

Abstract
The Strecker reaction is a three-component condensation of an aldehyde, an amine, and hydrogen cyanide, delivering an α-amino carbonitrile. Despite extensive investigations, the possibility to use amides instead of amines as one of the three condensation partners has been largely neglected. Nonetheless, the N-acylated α-aminocarbonitriles that are obtained in this way are of direct interest for drug discovery, because they make up a well-known class of mechanism-based inhibitors of serine- and cysteine-type hydrolases. In response, we have thoroughly explored the corresponding variant of the Strecker reaction, focusing on catalyst use, solvent, reaction time, and cyanide source. Optimized parameters were combined in a sequential one-pot protocol for which the scope was found to be compatible with library synthesis applications. Product yields ranged from 7 to 90%, and conditions were found to be mild and tolerant to a wide range of functional groups, including moieties that are typically present in druglike molecules.
Introduction
The Strecker reaction was originally reported in 1850 as a condensation between acetaldehyde, ammonia, and hydrogen cyanide.1 In the same paper, the corresponding reaction product (2-aminopropanenitrile) was submitted to acidic hydrolysis to produce racemic alanine. During the decades that followed, the reaction scope was demonstrated to be significantly broader, allowing the use of structurally diverse aldehydes and organic amines. As a result, the Strecker reaction became a standard approach for the synthesis of natural and non-natural α-amino acids.
This reaction is part of a larger group of multicomponent reactions (MCRs), in which an initially formed imine-type intermediate reacts with a nucleophile to deliver a final product.2 Other well-known representatives include the Mannich condensation, the Ugi- and Passerini-type reactions, and the so-called “A3 reaction”. MCRs are interesting synthetic tools for library synthesis, provided their functional group compatibility is sufficient to allow diversity-oriented approaches.3,4 In addition, the combined reaction of more than two molecular building blocks in one synthetic step is also interesting from a green chemistry perspective.5
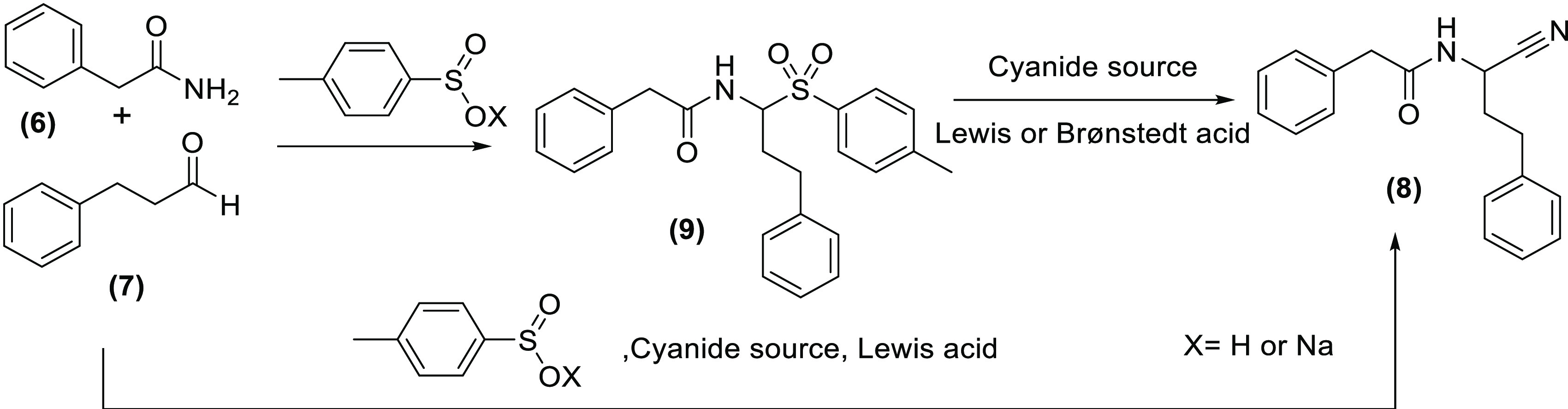
In this framework, it is not surprising that efforts on methodological optimization of the Strecker reaction are still ongoing. Relevant recent examples include the investigation of alternative catalysts, cyanide sources, and/or reaction protocols to maximize reaction yields.6−9 Equally importantly, development of enantioselective reaction variants continues to be pursued, especially with the aim of implementing the reaction in industrial settings. Currently, the antihypertensive methyldopa is the only compound that is produced industrially via the Strecker reaction. Other amino acid derivatives are generally still prepared by bacterial or enzymatic processes that are inherently enantiospecific.10,11 A scope aspect of the Strecker reaction that hitherto has largely been overlooked, however, is the possibility of using N-acylated amine building blocks in the condensation.12−14 Only a few successful applications were found in which carbamates are directly condensed with an aldehyde and a cyanide source15,16 (Figure 1, entry A). To the best of our knowledge, however, the same direct condensation involving a carboxamide instead of a carbamate has not been reported. In addition, limited documentation can be found in the literature for a related reaction type involving the use of sulfonamides.17 The under-representation of such reactions in the literature is likely to be related to the reduced nucleophilicity of acylated amines, implying a more sluggish formation of the N-acylimine intermediate of the reaction. Likewise, the relatively higher Gibbs free energy of an N-acylimine intermediate (compared to a classical imine) also contributes to the inherently lower reactivity of acylated amines in Strecker settings. Generalized strategies to increase rates/yields of reactions involving N-acylimine intermediates have been proposed by, among others, Petrini and co-workers. A first approach exploits the efficient stabilization of N-acylimines with para-tolylsulfinic acid in acidic media (Figure 1, entry B). The latter delivers a sulfone-type adduct that can be isolated and, in many cases, precipitates from the reaction media. In a separate acid- or base-catalyzed step, the sulfone equilibrates with the corresponding sulfinate and N-acylimine, which can then be trapped in situ by a variety of nucleophiles, including the cyanide anion. The latter, however, has only been superficially explored, again mainly using carbamates, although some individual examples with benzamides are present.13,14,18−21 Also, in the corresponding paper by Petrini, only aromatic aldehydes were found to be reliable reaction partners.13 Yields decreased with aliphatic aldehydes and these substrates were not further explored. An alternative but comparable strategy was reported by Katritzki and co-workers, who stabilized the N-acylimine intermediate with benzotriazole. As for the sulfinate-based approach, the benzotriazole adduct is transformed into the corresponding carbonitrile in a separate step.22
Figure 1.
Possible approaches to Strecker reactions involving acylimine-type intermediates: direct condensation of building blocks (entry A) vs strategies involving stabilization of the N-acylimine intermediate (entries B and C).
As part of our ongoing research, we were particularly interested in an efficient methodology allowing library synthesis of N-acylated aminocarbonitriles in which the acyl group is part of an amide function, preferably in a single synthetic step from commercially available starting materials. This compound family is an important class of inhibitors of serine- and cysteine-type proteases. Therefore, the molecules are of high interest to drug discovery and to the developing domain of disease biomarker research via activity-based protein profiling (ABPP). Since the turn of the century, several representatives of the class have entered clinical practice, including dipeptidyl peptidase IV-inhibitors vildagliptin (compound 1, Figure 2) and saxagliptin and the cathepsin K-inhibitor odanacatib (2), although the latter was withdrawn again in 2016 because it increased the risk of stroke (Figure 2). Additional relevant examples include the preclinical inhibitors of falcipain-2 and several cathepsins.23−25 In structural terms, all of these compounds share a pseudo-peptide architecture, in which the carbonitrile group functions as an electrophilic “warhead”, capable of forming covalent, reversible bonds with the catalytic machinery of the target serine or cysteine protease. In this way, the carbonitrile group strongly contributes to the inhibitor’s target affinity (Figure 3).
Figure 2.
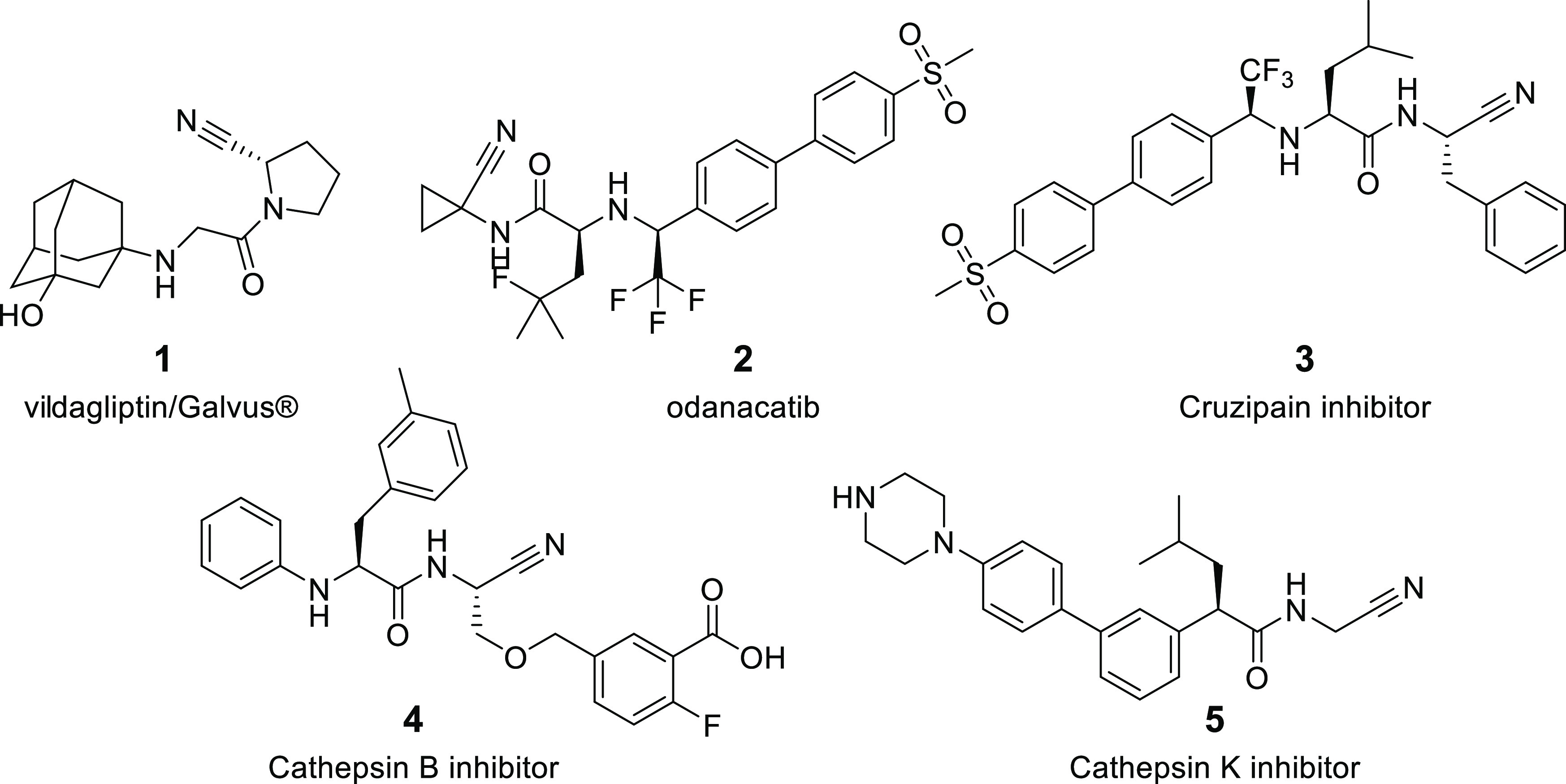
Examples of inhibitors with an N-acylated α-aminonitrile moiety.25−27
Figure 3.

Covalent adduct formation between a carbonitrile warhead and the catalytic nucleophile of a protease (exemplified for a cysteine protease).
Based on its clear potential for library synthesis of protease inhibitors, we decided to thoroughly explore variants of the Strecker reaction involving amide building blocks with the aim of delivering an optimized, broadly applicable reaction protocol.
Results and Discussion
For the investigation and optimization of experimental parameters in this study, we relied on the condensation of phenylacetamide, 3-phenylpropanal, and selected cyanide sources as a general model reaction. It is noteworthy that we specifically chose not to use a benzaldehyde-type aromatic aldehyde in this model reaction. While benzaldehyde and its derivatives generally perform very well in Strecker reactions, 3-phenylpropanal (in which the carbonyl group is flanked by a methylene group) was considered to be a more relevant model aldehyde, but also with more challenging properties under Strecker conditions. The latter is related to the well-known tendency of methylene-flanked imines to enolize and undergo aldol-type self-condensation, thereby forming polymeric products and significantly decreasing yields of the desired carbonitrile.28 Initial effort aimed at evaluating the possibility of directly condensating the amide, aldehyde, and cyanide source, relying on Lewis- or Brønstedt-type acids (Figure 4, entry A). While such condensations involving a carboxamide have, to the best of our knowledge, not been published, two literature examples exist that report a comparable reaction with a carbamate instead of an amide, relying, respectively, on BF3 catalysis and so-called “partially hydrolyzed titanium alkoxide” (PHTA).15 Applying the published protocols to our own model reaction, however, did not deliver the expected products. It is noteworthy that even repeating the reactions from the corresponding publications was not successful in our hands. Finally, Cu(OTf)2, InCl3, and trifluoroacetic acid (TFA) were also evaluated exploratorily as alternative catalysts for the same direct condensation, but these experiments were also not successful in producing an α-amino carbonitrile.15,16,29,30
Figure 4.
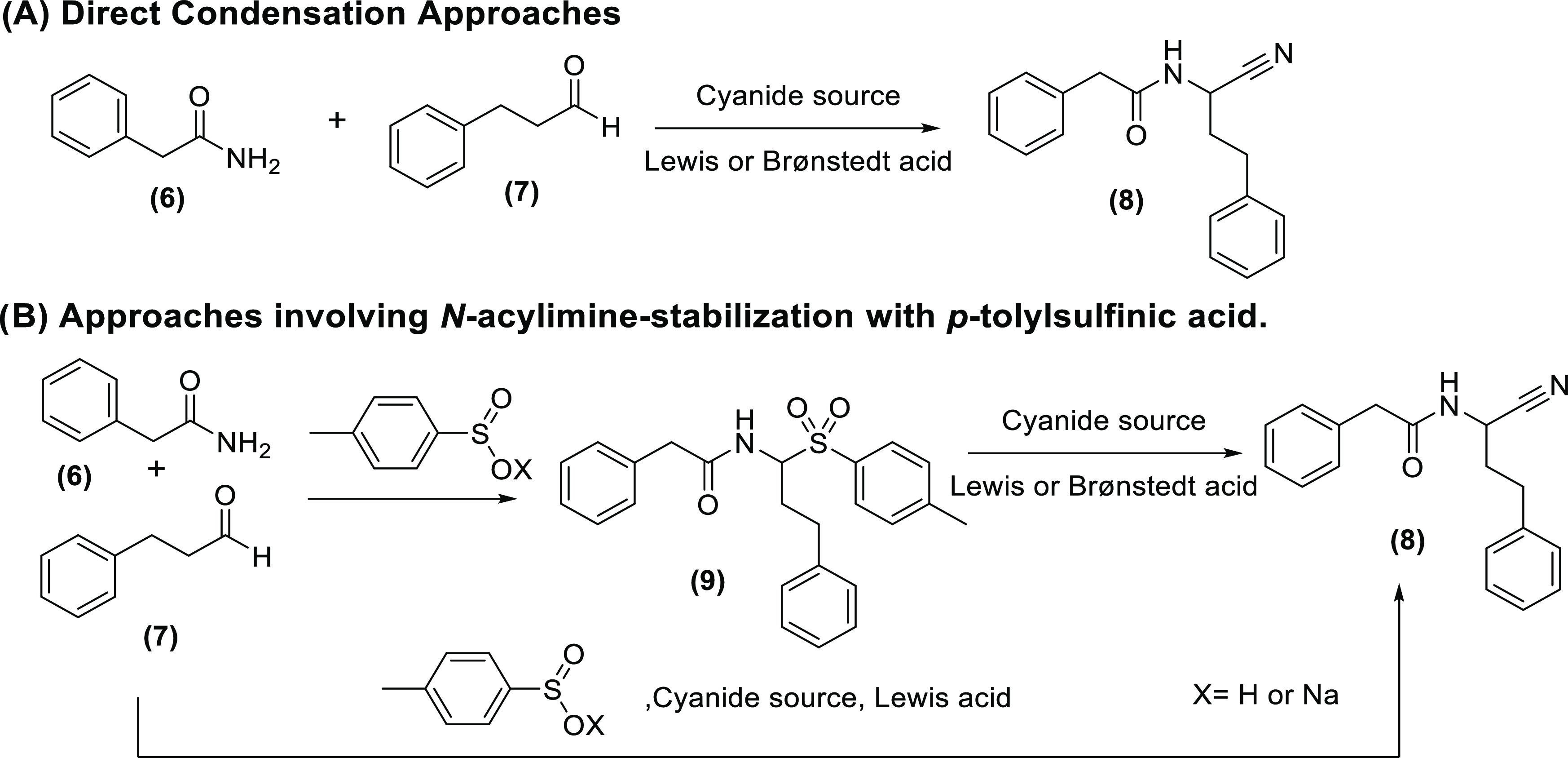
Overview of synthetic strategies explored in this study: (A) direct condensation approaches and (B) approaches involving N-acylimine stabilization.
In response, all direct condensation approaches were abandoned, and we decided to elaborate on strategies based on N-acylimine stabilization with phenylsulfinic acid (Figure 1, entry B). Two main routes were investigated in this context: (a) a two-step approach, in which the sulfone intermediate is prepared separately, isolated, and subsequently reacted with a cyanide source to obtain the carbonitrile product. (b) In addition, potentially more efficient protocols were explored in which all reagents are either mixed in one pot or brought to reaction in a two-step, “telescoping” manner (Figure 4, entry B). For systematicity reasons, it was decided to focus on the two-step approach first.
Because it is well documented in the literature, no extensive optimization effort was deemed required for the first, sulfone-yielding step (Scheme 1). The latter is mostly carried out by mixing an aldehyde, an amide, and a sulfinate salt with (super-)stoichiometric amounts of formic acid in aqueous methanol. This reference protocol in our hands delivered the sulfone in modest but reproducible yields, within ranges reported for other aldehydes. Variants of this protocol involving a sulfinic acid, an amide, and an aldehyde have also been reported. Although substantially less frequently applied, the latter protocol seems to produce comparable yield ranges in most organic solvents. A test reaction in tetrahydrofuran (THF), a general solvent with good solubility for many potential reaction partners, gave a yield comparable to that of the sulfinate-based protocol. In both cases, the pure sulfone was isolated after flash chromatography.
Scheme 1. Synthetic Preparation of Sulfone Intermediate 9 Relying on Literature Protocols.

Reagents and conditions: (a) sodium 4-tosylsulfinate (2 equiv), formic acid (20 equiv), methanol/water (1:2), 5 days, room-temperature (rt) (isolated yield: 43%) or (b) tosyl-4-sulfinic acid (1.2 equiv), THF, 1 day, and rt (isolated yield: 45%).
The second step of the transformation consists of the formal substitution of the sulfone part for cyanide, presumably passing via an acylimine intermediate (Scheme 2). A number of reaction protocols involving different potential catalysts, solvents, and cyanide sources were investigated for this step (Table 1). Catalysts were generally selected based on their reported efficacy in the related Strecker-type reactions of carbamates or sulfonamides (discussed earlier). These comprised the Lewis acids Cu(OTf)2, InCl3, BiBr3, and TiO2/rutile.6,17 The catalytic role of the latter could consist of activating the sulfone moiety for elimination and/or increasing the electrophilicity of the acylimine intermediate. Formic acid was included as a reference Brønstedt acid with potentially analogous catalytic roles as the Lewis acids. Although it has only been reported to promote “classical Strecker” reactions, formic acid is also present in one of the protocols for sulfone preparation (Scheme 1). In this respect, it was decided to anticipate the possibility of one-step, one-pot protocols (vide infra). In addition, the following organocatalysts were selected for evaluation: quinine and N-benzylquininium bromide.18,19,31 The latter has been proposed to increase the reactivity of acylimine intermediates via, a.o., hydrogen bond activation. The solvents investigated [N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), MeOH, acetonitrile (ACN), THF, dichloromethane (DCM), toluene, and water, water/DMF, MeOH/water] were selected either based on their status as general reference solvents or because they had been reported in the related Strecker-type reactions mentioned earlier. Finally, a number of frequently used cyanide sources were selected [KCN, trimethylsilyl cyanide (TMSCN), and acetone cyanohydrin], again based on their use in the related transformations. The argument of safety was not decisive in this selection, since all of these compounds are highly toxic, and extreme caution is required while handling them. Due to the number of possible combinations, we used the same combinations of cyanide source and catalyst as reported in the literature for Strecker-type reactions. The yields in Table 1 were determined using a ultra performance liquid chromatography–mass spectrometry (UPLC–MS) system, relying on calibrated UV-absorption spectrophotometry.
Scheme 2. Formal Substitution of the Sulfone Moiety for a Cyano Group.

Reagents and conditions: cyanide source, solvent, catalyst (specified in Table 2), rt, and 24 h.
Table 1. Reaction Conditions Investigated for the Second Step (Cyanide)a.
| catalyst | no catalyst | no catalyst | BiBr3 | BiBr3 | InCl3 | CuOTf2b | rutile | formic acidd | quinine | N-benzylquininium bromide |
|---|---|---|---|---|---|---|---|---|---|---|
| catalyst equivalents | 0.05 | 0.05 | 0.1 | 0.1 | 1 | 0.2 | 0.05 | 0.1 | ||
| cyanide sourcec | KCN | TMSCN | TMSCN | KCN | TMSCN | TMSCN | KCN | TMSCN | KCN | acetone cyanohydrin |
| ACN | 77% | nd | 42% | nd | 0% | 0% | nd | nd | <4% | 56% |
| THF | 82% | 0% | 0% | nd | <4% | 8% | nd | 0% | 53% | 72% |
| toluene | 74% | 0% | 39% | nd | 48% | 55% | nd | nd | 46% | 58% |
| DCM | 79% | 0% | 67% | 53% | <4% | 55% | nd | 0% | 25% | 65% |
| methanol | 73% | nd | 0% | nd | 0% | 28% | nd | nd | 57% | 0% |
| DMF | 81% | nd | 0% | nd | 0% | 0% | nd | 0% | 71% | 76% |
| DMSO | 93% | nd | 0% | 64% | 0% | 0% | nd | nd | 73% | 64% |
| water/DMF | 94% | nd | 0% | nd | 0% | nd | 49% | nd | 7% | 34% |
Reaction conditions: RT, 0.1M, and 24 h. nd = not determined
Na2SO4 anhydrous was added to ensure anhydrous conditions. Use of acetone cyanohydrin (with THF) produced no product.
2 equivalents.
Performed in a sealed tube.
A main observation when studying the data in Table 1 is that the reaction protocol involving KCN as the cyanide source and no catalyst addition is clearly optimal, with yields of up to 94% and very broad solvent tolerability. Conversely, not even trace amounts of the desired product are formed in three exploratory experiments where TMSCN was used instead of KCN under otherwise identical conditions. Surprisingly, however, the selected Lewis acids (but not the Brønstedt-type formic acid) are, in some cases, instrumental to activate TMSCN as a cyanide source. Nonetheless, even the best yields under these conditions remain considerably lower than those with the KCN/uncatalyzed protocol. Also, the efficiency of the catalyst seems to be strongly solvent dependent. Polar solvents with strong Lewis acid solvation properties are detrimental to yields in these reactions. More specifically, solvation of the Lewis acidic metal cation can reasonably be expected to prevent the latter from interacting with the other reactants and from activating them for the desired condensation reaction. A notable exception is Cu(OTf)2, which is still capable of promoting the conversion in methanol (albeit with only 28% yield). For the sake of completeness, the combination of KCN with Lewis acids was also superficially explored. The low yields that were obtained in experiments with BiBr3/KCN, however, were the decisive reason for not exhaustively exploring this possibility. In the same framework, the rutile/KCN/aqueous DMF was also investigated as published earlier but again found to be less performant than the corresponding uncatalyzed version. Overall, these findings suggest that considerable optimization may be possible for the published Lewis acid-catalyzed Strecker-type reactions involving carbamates or sulfonamides. To the best of our knowledge, these were never compared with uncatalyzed conditions side by side. Finally, the two organocatalyst protocols that were included involved either KCN or acetone cyanohydrin as the cyanide sources. Similar to what was already observed for the Lewis and Brønstedt acids, protocols involving organocatalysts were not able to deliver higher product yields. Nonetheless, both are clearly still superior to the Lewis acid-catalyzed protocols. Importantly, also, the protocol involving quinine catalysis by Reingruber et al. was published in the framework of enantioselective synthesis.19 In case sufficiently high enantiomeric excess (ee) can be obtained with this protocol (vide infra), it could be an argument to use this protocol for library synthesis of biologically active compounds.
Effort was then made toward combining both reaction steps (sulfone formation and carbonitrile formation) into a one-step, one-pot protocol. Based upon the above data, the most promising one-pot protocol seemed to be stirring a mixture of aldehyde, amide, KCN, and 4-tolylsulfinic acid in THF at room temperature without catalysts. However, no target product could be retrieved under these conditions, potentially explained by the occurrence of undesired competing reactions such as cyanohydrin formation and aldol-type condensation that prevent the formation of the desired product. In addition, the best-performing protocols involving a Lewis acid or an organocatalyst from Table 1 were also explored (Table 2, entries 1–9). Since, however, their potential effect on sulfone formation had never been reported, experiments were first run without the addition of a cyanide source, and sulfone formation was used as the read-out. None of the conditions in which the catalysts were combined with tolylsulfinate/formic acid was found to produce relevant amounts of the desired sulfone (based upon LC–MS analysis of the reaction mixture) (Table 2, entry 1–5). It is, however, unclear whether the absence of sulfone formation is indeed caused by the catalyst or, alternatively, by the corresponding reaction media (which are invariably different from the aqueous methanol used for the sulfinate-based condensation in Scheme 1). Given the absence of promising preliminary results, no eventual optimization measures were investigated. In addition, combinations of 4-tolylsulfinic acid with an organocatalyst or a Lewis acid were evaluated. (Table 2, entries 7–9). Of the two Lewis acids that were evaluated (entry 7 and 8), the presence of bismuth(III) bromide and the use of DCM as a solvent had a yield comparable with the reference reaction in Scheme 1. Rutile/aqueous DMF, however, was found not to promote sulfone formation. Finally, the combination of 4-tolylsulfinic acid with organocatalyst quinine in DMSO (entry 9) performed well in the substitution step.
Table 2. Screened Reaction Conditions for Sulfone Formation (as in Scheme 1)a.
| entry | reaction conditionsb | catalyst | solvent | time | yield (9) |
|---|---|---|---|---|---|
| 1.1 equiv | 0.05 equiv | ||||
| 1 | A | rutile | water/DMF (99:1) | 3 days | trace amountsc |
| 2 | A | indium(III) chloride | toluene | 8 days | 0% |
| 3 | A | bismuth(III) bromide | DCM | 8 days | trace amountsc |
| 4 | A | copper(II) triflate | DCM | 8 days | trace amountsc |
| 5 | A | N-benzylquininium bromide | DMF | 8 days | 0% |
| 6 | A | quinine | DMSO | 8 days | 0% |
| 7 | B | bismuth(III) bromide | DCM | 1 day | 50%d |
| 8 | B | rutile | water/DMF (99:1) | 7 days | 0% |
| 9 | B | quinine | DMSO | 7 days | 25%d |
In response, a telescoping protocol was devised, in which the aldehyde, the amide, and 4-tolylsulfinic acid in THF are allowed to react until chromatography indicated the reaction to be complete or stagnant, followed by addition of KCN to initiate the second step. The telescoping reaction was found to be successful, leading to the formation of (11) in an overall yield of 53%, but requiring three days for the first part and 7 days for the second part (Figure 5, (11)). The same telescoping reaction protocol was also applied on a larger scale (involving 6.5 mmol of the limiting reagent phenylpropanal) with a roughly comparable overall yield (41%). The protocol was subsequently applied to other aldehydes and carboxamides to define the scope of the reaction specifically considering the functional group compatibility of the reaction. A set of 14 additional compounds was synthesized (Figure 5) with commercially available reagents possessing moieties that are common in druglike and peptide-like molecules. Building blocks with unprotected nucleophilic moieties (e.g., free amines) were not considered, because they could interfere with both steps of the transformation. We aimed at obtaining the highest yields for each target molecule by adjusting the timing of cyanide addition based on LC–MS samples of the reaction. We observed highly variable reaction times of both parts of the telescoped reaction. One more adaptation was necessary for the cases where an acidic proton is present after the first step (Figure 5, (22–24)); potassium carbonate was added together with potassium cyanide to capture this proton. Molecules (11), (14), and (15) were obtained in good yields in 10–11 days; (13) was obtained with a comparable yield in four days of reaction time. Compound (12) was obtained in excellent yield, but the reaction time was 12 days. The adduct formation took eight days, probably due to the steric hindrance caused by the two phenyl moieties. The telescoping protocol is compatible with carbamate-protected amines, as shown with (16), (17), (20), and (21). In addition, Fmoc-protecting groups are also tolerated, a demonstration that the reaction conditions are mild enough to tolerate both acid- and base-labile protecting groups with acceptable yields. The reaction rates for (16) and (21) were fast when compared with (17) and (20): 3 and 11 days, respectively. Ketones are another interesting building block to explore; these molecules allow the formation of α,α-disubstituted amino acids. Thus, two of them (18) and (19) were produced; the yields were the lowest and the reaction times were the longest in the set. Finally, we explored the scope with succinamic acid, which contains a free carboxylate, and two building blocks with a pyridinyl moiety, nicotinamide, and 3-pyridinecarboxaldehyde. The three reactions needed the addition of potassium carbonate for the second reaction to occur. Compound (22) was obtained after 6 days; (23) and (24) were obtained in 6 and 4 days, respectively. During the expansion of the reaction scope, we also observed that some intermediate adducts were not stable when its isolation was attempted; similar observations were reported in the literature for other compounds.32,33
Figure 5.
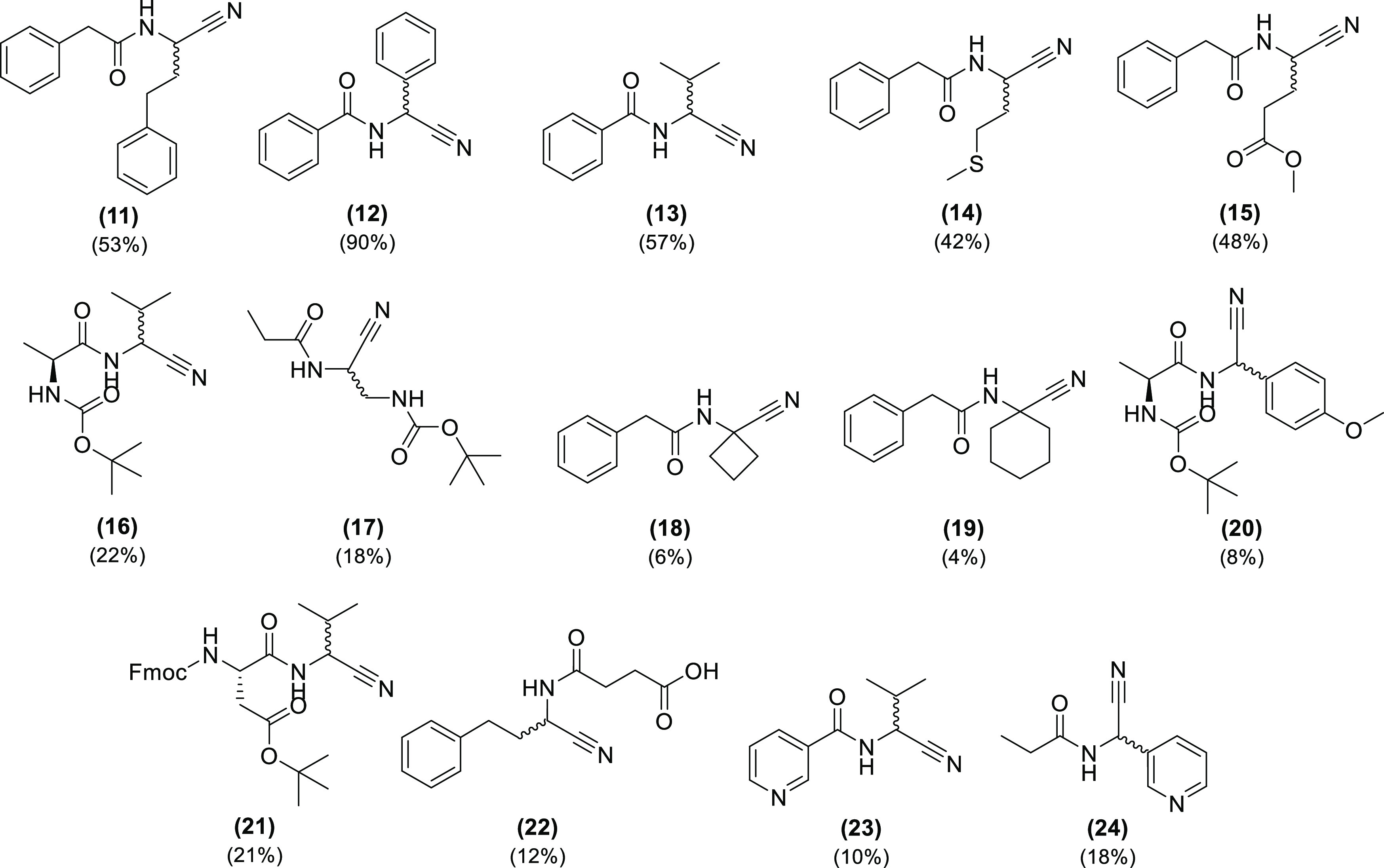
Library of compounds synthesized and respective yields.
Overall, the reaction yields obtained for compounds 11–24 are moderate to good. They can be considered acceptable in most cases when compared to the alternative synthetic approach (Strecker reaction, followed by N-acylation). The latter approach involves two reactions and, potentially, two purification steps. Furthermore, the classical Strecker reaction is characterized by highly variable yields. Nevertheless, the significant reaction time for the telescoping reaction, ranging from 3 to 14 days, was a point that we clearly wanted to optimize further. The possibility to reduce reaction times by increasing the temperature was investigated. This was originally not considered, taking into account the oxidation sensitivity of aldehydes and the possibility that undesired side reactions could also be promoted by increasing the temperature. To try the hypothesis of optimization with higher temperatures, (11) and (12) were resynthesized at 45 °C using a similar procedure, only cooling it down to add KCN. The yield for (12) decreased to 22%, possibly due to oxidation of benzaldehyde or benzoin formation. The reaction time also decreased considerably, from 12 to 3 days and 4 h. On the other hand, (11) not only had the reaction time reduced from 10 to 3 days, the yield also increased from 53 to 77%. These observations indicate that it is possible to further optimize each reaction and find the optimal temperature on a case-by-case basis (e.g., temperature and solvent).
In addition, we also attempted to further increase the atom efficiency of the process by using methylsulfinic acid, the smallest sulfinic acid possible, instead of tolylsulfinic acid. The reaction with (11) at room temperature was successful with comparable yield (43%) and reaction time (10 days).
Finally, the stereochemical aspects of the reaction were preliminarily probed, relying on chiral chromatography with (11), (13), and (21). Stereoselective reactions are of particular interest for the synthesis of drugs because the individual stereoisomers of a drug can have differing biological properties. Use of a specific stereoisomer can therefore be desirable, to reduce the probability of side effects or toxicity. As expected, in the absence of chiral catalysts or auxiliaries during their synthesis, (11) and (13) were found to occur as racemates. Analogously, the use of a chiral amide building block in the synthesis of (21) did not lead to diastereoselectivity, while the absolute configuration of the original stereocenter was maintained. Finally, quinine’s potential as an enantioselective catalyst was investigated, taking into account the earlier promising results of Reingruber and co-workers in carbamate condensations.19 This catalyst was already used during the screening (Tables 1 and 2) and it allowed the reaction to proceed in acceptable yields. Two protocols were tried: (a) adding the quinine initially and performing the telescoping reaction at −10 °C and adding the quinine with the cyanide and performing the cyanide substitution step at −10 °C. Only the second protocol seemed to yield the product with longer reaction times, but the reaction was found not to lead to enantiomeric excess of one optical isomer.
Conclusions
In summary, we have investigated diverse parameters for the reaction, solvent, catalyst, and temperature, and developed a simple telescoping reaction protocol that has the potential to assemble racemic, peptide-like N-acylated α-aminonitriles. The developed protocol has significant functional group compatibility and can be used for library synthesis of protease inhibitors. Further optimization of the general protocol that we have delivered seems difficult, but fine-tuning of reaction parameters on a case-by-case basis could be worthwhile, for example, in upscaling conditions for a particular molecule of interest. As shown, temperature is one of the factors that seems to have different optima for different reactant combinations. Other parameters might follow the same trend such as solvents.
Future work will include alternative acylimine-stabilizing strategies other than sulfone-adduct formation (such as benzotriazole-adduct formation, reported by Katritzky and co-workers), attempts to induce enantioselectivity with other catalysts, or use of a sulfonated solid-phase catalyst to facilitate product purification and enable catalyst recycling.22,34,35
Experimental Section
Caution! Acetone cyanohydrin, TMSCN, and KCN are extremely toxic and should be handled with caution in a well-ventilated fumehood. Cyanides in acidic pH produce HCN, an extremely toxic gas. The aqueous waste was brought to basic pH with a NaOH solution (1M) and then quenched with industrial-grade bleach (sodium hypochlorite 12%). The glassware and material in contact with cyanides were first washed with bleach. Bleach should not be mixed with acidic solutions; chlorine gas is formed, which is also extremely toxic.
General Information
Commercial reagents and solvents were used as received. All thin-layer chromatography experiments were performed using precoated silica gel 60 F254 plates. The LC–MS analysis was performed on a Waters UPLC–MS system equipped with a TUV and QDa detector; the column used is an Acquity UPLC BEH C18 (1.7 μm, 2.1 × 50 mm). Flash chromatography separations were carried out using a Biotage Isolera One purification system with silica gel columns (normal or reverse phase) from Büchi or Biotage. Preparative HPLC purifications were carried out using a Waters HPLC system equipped with a UV and MS detector and using an XBridge Prep C18 5 μm OBD column (19 ×100 mm). Melting points were measured on a Büchi Melting Point M-560. Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were recorded on a Bruker Alpha Platinium ATR. (Note: the nitrile band is missing in some spectra (low signal-to-noise ratio) for some compounds, which is a known phenomenon observed with ATR FTIR due to the high absorbance of diamond in that region.)361H and 13C{1H} nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Ultrashield 400 MHz NMR spectrometer (operating at 400 and 101 MHz, respectively) in CDCl3, acetone-d6, MeOD-d4, or DMSO-d6, and analyzed using MestreNova software. The chemical shifts (δ) reported are given in parts per million (ppm). The signal splitting patterns were described as s = singlet, d = doublet, t = triplet, q = quartet, p = pentuplet, dd = doublet of doublet, dt = doublet of triplet, td = triplet of doublet, tt = triplet of triplet, ddd = doublet of doublet of doublet, br = broad, and m = multiplet, with coupling constants (J) in hertz (Hz). High-resolution mass spectra (HRMS) were acquired using a Q-TOF II instrument (Waters, Manchester, U.K.) mass spectrometer. The MS was calibrated prior to use with a 0.1% H3PO4 solution.
General Method for Synthesis of Phenyl-N-(3-phenyl-1-tosylpropyl)acetamide (A)
Preparation is based on the literature.19
A mixture of 2-phenylacetamide (5 g, 37.0 mmol) and sodium 4-toluenesulfinate (13.18 g, 74.0 mmol) was suspended in a solution of methanol in water (1:2, 50 mL). Afterward, 3-phenylpropanal (7.37 mL, 55.5 mmol) was added in one portion, followed by formic acid (27.9 mL, 740 mmol). The resulting mixture was allowed to stir for 5 days at room temperature. The resulting white precipitate was filtered off and washed with water and diethyl ether to yield 2-phenyl-N-(3-phenyl-1-tosylpropyl)acetamide (6.3375 g, 15.55 mmol, 42% yield) as a white solid.
1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 8.2 Hz, 2H), 7.36–7.18 (m, 8H), 7.14–7.04 (m, 4H), 5.87 (d, J = 10.4 Hz, 1H), 5.24 (td, J = 10.7, 3.2 Hz, 1H), 3.32 (s, 2H), 2.67 (t, J = 7.5 Hz, 2H), 2.55 (dtd, J = 14.0, 7.7, 3.2 Hz, 1H), 2.41 (s, 3H), 1.98 (ddt, J = 18.3, 11.0, 6.9 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 170.2, 145.3, 140.00, 133.8, 133.4, 129.9, 129.4, 129.2, 129.1, 128.8, 128.5, 127.8, 126.6, 68.5, 43.4, 31.7, 28.5, 21.9.
General Method for Synthesis of Phenyl-N-(3-phenyl-1-tosylpropyl)acetamide (B)
A mixture of 2-phenylacetamide (0.216 g, 1.601 mmol), 3-phenylpropanal (0.213 mL, 1.601 mmol), and 4-toluenesulfinic acid (0.3 g, 1.921 mmol) was dissolved in THF (8 mL). The solution was stirred for 1 day at room temperature. Then, extraction was performed with ethyl acetate and water, the organic phase was washed with brine, dried with anhydrous sodium sulfate, and the solvent was evaporated. The obtained off-white solid was purified by normal-phase flash chromatography to yield 2-phenyl-N-(3-phenyl-1-tosylpropyl)acetamide (0.290 g, 0.712 mmol, 44.5% yield) as a white solid.
1H NMR (400 MHz, DMSO-d6) δ 9.00 (d, J = 9.6 Hz, 1H), 7.59–7.52 (m, 2H), 7.32–7.17 (m, 8H), 7.14–7.06 (m, 4H), 5.06–4.93 (m, 1H), 3.46–3.27 (m, 2H), 2.73–2.60 (m, 1H), 2.54–2.42 (m, 1H), 2.35–2.24 (m, 1H), 2.04–1.89 (m, 1H); 13C{1H} NMR (101 MHz, DMSO) δ 170.1, 144.4, 140.1, 135.4, 133.4, 129.5, 129.0, 128.8, 128.4, 128.3, 128.1, 126.4, 126.1, 68.2, 41.7, 30.5, 28.1, 21.1.
General Method for Synthesis of Phenyl-N-(3-phenyl-1-tosylpropyl)acetamide (“Screening”)
Benzene acetamide (1 equiv), sodium 4-toluenesulfinate, or 4-toluenesulfinic acid (2 equiv), and the catalyst (0.05 equiv) were suspended in a solvent (0.122 M). Phenylpropionaldehyde (1.5 equiv) was added and the mixture was stirred at room temperature. The reaction was probed by thin layer chromatography (TLC) or LC–MS every 24 h. In the cases where the product was isolated, extraction with ethyl acetate and water was performed, and flash chromatography was performed to yield the pure product.
General Method for Synthesis of N-(1-Cyano-3-phenylpropyl)-2-phenylacetamide (Screening)
The cyanide source (2 equiv) was added to a solution of 2-phenyl-N-(3-phenyl-1-tosylpropyl)acetamide (1 equiv) and the catalyst (0.1 equiv) in a solvent (0.122 M). The reaction was stirred for 24 h, when a sample for LC–MS was prepared and measured.
Preparation of 4-Tolylsulfinic acid
Sodium 4-tolylsulfinate was dissolved partially in a superstoichiometric amount of a 1M HCl aqueous solution. The suspension was extracted twice with ethyl acetate, and the organic phases were combined and dried with sodium sulfate anhydrous. The mixture was filtered, and the solvents were evaporated to yield a white powder, which was stored at −20 °C to avoid degradation.
Method for Synthesis of N-(1-Cyano-3-phenylpropyl)-2-phenylacetamide with Methylsulfinic Acid
Methylsulfinic acid was prepared by dissolving the required amount of sodium methylsulfinate (1 equiv) in HCl in dioxane (1 equiv). A suspension formed and it was added to the reaction.
General Method Used for “Library Synthesis”
An amide (1.1 equiv) and an aldehyde (1 equiv) were dissolved in the amount of THF required to obtain an aldehyde concentration of 0.165 M. 4-Tolylsulfinic acid (1.3 equiv) was added and the mixture was stirred for a certain period of time (Rt1) at room temperature. Once the starting materials were consumed or the reagents and intermediate quantities seemed to be stable, KCN (1.1 equiv) was added to the reaction and it was stirred for a certain period of time (Rt2) at room temperature. In the case of compounds (22), (23), and (24), K2CO3 (1 equiv) was added. Once the reaction was finished or was stable, filtration and washing with ethyl acetate were performed to remove the precipitate in some cases. Next, an extraction (ethyl acetate and water) was performed, the organic phase was dried with sodium sulfate, filtered, and the solvents were removed by evaporation under reduced pressure. The crude was then purified by flash chromatography and/or preparative TLC (gradient of n-heptane and ethyl acetate). In some cases, the compounds were further purified by preparative high performance liquid chromatography (HPLC) (gradient of water with formic acid and methanol).
N-(1-Cyano-3-phenylpropyl)-2-phenylacetamide (11)
This product was synthesized several times using different conditions. (1) In case the general library synthesis method was used, the reaction time was 10 days (=3 + 7 days) and the isolated yield was 53%. (2) In case the general library synthesis method was run at 45 °C (instead of ambient temperature), the reaction time was three days (=1 + 2 days) and the isolated yield was 77%. (3) In case the library synthesis protocol was used with methylsulfinic acid (instead of tolylsulfinic acid), the reaction time was 10 days (=6 + 4 days) and isolated yield was 43%. (4) Finally, the library synthesis method was scaled up. According to the general protocol, 6.5 mmol (872 mg) phenylpropanal, 7.15 mmol (943.8 mg) phenylacetamide, 8.45 mmol (1.45 g) tolylsulfinic acid, and 7.15 mmol (465 mg) potassium cyanide were used. The reaction time was 10 days (=3 + 7 days) and the isolated yield was 41% (2.7 mmol, 705 mg); white powder, mp 119 °C (degradation); 1H NMR (400 MHz, CDCl3) δ 7.44–7.21 (m, 8H), 7.17–7.13 (m, 2H), 5.54 (d, J = 8.6 Hz, 1H), 4.88 (dt, J = 8.5, 7.3 Hz, 1H), 3.60 (s, 2H), 2.77–2.71 (m, 2H), 2.15–1.98 (m, 2H); 1H NMR (400 MHz, DMSO-d6) δ 8.95 (d, J = 7.5 Hz, 1H), 7.38–7.13 (m, 11H), 4.63 (q, J = 7.5 Hz, 1H), 3.50 (s, 2H), 2.66 (t, J = 7.9 Hz, 2H), 2.15–1.91 (m, 2H); 13C{1H} NMR (101 MHz, DMSO-d6) δ 170.3, 139.9, 135.5, 129.0, 128.2, 126.6, 126.2, 119.3, 41.7, 39.5, 33.2, 30.9; HRMS (ESI) m/z: [M + H]+ calcd for C18H19N2O 279.1492, found 279.1499; UPLC–MS (generic method) 1.80 min, m/z 279.2 [M + H]+, 277.2 [M – H]−; IR (ATR-FTIR) ν (cm–1): 3337, 3254, 3060, 3027, 2923, and 2856.
N-(Cyano(phenyl)methyl)benzamide (12)
Reaction time (discriminated) = 12 (8 + 4) days; 3 days (4 h + 3 days) (reaction at 45 °C); 209 mg, 90%; 22% (reaction at 45 °C), white powder, mp 138 °C (degradation); 1H NMR (400 MHz, DMSO-d6) δ 9.78 (d, J = 8.0 Hz, 1H), 8.02–7.82 (m, 2H), 7.68–7.25 (m, 8H), 6.43 (d, J = 7.9 Hz, 1H); 13C{1H} NMR (101 MHz, DMSO-d6) δ 166.0, 134.5, 132.7, 132.1, 128.9, 128.8, 128.5, 127.6, 127.0, 118.5, 43.8; HRMS (ESI) m/z: [M + H]+ calcd for C15H13N2O 237.1022, found 237,1022; UPLC–MS (generic method) 1.65 min, m/z 237.2 [M + H]+, 235.1 [M – H]−; IR (ATR-FTIR) ν (cm–1): 3252, 3060, 2916, and 2246.
N-(1-Cyano-2-methylpropyl)benzamide (13)
Reaction time (discriminated) = 4 (1 + 3); 113 mg, 57%, off-white powder, mp 100 °C (degradation); 1H NMR (400 MHz, acetone-d6) δ 8.38 (br, NH, 1H), 7.99–7.90 (m, 2H), 7.64–7.55 (m, 1H), 7.55–7.45 (m, 2H), 4.94 (dd, J = 8.4, 7.7 Hz, 1H), 2.33–2.19 (m, 1H), 1.19 (d, J = 6.7 Hz, 3H), 1.12 (d, J = 6.7 Hz, 3H); 13C{1H} NMR (101 MHz, Acetone-d6) δ 167.3, 134.4, 132.7, 129.3, 128.4, 119.1, 47.9, 32.2, 19.1; HRMS (ESI) m/z: [M + H]+ calcd for C12H15N2O 203.1179, found 203.1187; UPLC–MS (generic method) 1.55 min, m/z 203.2 [M + H]+, 201.3 [M – H]−.
N-(1-Cyano-3-(methylthio)propyl)-2-phenylacetamide (14)
Reaction time (discriminated) = 11 (3 + 8) days; 103 mg, 42%, white powder, mp 66 °C (degradation); 1H NMR (400 MHz, CDCl3) δ 7.33–7.13 (m, 1H), 6.59 (d, J = 8.3 Hz, 0H), 4.98 (dt, J = 8.3, 6.8 Hz, 0H), 3.51 (s, 0H), 2.49 (t, J = 6.9 Hz, 0H), 2.01–1.88 (m, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 170.8, 133.8, 129.5, 129.2, 127.7, 118.1, 43.2, 40.0, 31.4, 29.8, 15.4; HRMS (ESI) m/z: [M + H]+ calcd for C13H17N2OS 249.1056, found 249.1049; UPLC–MS (generic method) 1.51 min, m/z 249.2 [M + H]+, 247.1 [M – H]−; IR (ATR-FTIR) ν (cm–1): 3300, 3063, 3027, 2954, 2913, and 2244.
Methyl 4-Cyano-4-(2-phenylacetamido)butanoate (15)
Reaction time (discriminated) = 11 (3 + 8) days; 124 mg, 48%, off-white solid, mp 77 °C (degradation); 1H NMR (400 MHz, CDCl3) δ 7.40–7.19 (m, 5H), 6.77 (d, J = 8.0 Hz, 1H), 4.90 (td, J = 7.8, 6.8 Hz, 1H), 3.66 (s, 3H), 3.56 (s, 2H), 2.55–2.36 (m, 2H), 2.13–2.01 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 172.7, 170.9, 133.8, 129.3, 129.1, 127.6, 118.0, 52.2, 43.0, 40.1, 29.7, 27.5; HRMS (ESI) m/z: [M + H]+ calcd for C14H17N2O3 261.1234, found 261.1236; UPLC–MS (generic method) 1.43 min, m/z 261.2 [M + H]+, 283.1 [M + Na]+, 259.1 [M – H]−; IR (ATR-FTIR) ν (cm–1): 3256.
tert-Butyl ((2S)-1-((1-Cyano-2-methylpropyl)amino)-1-oxopropan-2-yl)carbamate (16)
Reaction time (discriminated) = 3 (1 + 2) days; 58 mg, 22%, amorphous white solid; 1H NMR (400 MHz, Acetone-d6) δ 7.93 (s, 1H), 6.26 (s, 1H), 4.78–4.66 (m, 1H), 4.22–4.04 (m, 1H), 2.18–2.08 (m, 1H), 2.09–2.05 (m, 2H), 1.42 (d, J = 2.7 Hz, 12H), 1.37–1.29 (m, 4H), 1.10 (d, J = 6.7 Hz, 4H), 1.05 (d, J = 6.8 Hz, 4H); 13C{1H} NMR (101 MHz, Acetone-d6) δ 173.6, 156.3, 119.0, 79.5, 51.1, 50.9, 47.3, 32.2, 28.5, 18.6, 18.3, 18.0; HRMS (ESI) m/z: [M + H]+ calcd for C13H24N3O3 270.1812, found 270.1812; UPLC–MS (generic method) 1.52 min, m/z 292.2 [M + Na]+, 268.3 [M – H]−; IR (ATR-FTIR) ν (cm–1): 3336, 3300, 2969, 2936, 2878, and 2245.
tert-Butyl (2-Cyano-2-propionamidoethyl)carbamate (17)
Reaction time (discriminated) = 11 (3 + 8) days; 44 mg, 18%, white powder, mp 162 °C (degradation); 1H NMR (400 MHz, CDCl3) δ 7.22 (d, J = 6.5 Hz, 1H), 5.24 (s, 1H), 4.88–4.77 (m, 1H), 3.66–3.43 (m, 2H), 2.27 (q, J = 7.6 Hz, 2H), 1.46 (s, 9H), 1.17 (t, J = 7.6 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 173.8, 157.5, 117.1, 81.22, 43.3, 42.1, 29.2, 28.2, 9.3; HRMS (ESI) m/z: [M + H]+ calcd for C11H20N3O3 242.1499, found 242.1505; UPLC–MS (generic method) 1.27 min, m/z 264.3 [M + Na]+, 240.2 [M – H]−, 286.2 [M + 2Na + H]+; IR (ATR-FTIR) ν (cm–1): 3367, 3241, 2977, and 2940.
N-(1-Cyanocyclobutyl)-2-phenylacetamide (18)
Reaction time (discriminated) = 14 (7 + 7) days; 12 mg, 6%, pale brown powder; mp 148 °C (degradation); 1H NMR (400 MHz, CDCl3) δ 7.43–7.23 (m, 5H), 5.88 (s, 1H), 3.60 (s, 2H), 2.77–2.66 (m, 2H), 2.30–2.10 (m, 3H), 2.03 (d, J = 12.3 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 170.5, 134.0, 129.3, 129.2, 127.6, 120.6, 77.3, 77.0, 76.7, 47.6, 43.2, 34.0, 16.0; HRMS (ESI) m/z: [M + H]+ calcd for C13H15N2O 215.1179, found 215.1181; UPLC–MS (generic method) 1.37 min, m/z 215.2 [M + H]+, 237.2 [M + Na]+; IR (ATR-FTIR) ν (cm–1): 3239, 3028, 3000, 2953, 2922, 2853, and 2236.
N-(1-Cyanocyclohexyl)-2-phenylacetamide (19)
Reaction time (discriminated) = 13 (6 + 7) days; 10 mg, 4%, brown solid, mp 125 °C (degradation); 1H NMR (400 MHz, CDCl3) δ 7.44–7.22 (m, 5H), 5.40 (s, 1H), 3.63 (s, 2H), 2.38–2.19 (m, 2H), 1.71–1.50 (m, 8H); 13C{1H} NMR (101 MHz, CDCl3) δ 170.1, 134.2, 129.3, 129.3, 129.2, 127.7, 119.6, 43.8, 35.2, 24.6, 21.9; HRMS (ESI) m/z: [M + H]+ calcd for C15H19N2O 243.1492, found 243.1502; UPLC–MS (generic method) 1.63 min, m/z 243.2 [M + H]+, 265.2 [M + Na]+.
tert-Butyl (S)-(1-((Cyano(4-methoxyphenyl)methyl)amino)-1-oxopropan-2-yl)carbamate (20)
Reaction time (discriminated) = 11 (7 + 4) days; 25 mg, 8%, off-white powder, mp 111 °C (degradation); 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 8.6 Hz, 1H), 7.34–7.25 (m, 2H), 6.90–6.74 (m, 2H), 5.97–5.83 (m, 1H), 5.02 (dd, J = 15.3, 7.3 Hz, 1H), 4.14 (s, 1H), 3.74 (d, J = 3.7 Hz, 3H), 1.41–1.23 (m, 12H); 13C{1H} NMR (101 MHz, CDCl3) δ 172.1, 160.4, 155.8, 128.3, 128.3, 125.1, 117.6, 114.6, 114.6, 55.4, 50.0, 43.5, 43.5, 29.7, 28.2, 28.2, 17.4; HRMS (ESI) m/z: [M + H]+ calcd for C17H24N3O4 334.1761, found 334.1774; UPLC–MS (generic method) 1.52 min, m/z 332.2 [M – H]−; IR (ATR-FTIR) ν (cm–1): 3337, 3293, 2988, 2925, and 2849.
tert-Butyl (3S)-3-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-((1-cyano-2-methylpropyl)amino)-4-oxobutanoate (21)
Reaction time (discriminated) = 3 (1 + 2) days; 100 mg, 21%, white powder, mp 120 °C (degradation); 1H NMR (400 MHz, CDCl3) δ 7.77 (dt, J = 7.7, 0.9 Hz, 2H), 7.58 (dd, J = 7.8, 3.5 Hz, 2H), 7.41 (t, J = 7.5 Hz, 2H), 7.37–7.28 (m, 2H), 7.22 (br, 1H), 6.00 (dd, J = 15.5, 8.0 Hz, 1H), 4.80–4.67 (m, 1H), 4.54 (br, 1H), 4.44 (d, J = 6.9 Hz, 2H), 4.29–4.17 (m, 1H), 2.99–2.80 (m, 1H), 2.72–2.53 (m, 1H), 2.12–1.94 (m, 1H), 1.46 (s, 9H), 1.16–0.96 (m, 6H); 13C{1H} NMR (101 MHz, CDCl3) δ 171.5, 171.1, 170.4, 156.3, 143.7, 141.4, 128.0, 127.9, 127.2, 127.2, 125.1, 125.0, 120.2, 117.5, 82.5, 67.5, 67.5, 51.0, 50.9, 47.2, 47.2, 46.8, 46.8, 37.5, 37.2, 31.7, 31.6, 28.1, 28.1, 18.6, 18.5, 18.1, 18.1; HRMS (ESI) m/z: [M + H]+ calcd for C28H34N3O5 492.2493, found 492.2503; UPLC–MS (generic method) 2.31 min, m/z 514.3 [M + Na]+, 259.1 [M – H]−; IR (ATR-FTIR) ν (cm–1): 3281, 2971, and 2932.
4-((1-Cyano-3-phenylpropyl)amino)-4-oxobutanoic Acid (22)
Reaction time (discriminated) = 6 (3 + 3) days; 30 mg, 12%, white powder, mp 175 °C (degradation); 1H NMR (400 MHz, methanol-d4) δ 7.33–7.10 (m, 5H), 4.69 (t, J = 7.6 Hz, 1H), 2.79 (td, J = 7.3, 1.8 Hz, 2H), 2.73–2.61 (m, 1H), 2.55–2.42 (m, 2H), 2.20–2.08 (m, 2H), 2.02–1.80 (m, 1H); 13C{1H} NMR (101 MHz, methanol-d4) δ 180.6, 175.5, 141.1, 129.6, 129.5, 129.4, 127.4, 120.0, 41.1, 35.2, 34.0, 33.3, 32.6; HRMS (ESI) m/z: [M + H]+ calcd for C14H17N2O3 261.1234, found 261.1241; UPLC–MS (generic method) 1.36 min, m/z 261.2 [M + H]+, 283.1 [M + Na]+, 259.1 [M – H]−; IR (ATR-FTIR) ν (cm–1): 3294, 3061, 3029, 2929, and 2246.
N-(1-Cyano-2-methylpropyl)nicotinamide (23)
Reaction time (discriminated) = 11 (7 + 4) days; 20 mg, 10%, pale brown powder, mp 100 °C (degradation); 1H NMR (400 MHz, CDCl3) δ 8.98 (d, J = 1.9 Hz, 1H), 8.69 (dd, J = 4.9, 1.7 Hz, 1H), 8.13 (ddd, J = 7.9, 2.3, 1.6 Hz, 1H), 7.38 (ddd, J = 7.9, 4.9, 0.9 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H), 4.96 (m, 1H), 2.13 (m, 1H), 1.08 (m, 6H); 13C{1H} NMR (101 MHz, CDCl3) δ 165.0, 152.7, 147.9, 136.0, 129.0, 123.9, 117.8, 47.2, 31.8, 18.8, 18.2; HRMS (ESI) m/z: [M + H]+ calcd for C11H14N3O 204.1131, found 204.1124; UPLC–MS (generic method) 1.08 min, m/z 204.2 [M + H]+, 202.1 [M – H]−.
N-(Cyano(pyridin-3-yl)methyl)propionamide (24)
Reaction time (discriminated) = 4 (3 + 1) days; 33 mg, 18%, yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.71–8.66 (m, 1H), 8.64 (d, J = 4.8 Hz, 1H), 7.86–7.82 (m, 1H), 7.43–7.37 (m, 1H), 7.14 (d, J = 8.5 Hz, 1H), 6.25 (d, J = 8.4 Hz, 1H), 2.33 (qd, J = 7.6, 1.9 Hz, 2H), 1.20 (t, J = 7.5 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 173.2, 150.5, 148.1, 135.1, 129.9, 124.1, 116.7, 76.7, 41.9, 31.0, 29.1, 9.3; HRMS (ESI) m/z: [M + H]+ calcd for C10H12N3O 190.0975, found 190.0970; UPLC–MS (purity method) 0.51 min, m/z 190.2 [M + H]+.
General Method for Synthesis at 45 °C
A mixture of 2-phenylacetamide (0.1 g, 0.74 mmol), 3-phenylpropanal (98 μl, 0.74 mmol), and 4-toluenesulfinic acid (0.150 g, 0.962 mmol) was dissolved in THF (4.48 mL). The solution was stirred for 14 h at 45 °C (heating source: stirrer plate and Asynt DrySyn). Once the starting materials were consumed or the reagents and intermediate quantities seemed to be stable, the reaction was cooled to room temperature, KCN (52 mg, 0.814 mmol) was added to the reaction, and it was stirred at 45 °C for two days (heating source: stirrer plate and Asynt DrySyn). Once the reaction was finished or was stable, an extraction (ethyl acetate/water) was performed, the organic phase was dried with sodium sulfate, filtered, and the solvents were removed by evaporation under reduced pressure. The crude was then purified by flash chromatography to obtain the product in 77% yield.
In the case of compound (12), the protocol was similar, with the only difference in the time of reaction, 4 h for the first part and three days for the second. After purification, the product was obtained in 53% yield.
Method for Synthesis of N-(1-Cyano-3-phenylpropyl)-2-phenylacetamide with Quinine
Two different protocols were tried, but only one allowed us to obtain the product. The protocol where quinine was added at the beginning and the reaction was maintained at −10 °C all of the time was not successful; thus, it will not be described in detail.
The protocol where quinine was added with potassium cyanide and the reaction was cooled down to −10 °C yielded a racemic mixture of the product.
Chiral Chromatography
Certain reaction conditions could induce enantioselectivity to a certain degree. Thus, four compounds were analyzed by chiral chromatography.
Chiral SCF
A Waters Thar SFC system equipped with a Daicel Chiralpak IB column (5 μm particle size, 4.6 × 250 mm) was used. An isocratic method was used (5% methanol/95% supercritical CO2, 3 mL/min flow, 40 min of elution at 25 °C). A chiral standard was synthesized to compare the retention time (see the Supporting Information); the separation of compound (11) was not optimal but sufficient to notice that the reaction is not enantioselective.
Chiral HPLC
Compounds (9), (11), (13), and (21) were analyzed by chiral chromatography to confirm that the reaction is not enantioselective. A Waters HPLC system equipped with a Jasco X-LC 3195CD detector, a PDA detector, an MS detector, and a Daicel Chiralpak IA column (5 μm particle size, 4.6 × 150mm) was used. The gradient used was n-hexane/isopropanol.
Acknowledgments
Sophie Lyssens is thanked for the SFC analysis, Elke Martin for the help with FTIR, and Prof. Filip Lemière for recording HRMS spectra. The Fund for Scientific Research—Flanders (FWO—Vlaanderen) is acknowledged for supporting this work with a research grant.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04908.
1H and 13C NMR spectra; SFC-MS and LC–MS chromatograms; HRMS results of synthesized compounds; FTIR spectra of 10 compounds; and two examples of unstable 4-tolylsulfonyl adducts (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Strecker A. Ueber Die Künstliche Bildung Der Milchsäure Und Einen Neuen, Dem Glycocoll Homologen Körper. Ann. Chem. Pharm. 1850, 75, 27–45. 10.1002/jlac.18500750103. [DOI] [Google Scholar]
- Dömling A.; Ugi I. Multicomponent Reactions with Isocyanides. Angew. Chem. 2000, 39, 3168–3210. . [DOI] [PubMed] [Google Scholar]
- Weber L. The Application of Multi-Component Reactions in Drug Discovery. Curr. Med. Chem. 2002, 9, 2085–2093. 10.2174/0929867023368719. [DOI] [PubMed] [Google Scholar]
- Nielsen T. E.; Schreiber S. L. Towards the Optimal Screening Collection: A Synthesis Strategy. Angew. Chem., Int. Ed. 2008, 47, 48–56. 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioc R. C.; Ruijter E.; Orru R. V. A. Multicomponent Reactions: Advanced Tools for Sustainable Organic Synthesis. Green Chem. 2014, 16, 2958–2975. 10.1039/C4GC00013G. [DOI] [Google Scholar]
- Kouznetsov V. V.; Galvis C. E. P. Strecker Reaction and α-Amino Nitriles: Recent Advances in Their Chemistry, Synthesis, and Biological Properties. Tetrahedron 2018, 74, 773–810. 10.1016/j.tet.2018.01.005. [DOI] [Google Scholar]
- Merino P.; Marqués-López E.; Tejero T.; Herrera R. P. Organocatalyzed Strecker Reactions. Tetrahedron 2009, 65, 1219–1234. 10.1016/j.tet.2008.11.020. [DOI] [Google Scholar]
- Wang J.; Liu X.; Feng X. Asymmetric Strecker Reactions. Chem. Rev. 2011, 111, 6947–6983. 10.1021/cr200057t. [DOI] [PubMed] [Google Scholar]
- Cai X.-H.; Xie B. Recent Advances on Asymmetric Strecker Reactions. Arkivoc 2014, 1, 205–248. 10.3998/ark.5550190.p008.487. [DOI] [Google Scholar]
- Drauz K.; Grayson I.; Kleemann A.; Krimmer H.-P.; Leuchtenberger W.; Weckbecker C.. Amino Acids. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. [Google Scholar]
- Grabowski E. J. J. Enantiopure Drug Synthesis: From Methyldopa to Imipenem to Efavirenz. Chirality 2005, 17, S249–S259. 10.1002/chir.20143. [DOI] [PubMed] [Google Scholar]
- Petrini M. α-Amido Sulfones as Stable Precursors of Reactive N- Acylimino Derivatives. Chem. Rev. 2005, 105, 3949–3977. 10.1021/cr050528s. [DOI] [PubMed] [Google Scholar]
- Marcantoni E.; Palmieri A.; Petrini M. Recent Synthetic Applications of α-Amido Sulfones as Precursors of N -Acylimino Derivatives. Org. Chem. Front. 2019, 6, 2142–2182. 10.1039/C9QO00196D. [DOI] [Google Scholar]
- Banphavichit V.; Chaleawlertumpon S.; Bhanthumnavin W.; Vilaivan T. A Convenient Synthesis of N-Boc-Protected α-Aminonitriles from α-Amidosulfones. Synth. Commun. 2004, 34, 3147–3160. 10.1081/SCC-200028592. [DOI] [Google Scholar]
- Seayad A. M.; Ramalingam B.; Yoshinaga K.; Nagata T.; Chai C. L. L. Highly Enantioselective Titanium-Catalyzed Cyanation of Imines at Room Temperature. Org. Lett. 2010, 12, 264–267. 10.1021/ol902540h. [DOI] [PubMed] [Google Scholar]
- Meester W. J. N. Scope and Limitations of the Solution and Solid Phase Synthesis of Homoallylic Amines via N-Acyliminium Ion Reactions. Arkivoc 2003, 2004, 122. 10.3998/ark.5550190.0005.209. [DOI] [Google Scholar]
- Costantini N. V.; Bates A. D.; Haun G. J.; Chang N. M.; Moura-Letts G. Rutile Promoted Synthesis of Sulfonylamidonitriles from Simple Aldehydes and Sulfonamides. ACS Sustainable Chem. Eng. 2016, 4, 1906–1911. 10.1021/acssuschemeng.6b00241. [DOI] [Google Scholar]
- Ooi T.; Uematsu Y.; Fujimoto J.; Fukumoto K.; Maruoka K. Advantage of in Situ Generation of N-Arylsulfonyl Imines from α-Amide Sulfones in the Phase-Transfer-Catalyzed Asymmetric Strecker Reaction. Tetrahedron Lett. 2007, 48, 1337–1340. 10.1016/j.tetlet.2006.12.122. [DOI] [Google Scholar]
- Reingruber R.; Baumann T.; Dahmen S.; Bräse S. Use of the Chiral Pool - Practical Asymmetric Organocatalytic Strecker Reaction with Quinine. Adv. Synth. Catal. 2009, 351, 1019–1024. 10.1002/adsc.200800798. [DOI] [Google Scholar]
- Petrini M.; Seri M. α-Amido Sulfones from Natural α-Amino Acids and Their Reaction with Carbon Nucleophiles. Tetrahedron 2006, 62, 960–967. 10.1016/j.tet.2005.10.033. [DOI] [Google Scholar]
- Kadam S. T.; Thirupathi P.; Kim S. S. Synthetic Application of in Situ Generation of N-Acyliminium Ions from α-Amido p-Tolylsulfones for the Synthesis of α-Amino Nitriles. Tetrahedron 2010, 66, 1684–1688. 10.1016/j.tet.2010.01.010. [DOI] [Google Scholar]
- Katritzky A. R.; Urogdi L. Benzotriazole-Assisted Synthesis of α-Acylaminonitriles and a Conceptually Novel Method for Peptide Elongation. J. Chem. Soc., Perkin Trans. 1 1990, 7, 1853–1857. 10.1039/P19900001853. [DOI] [Google Scholar]
- Ehmke V.; Kilchmann F.; Heindl C.; Cui K.; Huang J.; Schirmeister T.; Diederich F. Peptidomimetic Nitriles as Selective Inhibitors for the Malarial Cysteine Protease Falcipain-2. MedChemComm 2011, 2, 800. 10.1039/c1md00115a. [DOI] [Google Scholar]
- Bondebjerg J.; Fuglsang H.; Valeur K. R.; Pedersen J.; Nærum L. Dipeptidyl Nitriles as Human Dipeptidyl Peptidase I Inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 3614–3617. 10.1016/j.bmcl.2006.01.102. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Stirnberg M.; Sisay M.; Gutschow M. Development of Nitrile-Based Peptidic Inhibitors of Cysteine Cathepsins. Curr. Top. Med. Chem. 2010, 10, 294–322. 10.2174/156802610790725452. [DOI] [PubMed] [Google Scholar]
- Ndao M.; Beaulieu C.; Black W. C.; Isabel E.; Vasquez-Camargo F.; Nath-Chowdhury M.; Massé F.; Mellon C.; Methot N.; Nicoll-Griffith D. A. Reversible Cysteine Protease Inhibitors Show Promise for a Chagas Disease Cure. Antimicrob. Agents Chemother. 2014, 58, 1167–1178. 10.1128/AAC.01855-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan P. D.; Clark K. L.; Cowen S. D.; McQuire L. W.; Tommasi R. A.; Farley D. L.; Quadros E.; Coppa D. E.; Du Z.; Fang Z.; Zhou H.; Doughty J.; Toscano K. T.; Wigg A. M.; Zhou S. N-Arylaminonitriles as Bioavailable Peptidomimetic Inhibitors of Cathepsin B. Bioorg. Med. Chem. Lett. 2003, 13, 4121–4124. 10.1016/j.bmcl.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Tehrani K. A.; De Kimpe N.. Category 4. Compounds with Two Carbon Heteroatom Bonds. In Science of Synthesis; Padwa A.; Bellus D., Eds.; Georg Thieme Verlag: Stuttgart, 2005; pp 245–312. [Google Scholar]
- Paraskar A. S.; Sudalai A. Cu(OTf)2 or Et3N-Catalyzed Three-Component Condensation of Aldehydes, Amines and Cyanides: A High Yielding Synthesis of α-Aminonitriles. Tetrahedron Lett. 2006, 47, 5759–5762. 10.1016/j.tetlet.2006.06.008. [DOI] [Google Scholar]
- Das B.; Damodar K.; Shashikanth B.; Srinivas Y.; Kalavathi I. A Mild and Efficient Catalytic Strecker Reaction of N -Alkoxycarbonylamino Sulfones with Trimethylsilyl Cyanide Using Indium(III) Chloride: A Facile Synthesis of α-Aminonitriles. Synlett 2008, 2008, 3133–3136. 10.1055/s-0028-1087277. [DOI] [Google Scholar]
- Ooi T.; Uematsu Y.; Maruoka K. Asymmetric Strecker Reaction of Aldimines Using Aqueous Potassium Cyanide by Phase-Transfer Catalysis of Chiral Quaternary Ammonium Salts with a Tetranaphthyl Backbone. J. Am. Chem. Soc. 2006, 128, 2548–2549. 10.1021/ja058066n. [DOI] [PubMed] [Google Scholar]
- Engberts J. B. F. N.; Strating J. The Mannich Condensation of Sulfinic Acids, Aldehyde, and Ethyl Carbamate II.: The Use of Higher Aldehydes. Recl. Trav. Chim. Pays-Bas 1965, 84, 942–950. 10.1002/recl.19650840714. [DOI] [Google Scholar]
- Engberts J. B. F. N.; Olijnsma T.; Strating J. The Mannich Condensation of Sulfinic Acids, Aldehyde, and Ethyl Carbamate: III. The Use of Ethyl N-Monosubstituted Carbamates, Ethyl Monothiocarbamates and Ethyl Dithiocarbamates. Recl. Trav. Chim. Pays-Bas 1966, 85, 1211–1222. 10.1002/recl.19660851207. [DOI] [Google Scholar]
- Maleki A.; Akhlaghi E.; Paydar R. Design, Synthesis, Characterization and Catalytic Performance of a New Cellulose-Based Magnetic Nanocomposite in the One-Pot Three-Component Synthesis of α-Aminonitriles. Appl. Organomet. Chem. 2016, 30, 382–386. 10.1002/aoc.3443. [DOI] [Google Scholar]
- Maleki A.; Rahimi J.; Hajizadeh Z.; Niksefat M. Synthesis and Characterization of an Acidic Nanostructure Based on Magnetic Polyvinyl Alcohol as an Efficient Heterogeneous Nanocatalyst for the Synthesis of α-Aminonitriles. J. Organomet. Chem. 2019, 881, 58–65. 10.1016/j.jorganchem.2018.12.002. [DOI] [Google Scholar]
- Larkin P. J.Instrumentation and Sampling Methods. In Infrared and Raman Spectroscopy; Elsevier, 2018; pp 29–61. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.