

Abstract
Objective
To develop a model and methodology for predicting the risk of Gleason upgrading in patients with prostate cancer on active surveillance (AS) and using the predicted risks to create risk‐based personalised biopsy schedules as an alternative to one‐size‐fits‐all schedules (e.g. annually). Furthermore, to assist patients and doctors in making shared decisions on biopsy schedules, by providing them quantitative estimates of the burden and benefit of opting for personalised vs any other schedule in AS. Lastly, to externally validate our model and implement it along with personalised schedules in a ready to use web‐application.
Patients and Methods
Repeat prostate‐specific antigen (PSA) measurements, timing and results of previous biopsies, and age at baseline from the world’s largest AS study, Prostate Cancer Research International Active Surveillance (PRIAS; 7813 patients, 1134 experienced upgrading). We fitted a Bayesian joint model for time‐to‐event and longitudinal data to this dataset. We then validated our model externally in the largest six AS cohorts of the Movember Foundation’s third Global Action Plan (GAP3) database (>20 000 patients, 27 centres worldwide). Using the model predicted upgrading risks; we scheduled biopsies whenever a patient’s upgrading risk was above a certain threshold. To assist patients/doctors in the choice of this threshold, and to compare the resulting personalised schedule with currently practiced schedules, along with the timing and the total number of biopsies (burden) planned, for each schedule we provided them with the time delay expected in detecting upgrading (shorter is better).
Results
The cause‐specific cumulative upgrading risk at the 5‐year follow‐up was 35% in PRIAS, and at most 50% in the GAP3 cohorts. In the PRIAS‐based model, PSA velocity was a stronger predictor of upgrading (hazard ratio [HR] 2.47, 95% confidence interval [CI] 1.93–2.99) than the PSA level (HR 0.99, 95% CI 0.89–1.11). Our model had a moderate area under the receiver operating characteristic curve (0.6–0.7) in the validation cohorts. The prediction error was moderate (0.1–0.2) in theGAP3 cohorts where the impact of the PSA level and velocity on upgrading risk was similar to PRIAS, but large (0.2–0.3) otherwise. Our model required re‐calibration of baseline upgrading risk in the validation cohorts. We implemented the validated models and the methodology for personalised schedules in a web‐application (http://tiny.cc/biopsy).
Conclusions
We successfully developed and validated a model for predicting upgrading risk, and providing risk‐based personalised biopsy decisions in AS of prostate cancer. Personalised prostate biopsies are a novel alternative to fixed one‐size‐fits‐all schedules, which may help to reduce unnecessary prostate biopsies, while maintaining cancer control. The model and schedules made available via a web‐application enable shared decision‐making on biopsy schedules by comparing fixed and personalised schedules on total biopsies and expected time delay in detecting upgrading.
Keywords: active surveillance, biopsies, personalised medicine, prostate cancer, shared decision‐making
Introduction
Patients with low‐ and very‐low‐risk screening‐detected localised prostate cancer are usually recommended for active surveillance (AS), instead of immediate radical treatment [1]. In AS, cancer progression is monitored routinely via PSA measurement, DRE, repeat biopsies, and recently, MRI. Among these, the strongest indicator of cancer‐related outcomes is the biopsy Gleason Grade Group (GGG) [2]. When it increases from GGG1 (Gleason 3 + 3) to GGG2 (Gleason 3 + 4) or higher, it is called ‘upgrading’ [3]. Upgrading is an important endpoint in AS upon which patients’ are commonly advised curative treatments [4].
Biopsies in AS are always conducted with a time gap between them. Consequently, upgrading is always detected with a time delay (Fig. 1) that cannot be measured directly. In this regard, to detect upgrading in a timely manner, many patients are prescribed fixed and frequent biopsies, most often annually [5]. However, such one‐size‐fits‐all schedules lead to unnecessary biopsies in slow/non‐progressing patients. Biopsies are invasive, may be painful, and are prone to medical complications such as bleeding and septicaemia [6]. Thus, biopsy burden and patient non‐compliance to frequent biopsies [7] have raised concerns regarding the optimal biopsy schedule [8, 9] in AS.
Fig. 1.
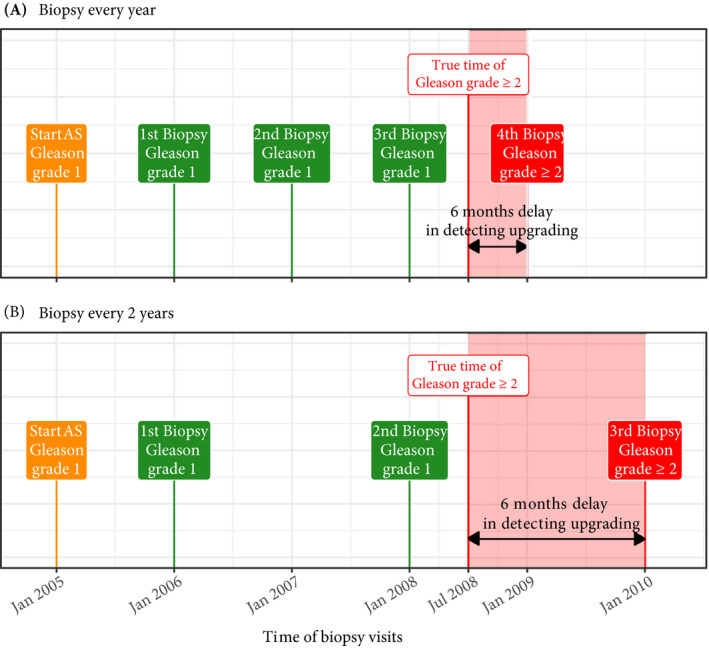
Trade‐off between the timing and number of biopsies (burden) and time delay in detecting Gleason upgrading (shorter is better): the true time of Gleason upgrading (increase in GGG1 to GGG≥2) for the patient in this figure is July 2008. When biopsies are scheduled annually (Panel A), upgrading is detected in January 2009 with a time delay of 6 months, and a total of four biopsies are scheduled. When biopsies are scheduled biennially (Panel B), upgrading is detected in January 2010 with a time delay of 18 months, and a total of three biopsies are scheduled. As biopsies are conducted periodically, the time of upgrading is observed as an interval. For example, between January 2008 and January 2009 in Panel A and between January 2008 and January 2010 in Panel B. The phrase ‘Gleason Grade Group’ is shortened to ‘Gleason grade’ for brevity.
Except for the confirmatory biopsy at 1 year of AS [7], opinions and practice regarding the timing of remaining biopsies lack consensus [10]. Some AS programmes utilise patients’ observed PSA level, DRE, previous biopsy Gleason Grade, and lately, MRI results to decide whether to take biopsies [4, 10, 11]. In contrast, others discourage schedules based on clinical data and MRI results [5, 12], and instead support periodical one‐size‐fits‐all biopsy schedules. Furthermore, some suggest replacing frequent periodical schedules with infrequent ones (e.g. biennially) [8, 13]. Each of these approaches has limitations. For example, one‐size‐fits‐all schedules can lead to many unnecessary biopsies because of differences in baseline upgrading risk across cohorts [8]. Whereas, as observed clinical data have measurement error (e.g. PSA fluctuations) a flaw in using it directly is that it may lead to poor decisions. Also, decisions based on clinical data typically rely only on the latest data point and ignore previous repeated measurements. A novel alternative that counters these drawbacks is first processing patient data via a statistical model, and subsequently using model‐predicted upgrading risks to create personalised biopsy schedules [10] (Fig. 2). While, upgrading‐risk calculators are not new [14, 15, 16, 17], not all are personalised either. Besides, they do not specify how risk predictions can be exploited to create a schedule.
Fig. 2.
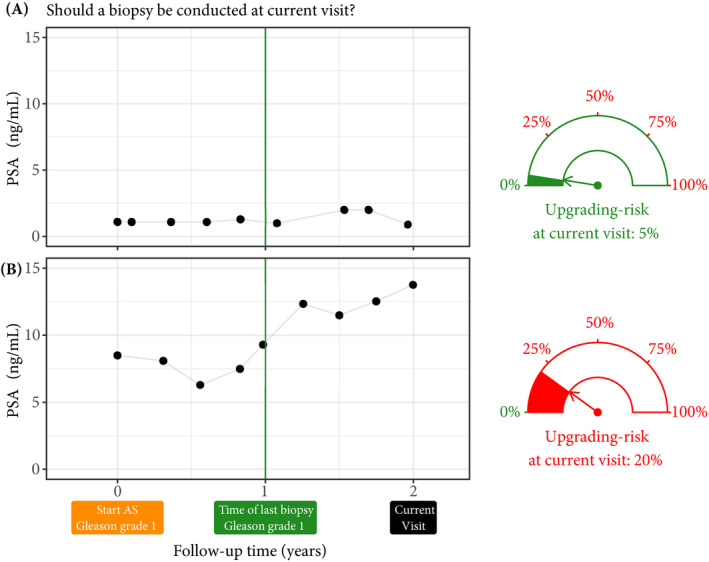
Motivation for upgrading risk based personalised biopsy decisions: to utilise patients’ complete longitudinal data and results from previous biopsies in making biopsy decisions. For this purpose, we first process data using a statistical model and then utilise the patient‐specific predictions for risk of Gleason upgrading to schedule biopsies. For example, Patient A (Panel A) and B (Panel B) had their latest biopsy at 1 year of follow‐up (green vertical line). Patient A’s PSA profile remained stable until his current visit at 2 years, whereas patient B’s profile has shown a rise. Consequently, patient B’s upgrading risk at the current visit (2 years) is higher than that of patient A. This makes patient B a more suitable candidate for biopsy than patient A. Risk estimates in this figure are only illustrative.
The present work was motivated by the problem of scheduling biopsies in AS. We had two goals. First, we wanted to assist practitioners in using clinical data in biopsy decisions in a statistically sound manner. To this end, we planned to develop a robust, generalisable statistical model that provides reliable individual upgrading risk in AS. Subsequently, we would employ these predictions to derive risk‐based personalised biopsy schedules. Our second goal was to enable shared decision‐making on biopsy schedules. We intended to achieve this by allowing patients and doctors to compare the burden and benefit (Fig. 1) of opting for personalised schedules vs periodical schedules vs schedules based on clinical data. Specifically, we proposed timing and number of planned biopsies (more/frequent are burdensome), and the expected time delay in detecting upgrading (shorter is beneficial) for any given schedule. While fulfilling our goals, we wanted to capture the maximum possible information from the available data. Hence, we used all repeated PSA measurements of patients, previous biopsy results, and baseline characteristics. To fit this model, we utilised data of the world’s largest AS study, Prostate Cancer Research International Active Surveillance (PRIAS). To evaluate our model, we externally validated it in the largest six AS cohorts from the Movember Foundation’s third Global Action Plan (GAP3) database [18]. Last, we aimed to implement the validated model and methodology in a web‐application.
Patients and Methods
Study Cohort
For developing a statistical model to predict upgrading risk, we used the world’s largest AS dataset, PRIAS [4], dated April 2019 (Table 1). In PRIAS, biopsies were scheduled at 1, 4, 7, and 10 years, and additional yearly biopsies were scheduled when PSA doubling time was between zero and 10 years. We selected all 7813 patients who had GGG1 at inclusion in AS. Our primary event of interest was an increase in this GGG observed upon repeat biopsy, called ‘upgrading’ (1134 patients). Upgrading is a trigger for treatment advice in PRIAS. Some examples of treatment options in AS are radical prostatectomy, brachytherapy, definitive radiation therapy, and other alternative local treatments such as cryosurgery, high‐intensity focussed ultrasound, and external beam radiation therapy. Comprehensive details on treatment options and their side‐effects are available in European Association of Urology (EAU)‐European Society for Radiotherapy and Oncology (ESTRO)‐International Society of Geriatric Oncology (SIOG) guidelines on prostate cancer [19]. In PRIAS, 2250 patients were provided treatment based on their PSA level, the number of biopsy cores with cancer, or anxiety/other reasons. However, our reason for focussing solely on upgrading was that upgrading is strongly associated with cancer‐related outcomes, and other treatment triggers vary between cohorts [10].
Table 1.
Summary of the PRIAS dataset as of April 2019.
| Characteristic | Value |
|---|---|
| Total patients, n | 7813 |
| Upgrading (primary event), n | 1134 |
| Treatment, n | 2250 |
| Watchful waiting, n | 334 |
| Loss to follow‐up, n | 249 |
| Death (unrelated to prostate cancer), n | 95 |
| Death (related to prostate cancer), n | 2 |
| Age at diagnosis, years, median (IQR) | 66 (61–71) |
| Maximum follow‐up per patient, years, median (IQR) | 1.8 (0.9–4.0) |
| Total PSA measurements, n | 67 578 |
| Number of PSA measurements per patient, median (IQR) | 6 (4–12) |
| PSA level, ng/mL, median (IQR) | 5.7 (4.1–7.7) |
| Total biopsies, n | 15 686 |
| Number of biopsies per patient, median (IQR) | 2 (1–2) |
IQR, interquartile range.
The primary event of interest is upgrading, that is, increase from GGG1 to GGG≥2 [2]. Study protocol URL: https://www.prias‐project.org.
For externally validating our model’s predictions, we selected the following largest (by the number of repeated measurements) six cohorts from Movember Foundation’s GAP3 database [18], version 3.1, covering nearly 73% of the GAP3 patients: the University of Toronto AS (Toronto), Johns Hopkins AS (Hopkins), Memorial Sloan Kettering Cancer Center AS (MSKCC), King’s College London AS (KCL), Michigan Urological Surgery Improvement Collaborative AS (MUSIC), and University of California San Francisco AS (UCSF, version 3.2). Only patients with a GGG1 at the time of inclusion in these cohorts were selected. Summary statistics are presented in Appendix S1.2.
Choice of Predictors
In our model, we used all repeated PSA measurements, the timing of the previous biopsy and Gleason Grade, and age at inclusion in AS. Other predictors, e.g. prostate volume and MRI results, can also be important. MRI is utilised already for targeting biopsies, but regarding its use in deciding the time of biopsies, there are arguments both for and against it [11, 12, 20]. MRI is still a recent addition in most AS protocols. Consequently, repeated MRI data were very sparsely available in both PRIAS and GAP3 databases to make a stable prediction model. Prostate volume data was also sparsely available, especially in the validation cohorts. Based on these reasons, we did not include them in our model.
Statistical Model
Modelling an AS dataset such as PRIAS, posed certain challenges. First, PSA level was measured longitudinally, and over follow‐up time it did not always increase linearly. Consequently, we expect that PSA measurements of a patient are more similar to each other than of another patient. In other words, we needed to accommodate the within‐patient correlation for PSA level. Second, the PSA level was available only until a patient observed upgrading. Thus, we also needed to model the association between the Gleason Grades and PSA profiles of a patient, and handle missing PSA measurements after a patient had upgrading. Third, as the PRIAS biopsy schedule uses PSA, a patient’s observed time of upgrading was also dependent on their PSA. Thus, the effect of PSA on the upgrading risk needed to be adjusted for the effect of PSA on the biopsy schedule. Fourth, many patients obtained treatment and watchful waiting before observing upgrading. As we considered events other than upgrading as censoring, the model needed to account for patients’ reasons for treatment or watchful waiting (e.g. age, treatment based on observed data). A model that handles these challenges in a statistically sound manner is the joint model for time‐to‐event and longitudinal data [14, 21, 22].
Our joint model consisted of two sub‐models; namely, a linear mixed‐effects sub‐model [23] for longitudinally measured PSA (log‐transformed) and a relative‐risk sub‐model (similar to the Cox model) for the interval‐censored time of upgrading. Patient age was used in both sub‐models. Results and timing of the previous negative biopsies were used only in the risk sub‐model. To account for PSA fluctuations [24], we assumed t‐distributed PSA measurement errors. The correlation between PSA measurements of the same patient was established using patient‐specific random‐effects. We fitted a unique curve to the PSA measurements of each patient (Panel A, Fig. 3 [2]). Subsequently, we calculated the mathematical derivative of the patient’s fitted PSA profile (Appendix S1, Equation 2), to obtain his follow‐up time‐specific instantaneous PSA velocity (Panel B, Fig. 3). This instantaneous velocity is a stronger predictor of upgrading than the widely used average PSA velocity [25].
Fig. 3.
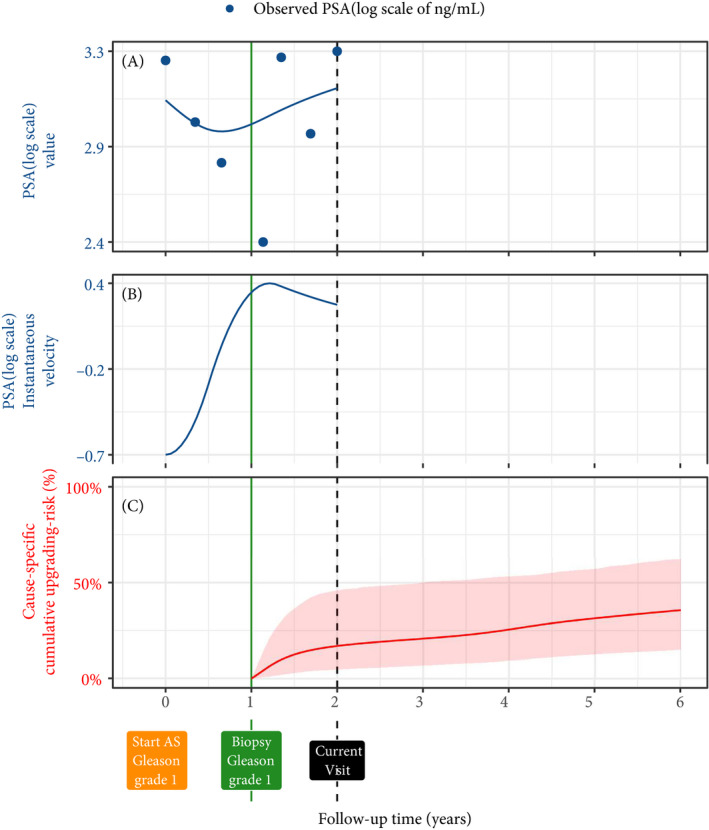
Illustration of the joint model on a real PRIAS patient. Panel A: Observed PSA (blue dots) and fitted PSA (solid blue line), log‐transformed from ng/mL. Panel B: Estimated instantaneous velocity of PSA (log‐transformed). Panel C: Predicted cause‐specific cumulative upgrading risk (95% credible interval shaded). Upgrading is defined as an increase in the GGG1 to GGG≥2 [2]. This upgrading risk is calculated starting from the time of the latest negative biopsy (vertical green line at 1 year of follow‐up). The joint model estimated it by combining the fitted PSA (log scale) level and instantaneous velocity, and time of the latest negative biopsy. Black dashed line at 2 years denotes the time of current visit.
We modelled the impact of PSA on upgrading risk by employing fitted PSA level and instantaneous velocity as predictors in the risk sub‐model (Panel C, Fig. 3). We adjusted the effect of PSA on upgrading risk for the PSA‐dependent PRIAS biopsy schedule by estimating parameters using a full likelihood method (proof in Appendix S1). This approach also accommodates watchful waiting and treatment protocols that are also based on patient data. Specifically, the parameters of our two sub‐models were estimated jointly under the Bayesian paradigm (Appendix S1) using the R package JMbayes [26].
Risk Prediction and Model Validation
Our model provides predictions for upgrading risk over the entire future follow‐up period of a patient (Panel C, Fig. 3). However, we recommend using predictions only after 1 year. This is because most AS programmes recommend a confirmatory biopsy at 1 year, especially to detect patients who may be misdiagnosed as low grade at inclusion in AS. The risk predictions for a patient are not calculated only once. Rather, as illustrated in Fig. S5 of Appendix S2, risk‐predictions update over the follow‐up, to account for additional patient data (e.g. new biopsy results, PSA measurements) that become available. We validated our model internally in the PRIAS cohort, and externally in the largest six GAP3 database cohorts. We employed calibration plots [27, 28] and follow‐up time‐dependent mean absolute risk prediction error (MAPE) [29] to graphically and quantitatively evaluate our model’s risk prediction accuracy, respectively. We assessed our model’s ability to discriminate between patients who experience/do not experience upgrading via the time‐dependent area under the receiver operating characteristic curve (AUC) [29].
The aforementioned time‐dependent AUC and MAPE [29] are temporal extensions of their standard versions [28] in a longitudinal setting. Specifically, at every 6 months of follow‐up, we calculated a unique AUC and MAPE for predicting upgrading risk in the subsequent 1 year (Appendix S2.1). For emulating a realistic situation, we calculated the AUC and MAPE at each follow‐up using only the validation data available until that follow‐up. For example, calculations for AUC and MAPE for the time interval 2–3 years do not utilise data of patients who progressed before 2 years. Last, to resolve any potential model miscalibration in the validation cohorts, we aimed to re‐calibrate our model’s baseline hazard of upgrading (Appendix S2.1), individually for each cohort.
Results
The cause‐specific cumulative upgrading risk at the 5‐year follow‐up was 35% in PRIAS and at most 50% in the validation cohorts (Panel B, Fig. 4). In the fitted PRIAS model, the adjusted hazard ratio (aHR) of upgrading for an increase in patient age from 61 to 71 years (25th to 75th percentile) was 1.45 (95% CI 1.30–1.63). For an increase in fitted PSA level from 2.36 to 3.07 ng/mL (25th to 75th percentile, log scale), the aHR was 0.99 (95% CI 0.89–1.11). The strongest predictor of upgrading risk was instantaneous PSA velocity, with an increase from −0.09 to 0.31 (25th to 75th percentile), giving an aHR of 2.47 (95% CI 1.93–2.99). The aHR for PSA level and velocity was different in each GAP3 cohort (Appendix S1.3: Table S8).
Fig. 4.
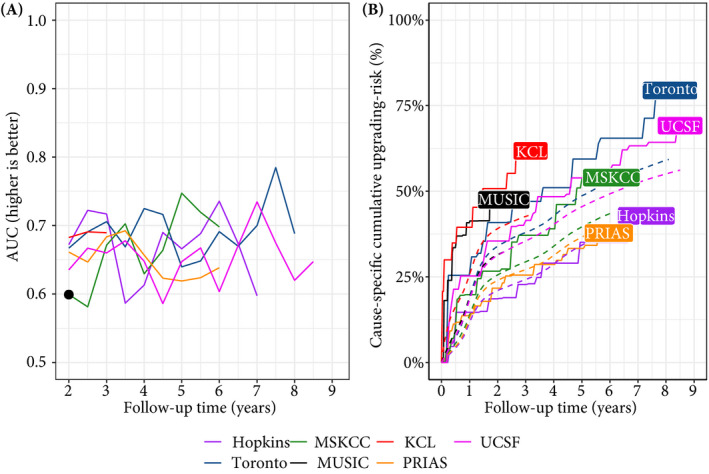
Model validation results. Panel A: time‐dependent AUC (measure of discrimination). AUC at 1 year is not shown because we do not intend to replace the confirmatory biopsy at 1 year. Panel B: calibration‐at‐large indicates model miscalibration. This is because solid lines depicting the non‐parametric estimate of the cause‐specific cumulative upgrading risk [30], and dashed lines showing the average cause‐specific cumulative upgrading risk obtained using the joint model fitted to the PRIAS dataset, are not overlapping. Re‐calibrating the baseline hazard of upgrading resolved this issue (Appendix S2.1: Fig. S6). Full names of cohorts are :PRIAS, Prostate Cancer International Active Surveillance; Toronto, University of Toronto Active Surveillance; Hopkins, Johns Hopkins Active Surveillance; MSKCC, Memorial Sloan Kettering Cancer Center Active Surveillance; KCL, King’s College London Active Surveillance; MUSIC, Michigan Urological Surgery Improvement Collaborative Active Surveillance; UCSF, University of California San Francisco AS.
The time‐dependent AUC, calibration plot, and time‐dependent MAPE of our model are shown in Fig. 4, and Appendix S2.1: Fig. S8. In all cohorts, time‐dependent AUC was moderate (0.6–0.7) over the whole follow‐up period. Time‐dependent MAPE was moderate (0.1–0.2) in those cohorts where the impact of PSA on upgrading risk was similar to PRIAS (e.g. Hopkins cohort, Appendix S1.3: Table S8), and large (0.2–0.3) otherwise. Our model was miscalibrated for the validation cohorts (Panel B, Fig. 4) because cohorts had differences in inclusion criteria (e.g. PSA density) and follow‐up protocols [18], which were not accounted for in our model. Consequently, the PRIAS‐based model’s fitted baseline hazard did not correspond to the baseline hazard in the validation cohorts. To solve this problem, we re‐calibrated the baseline hazard of upgrading in the validation cohorts (Appendix S2.1: Fig. S6). We compared risk predictions from the re‐calibrated models, with predictions from separately fitted cohort‐specific joint models (Appendix S2.1: Fig. S7). The difference in predictions was lowest in the Hopkins cohort (impact of PSA on upgrading risk similar to PRIAS). Comprehensive results are in Appendix S1.3 and Appendix S2.
Personalised Biopsy Schedules
We used the PRIAS‐based fitted model to create personalised biopsy schedules for real PRIAS patients. Particularly, first using the model and patient’s observed data; we predicted his cumulative upgrading risk (Fig. 5) on all of his future follow‐up visits (biannually in PRIAS). Subsequently, we planned biopsies on those future visits where his conditional cumulative upgrading risk was more than a certain threshold (see Appendix S3 for mathematical details). The choice of this threshold dictates the timing of biopsies in a risk‐based personalised schedule. For example, personalised schedules based on 5% and 10% risk thresholds are shown in Fig. 5, and in Appendix S3.1: Figs S10–S12.
Fig. 5.
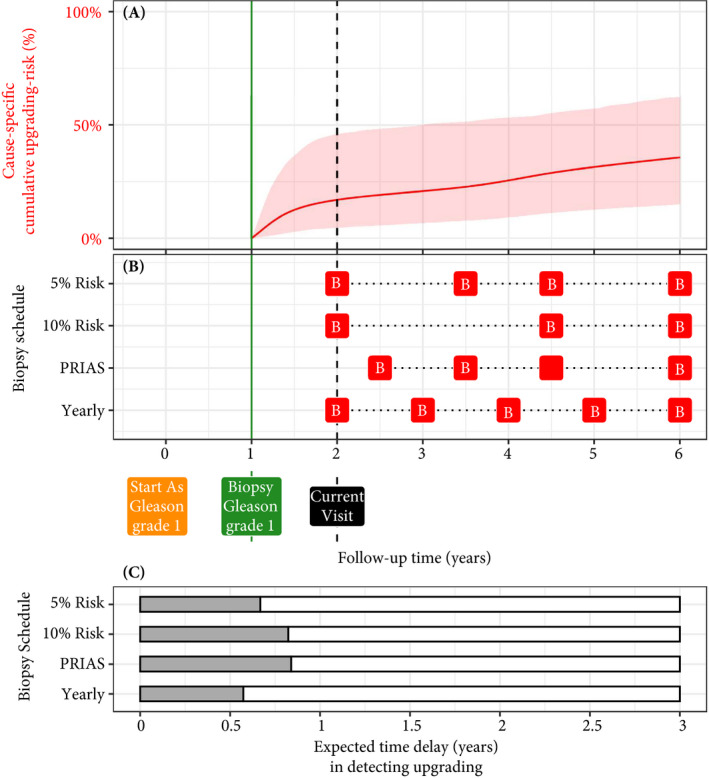
Illustration of personalised and fixed schedules of biopsies for the patient from Figure 3. Panel A: Predicted cumulative upgrading risk (95% credible interval shaded). Panel B: Different biopsy schedules with a red ‘B’ indicating a future biopsy. Risk: 5% and Risk: 10% are personalised schedules in which a biopsy is planned whenever the conditional cause‐specific cumulative upgrading risk is above 5% or 10% risk, respectively. Smaller risk thresholds lead to more frequently planned biopsies. Green vertical line at 1 year is the time of the latest negative biopsy. Black dashed line at 2 years denotes the time of the current visit. Panel C: Expected time delay in detecting upgrading (years) if patient progresses before 6 years. The maximum time delay with limited adverse consequences is 3 years [13]. A compulsory biopsy was scheduled at 6 years (maximum biopsy scheduling time in PRIAS, Appendix S3) in all schedules for a meaningful comparison between delays.
To facilitate the choice of a risk threshold, and for comparing the consequences of opting for a risk‐based schedule vs any other schedule (e.g. annual, PRIAS), we predict expected time delay in detecting upgrading for following a schedule. We were able to predict this delay for any schedule. For example, in Panel C of Fig. 5, the annual schedule has the least expected delay. In contrast, a personalised schedule based on a 10% risk threshold has a slightly larger expected delay, but it also schedules much fewer biopsies. An important aspect of this delay is that it is personalised as well. That is, even if two different patients are prescribed the same biopsy schedule, their expected delays will be different. This is because delay is estimated using all available clinical data of the patient (Appendix S3). While the timing and the total number of planned biopsies denote the burden of a schedule, a shorter expected time delay in detecting upgrading can be a benefit. These two, along with other measures such as a patient’s comorbidities, anxiety, etc., can help to make an informed biopsy decision.
Web‐application
We implemented the PRIAS‐based model, re‐calibrated models for GAP3 cohorts, and personalised schedules in a user‐friendly web‐application https://emcbiostatistics.shinyapps.io/prias_biopsy_recommender/ . This application works on both desktop and mobile devices. Users must first choose the cohort to which the patient belongs (left panel), and then upload patient data in Microsoft Excel format. Internally, the web‐application loads the appropriate validated and re‐calibrated model for that cohort. The maximum follow‐up time up to which predictions can be obtained depends on each cohort (Appendix S2.1: Table S9). The web‐application supports personalised, annual, and PRIAS schedules. For personalised schedules, users can control the choice of risk threshold. The web‐application also compares the resulting risk‐based schedule’s timing of biopsies, and expected time delay in detecting upgrading, with annual and PRIAS schedules, to enable sharing biopsy decision‐making.
Discussion
We successfully developed and externally validated a statistical model for predicting upgrading risk [3] in prostate cancer AS, and providing risk‐based personalised biopsy decisions. Our present work has four novel features over earlier risk calculators [14, 15]. First, our model was fitted to the world’s largest AS dataset, PRIAS, and externally validated in the largest six cohorts of the Movember Foundation’s GAP3 database [18]. Second, the model predicts a patient’s current and future upgrading risk in a personalised manner. Third, using the predicted risks, we created personalised biopsy schedules. We also calculated the expected time delay in detecting upgrading (less is beneficial) for following any schedule. Thus, patients/doctors can compare schedules before making a choice. Fourth, we implemented our methodology in a user‐friendly web‐application (https://emcbiostatistics.shinyapps.io/prias_biopsy_recommender/) for both PRIAS and the validated cohorts.
Our present model and methods can be useful for numerous patients from PRIAS and the validated GAP3 cohorts (nearly 73% of all GAP3 patients). The model utilises all repeated PSA measurements, results of previous biopsies, and baseline characteristics of a patient. We could not include MRI and PSA density because of sparsely available data in both the PRIAS and GAP3 databases. But, our present model is extendable to include them in the near future. A benefit of our present model is that it allows the biopsy schedule, schedule of longitudinal measurements, and loss to follow‐up in each cohort to depend on patient age and PSA characteristics. Consequently, in future, when MRI data is included in the model, our model will also accommodate biopsy schedules dependent on MRI data or MRI schedules dependent on previous biopsy results, PSA characteristics, and age of the patient (mathematical proof in Appendix S1.2). An additional advantage of our present model and resulting personalised schedules is that they update as more patient data becomes available over follow‐up. The current discrimination ability of our model, exhibited by the time‐dependent AUC, was between 0.6 and 0.7 over‐follow. While this is moderate, it is also so because unlike the standard AUC [28] the time‐dependent AUC is more conservative, as it utilises only the validation data available until the time at which it is calculated. The same holds for the time‐dependent MAPE, although MAPE varied much more between cohorts than the AUC. In cohorts where the effect size for the impact of PSA level and velocity on upgrading risk was similar to that for PRIAS (e.g. Hopkins cohort), the MAPE was moderate. Otherwise, the MAPE was large (e.g. KCL and MUSIC cohorts). We required re‐calibration of our model’s baseline hazard of upgrading for all the validation cohorts.
The clinical implications of our present work are as follows. First, the cause‐specific cumulative upgrading risk at the 5‐year follow‐up was at most 50% in all cohorts (Panel B, Fig. 4). That is, many patients may not require some of the biopsies planned in the first 5 years of AS. Given the non‐compliance and burden of frequent biopsies [7], the availability of our methodology as a web‐application may encourage patients/doctors to consider upgrading risk‐based personalised schedules instead. Despite the moderate predictive performance, we expect the overall impact of our model to be positive. There are two reasons for this. First, the risk of adverse outcomes because of personalised schedules is quite low because of the low rate of metastases and prostate cancer specific mortality in AS patients (Table 1). Second, studies [8, 31] have suggested that after the confirmatory biopsy at 1 year of follow‐up, biopsies may be done as infrequently as every 2–3 years, with limited adverse consequences. In other words, longer delays in detecting upgrading may be acceptable after the first negative biopsy. To evaluate the potential harm of personalised schedules, we compared them with fixed schedules in a realistic and extensive simulation study [32]. We concluded that personalised schedules plan, on average, six fewer biopsies compared to annual schedule and two fewer biopsies than the PRIAS schedule in slow/non‐progressing AS patients, while maintaining almost the same time delay in detecting upgrading as the PRIAS schedule. Personalised schedules with different risk thresholds indeed have different performances across cohorts. Thus, to assist patients/doctors in choosing between fixed schedules and personalised schedules based on different risk thresholds, the web‐application provides a patient‐specific estimate of the expected time delay in detecting upgrading, for both personalised and fixed schedules. We hope that access to these estimates will objectively address patient apprehensions regarding adverse outcomes in AS. Last, we note that our web‐application should only be used to decide biopsies after the compulsory confirmatory biopsy at 1 year of follow‐up.
This work has certain limitations. Predictions for upgrading risk and personalised schedules are available only for a currently limited, cohort‐specific, follow‐up period (Appendix S2.1: Table S9). This problem can be mitigated by re‐fitting the model with new follow‐up data in the future. Recently, some cohorts started utilising MRI to explore the possibility of targeting visible lesions by biopsy. Presently, the GAP3 database has limited PSA density and MRI follow‐up data available. As PSA density is used as an entry criterion in some AS studies, including it as a predictor can improve the model. In this regard, the present model can be extended to include both MRI and PSA density data as predictors when they become available in the future. Our model schedules biopsies in a personalised manner, but the patient burden can be decreased even more if we also personalise the schedule of PSA measurements. A caveat of doing so is that reduction in the number of PSA measurements can also lead to an increase in the variance of risk estimates, and also affect the performance of personalised schedules. Although we have done a simulation study to conclude that personalised schedules may not be any more harmful than PRIAS or an annual schedule [32], with an infrequent PSA schedule, these conclusions may not hold. Hence, we do not recommend any changes in the schedule of PSA measurements from the current protocol of PSA measurements every 6 months. At the same time, personalising the schedule of both biopsies and PSA measurements together is a research problem we intend to tackle in the near future. We scheduled biopsies using cause‐specific cumulative upgrading risk, which ignores competing events such as treatment based on the number of positive biopsy cores. Employing a competing‐risk model may lead to improved personalised schedules. Upgrading is susceptible to inter‐observer variation too. Models that account for this variation [14, 33] will be interesting to investigate further. Even with an enhanced risk‐prediction model, the methodology for personalised scheduling and calculation of expected time delay (Appendix S3) need not change. Last, our web application only allows uploading patient data in Microsoft Excel format. Connecting it with patient databases can increase usability.
Conclusions
We successfully developed a statistical model and methodology for predicting upgrading risk, and providing risk‐based personalised biopsy decisions, in prostate cancer AS. We externally validated our model, covering nearly 73% patients from the Movember Foundations’ GAP3 database. The model, made available via a user‐friendly web‐application (https://emcbiostatistics.shinyapps.io/prias_biopsy_recommender/), enables shared decision‐making of biopsy schedules by comparing fixed and personalised schedules on total biopsies and expected time delay in detecting upgrading. Novel biomarkers and MRI data can be added as predictors in the model to improve predictions in the future. Re‐calibration of baseline upgrading risk is advised for cohorts not validated in this work.
Author Contributions
Anirudh Tomer had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Tomer, Nieboer, Roobol, Bjartell, and Rizopoulos. Acquisition of data: Tomer, Nieboer, and Roobol. Analysis and interpretation of data: Tomer, Nieboer, and Rizopoulos. Drafting of the manuscript: Tomer, and Rizopoulos. Critical revision of the manuscript for important intellectual content: Tomer, Nieboer, Roobol, Bjartell, Steyerberg, and Rizopoulos. Statistical analyses: Tomer, Nieboer, Steyerberg, and Rizopoulos. Obtaining funding: Roobol, Steyerberg, and Rizopoulos. Administrative, technical or material support: Nieboer. Supervision: Roobol, and Rizopoulos. Other: none.
Conflicts of Interest
The authors do not report any conflict of interest, and have nothing to disclose.
Abbreviations
- AS
active surveillance
- AUC
area under the receiver operating characteristic curve
- GAP3
Movember Foundation’s third Global Action Plan
- GGG
Gleason Grade Group
- (a)HR
(adjusted) hazard ratio
- KCL
King’s College London
- MAPE
mean absolute risk prediction error
- MSKCC
Memorial Sloan Kettering Cancer Center
- MUSIC
Michigan Urological Surgery Improvement Collaborative
- PRIAS
Prostate Cancer Research International Active Surveillance
- UCSF
University of California San Francisco
Supporting information
Appendix S1. A joint model for the longitudinal PSA, and time to Gleason upgrading.
Appendix S1.1. Assumption of t‐distributed (d.f. = 3) error terms.
Appendix S1.2. PSA‐dependent biopsy schedule of PRIAS, and competing risks.
Appendix S1.3. Results.
Appendix S2. Risk predictions for upgrading.
Appendix S2.1. Validation of risk predictions.
Appendix S3. Personalised biopsies based on cause‐specific cumulative upgrading risk.
Appendix S3.1. Expected time delay in detecting upgrading.
Appendix S4. Web‐application for practical use of personalised schedule of biopsies.
Appendix S5. Source code.
Appendix S5.1. Fitting the joint model to the PRIAS dataset.
Appendix S5.2. Validation of predictions of upgrading.
Appendix S5.3. Creating personalised schedules of biopsies.
Appendix S5.4. Source code for web application.
Acknowledgements
We thank Jozien Helleman from the Department of Urology, Erasmus University Medical Center, for coordinating the project. The first and last authors would like to acknowledge support by Nederlandse Organisatie voor Wetenschappelijk Onderzoek (the National Research Council of the Netherlands) VIDI grant nr. 016.146.301, and Erasmus University Medical Center funding. Part of this work was carried out on the Dutch national einfrastructure with the support of SURF Cooperative. The authors also thank the Erasmus University Medical Center’s Cancer Computational Biology Center for giving access to their IT‐infrastructure and software that was used for the computations and data analysis in this study. The PRIAS website is funded by the Prostate Cancer Research Foundation, Rotterdam (SWOP). We would like to thank the PRIAS consortium for enabling this research project. This work was supported by the Movember Foundation. The funder did not play any role in the study design, collection, analysis or interpretation of data, or in the drafting of this paper.
Appendix A.
Members of The Movember Foundation’s Global Action Plan Prostate Cancer Active Surveillance (GAP3) consortium
Principle Investigators
Bruce Trock (Johns Hopkins University, The James Buchanan Brady Urological Institute, Baltimore, USA), Behfar Ehdaie (Memorial Sloan Kettering Cancer Center, New York, USA), Peter Carroll (University of California San Francisco, San Francisco, USA), Christopher Filson (Emory University School of Medicine, Winship Cancer Institute, Atlanta, USA), Jeri Kim/Christopher Logothetis (MD Anderson Cancer Centre, Houston, USA), Todd Morgan (University of Michigan and Michigan Urological Surgery Improvement Collaborative (MUSIC), Michigan, USA), Laurence Klotz (University of Toronto, Sunnybrook Health Sciences Centre, Toronto, Ontario, Canada), Tom Pickles (University of British Columbia, BC Cancer Agency, Vancouver, Canada), Eric Hyndman (University of Calgary, Southern Alberta Institute of Urology, Calgary, Canada), Caroline Moore (University College London & University College London Hospital Trust, London, UK), Vincent Gnanapragasam (University of Cambridge & Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK), Mieke Van Hemelrijck (King’s College London, London, UK & Guy’s and St Thomas’ NHS Foundation Trust, London, UK), Prokar Dasgupta (Guy’s and St Thomas’ NHS Foundation Trust, London, UK), Chris Bangma (Erasmus Medical Center, Rotterdam, The Netherlands/representative of Prostate cancer Research International Active Surveillance (PRIAS) consortium), Monique Roobol (Erasmus Medical Center, Rotterdam, The Netherlands/representative of Prostate cancer Research International Active Surveillance (PRIAS) consortium), The PRIAS study group, Arnauld Villers (Lille University Hospital Center, Lille, France), Antti Rannikko (Helsinki University and Helsinki University Hospital, Helsinki, Finland), Riccardo Valdagni (Department of Oncology and Hemato‐oncology, Università degli Studi di Milano, Radiation Oncology 1 and Prostate Cancer Program, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy), Antoinette Perry (University College Dublin, Dublin, Ireland), Jonas Hugosson (Sahlgrenska University Hospital, Göteborg, Sweden), Jose Rubio‐Briones (Instituto Valenciano de Oncologia, Valencia, Spain), Anders Bjartell (Skåne University Hospital, Malmö, Sweden), Lukas Hefermehl (Kantonsspital Baden, Baden, Switzerland), Lee Lui Shiong (Singapore General Hospital, Singapore, Singapore), Mark Frydenberg (Monash Health; Monash University, Melbourne, Australia), Yoshiyuki Kakehi/Mikio Sugimoto (Kagawa University Faculty of Medicine, Kagawa, Japan), Byung Ha Chung (Gangnam Severance Hospital, Yonsei University Health System, Seoul, Republic of Korea).
Pathologist
Theo van der Kwast (Princess Margaret Cancer Centre, Toronto, Canada).
Technology Research Partners
Henk Obbink (Royal Philips, Eindhoven, the Netherlands), Wim van der Linden (Royal Philips, Eindhoven, the Netherlands), Tim Hulsen (Royal Philips, Eindhoven, the Netherlands), Cees de Jonge (Royal Philips, Eindhoven, the Netherlands).
Advisory Regional Statisticians
Mike Kattan (Cleveland Clinic, Cleveland, Ohio, USA), Ji Xinge (Cleveland Clinic, Cleveland, Ohio, USA), Kenneth Muir (University of Manchester, Manchester, UK), Artitaya Lophatananon (University of Manchester, Manchester, UK), Michael Fahey (Epworth Health‐Care, Melbourne, Australia), Ewout Steyerberg (Erasmus Medical Center, Rotterdam, The Netherlands), Daan Nieboer (Erasmus Medical Center, Rotterdam, The Netherlands); Liying Zhang (University of Toronto, Sunnybrook Health Sciences Centre, Toronto, Ontario, Canada).
Executive Regional Statisticians
Ewout Steyerberg (Erasmus Medical Center, Rotterdam, The Netherlands), Daan Nieboer (Erasmus Medical Center, Rotterdam, The Netherlands); Kerri Beckmann (King’s College London, London, UK & Guy’s and St Thomas’ NHS Foundation Trust, London, UK), Brian Denton (University of Michigan, Michigan, USA), Andrew Hayen (University of Technology Sydney, Australia), Paul Boutros (Ontario Institute of Cancer Research, Toronto, Ontario, Canada).
Clinical Research Partners’ IT Experts
Wei Guo (Johns Hopkins University, The James Buchanan Brady Urological Institute, Baltimore, USA), Nicole Benfante (Memorial Sloan Kettering Cancer Center, New York, USA), Janet Cowan (University of California San Francisco, San Francisco, USA), Dattatraya Patil (Emory University School of Medicine, Winship Cancer Institute, Atlanta, USA), Emily Tolosa (MD Anderson Cancer Centre, Houston, Texas, USA), Tae‐Kyung Kim (University of Michigan and Michigan Urological Surgery Improvement Collaborative, Ann Arbor, Michigan, USA), Alexandre Mamedov (University of Toronto, Sunnybrook Health Sciences Centre, Toronto, Ontario, Canada), Vincent LaPointe (University of British Columbia, BC Cancer Agency, Vancouver, Canada), Trafford Crump (University of Calgary, Southern Alberta Institute of Urology, Calgary, Canada), Vasilis Stavrinides (University College London & University College London Hospital Trust, London, UK), Jenna Kimberly‐Duffell (University of Cambridge & Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK), Aida Santaolalla (King’s College London, London, UK & Guy’s and St Thomas’ NHS Foundation Trust, London, UK), Daan Nieboer (Erasmus Medical Center, Rotterdam, The Netherlands), Jonathan Olivier (Lille University Hospital Center, Lille, France), Tiziana Rancati (Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Milan, Italy), Helén Ahlgren (Sahlgrenska University Hospital, Göteborg, Sweden), Juanma Mascarós (Instituto Valenciano de Oncología, Valencia, Spain), Annica Löfgren (Skåne University Hospital, Malmö, Sweden), Kurt Lehmann (Kantonsspital Baden, Baden, Switzerland), Catherine Han Lin (Monash University and Epworth HealthCare, Melbourne, Australia), Hiromi Hirama (Kagawa University, Kagawa, Japan), Kwang Suk Lee (Yonsei University College of Medicine, Gangnam Severance Hospital, Seoul, Korea).
Research Advisory Committee
Guido Jenster (Erasmus MC, Rotterdam, the Netherlands), Anssi Auvinen (University of Tampere, Tampere, Finland), Anders Bjartell (Skåne University Hospital, Malmö, Sweden), Masoom Haider (University of Toronto, Toronto, Canada), Kees van Bochove (The Hyve B.V. Utrecht, Utrecht, the Netherlands), Ballentine Carter (Johns Hopkins University, Baltimore, USA – until 2018).
Management Team
Sam Gledhill (Movember Foundation, Melbourne, Australia), Mark Buzza/Michelle Kouspou (Movember Foundation, Melbourne, Australia), Chris Bangma (Erasmus Medical Center, Rotterdam, The Netherlands), Monique Roobol (Erasmus Medical Center, Rotterdam, The Netherlands), Sophie Bruinsma/Jozien Helleman (Erasmus Medical Center, Rotterdam, The Netherlands).
Contributor Information
Anirudh Tomer, Email: anirudhtomer@gmail.com.
the Movember Foundation’s Global Action Plan Prostate Cancer Active Surveillance (GAP3) consortium:
Bruce Trock, Behfar Ehdaie, Peter Carroll, Christopher Filson, Jeri Kim, Christopher Logothetis, Todd Morgan, Laurence Klotz, Tom Pickles, Eric Hyndman, Caroline Moore, Vincent Gnanapragasam, Mieke Van Hemelrijck, Prokar Dasgupta, Chris Bangma, Arnauld Villers, Antti Rannikko, Riccardo Valdagni, Antoinette Perry, Jonas Hugosson, Jose Rubio‐Briones, Lukas Hefermehl, Lee Lui Shiong, Mark Frydenberg, Yoshiyuki Kakehi, Mikio Sugimoto, Theo van der Kwast, Henk Obbink, Wim van der Linden, Tim Hulsen, Cees de Jonge, Mike Kattan, Ji Xinge, Kenneth Muir, Artitaya Lophatananon, Michael Fahey, Liying Zhang, Kerri Beckmann, Brian Denton, Andrew Hayen, Paul Boutros, Wei Guo, Nicole Benfante, Janet Cowan, Dattatraya Patil, Emily Tolosa, Tae‐Kyung Kim, Alexandre Mamedov, Vincent LaPointe, Trafford Crump, Vasilis Stavrinides, Jenna Kimberly‐Duffell, Aida Santaolalla, Jonathan Olivier, Tiziana Rancati, Helén Ahlgren, Juanma Mascarós, Annica Löfgren, Kurt Lehmann, Catherine Han Lin, Hiromi Hirama, Kwang Suk Lee, Guido Jenster, Anssi Auvinen, Masoom Haider, Kees van Bochove, Ballentine Carter, Sam Gledhill, Mark Buzza, Michelle Kouspou, Sophie Bruinsma, and Jozien Helleman
References
- 1. Briganti A, Fossati N, Catto JW et al. Active surveillance for low‐risk prostate cancer: the European Association of Urology position in 2018. Eur Urol 2018; 74: 357–68 [DOI] [PubMed] [Google Scholar]
- 2. Epstein JI, Egevad L, Amin MB, Delahunt B, Srigley JR, Humphrey PA. The 2014 international society of urological pathology (ISUP) consensus conference on Gleason grading of prostatic carcinoma. Am J Surg Pathol 2016; 40: 244–52 26492179 [Google Scholar]
- 3. Bruinsma SM, Roobol MJ, Carroll PR et al. Expert consensus document: semantics in active surveillance for men with localized prostate cancer—results of a modified Delphi consensus procedure. Nat Rev Urol 2017; 14: 312–22 [DOI] [PubMed] [Google Scholar]
- 4. Bul M, Zhu X, Valdagni R et al. Active surveillance for low‐risk prostate cancer worldwide: the PRIAS study. Eur Urol 2013; 63: 597–603 [DOI] [PubMed] [Google Scholar]
- 5. Loeb S, Carter HB, Schwartz M, Fagerlin A, Braithwaite RS, Lepor H. Heterogeneity in active surveillance protocols worldwide. Rev Urol 2014; 16: 202–3 [PMC free article] [PubMed] [Google Scholar]
- 6. Loeb S, Vellekoop A, Ahmed HU et al. Systematic review of complications of prostate biopsy. Eur Urol 2013; 64: 876–92 [DOI] [PubMed] [Google Scholar]
- 7. Bokhorst LP, Alberts AR, Rannikko A et al. Compliance rates with the Prostate Cancer Research International Active Surveillance (PRIAS) protocol and disease reclassification in noncompliers. Eur Urol 2015; 68: 814–21 [DOI] [PubMed] [Google Scholar]
- 8. Inoue LY, Lin DW, Newcomb LF et al. Comparative analysis of biopsy upgrading in four prostate cancer active surveillance cohorts. Ann Intern Med 2018; 168: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bratt O, Carlsson S, Holmberg E et al. The study of active monitoring in Sweden (SAMS): a randomized study comparing two different follow‐up schedules for active surveillance of low‐risk prostate cancer. Scand J Urol 2013; 47: 347–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nieboer D, Tomer A, Rizopoulos D, Roobol MJ, Steyerberg EW. Active surveillance: a review of risk‐based, dynamic monitoring. Transl Androl Urol 2018; 7: 106–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kasivisvanathan V, Giganti F, Emberton M, Moore CM. Magnetic resonance imaging should be used in the active surveillance of patients with localised prostate cancer. Eur Urol 2020; 77: 318–9 [DOI] [PubMed] [Google Scholar]
- 12. Chesnut GT, Vertosick EA, Benfante N et al. Role of changes in magnetic resonance imaging or clinical stage in evaluation of disease progression for men with prostate cancer on active surveillance. Eur Urol 2020; 77: 501–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Carvalho TM, Heijnsdijk EA, de Koning HJ. Estimating the risks and benefits of active surveillance protocols for prostate cancer: a microsimulation study. BJU Int 2017; 119: 560–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coley RY, Zeger SL, Mamawala M, Pienta KJ, Carter HB. Prediction of the pathologic Gleason score to inform a personalized management program for prostate cancer. Eur Urol 2017; 72: 135–41 [DOI] [PubMed] [Google Scholar]
- 15. Ankerst DP, Xia J, Thompson IM Jr et al. Precision medicine in active surveillance for prostate cancer: development of the canary–early detection research network active surveillance biopsy risk calculator. Eur Urol 2015; 68: 1083–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Partin AW, Yoo J, Carter HB et al. The use of prostate specific antigen, clinical stage and Gleason score to predict pathological stage in men with localized prostate cancer. J Urol 1993; 150: 110–4 [DOI] [PubMed] [Google Scholar]
- 17. Makarov DV, Trock BJ, Humphreys EB et al. Updated nomogram to predict pathologic stage of prostate cancer given prostate‐specific antigen level, clinical stage, and biopsy Gleason score (Partin tables) based on cases from 2000 to 2005. Urology 2007; 69: 1095–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bruinsma SM, Zhang L, Roobol MJ et al. The Movember foundation’s GAP3 cohort: a profile of the largest global prostate cancer active surveillance database to date. BJU Int 2018; 121: 737–44 [DOI] [PubMed] [Google Scholar]
- 19. Mottet N, Bellmunt J, Bolla M et al. EAU‐ESTRO‐SIOG guidelines on prostate cancer. part 1: screening, diagnosis, and local treatment with curative intent. Eur Urol 2017; 71: 618–29 [DOI] [PubMed] [Google Scholar]
- 20. Schoots IG, Petrides N, Giganti F et al. Magnetic resonance imaging in active surveillance of prostate cancer: a systematic review. Eur Urol 2015; 67: 627–36 [DOI] [PubMed] [Google Scholar]
- 21. Tomer A, Nieboer D, Roobol MJ, Steyerberg EW, Rizopoulos D. Personalized schedules for surveillance of low‐risk prostate cancer patients. Biometrics 2019; 75: 153–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rizopoulos D. Joint Models for Longitudinal and Time‐to‐Event Data: With Applications in R. Boca Raton, FL, USA: CRC Press, 2012. ISBN 9781439872864. [Google Scholar]
- 23. Laird NM, Ware JH. Random‐effects models for longitudinal data. Biometrics 1982; 38: 963–74 [PubMed] [Google Scholar]
- 24. Nixon RG, Wener MH, Smith KM, Parson RE, Strobel SA, Brawer MK. Biological variation of prostate specific antigen levels in serum: an evaluation of day‐to‐day physiological fluctuations in a well‐defined cohort of 24 patients. J Urol 1997; 157: 2183–90 [DOI] [PubMed] [Google Scholar]
- 25. Cooperberg MR, Brooks JD, Faino AV et al. Refined analysis of prostate‐specific antigen kinetics to predict prostate cancer active surveillance outcomes. Eur Urol 2018; 74: 211–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rizopoulos D. The R package JMbayes for fitting joint models for longitudinal and time‐to‐event data using MCMC. J Stat Softw 2016; 72: 1–46 [Google Scholar]
- 27. Royston P, Altman DG. External validation of a Cox prognostic model: principles and methods. BMC Med Res Methodol 2013; 13: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steyerberg EW, Vickers AJ, Cook NR et al. Assessing the performance of prediction models: a framework for some traditional and novel measures. Epidemiology 2010; 21: 128–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rizopoulos D, Molenberghs G, Lesaffre EM. Dynamic predictions with time‐dependent covariates in survival analysis using joint modeling and landmarking. Biom J 2017; 59: 1261–76 [DOI] [PubMed] [Google Scholar]
- 30. Turnbull BW. The empirical distribution function with arbitrarily grouped, censored and truncated data. J Roy Stat Soc: Ser B (Methodol) 1976; 38: 290–5 [Google Scholar]
- 31. de Carvalho TM, Heijnsdijk EA, de Koning HJ. When should active surveillance for prostate cancer stop if no progression is detected? Prostate 2017; 77: 962–9 [DOI] [PubMed] [Google Scholar]
- 32. Tomer A, Rizopoulos D, Nieboer D, Drost FJ, Roobol MJ, Steyerberg EW. Personalized decision making for biopsies in prostate cancer active surveillance programs. Med Decis Making 2019; 39: 499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Balasubramanian R, Lagakos SW. Estimation of a failure time distribution based on imperfect diagnostic tests. Biometrika 2003; 90: 171–82 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. A joint model for the longitudinal PSA, and time to Gleason upgrading.
Appendix S1.1. Assumption of t‐distributed (d.f. = 3) error terms.
Appendix S1.2. PSA‐dependent biopsy schedule of PRIAS, and competing risks.
Appendix S1.3. Results.
Appendix S2. Risk predictions for upgrading.
Appendix S2.1. Validation of risk predictions.
Appendix S3. Personalised biopsies based on cause‐specific cumulative upgrading risk.
Appendix S3.1. Expected time delay in detecting upgrading.
Appendix S4. Web‐application for practical use of personalised schedule of biopsies.
Appendix S5. Source code.
Appendix S5.1. Fitting the joint model to the PRIAS dataset.
Appendix S5.2. Validation of predictions of upgrading.
Appendix S5.3. Creating personalised schedules of biopsies.
Appendix S5.4. Source code for web application.