol Neurobiol 2019;56:5608‐5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu F, Lou YL, Wu J, Ruan QF, Xie A, Guo F, et al. Upregulation of microRNA‐210 regulates renal angiogenesis mediated by activation of VEGF signaling pathway under ischemia/perfusion injury in vivo and in vitro . Kidney Blood Press Res 2012;35:182‐191. [DOI] [PubMed] [Google Scholar]
- 18. Diao H, Liu B, Shi Y, Song C, Guo Z, Liu N, et al. MicroRNA‐210 alleviates oxidative stress‐associated cardiomyocyte apoptosis by regulating BNIP3. Biosci Biotechnol Biochem 2017;81:1712‐1720. [DOI] [PubMed] [Google Scholar]
- 19. Arif M, Pandey R, Alam P, Jiang S, Sadayappan S, Paul A, et al. MicroRNA‐210‐mediated proliferation, survival, and angiogenesis promote cardiac repair post myocardial infarction in rodents. J Mol Med (Berl) 2017;95:1369‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan W, Wang L, Zhang XF, Zhang H, Zhang J, Wang G, et al. Hypoxia‐induced microRNA‐191 contributes to hepatic ischemia/reperfusion injury through the ZONAB/Cyclin D1 axis. Cell Death Differ 2019;26:291‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang H, Wang G, Zhang L, Zhang J, Zhang J, Wang Q, et al. ADAR1 suppresses the activation of cytosolic RNA‐sensing signaling pathways to protect the liver from ischemia/reperfusion injury. Sci Rep 2016;6:20248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu J, Zhu XH, Zhang XJ, Wang PX, Zhang R, Zhang P, et al. Targeting TRAF3 signaling protects against hepatic ischemia/reperfusions injury. J Hepatol 2016;64:146‐159. [DOI] [PubMed] [Google Scholar]
- 23. Suresh PS, Ma S, Migliaccio A, Chen G. Protein‐tyrosine phosphatase H1 increases breast cancer sensitivity to antiestrogens by dephosphorylating estrogen receptor at Tyr537. Mol Cancer Ther 2014;13:230‐238. [DOI] [PubMed] [Google Scholar]
- 24. Munoz‐Felix JM, Gonzalez‐Nunez M, Martinez‐Salgado C, Lopez‐Novoa JM. TGF‐β/BMP proteins as therapeutic targets in renal fibrosis. Where have we arrived after 25 years of trials and tribulations? Pharmacol Ther 2015;156:44‐58. [DOI] [PubMed] [Google Scholar]
- 25. Zhong X, Chung AC, Chen HY, Dong Y, Meng XM, Li R, et al. miR‐21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia 2013;56:663‐674. [DOI] [PubMed] [Google Scholar]
- 26. Chung AC, Huang XR, Meng X, Lan HY. miR‐192 mediates TGF‐beta/Smad3‐driven renal fibrosis. J Am Soc Nephrol 2010;21:1317‐1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Di YF, Li DC, Shen YQ, Wang CL, Zhang DY, Shang AQ, et al. MiR‐146b protects cardiomyocytes injury in myocardial ischemia/reperfusion by targeting Smad4. Am J Transl Res 2017;9:656‐663. [PMC free article] [PubMed] [Google Scholar]
- 28. Li Y, Du Y, Cao J, Gao Q, Li H, Chen Y, et al. MiR‐130a inhibition protects rat cardiac myocytes from hypoxia‐triggered apoptosis by targeting Smad4. Kardiol Pol 2018;76:993‐1001. [DOI] [PubMed] [Google Scholar]
- 29. Fasanaro P, D'Alessandra Y, Magenta A, Pompilio G, Capogrossi MC. microRNAs: promising biomarkers and therapeutic targets of acute myocardial ischemia. Curr Vasc Pharmacol 2015;13:305‐315.23713865 [Google Scholar]
- 30. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006;441:437‐443. [DOI] [PubMed] [Google Scholar]
- 31. Fasanaro P, Greco S, Ivan M, Capogrossi MC, Martelli F. microRNA: emerging therapeutic targets in acute ischemic diseases. Pharmacol Ther 2010;125:92‐104. [DOI] [PubMed] [Google Scholar]
- 32. Feng S, He A, Wang D, Kang B. Diagnostic significance of miR‐210 as a potential tumor biomarker of human cancer detection: an updated pooled analysis of 30 articles. Onco Targets Ther 2019;12:479‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim JH, Park SG, Song SY, Kim JK, Sung JH. Reactive oxygen species‐responsive miR‐210 regulates proliferation and migration of adipose‐derived stem cells via PTPN2. Cell Death Dis 2013;4:e588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang W, Sun T, Cao J, Liu F, Tian Y, Zhu W. Downregulation of miR‐210 expression inhibits proliferation, induces apoptosis and enhances radiosensitivity in hypoxic human hepatoma cells in vitro . Exp Cell Res 2012;318:944‐954. [DOI] [PubMed] [Google Scholar]
- 35. Ying Q, Liang L, Guo W, Zha R, Tian Q, Huang S, et al. Hypoxia‐inducible microRNA‐210 augments the metastatic potential of tumor cells by targeting vacuole membrane protein 1 in hepatocellular carcinoma. Hepatology 2011;54:2064‐2075. [DOI] [PubMed] [Google Scholar]
- 36. Huang L, Ma Q, Li Y, Li B, Zhang L. Inhibition of microRNA‐210 suppresses pro‐inflammatory response and reduces acute brain injury of ischemic stroke in mice. Exp Neurol 2018;300:41‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wharton K, Derynck R. TGFbeta family signaling: novel insights in development and disease. Development 2009;136:3691‐3697. [DOI] [PubMed] [Google Scholar]
- 38. Attisano L, Wrana JL. Signal transduction by the TGF‐beta superfamily. Science 2002;296:1646‐1647. [DOI] [PubMed] [Google Scholar]
- 39. Euler G. Good and bad sides of TGFbeta‐signaling in myocardial infarction. Front Physiol 2015;6:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y, Fei M, Xue G, Zhou Q, Jia Y, Li L, et al. Elevated levels of hypoxia‐inducible microRNA‐210 in pre‐eclampsia: new insights into molecular mechanisms for the disease. J Cell Mol Med 2012;16:249‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4