

Abstract
The natural function of cellobiose dehydrogenase (CDH) to donate electrons from its catalytic flavodehydrogenase (DH) domain via its cytochrome (CYT) domain to lytic polysaccharide monooxygenase (LPMO) is an example of a highly efficient extracellular electron transfer chain. To investigate the function of the CYT domain movement in the two occurring electron transfer steps, two CDHs from the ascomycete Neurospora crassa (NcCDHIIA and NcCDHIIB) and five chimeric CDH enzymes created by domain swapping were studied in combination with the fungus’ own LPMOs (NcLPMO9C and NcLPMO9F). Kinetic and electrochemical methods and hydrogen/deuterium exchange mass spectrometry were used to study the domain movement, interaction, and electron transfer kinetics. Molecular docking provided insights into the protein–protein interface, the orientation of domains, and binding energies. We find that the first, interdomain electron transfer step from the catalytic site in the DH domain to the CYT domain depends on steric and electrostatic interface complementarity and the length of the protein linker between both domains but not on the redox potential difference between the FAD and heme b cofactors. After CYT reduction, a conformational change of CDH from its closed state to an open state allows the second, interprotein electron transfer (IPET) step from CYT to LPMO to occur by direct interaction of the b-type heme and the type-2 copper center. Chimeric CDH enzymes favor the open state and achieve higher IPET rates by exposing the heme b cofactor to LPMO. The IPET, which is influenced by interface complementarity and the heme b redox potential, is very efficient with bimolecular rates between 2.9 × 105 and 1.1 × 106 M–1 s–1.
Keywords: cellobiose dehydrogenase, chimeric enzyme, domain swapping, electron transfer, lytic polysaccharide monooxygenase
Introduction
The catalytic activity of lytic polysaccharide monooxygenase (LPMO) and its interaction with cellobiose dehydrogenase (CDH) have been reported to increase the rate of cellulose hydrolysis from the recalcitrant biomass and to increase the overall efficiency of enzymatic cocktails.1−5 In contrast to electron-donating, low-molecular weight reductants of LPMO such as gallate or ascorbate, CDH is specific for LPMO and shows a fast electron transfer at physiological concentrations.6,7 CDH is an extracellular flavocytochrome and contains FAD and a b-type heme in the flavodehydrogenase (DH) and cytochrome (CYT) domains, respectively, which are connected via a flexible linker. The electron transfer between the domains is pH dependent and has been studied by Igarashi and coworkers in detail.8 Recently, the structure of the full-length protein has been elucidated and two conformations (closed- and open state) of the CYT domain were observed, which are supposed to play a role in interdomain electron transfer (IDET) and interprotein electron transfer (IPET).9
LPMO activation by CDH comprises three steps: (i) catalytic cellobiose oxidation in the DH active site leads to the formation of the reduced FAD cofactor, (ii) interaction of CYT with DH in the closed state results in the subsequent one-electron IDET, and (iii) interaction of CYT in the open state with LPMO results in the one-electron IPET. In the closed state of CDH, the FAD and heme b cofactors are in close proximity (∼0.9 nm), which should favor IDET, whereas IPET depends on the interaction of the heme b with LPMO,9 which should be favored in the open state. The structure of the linker in the open- or closed states could not be fully determined in crystal structures, which indicates its high flexibility.
The two CDHs encoded in the genome of Neurospora crassa provide a good basis to study the influence of the CYT mobility on electron transfer because of several reasons. First, the structure of NcCDHIIA has been elucidated (PDB ID: 4QI7), and second, a comparison of the steady-state kinetic constants of the two CDHs in a previous study found a 3.5-fold faster IDET rate for NcCDHIIA at pH 6.0 compared to NcCDHIIB despite the ∼50 mV higher redox potential of the heme b cofactor.7 The independence of the IDET rate from the electrochemical driving force suggests a different function of both enzymes’ CYT domains, possibly an adaptation to the copper center redox potentials of different LPMOs.7 Structural features of the domains and surface charge distribution have been shown to influence the CDH domain interaction kinetics.10,11 SAXS and SANS studies showed that the oxidized form of CDH populates a variety of conformational states between the closed- and fully open state and that pH, presence of divalent cations, and the presence of LPMO modulate the occupation of the closed- and open states.12,13 Fast scanning AFM studies showed a preference of the open state in the reduced form of CDH.14 These observations raise the question of how CYT interacts with either DH or LPMO and which structural and kinetic determinants govern this interaction.
Based on sequence alignment and the elucidated crystal structures, we created chimeric CDH enzymes by exchanging linker, CYT, and DH domains of the two NcCDHs to study the role of the CYT–DH interface, the effect of different cofactor redox potentials, and the influence of the linker length on the protein–protein interaction and IDET. CYT–LPMO interaction was also studied by hydrogen/deuterium exchange mass spectrometry (HDX-MS) measurements and transient-state kinetics to determine the interaction site of CDH–LPMO. We also evaluated the structural and kinetic determinants of the domain interaction to test recent results obtained by Courtade et al., who showed the binding of CDH and CYT to the LPMO active site by means of 15N-HSQC and 13C-aromatic-HSQC,15 and by Laurent et al. who modeled the interaction between both enzymes.16
To study the effect of (i) the surface complementarity at the protein–protein interface, (ii) differences in the redox potentials of the cofactors, and (iii) the linker length on the domain interaction and the electron transfer rate, a domain swapping strategy was applied to create chimeric enzymes of the two N. crassa CDHs by exchanging CYT and linkers with different structural and physical properties. The chimeric CDHs were studied by steady-state and presteady-state kinetics, electrochemical methods, and molecular modeling in combination with two N. crassa LPMOs.
Results
Construction and Properties of Chimeric CDH Variants
A domain swapping strategy was applied to exchange linkers and CYT domains of the two N. crassa CDHs (Figure 1A). The sequence alignment of NcCDHIIA (UniProt: Q7RXM0) with NcCDHIIB (UniProt: Q7S0Y1) gives a sequence identity of 53% and was used together with the crystal structure of NcCDHIIA (PBD ID: 4QI7) and a homology model of NcCDHIIB to define the individual CDH domains. The end of the N-terminal CYT domain is defined by a cysteine residue forming a disulfide bond (CYTA: Q1–C211, CYTB: Q1–C216, for brevity, we denote the domains and the linker of NcCDHIIA by A and NcCDHIIB by B). This disulfide bond in CYT is found in several CDHs and possibly evolved to stabilize the C-terminus against mechanical stress exerted by the linker. After this cysteine, the linker sequence starts (linkerA: S212–S229, linkerB: S217–T250). The DH domain starts with the first amino acid firmly connected with DH and ends with the C-terminus (DHA: F230–V772, DHB: Y251–R805). The C-terminus of NcCDHIIA features an additional family 1 carbohydrate-binding module (CBM1, P773–V806), which is not present in NcCDHIIB. Because in this study the binding of CDHs to cellulose is not interfering with the experiments, the CBM1 was not removed. It is present in all chimeric CDHs with a DHA domain. The sequence identities of individual linkers, CYT and DH domains deviate considerably from the global sequence identity (Figure 1B). The catalytically active DH domains are most conserved, the linkers least. The linkers of both enzymes are rich in serine, threonine, and proline but differ substantially in length. LinkerA consists of 17 amino acids, while linkerB is twice as long and consists of 33 amino acids. The evolutionary divergence of the CYT domains and linkers points toward different mechanistic properties, physiological functions, and interacting LPMOs.
Figure 1.

Properties of chimeric enzymes. (A) Domain architecture. The two N. crassa wild-type CDHs (CDHIIA denoted CDHAAA and CDHIIB denoted CDHBBB) consist of an N-terminal CYT domain, C-terminal DH domain, and a protein linker connecting the two domains. Four chimeric CDHs (CDHBBA, CDHBAA, CDHAAB, and CDHABB) were created by domain swapping. (B) Linker sequence and position in CDH. The alignment shows sequence identities and the N- and C-terminal ends of the linkers. (C) Purified wild-type and chimeric CDHs. The measured and calculated molecular weights differ due to glycosylation.
Production and Purification of Enzymes
Wild-type N. crassa CDHs (NcCDHIIA denoted CDHAAA and NcCDHIIB denoted CDHBBB) and five chimeric CDHs (CDHAAB, CDHABB, CDHBBA, CDHBAA, and CDHABA) were recombinantly produced in Pichia pastoris and chromatographically purified (Figure S1 and Table S1). LPMO9C and LPMO9F from N. crassa were also produced in P. pastoris and chromatographically purified. The molecular weight of the individual domains and linkers can be calculated from the amino acid sequence and summed up to obtain molecular weights for full-length CDHs (Figure 1C). Similar molecular weights for the two wild-type enzymes CDHAAA and CDHBBB are predicted, and also between the smallest and largest chimeric enzymes (CDHAAB and CDHBBA, respectively) the mass difference is only 4484 Da. The molecular weights of the six purified CDHs determined by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) differ from the calculated values. The observed molecular weights are 12–39% larger and a result of posttranslational N-glycosylation17 and O-glycosylation.18 Considerable differences in the glycosylation, even between structurally quite similarly built chimeric CDHBBA and CDHBAA, point toward batch-to-batch variations between fermentations or differences in the post-translational processing of the chimeric CDHs. This heterogeneity of glycoforms is also known from homologously secreted CDHs and cannot be avoided so far. A deglycosylation of CDH results in low stability and solubility. The effects of differences in the O-glycosylation of the linker are unknown but might influence its flexibility. Bivariate correlation analysis of the mass percentage of each CDH’s glycosylation shows no correlation with the domain composition in wild-type or chimeric CDH and also no correlation with the observed catalytic- or electron transfer rates later reported (Figure S7, last column). The UV–visible (UV–vis) spectra of the oxidized and reduced chimeric CDHs reveal that the FAD and heme b cofactors are properly incorporated (Figure S2).
Catalytic Performance of DH Domains in Chimeric CDHs
In the reductive half-reaction, the oxidation of cellobiose in the active site of the DH domain results in the formation of cellobiono-δ-lactone and the reduced cofactor. In the oxidative half-reaction, the two electrons stored at the FADH2 are then transferred to the heme b cofactor in the CYT domain in two separate, one-electron transfer steps to provide electrons for LPMO reduction.7 Alternatively, the electrons can be transferred from FADH2 to a two-electron acceptor such as 2,6-dichloroindophenol, which allows the assessment of catalysis without the contribution of the subsequent electron transfer step to the CYT domain. To investigate if the exchange of the CYT domain influences catalysis in the DH domain, we determined the pH optima, steady-state catalytic constants, and presteady-state rates for the two wild-type CDHs and the four chimeric CDHs.
The pH-dependence of the catalytic reaction with cellobiose and 2,6-dichloroindophenol resulted in bell-shaped pH profiles with optima between 4.5 and 5.5 (Figure 2A,B). In comparison with the wild-type enzymes, the chimeric CDHs show a slight shift of the pH optimum, narrower peaks, and a reduced activity above pH 7. The catalytic constants and presteady-state rates were determined at pH 6.0 (Figure 2C and Table 1). At this pH, the optimal CDH–LPMO interaction was observed,6 which is important for IPET experiments. The determined KM and kcat of CDHAAA for cellobiose are both about four times higher than that of CDHBBB, which results in the same catalytic efficiency. In the presence of CYTB, the KM and kcat of DHA are slightly lower compared to CDHAAA, whereas the presence of CYTA has no significant effect on the KM and kcat of DHB compared to CDHBBB. Statistical analysis shows, as expected, a strong correlation between the type of the DH domain and the kcat for cellobiose, whereas no correlation is found for the influence of the CYT domain on kcat (Figure S7). The catalytic efficiencies at pH 6.0 are similar for all wild-type and chimeric CDHs, which renders this pH suitable for studying the subsequent IDET and IPET steps.
Figure 2.

Effects of domain swapping on catalytic rates in the DH domain. (A) pH optima of cellobiose conversion in CDHs with a DHA when using the two-electron acceptor 2,6-dichloroindophenol. (B) pH optima of CDHs with a DHB using the same substrate and cosubstrate as in (A). (C) FAD reduction rate in all CDHs measured at 449 nm (kobs449) for increasing cellobiose concentrations.
Table 1. Catalytic Constants, Transient Rates, and FAD Redox Potentials of CDHsa.
| enzyme | kcat [s–1] | KM [mM] | kcat/KM [M–1 s–1] | klim449 [s–1] | E vs SHE [mV] |
|---|---|---|---|---|---|
| CDHAAA | 17.8 ± 0.4 | 0.105 ± 0.003 | 1.7 × 105 | 81.8 ± 2.2 | 33 ± 5 |
| CDHBAA | 9.0 ± 0.3 | 0.057 ± 0.006 | 1.6 × 105 | 82.2 ± 2.6 | 24 ± 5 |
| CDHBBA | 14.2 ± 0.4 | 0.075 ± 0.005 | 1.9 × 105 | 79.8 ± 2.4 | 31 ± 1 |
| CDHABA | 9.1 ± 0.2 | n.d. | n.d. | 89.6 ± 2.3 | n.d. |
| CDHBBB | 4.6 ± 0.1 | 0.027 ± 0.002 | 1.7 × 105 | 33.5 ± 0.4 | 43 ± 15 |
| CDHABB | 5.0 ± 0.1 | 0.026 ± 0.003 | 1.9 × 105 | 32.3 ± 0.4 | 33 ± 23 |
| CDHAAB | 4.5 ± 0.2 | 0.046 ± 0.003 | 1.0 × 105 | 30.4 ± 0.6 | n.d. |
The steady-state catalytic constants of the DH domains in wild-type and chimeric CDHs were determined for cellobiose as substrate and 2,6-dichloroindophenol as saturating co-substrate. Transient FAD reduction rates (kobs449) measured in a stopped-flow spectrophotometer at different cellobiose concentrations were used to extrapolate the maximal reduction rate of FAD for an infinite cellobiose concentration (klim449). The midpoint redox potentials (E1/2) of the FAD cofactor in regard to the SHE was determined in a spectroelectrochemical cell is given in the last column. All measurements were performed at pH 6.0. n.d.: not determined because of too small amount of chimeric CDH.
The presteady-state reduction rates of FAD at 449 nm (Figure 2C, kobs449) and the extrapolated limiting rates for infinite substrate concentrations (Table 1, klim449) show that enzymes with a DHA domain oxidized cellobiose ∼2.5 times faster than enzymes with a DHB domain but no influence of the swapped CYT domains is observed. A plot of the kobs449 versus the cellobiose concentration indicates a higher substrate affinity of the DHB active site, which is in agreement with the results from steady-state analysis. The performed experiments show that the reductive-half reaction of DH is not affected by a swap of the CYT domain. No bivariate correlation was found between CYT-type and kobs449 in contrast to the high correlation between DH-type and kobs (Figure S7).
Cofactor Redox Potentials in Chimeric CDH
The FAD and heme b cofactors in CDH make close contact (∼0.9 nm edge-to-edge distance) in the enzyme’s closed state. To determine if a domain swap influences the redox properties of FAD (Table 1) and heme b (Table 2), the midpoint redox potentials of the wild-type and chimeric CDHs were measured. However, no significant change was found. The midpoint redox potentials of FAD in all CDHs were between 24 and 43 mV versus standard hydrogen electrode (SHE). The spectroelectrochemical measurement of the relatively low FAD absorbance in the presence of the strong CYT Soret-band resulted in bigger errors for enzyme solutions with a lower enzyme concentration. The low amount of purified CDHAAB and CDHABA did not allow the determination of its FAD redox potential. In contrast to the similar redox potential of FAD in DHA and DHB, the heme b redox potential in the CYT domains differ by about 60 mV. While CYTA showed little modulation of its redox potential in wild-type and chimeric CDHs (∼97–110 mV vs SHE), the CYTB redox potentials were slightly increased (169 and 172 mV vs SHE) in the chimeric CDHs compared to wild-type CDHBBB (158 mV vs SHE). However, statistical analysis shows no significant correlation between the type of the DH domain and the CYT midpoint potential (Figure S7).
Table 2. Steady-State and Transient-State IDET Rates and Heme b Redox Potentialsa.
| enzyme | TN@pH 6 [s–1] | IDET [s–1] | E vs SHE [mV] |
|---|---|---|---|
| CDHAAA | 6.14 ± 0.12 | 50.00 ± 0.10 | 102 ± 4 |
| CDHBAA | 0.50 ± 0.03 | 0.40 ± 0.02 | 172 ± 5 |
| CDHBBA | 0.04 ± 0.01 | 0.02 ± 0.02 | 169 ± 5 |
| CDHABA | 2.05 ± 0.01 | 8.42 ± 0.23 | 110 ± 2 |
| CDHBBB | 1.93 ± 0.03 | 4.00 ± 0.01 | 158 ± 2 |
| CDHABB | 0.48 ± 0.01 | 0.40 ± 0.01 | 97 ± 4 |
| CDHAAB | 0.52 ± 0.01 | 0.40 ± 0.02 | 103 ± 4 |
Comparison of cytochrome c reduction rates (turnover numbers, TN) as an indicator of IDET with transient rates (kobs563) at pH 6.0, along the heme b midpoint redox potentials (E vs SHE).
IDET in Chimeric CDHs
Cellobiose oxidation in the DH domain is followed by IDET from the FADH2 or FAD semiquinone to the oxidized heme b. Steady-state kinetic measurements with cellobiose and the one-electron acceptor cytochrome c, which interacts only with CYT but not with the DH domain, were used to compare wild-type and chimeric CDHs (Figure 3A and Table 2). The cytochrome c turnover number (TN) of CDHAAA was about 3.5-fold higher than that of CDHBBB, which corresponds to the faster catalytic turnover found for DHA. All chimeric CDHs exhibit lower cytochrome c TNs than the wild-type CDHs, but it is surprising that for four out of the five chimeric enzymes the decrease is only 3–12-fold. Only CDHBBA showed an almost complete shutdown but still had a measurable IDET. This indicates two points: (i) the relatively good compatibility of the DH domains with unfamiliar CYT domains despite their low sequence identity of 42% and (ii) the influence of the linker on the CYT–DH interaction, which is demonstrated by the reduced IDET of chimeric CDHs featuring the longer linkerB. In CDHABA, the longer linker reduced the steady-state turnover of cytochrome c by a factor of 3 and the IDET rate by a factor of 19. The pH optimum of the IDET was partially influenced by the domain swapping. The wild-type CDHBBB has a lower pH optimum (pH 4.5) than CDHAAA (pH 5.5) but exhibits a plateau until pH 8.0. The pH optima of chimeric CDHs are identical or close to that of the respective DH domain, which can be explained by the isoelectric points of the individual domains. The CYT domains in CDH typically have a very low pI of ∼3, whereas the DH domains have a pI of ∼5.19 The deprotonation of acidic amino acid residues on the DH domain close to the CYT–DH interface generates electrostatic repulsion of the strongly negatively charged CYT domain. Interestingly, the plateau observed for CDHBBB is also found in CDHBBA and CDHBAA and thus seems to be a feature of CYTB.
Figure 3.

Effects of domain swapping on IDET. (A) pH optima of cytochrome c turnover numbers for wild-type and chimeric CDHs. (B) IDET rates (kobs563) of DHA to different CYT domains measured for increasing cellobiose concentrations. (C) IDET rates of DHB to different CYT domains measured for increasing cellobiose concentrations.
Although the IPET between CYT and cytochrome c is very fast,20−22 it still could influence the observed IDET rate. Therefore, we also measured the transient reduction rate of the heme b cofactor to avoid a possible rate-limiting step. This direct measurement of IDET in CDH was performed by stopped-flow spectrophotometry at 563 nm to observe the reduction of the heme b α-peak (Figure 3B,C and Table 2). The observed transient rates are consistent with the trend of the steady-state rates with the wild-type CDHs having the most efficient IDET. The data also show that the cytochrome c assay provides a good estimate for the IDET rate in chimeric CDHs but not for the faster wild-type CDHs. As expected, the IDET rate (kobs563) of all CDHs is slower than the respective FAD reduction rates (kobs449). However, in the case of CDHAAA, kobs563 is 50 s–1 and very close to kobs449 (80 s–1). In this case, IDET is limited at low cellobiose concentrations (<1 mM). For CDHBBB and all chimeric CDHs, a much slower IDET was observed and, therefore, a limitation was found only for substrate concentrations below 50 μM. Considering that the redox potential difference between CYTA and DHA (∼102 mV) is lower than for CYTB and DHA or DHB (∼158 mV), the thermodynamic driving force between the cofactors is obviously irrelevant for the IDET rate. Also, no statistical correlation was observed between the midpoint redox potential and IDET rate (Figure S7). This exciting observation was further investigated by calculating the electron transfer rate based on the Marcus theory of electron tunneling. A modified version used by Dutton and coworkers23,24 was applied using reported maximum, average, and minimum values for the quantum mechanical constants (λ, B, E) for the calculation of the corresponding distance-dependent electron transfer rates. The average edge-to-edge distance between the FAD and the heme b propionate A in docking models of CDHAAA and CDHBBB was found to be 0.9 nm, respectively (Figure 4). This corresponds to theoretical IDET rates in the order of 105 to 106 s–1, which are at least four orders of magnitude faster than the measured IDET rates.
Figure 4.
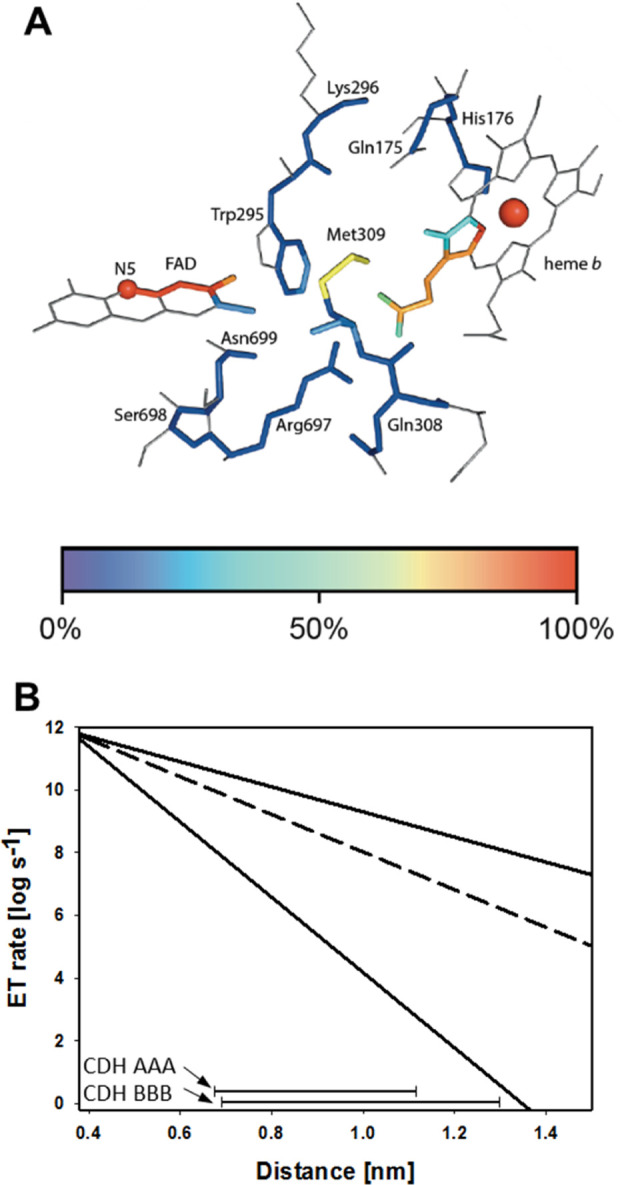
Electron transfer in CDH (IDET). (A) Detail of the crystal structure of MtCDHIIA (PDB: 4QI6) featuring the closed-state conformation. The edge-to-edge distance between the FAD and heme cofactors is 0.9 nm. (B) Electron transfer rate plotted against cofactor distance for CDH (lower and upper limit, solid lines; most probable parameters, dashed line). The bars at the bottom indicate the observed IDET rates and edge-to-edge distances for CDHAAA and CDHBBB in docking calculations.
Considering the observed mobility of the linker and CYT domain in CDH, we postulate that this large difference between the calculated and the measured rates is because of conformational changes: the transition between the open- and closed states of the CDH. The optimal, closest possible distance between the FAD and heme b cofactors depends on the correct orientation of the CYT domain at its DH domain interface. The open-state distance between the cofactors can easily exceed 1.5 nm and shut down IDET. With IDET depending on the closed state or at least very close proximity between DH and CYT, a steric mismatch between the domain surfaces, repulsive electrostatic interactions, or a linker that provides too much mobility will reduce IDET. This is supported by the inspection of the kobs563 rates for both evolved wild-type CDHs and the chimeric CDHs (Table 2). The IDET for the constructed chimeric enzymes decreased by one order of magnitude for CDHAAB, CDHABB, and CDHABA, two orders of magnitude for CDHBAA, and three orders of magnitude for CDHBBA. In the case of CDHABA, in which the CYTA–DHA interface is not altered, the longer linker results in an only 19 times lower IDET compared to the 125 times reduction of CDHBAA, in which the CYT domain is swapped.
Based on the steady-state catalytic constants and kobs563, a limiting substrate concentration, above which the IDET confines the catalytic rate, can be calculated. For the naturally occurring NcCDHAAA and NcCDHBBB, already low cellobiose concentrations (55 and 35 μM, respectively) ensure that both CDHs reach their maximum IDET rate, which is the prerequisite of efficient LPMO reduction.
Evaluation of the DH–CYT Interaction Site by Docking
The program HADDOCK25,26 was used to determine the interface of the four possible CYT–DH combinations found in the wild-type and chimeric enzymes by ambiguous restraint driven docking. A sample size of 200 docking poses for each CYT–DH pair was used for analysis. A “rotation” angle is used to define the rotation of CYT around a defined interdomain axis (Figure 5A and Table S2) in regard to DH, relative to the corresponding angle observed in the closed state of the Myriococcum thermophilum CDH structure (PDB ID: 4QI6). Similarly, we used the terms “declination” to describe the vertical offset angle and “inclination” to describe the horizontal offset angle of the docked CYT domain relative to the DH domain. The feasibility of docking poses was further assessed by considering the maximal extension of linkerA and linkerB, which was estimated to be 6 and 11 nm, respectively. By using the distance field reaction coordinate as implemented in GROMOS++ software,27,28 the shortest curved distance between the C-terminus of the CYT domain and the N-terminus of the DH domain not passing through the protein was computed. Docking poses, in which this distance was longer than the maximal extension of the linker, were excluded from subsequent analysis (Figure 5B, grey squares). The pH-dependent surface charges of the domains were calculated from pH 4–8 (Figure S3) and the protonation states corresponding to pH 6.0 were used for the docking. For this pH, the contribution of the van der Waals energy to the protein–protein interaction (−158.14 ± 62.2 kJ mol–1) is generally 4–5 times higher than the electrostatic energy (−36.79 ± 20.9 kJ mol–1), which indicates the importance of structurally complementary domain surfaces. A comparison of CDHAAA and CDHBBB shows that the declination and inclination angles of the 200 docking positions are narrower for CDHAAA, which is indicative of a sterically more defined CYT–DH interaction. In CDHAAA, the CYT rotation around the rotation axis is well defined by two groups with angles at −5 ± 15 and 25 ± 15°. The rotational position at 19.5° is preferred because it exhibits the strongest van der Waals and electrostatic interaction energies. In CDHBBB, the docked rotational positions fall further apart (40 ± 50, 110 ± 20°), indicating a less directed interaction and a lower complementarity of the domain surfaces. The interaction energies are less favorable than in CDHAAA. Interestingly, the energetically most favorable docking position of CYT and DH in CDHBBB is not feasible because of the restricting length of the linker. This particular position with a rotation angle of −144.5° corresponds to an almost 180° rotation of the allowed rotational position with the second lowest van der Waals energy.
Figure 5.

Orientation of CYT to DH in docking poses. (A) Schematic representation of evaluated angles. (B) From a total of 200 docking poses for each CYT–linker–DH pair the angle of rotation, declination, and inclination were measured in regard to its deviation from the crystal structure of the closed-state conformation of M. thermophilum CDH (PDB ID: 4QI6). The electrostatic (red) and van der Waals (blue) binding energies for each pose are given in kJ mol–1. Docking poses in wild-type CDHAAA and wild-type CDHBBB are compared to docking poses of chimeric CDHBAA, CDHABB, CDHBBA, and CDHAAB.
In chimeric CDHs, the linker plays an important role. The shorter linkerA restricts the angular CYT orientation in CDHBAA and CDHAAB much more than the longer linkerB in CDHBBA and CDHABB. Only one angular orientation at 45 ± 25° is allowed by the length of linkerA, while the longer linkerB allows for rotational positions between 45 ± 25 and 110 ± 40°. In the case of CDHBBA (kobs563 = 0.04 s–1), the rotational orientation of CYTB against DHA at 126.3° is strongly preferred in terms of interaction energies (EvdW: −300.7 kJ mol–1; EElec −40.4 kJ mol–1) over rotational position at 32.7° (which is the IDET competent orientation in CDHBAA), which has less favorable interaction energies (EvdW: −176.8 kJ mol–1; EElec: −22.8 kJ mol–1). The steric restriction provided by linkerA prevents the CYT in CDHBAA to bind in a noncompetent position and thereby increases IDET (kobs563 = 0.4 s–1).
The average contact surface area for all possible complexes was calculated and averaged for each CYT–DH combination as well as the binding affinity using PRODIGY29−31 (Table S3). The averaged contact surface areas correspond to ∼4% of the total DH surface area and ∼9% of the total CYT surface area. The small interaction site and low calculated affinities of the CYT–DH complexes (KD = 3.2–47 μM) suggest a relatively transient and reversible interaction when compared to other redox proteins.32
Interaction Site of CYT with LPMO
Two interaction sites on CDH’s CYT domain with LPMO have been proposed in the literature based on computational docking. One potential interaction site has been proposed to be opposite to LPMO’s type-2 copper center around a conserved Pro-Gly-Pro patch,33 which requires long-range electron transfer through LPMO but would allow the reduction of the substrate-bound LPMO. Another study suggested a direct interaction of heme b in CYT with the copper center of LPMO, which would necessitate the desorption of LPMO from its polymeric substrate. This mode would require no long-range electron transfer through the LPMO molecule.9 To experimentally determine the protein–protein interaction site of CDH and LPMO in solution, H/D exchange kinetics were followed by mass spectrometry for CDHAAA and N. crassa LPMO9F. This particular LPMO is well suited for such an analysis because it is relatively small (24.8 kDa) and lacks N-glycosylation, a C-terminal CBM1, and the linker region, which is often heavily O-glycosylated.34
Both proteins alone or in a mixture were subjected to H/D exchange followed by online digestion with pepsin and the resulting fragments were analyzed as described previously.32 No detectable difference in the deuteration was observed on CDHAAA. This could be caused by a combination of several factors: (i) a very short-lived or weak interaction of both enzymes, (ii) the protruding heme propionate-A group being the most prominent interaction partner leading to little involvement of other CYT residues, or (iii) the subsequent CYT–DH interaction interfering with the CYT deuteration.
For LPMO9F, on the other hand, protein backbone deprotection was observed in several peptide fragments when CDH was present in the solution. Visualization on the crystal structure (PDB ID: 4QI8)9 shows that the perturbed protein regions occur in three loops surrounding the active site copper center (Figure 6). Although deprotection by the interaction is not the most common scenario in H/D exchange, it has been recognized as one of the possible biologically relevant outcomes.35−37 In the case of CDH–LPMO interaction, it likely reflects the transient nature of the complex, where a short-lived interaction with the heme b in CYT leads to the local loosening of the structure around the copper center of LPMO and/or destabilization of the hydrogen bonding network in this region. Finally, no deuteration changes of any kind were observed around the conserved patch 207Pro-Gly-Pro209 (Figure 6) close to the C-terminus.
Figure 6.

Structure dynamics arising from N. crassa LPMO9F and CDHAAA interaction detected by H/D exchange. Structural differences between free LPMO and LPMO in the presence of CDHAAA were visualized using a difference heat map (A) (http://peterslab.org/MSTools/). Deuteration levels of the protein alone were subtracted from those observed for the protein in the presence of CDHAAA. Increased deuteration (deprotection) upon interaction is shown by red colors while protection is in blue (scale bar is at the bottom of the panel). Secondary structure elements, loops, and copper coordinating residues (green) and ProGlyPro patch (orange) are depicted above the heat map. Individual exchange times are shown on the right. Two selected time points (30 min and 3 h, indicated by arrowhead) were visualized on the LPMO structure (PDB ID: 4QI8) (B). The coloring scale follows the one in panel A. The central copper atom is shown in green and the side chains of the histidine brace residues and Pro-Gly-Pro patch are shown as sticks. The structure on the left visualizes histidine brace (green) and Pro-Gly-Pro patch (orange) residues.
Heterogenous Electron Transfer
CDH is recognized for its ability to directly transfer electrons to electrode surfaces via its CYT domain.38 The heterogeneous electron transfer of wild-type and chimeric CDHs to a self-assembled monolayer (SAM) of thioglycerol on gold electrodes7 was investigated for two reasons: (i) to verify that all produced enzymes and their domains are in their native, electron transfer competent conformation and (ii) to study the effect of swapped linkers and CYT domains on the direct electron transfer to an electrode. Unfortunately, CDHBBA was not available in sufficient amounts for these experiments. In the presence of 20 mM cellobiose, catalytic currents were observed for all variants (Figure S4). The onset potentials of the anodic waves correlate well with the corresponding, spectroelectrochemically determined CYT redox potentials. Current densities were extracted at an overpotential of 200 mV, above the midpoint potential of the CYT domain (CYTA at 300 mV, CYTB at 360 mV vs SHE) and a scan rate of 15 mV s–1. The highest current density was found for CDHAAA (11.3 ± 1.8 μA cm–2), followed by CDHABB (3.7 ± 1.5 μA cm–2), CDHBBB (2.6 ± 1.7 μA cm–2), CDHBBAA (1.9 ± 0.3 μA cm–2), and CDHAAB (1.2 ± 1.0 μA cm–2). Every CDH clearly showed direct electron transfer to the electrode and thereby verified the integrity of the electron transfer route.
Anodic and cathodic peak currents were obtained for all CDHs over a range of scan rates (3–150 mV s–1). The plot of the peak currents versus the square root of the scan rates is linear for all enzymes and indicate a freely diffusing redox species and no adsorption onto the electrode (Figure S5). The peak separation of the anodic and cathodic peak increased with increasing scan rates (Figure S6). The heterogeneous electron transfer is reversible at very low scan rates and quasi-reversible at scan rates above 5 mV s–1, pointing toward a fast electron transfer compared to mass transport. This allows the calculation of the heterogeneous electron transfer constant (k0) according to the method of Nicholson and Shain for the quasi-reversible electron transfer regime.39 All wild-type and chimeric CDHs show a similar k0 between 7.7 and 17.7 × 10–4 cm s–1 at the most relevant scan speed for comparison (50 mV s–1, Figure 7), which demonstrates that there is no restrained interaction of any CYT with the thioglycerol monolayer on the gold electrode and all CDH variants are functional. This is comparable with a k0 of ca. 10–3 to 10–4 cm s–1 for cytochrome c on gold electrodes.40 CDHBBB with the lowest k0 has the least efficient electron transfer of its CYT domain with the electrode.
Figure 7.
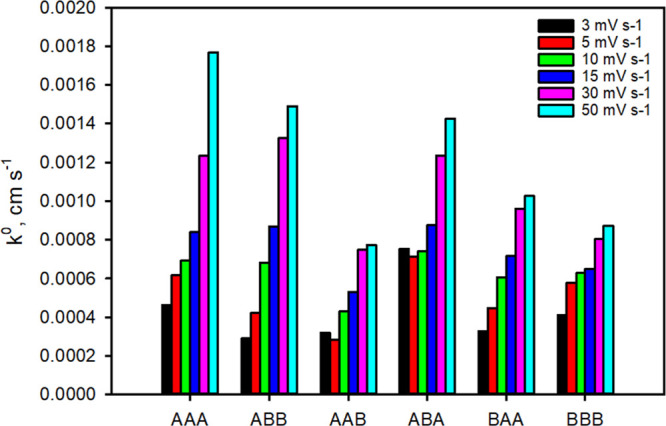
Heterogeneous electron transfer rates (k0). k0 was calculated from the peak separation of the anodic and cathodic wave observed from cyclic voltammograms measured at different scan rates (3–50 mV s–1) according to Nicholson–Shain. Data from scan rates above 50 mV s–1 could not be used because the increased capacitive current did not allow the exact determination of the peak maxima. The data (peak separation vs scan rate) are given in Figure S6.
IPET Kinetics
After verifying that the wild-type and chimeric CDHs are all electron transfer competent, we investigated the final electron transfer from CYT to LPMO (IPET). In sequential stopped-flow experiments, CDHs were prereduced by a stoichiometric amount of cellobiose. After 90 s in the aging loop, oxygen had fully reoxidized the FADH2, which was necessary to prevent any interfering IDET to CYT. Then, the CDH with the reduced CYT was shot against an equimolar, 3-, 10-, and a 50-fold molar ratio of NcLPMO9C to measure the IPET rate. A linear dependence of kobs563 on LPMO concentrations was found (Figure 8), which indicates that the electron transfer between both enzymes is fast enough to show no saturation even for the highest measured LPMO concentration.21 The bimolecular IPET rate was calculated from the slope of kobs563 versus the LPMO concentration. The determined rates are all within the same order of magnitude, which indicates that the interaction mechanism is not evolved to recognize and favor specific CDH–LPMO combinations but is based on a universal recognition mechanism which depends little on surface complementarity. The observed differences in IPET show that CYTB, which is present in three measured CDHs, exhibit a two times faster bimolecular rate with NcLPMO9C (7.4–8.8 × 105 M–1 s–1) than most CDHs with CYTA (2.9–5.1 × 105 M–1 s–1) with the exception of CDHABA, which exhibits the highest IPET rate. This points toward the importance of the linker and its influence on the closed- and open-state conformation. CDHAAA with the short linker, the fastest IDET, and the slowest IPET prefers the closed-state conformation, whereas CDHBBA or CDHABA with reduced IDET and fast IPET prefer the open-state conformation.
Figure 8.

Effects of domain swapping on IPET. Stopped-flow measurements of the electron transfer from prereduced CYT to LPMO at 563 nm at for increasing LPMO concentrations show a linear relation from which bimolecular rates were calculated.
In contrast, a higher redox potential difference between the CDH and LPMO cofactors has not the expected, rate enhancing effect on the IPET rate (Table 2, Figure 8). CYTB with its ∼60 mV higher midpoint potential compared to CYTA has a comparatively lower driving force for electron transfer between the heme b and LPMO’s type-2 copper but shows similar IPET rates to CYTA. This indicates that CYT–DH combinations of poor surface complementarity or with an unsuitable linker preferably populate the IPET competent open-state conformation. We conclude that the closed- and open-state distribution of CDH populations define the electron transfer rates of CYT in IDET and IPET.
Multivariate Analysis
Mixed factor principal component analysis (PCA), including the quantitative variables (kcat, kobs, IDET, IPET, CYT midpoint redox potential, and glycosylation) and qualitative variables [DH-, CYT-, and linker-type (Tables S5 and Figure S9)], has been performed on the data set from wild-type and chimeric CDHs to explore intercorrelation. Glycosylation shows the smallest effect of all quantitative variables, while the kinetic variables cluster as expected from bivariate analysis.
Discussion
The two-domain structure of CDH has been recognized soon after its discovery, by observing the spectral features of its two cofactors, proteolytic cleavage into the separated domains, and distinct catalytic properties of the full-length CDH and its DH domain. The domain organization became evident with the first isolated CDH sequence of Phanerochaete chrysosporium,41 but the purpose of the CYT domain remained unknown. Crystallization experiments in which only the separated, proteolytically generated CYT and DH domains formed crystals indicated the high mobility of the linker and CYT domain.18,42 At the same time, Igarashi et al. investigated the pH dependence of the IDET between the DH and CYT domains of P. chrysosporium CDH in a presteady-state kinetic study. He also determined the redox potentials of the heme b and FAD cofactor, which can influence IDET.10
However, the physiological function of the CYT domain and the highly variable length of the linker in CDHs (16–40 amino acids) remained enigmatic. Also, the considerable length of linkerB in comparison to other flavocytochromes, for example, the flavocytochrome b2’s hinge (linker) region consisting of only 15 amino acids,43 is unusual. In the two CDHs from N. crassa, linkerB is almost twice as long as linkerA. Both enzymes have the lowest sequence identity (29%) among CDH’s structural elements despite sharing two common features: a high percentage of serine, threonine, and proline residues and a conserved Pro-Val-Pro motif. Likewise, the sequence identity among the CYT domains (43%) is low compared to that of the DH domains (60%). The higher diversity of the linker and CYT sequences is observed for all CDHs and suggests an evolutionary adaptation to contact various redox partner proteins, while the DH domain serves as a source of electrons.
With the discovery of LPMO in 2010,44 the physiological redox partner of CDH was finally revealed, which gives us the opportunity to study the CYT domain’s IDET and IPET mechanism as part of a natural, extracellular electron transfer chain. This framework allows the testing of hypotheses on CDH’s molecular, catalytic, and electron transfer properties. Swapping domains between the structurally, catalytically and electrochemically different CDHs of one organism allows differentiating between the functions of the involved domains and linkers in the electron transfer route from CDH’s FADH2 to LPMO’s type-2 copper center. Two wild-type and five chimeric CDHs could be recombinantly expressed in P. pastoris and all enzymes except CDHAAB and CDHBBA could be produced in quantities above 10 mg, sufficient for a full set of analysis. The specific activities of the purified chimeric enzymes and their absorption spectra are in consonance with the wild-type CDHs and, therefore, these enzymes are properly folded. However, a difference in the extent of glycosylation of the wild-type and chimeric CDHs was found. This variation is inevitable with the chosen yeast expression system, which is known to produce various glycoforms. However, the N-glycosylation sites are not located at the DH–CYT interface and thus should not affect the experiments. O-Glycosylation of the linker was previously reported,45 but we lacked the resources to determine if this minor fraction of glycosides varied between the produced CDHs. However, the determined heterogeneous electron transfer rates for all CDHs were relatively similar and indicated no significant influence of the glycosylation on the interaction with the thioglycerol-modified gold electrode.
Transient kinetic studies of the catalytic reaction of the DH domain showed no change of the reductive half-reaction in chimeric CDHs but showed an effect of the swapped CYT domains on the oxidative half-reaction by shifting the pH optima for the two-electron acceptor 2,6-dichloroindophenol. The pH optima of CDHAAA and CDHBBB are identical to previous data.7 Because the pH optimum of the catalytic reaction in CDH generally depends on the electron acceptor,19,46 this indicates an impact of the CYT domain on the oxidative catalytic half-reaction. These results support the previously observed effect of the CYT domain on the catalytic step in the DH domain of Crassicarpon hotsonii (syn. M. thermophilum) CDH.11 At pH 6.0, which is also the pH optimum of the CDH–LPMO interaction,6 only small differences between the catalytic efficiencies were observed between the wild-type and chimeric CDHs, rendering this pH as suitable to study the subsequent electron transfer steps. The presteady-state reduction rates of FAD by cellobiose at 449 nm (kobs449) show a clear separation between enzymes with a DHA domain (80–89 s–1) and a DHB domain (30–33 s–1) but no effect of a CYT swap on the rate of the reductive-half reaction.
While the effects of the domain swap on the catalysis of the chimeric enzymes were moderate, the IDET between DH and CYT was strongly affected. Steady-state experiments showed different pH optima and 3–12 times (except for CDHBBA) reduced TN’s of the chimeric CDHs with cytochrome c. These findings were corroborated by transient-state data. The highest IDET rates were measured for wild-type enzymes, which had a 19 times (CDHAAA) or 10 times (CDHBBB) higher IDET rate than the successive chimeric CDHs. Modeling studies showed the importance of surface complementarity and the degree of orientational freedom provided by the linker. Given the varying length of linkerA (7 nm) and linkerB (11 nm), it can be expected that in the open state the distance limit for a reasonable fast electron transfer (∼1.5 nm) between CYT and DH is often exceeded. It was also found that the redox potential difference between CYT and DH is not the dominant driving force for IDET because CDHs with a CYTA have a lower ΔE (64–104 mV) between the cofactors than CYTB carrying CDHs (158–172 mV) but similar or faster IDET rates. The reason is the close edge-to-edge distance between the FAD and heme. For the typical distance of ∼0.9 nm in N. crassa CDH’s closed state, the electron transfer rates are 105 times higher than the measured rates. This suggests that the mobility of the CYT domain and its shift between closed- and open-state conformations is the rate-limiting factor of IDET, rather than the electron transfer event itself. A shorter linker (linkerA) and a higher complementarity at the CYT–DH interface increases IDET by supporting the closed state of the CDH.
The efficiency of the subsequent electron transfer step from CYT to LPMO, the IPET, is most important for the efficiency of the process and determines the rate of LPMO reduction. A specific and fast IPET saves valuable resources for the metabolism of the cellulolytic organism (less enzymatic consumption of cellobiose, less secreted CDH needed) and prevents futile electron transfer to other electron acceptors or scavengers, which reduces not only the efficiency of the extracellular electron transfer system but could also produce degradation products detrimental to the organism’s growth. The HDX-MS experiments showed the interaction of CYT and LPMO to happen via direct contact between their active centers. This is in agreement with NMR and docking studies.15,16 No alternative interaction site of CYT–LPMO has experimentally found so far. The reported electron transfer between the active site copper and amino acids within LPMO resulting in tyrosyl- or tryptophanyl radicals indicates the presence of electron transfer pathways, which have been attributed to the protection of the active site during uncoupled turnover.47,48 The observed interaction is relatively weak, underlining the formation of a flexible and transient complex. This observation aligns well with the necessity of the LPMO to detach from the cellulose in order to receive an electron from CDH, which fits very well to the proposed peroxide-dependent catalytic mechanism of LPMO.49 The apparent transient interaction also complements previous findings,45 hinting at a very dynamic system which depends on the interplay between electrostatic forces of its cofactors and thermodynamic forces governing domain movements.
The measured IPET rates all have the same order of magnitude, which indicates that the interaction mechanism did not evolve to recognize and favor specific CDH–LPMO combinations but is based on a universal recognition mechanism between the heme b propionate A and the copper center, which depends little on surface complementarity.16 This is corroborated by reports on the interaction of different CDHs and LPMOs from N. crassa,6 CDH and LPMO from different fungi1,2,50 and even from fungal CDH to bacterial LPMOs.51 The measured bimolecular rates for the final electron transfer step from CYT to LPMO were found to be very fast with values between 2.9 × 105 and 1.1 × 106 M–1 s–1. A similar rate was also found for the very fast CYT–cytochrome c interaction (106 M–1 s–1).21 We conclude that the rate-determining driving force for IPET is not the redox potential difference between LPMO and CYT because CDHs with a CYTB have a lower redox potential difference to LPMO but exhibit, in most cases, slightly faster IPET rates than CYTA featuring CDHs. Likewise, the IPET rates do not show a preferred complementarity of either the CYTA or CYTB surface with NcLPMO9C. This supports the previously published modeling of the CYT–LPMO interface, which shows that only a very small surface area of the domains besides the cofactors is involved in the recognition and interaction.16 Hence, the most important factor for the higher IPET rates of chimeric CDHs over the wild-type CDHs seems to stem from their preference of the open-state conformation induced by an unfitting CYT–DH interface or linker.
Experimental Procedures
Molecular Biology, Expression, and Purification
Genetic constructs of cdhIIA (NCU00206) and cdhIIB (NCU05923) were described previously7 and used for this study. A silent mutation (C456T) was introduced to the gene NCU05923 to delete the BstBI (Bsp119I) restriction site. Alignments using MEGA 652 applying the BLOSSOM 62 algorithm together with the 3D structure analysis of NCU00206 (PDB ID: 4QI7) were applied to define exact borders of the individual domains (Table S4). Fragments of the individual domains were generated and joined to create four chimeric constructs by overlap extension PCR. The genetic integrity of the amplicons encoding chimeric CDHs was checked by DNA sequencing at Microsynth (Wolfurt, Austria). Following established methods,53 the constructs were expressed in a P. pastoris expression system (KM71H, Invitrogen). Best producing variants were preselected,54 cultivated in 500 mL scale fermentations (Figure S1), and purified by hydrophobic interaction (PHE-Sepharose Fast Flow, GE Healthcare) and anion exchange chromatography (Source 15Q, GE Healthcare). Two chimeric CDHs (CDHAAB and CDHBBA) still contained minor impurities after these two steps and were subjected to additional size exclusion chromatography (Superdex 75). The SDS-PAGE analysis of all preparations used in this study is displayed in Figure 1.
Enzyme Activity Assays and Protein Quantitation
The activity of CDHs was determined in 1 mL assays by following the reduction of either 0.3 mM 2,6-dichloroindophenol (DCIP, ε520 = 6.8 mM–1 cm–1) or 50 μM cytochrome c from equine heart (ε550 = 19.6 mM–1 cm–1). Assays were buffered with 100 mM sodium citrate-phosphate buffer according to ref (55) at the indicated pH. The pH-dependent activity was measured with 30 mM lactose as saturating substrate. Assay reactions were monitored for 180 s at 30 °C at the indicated wavelengths in a LAMBDA 35 UV–vis spectrophotometer equipped with a temperature-controlled 8-cell changer (PerkinElmer). The protein concentration of wild-type and chimeric CDHs was determined via the absorbance at 280 nm and the theoretical molar absorption coefficient ε280 calculated with the Expasy Prot-Param program56 using the mature amino acid sequence.
Spectroelectrochemistry
Spectroelectrochemical experiments were performed using 500 μL samples containing around 50 μM wild-type or chimeric CDH, 100 mM KCl, 100 mM potassium phosphate buffer pH 6.0, and a redox mediator mixture comprising anthraquinone-1,5-disulfonate, 2-hydroxy-1,4-naphthoquinone, indigo carmine, indigo trisulfonate, duroquinone, methylene blue, phenazine methosulfate, 1,2-naphthoquinone and N,N,N′,N′-tetramethyl-p-phenylenediamine (all 3 μM), and methyl viologen (150 μM). All experiments were carried out under anaerobic conditions at 25 °C in a thin-layer (d = 0.05 mm) spectroelectrochemical cell (BASi, West Lafayette, IN; USA) with a standard three-electrode setup comprising a Ag|AgCl—reference electrode (BASi), a platinum gauze (Goodfellow Cambridge Ltd., Huntington, England, UK) as the working electrode, and a platinum wire (Goodfellow Cambridge Ltd.) as the auxiliary electrode. Potentials were applied using a Gamry Series G 300 Potentiostat/Galvanostat/ZRA (Gamry Instruments, Warminster, PA, USA). A Whitley DG 250 Anaerobic Workstation (Don Whitley Scientific Ltd., Shipley, England, UK) was used to work under oxygen-free conditions. The reference electrode was calibrated against a saturated calomel electrode. All potentials are reported relative to the SHE. Nernst plots consisted of at least 5 data points, showed linear behavior, and were consistent with a one-electron redox process in the case of the heme b and a two-electron process in the case of the FAD.
Voltammetry
Preparation of enzyme-modified electrodes started with the cleaning of gold disk-electrodes (d = 1.6 mm, BASi, West Lafayette, IN, USA) by dipping in acidic piranha solution [H2SO4/H2O2 = 3:1 (v/v)] for 5 min, cycling in 0.1 M NaOH (−0.205 to −1.205 V vs Ag|AgCl, 10 cycles, 100 mV s–1), polishing to mirror finish with aqueous alumina particles (0.05 μm) on a MicroCloth (Buehler, Lake Bluff, IL, USA), ultrasonication to remove residual polishing particles and cycling in 0.5 M H2SO4 (−0.205 to +1.705 V vs Ag|AgCl, 20 cycles, 200 mV s–1). After rinsing with ultrapure water and drying with nitrogen gas the electrodes were immersed overnight in 10 mM 1-thioglycerol dissolved in absolute ethanol for SAM formation. The electrodes were then washed with 20% ethanol to remove unbound thioglycerol, ultrapure water and dried over a stream of nitrogen gas. A custom-made Teflon holder was put over the electrode surface, leaving a cylindrical cavity with a volume of ∼20 μL above the thioglycerol-modified gold surface. Then, a 100 μM CDH solution in 100 mM McIlvaine buffer, pH 6.0, was applied to the cavity. The assembly was covered with a dialysis membrane (45 kDa cut-off) held in place via a rubber O-ring.
Electrochemical experiments were carried out using a PGSTAT204 potentiostat/galvanostat (Metrohm Inula GmbH, Vienna, Austria) with a standard three-electrode setup comprising the enzyme-modified gold electrode as working electrode, a platinum wire as the counter electrode, and a Ag|AgCl electrode as the reference electrode. The 100 mM McIlvaine buffer, pH 6.0 contained 0.1 M KCl as the supporting electrolyte. A typical set of experiments comprised cyclic voltammetry of the thioglycerol-modified electrode (blank), the enzyme-modified thioglycerol-electrode, the enzyme-modified thioglycerol-electrode in the presence of 20 mM cellobiose, and the enzyme-modified thioglycerol-electrode in the presence of 20 mM cellobiose and 50 μM ferrocenemethanol. The applied potential window ranged from 5 to 550 mV versus SHE. Scan rates were varied from 3 to 500 mV s–1. Before the start of the experiment, the electrochemical cell containing buffer and the electrode setup was deoxygenated by purging with argon gas for 15 min. The bulk solution was not agitated during the measurement which was performed at 25 °C.
Cyclic voltammograms were analyzed using NOVA software (Metrohm) and Microsoft Excel. To evaluate whether freely diffusing or adsorbed CDH species dominate the electrochemical process, the linearity of plots of peak current versus the square root of the scan rate was analyzed. Reversibility, quasi-reversibility, or irreversibility of the electron transfer process was assessed by the shape of the voltammograms and the peak-to-peak separation. Standard heterogeneous electron transfer rate constants k0 were calculated using the model for quasi-reversible processes described by Nicholson & Shain57 and Matsuda & Ayabe58 with a transfer coefficient of α = 0.5 and interpolated values [Ψ = 1/(−2.46 + 0.041)*dEp] of the kinetic parameter Ψ for the scan rate-dependent peak potential separation. Diffusion coefficients for CDH were calculated from the slope of the linear correlation of the anodic or cathodic peak currents, the square root of the scan rate, the active electrode surface area (A = 0.0177 cm2), and an enzyme concentration of 100 μM (10–7 mol cm–3) applying the Randles–Sevcik equation.59,60 Peak currents were assessed by applying Nicholson’s empiric equation Ipa/Ipc = (Ipa)0/Ipc + 0.485(Isp)0/Ipc + 0.086.39
Presteady-State Kinetic Studies
The rapid spectral changes induced by substrate oxidation and the resulting change of the redox state of the CDH cofactors were followed with a SX-20 stopped-flow instrument (Applied Photophysics, Leatherhead, UK) equipped with a photomultiplier tube (AP/PMT.R928). The redox state of the FAD cofactor was monitored at the appropriate isosbestic point (449 nm) of the heme b cofactor, which itself was monitored at 563 nm. The observed rates (kobs) for the indicated cellobiose concentrations were estimated by fitting the data to a single exponential function. The reduction of NcLPMO9C by CDH was studied using a UV–vis photodiode array detector (AP/SXPDAUV) in the sequential mixing mode. CDH was fully reduced in the first step by mixing with an appropriate concentration of cellobiose in air-saturated buffer. The reaction was held in an ageing loop until full reoxidation of the FAD cofactor occurred via its weak oxidase activity. Approx. 70% of the heme b remained reduced because of its slower interaction with O2. The partially reoxidized CDH was rapidly mixed with NcLPMO9C. The observed rates of transfer were estimated by following the redox state of the CYT domain of CDH and fitting the data of A563 to a single exponential curve. All presteady-state experiments were performed in 100 mM sodium citrate-phosphate buffer, pH 6.0 at 30 °C.
Modeling of CDH Chimeras
SWISS-MODEL61−63 was used to generate structure-guided homology models of the CYT and dehydrogenase (DH) domains of NcCDHIIB (ORF: NCU05923) using the crystal structure of NcCDHIIA (PDB ID: 4QI7)9 as a template. Steepest descent energy minimization with 2500 steps (initial step size of 0.1 nm) was performed with the GROMOS software package for molecular simulation64 using the 54a7 force field65,66 as a further refinement for the resulting homology models. Subsequently, the complexes CYTA–DHA, CYTA–DHB, CYTB–DHA, and CYTB–DHB have been modeled using HADDOCK 2.225,26 with interaction restraints between heme b and the Arg697 and Arg719 for NcCDHIIA and NcCDHIIB, respectively. The number of starting structures was set to 1000 and refined to 200 structures. Nonbonded energy values (i.e., van der Waals and electrostatic energies) were taken from the HADDOCK output and the angles of the CYT domain relative to the DH domain around three axes defined by two (virtual) atoms j and k was measured by computing the dihedral angle i–j–k–l with the (virtual) atoms, as listed in Table S2. The distance-field reaction coordinate28 was used to estimate the shortest distance between the linker anchor points along a path that does not pass through the protein domains. Electrostatic surface representations, as well as protonation states, were computed with PROPKA 3.1,67,68 PDB2PQR69 and the PyMOL APBS plug-in.70−74 Binding affinities were predicted with PRODIGY.75−77
H/D Exchange Mass Spectrometry
Prior to the mass spectrometric analyses, NcCDHIIA was deglycosylated under nondenaturing conditions as utilized previously for the analyses of CDH from M. thermophilum.35 CDH was incubated overnight with 15 U Endo Hf (New England Biolabs, USA) per 1 μg of protein at 37 °C in 50 mM sodium acetate buffer pH 5.75 to detach the N-glycans. The deglycosylated CDH was preincubated alone or in a mixture with NcLPMO9F (1:3 and 3:1 M ratios) in H2O-based 50 mM sodium acetate buffer pH 5.75 for 30 min. After preincubation, the deuterium labeling was started by a 10-fold dilution of the protein samples into a deuterated buffer (50 mM sodium acetate pD 5.75). The final protein concentration during the labeling was 5 μM for the examined protein and 15 μM for the interaction partner. The deuteration reaction proceeded at 21 °C and 50 μL aliquots were removed after 0.33, 1, 3, 10, 30, 60, 180, and 300 min. The rest of the HDX-MS workflow, including the stopping of the exchange in the aliquots, denaturation of samples, and their online enzymatic digestion by immobilized porcine pepsin, LC–MS analysis by Fourier transform ion cyclotron resonance mass spectrometry and data processing, was performed exactly as optimized for M. thermophilum CDH as described elsewhere.35
Statistical Analysis
A statistical evaluation of the data set, which aimed to identify significant correlations of individual variables using bivariate correlation analysis and PCA has been performed using the software R Studio (MA, USA) and the packages FactoMiner78 and Psych.79 Selected code snippets describing the libraries used, the intermediate data produced, as well as the code generating tables and plots are presented in the statistical analysis section (SA) in the Supporting Information. The full R script and data set has been released in a public repository (https://doi.org/10.5281/zenodo.4297843).
Acknowledgments
Daniela Gamperl is acknowledged for her fantastic work to produce chimeric CDHABA.
Glossary
Abbreviations
- AFM
atomic force microscopy
- CDH
cellobiose dehydrogenase
- CYT
cytochrome domain
- DCIP
2,6-dichloroindophenol
- DH
dehydrogenase domain
- FAD
flavin adenine dinucleotide
- HDX
hydrogen/deuterium exchange
- IDET
interdomain electron transfer
- IPET
interprotein electron transfer
- LPMO
lytic polysaccharide monooxygenase
- MS
mass spectrometry
- SAM
self-assembled monolayer
- SANS
small-angle neutron scattering
- SAXS
small-angle X-ray scattering
- SHE
standard hydrogen electrode
- TN
turnover number
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c05294.
Protein production, electrochemical characterization, modeling, and statistical analysis (PDF)
Author Contributions
A.K.G.F. wrote the first draft of the manuscript and performed together with Christian Schuster and Christoph Sygmund the production and purification of chimeric CDHs and steady-state kinetic measurements. C.V.F.P.L. and C.O. performed and interpreted docking and modeling studies. A.K., F.F., P.M., and P.H. performed and analyzed H/D exchange experiments, A.K.G.F. and D.K. performed and analyzed fast-kinetic experiments, S.S., A.K.G.F, L.S., and F.S. performed and analyzed electrochemical experiments. R.L. initiated the study, designed experiments, evaluated and interpreted data, and wrote the final draft of the manuscript together with C.O, P.M., and P.H.
This work was funded by the Austrian Science Fund (project I2385-N28) and the Czech Science Foundation (projects 16-34818L) and the Europeans Union’s Horizon 2020 research and innovation programme (ERC Consolidator Grant OXIDISE) under grant agreement no. 726396. A.K.G.F. was supported by a scholarship of the Austrian Academy of Sciences (DOC scholarship), S.S. by the MBW FM (Austrian Federal Ministry of Science, Research and Economy) International Graduate School BioNano Technology (IGS BioNano Tech) and C.V.F.P.L., L.S., F.S., and D.K. by the doctoral programme BioToP (W1224) funded by the Austrian Science Fund. A.K.G.F. was supported by a Doc fellowship of the Austrian Academy of Science. Access to the MS facility was enabled by MEYS CR (CZ.1.05/1.1.00/02.0109, LQ1604 and LM2015043 CIISB) funding.
The authors declare no competing financial interest.
Supplementary Material
References
- Langston J. A.; Shaghasi T.; Abbate E.; Xu F.; Vlasenko E.; Sweeney M. D. Oxidoreductive Cellulose Depolymerization by the Enzymes Cellobiose Dehydrogenase and Glycoside Hydrolase 61. Appl. Environ. Microbiol. 2011, 77, 7007–7015. 10.1128/aem.05815-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips C. M.; Beeson W. T.; Cate J. H.; Marletta M. A. Cellobiose Dehydrogenase and a Copper-Dependent Polysaccharide Monooxygenase Potentiate Cellulose Degradation by Neurospora crassa. ACS Chem. Biol. 2011, 6, 1399–1406. 10.1021/cb200351y. [DOI] [PubMed] [Google Scholar]
- Phillips C. M.; Iavarone A. T.; Marletta M. A. Quantitative Proteomic Approach for Cellulose Degradation by Neurospora crassa. J. Proteome Res. 2011, 10, 4177–4185. 10.1021/pr200329b. [DOI] [PubMed] [Google Scholar]
- Harris P. V.; Welner D.; McFarland K. C.; Re E.; Navarro Poulsen J.-C.; Brown K.; Salbo R.; Ding H.; Vlasenko E.; Merino S.; Xu F.; Cherry J.; Larsen S.; Lo Leggio L. Stimulation of Lignocellulosic Biomass Hydrolysis by Proteins of Glycoside Hydrolase Family 61: Structure and Function of a Large, Enigmatic Family. Biochemistry 2010, 49, 3305–3316. 10.1021/bi100009p. [DOI] [PubMed] [Google Scholar]
- Quinlan R. J.; Sweeney M. D.; Lo Leggio L.; Otten H.; Poulsen J.-C. N.; Johansen K. S.; Krogh K. B. R. M.; Jorgensen C. I.; Tovborg M.; Anthonsen A.; Tryfona T.; Walter C. P.; Dupree P.; Xu F.; Davies G. J.; Walton P. H. Insights into the Oxidative Degradation of Cellulose by a Copper Metalloenzyme That Exploits Biomass Components. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 15079–15084. 10.1073/pnas.1105776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kracher D.; Scheiblbrandner S.; Felice A. K. G.; Breslmayr E.; Preims M.; Ludwicka K.; Haltrich D.; Eijsink V. G. H.; Ludwig R. Extracellular Electron Transfer Systems Fuel Cellulose Oxidative Degradation. Science 2016, 352, 1098–1101. 10.1126/science.aaf3165. [DOI] [PubMed] [Google Scholar]
- Sygmund C.; Kracher D.; Scheiblbrandner S.; Zahma K.; Felice A. K. G.; Harreither W.; Kittl R.; Ludwig R. Characterization of the Two Neurospora crassa Cellobiose Dehydrogenases and Their Connection to Oxidative Cellulose Degradation. Appl. Environ. Microbiol. 2012, 78, 6161–6171. 10.1128/aem.01503-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi K.; Yoshida M.; Matsumura H.; Nakamura N.; Ohno H.; Samejima M.; Nishino T. Electron Transfer Chain Reaction of the Extracellular Flavocytochrome Cellobiose Dehydrogenase from the Basidiomycete Phanerochaete chrysosporium. FEBS J. 2005, 272, 2869–2877. 10.1111/j.1742-4658.2005.04707.x. [DOI] [PubMed] [Google Scholar]
- Tan T.-C.; Kracher D.; Gandini R.; Sygmund C.; Kittl R.; Haltrich D.; Hällberg B. M.; Ludwig R.; Divne C. Structural Basis for Cellobiose Dehydrogenase Action during Oxidative Cellulose Degradation. Nat. Commun. 2015, 6, 7542. 10.1038/ncomms8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi K.; Momohara I.; Nishino T.; Samejima M. Kinetics of Inter-Domain Electron Transfer in Flavocytochrome Cellobiose Dehydrogenase from the White-Rot Fungus Phanerochaete chrysosporium. Biochem. J. 2002, 365, 521. 10.1042/bj20011809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kracher D.; Zahma K.; Schulz C.; Sygmund C.; Gorton L.; Ludwig R. Inter-Domain Electron Transfer in Cellobiose Dehydrogenase: Modulation by pH and Divalent Cations. FEBS J. 2015, 282, 3136–3148. 10.1111/febs.13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenheimer A. M.; O’Dell W. B.; Oliver R. C.; Qian S.; Stanley C. B.; Meilleur F. Structural Investigation of Cellobiose Dehydrogenase IIA: Insights from Small Angle Scattering into Intra- and Intermolecular Electron Transfer Mechanisms. Biochim. Biophys. Acta, Gen. Subj. 2018, 1862, 1031–1039. 10.1016/j.bbagen.2018.01.016. [DOI] [PubMed] [Google Scholar]
- Bodenheimer A. M.; O’Dell W. B.; Stanley C. B.; Meilleur F. Structural Studies of Neurospora crassa LPMO9D and Redox Partner CDHIIA Using Neutron Crystallography and Small-Angle Scattering. Carbohydr. Res. 2017, 448, 200–204. 10.1016/j.carres.2017.03.001. [DOI] [PubMed] [Google Scholar]
- Harada H.; Onoda A.; Uchihashi T.; Watanabe H.; Sunagawa N.; Samejima M.; Igarashi K.; Hayashi T. Interdomain Flip-Flop Motion Visualized in Flavocytochrome Cellobiose Dehydrogenase Using High-Speed Atomic Force Microscopy during Catalysis. Chem. Sci. 2017, 8, 6561–6565. 10.1039/c7sc01672g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtade G.; Wimmer R.; Røhr Å. K.; Preims M.; Felice A. K. G.; Dimarogona M.; Vaaje-Kolstad G.; Sørlie M.; Sandgren M.; Ludwig R.; Eijsink V. G. H.; Aachmann F. L. Interactions of a Fungal Lytic Polysaccharide Monooxygenase with β-Glucan Substrates and Cellobiose Dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 5922–5927. 10.1073/pnas.1602566113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent C. V. F. P.; Breslmayr E.; Tunega D.; Ludwig R.; Oostenbrink C. Interaction between Cellobiose Dehydrogenase and Lytic Polysaccharide Monooxygenase. Biochemistry 2019, 58, 1226–1235. 10.1021/acs.biochem.8b01178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz R.; Matsumura H.; Tasca F.; Zahma K.; Samejima M.; Igarashi K.; Ludwig R.; Gorton L. Effect of Deglycosylation of Cellobiose Dehydrogenases on the Enhancement of Direct Electron Transfer with Electrodes. Anal. Chem. 2012, 84, 10315–10323. 10.1021/ac3022899. [DOI] [PubMed] [Google Scholar]
- Hallberg B. M.; Henriksson G.; Pettersson G.; Divne C. Crystal Structure of the Flavoprotein Domain of the Extracellular Flavocytochrome Cellobiose Dehydrogenase. J. Mol. Biol. 2002, 315, 421–434. 10.1006/jmbi.2001.5246. [DOI] [PubMed] [Google Scholar]
- Zamocky M.; Ludwig R.; Peterbauer C.; Hallberg B.; Divne C.; Nicholls P.; Haltrich D. Cellobiose Dehydrogenase-a Flavocytochrome from Wood-Degrading, Phytopathogenic and Saprotropic Fungi. Curr. Protein Pept. Sci. 2006, 7, 255–280. 10.2174/138920306777452367. [DOI] [PubMed] [Google Scholar]
- Mason M. G.; Wilson M. T.; Ball A.; Nicholls P. Oxygen Reduction by Cellobiose Oxidoreductase: The Role of the Haem Group. FEBS Lett. 2002, 518, 29–32. 10.1016/s0014-5793(02)02633-9. [DOI] [PubMed] [Google Scholar]
- Mason M. G.; Nicholls P.; Divne C.; Hallberg B. M.; Henriksson G.; Wilson M. T. The Heme Domain of Cellobiose Oxidoreductase: A One-Electron Reducing System. Biochim. Biophys. Acta, Rev. Bioenerg. 2003, 1604, 47–54. 10.1016/s0005-2728(03)00023-9. [DOI] [PubMed] [Google Scholar]
- Samejima M.; Phillips R. S.; Eriksson K.-E. L. Cellobiose Oxidase from Phanerochaete chrysosporium Stopped-Flow Spectrophotometric Analysis of pH-Dependent Reduction. FEBS Lett. 1992, 306, 165–168. 10.1016/0014-5793(92)80991-o. [DOI] [PubMed] [Google Scholar]
- Moser C. C.; Keske J. M.; Warncke K.; Farid R. S.; Dutton P. L. Nature of Biological Electron Transfer. Nature 1992, 355, 796–802. 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- Page C. C.; Moser C. C.; Chen X.; Dutton P. L. Natural Engineering Principles of Electron Tunnelling in Biological Oxidation–Reduction. Nature 1999, 402, 47–52. 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- Dominguez C.; Boelens R.; Bonvin A. M. J. J. HADDOCK: A Protein–Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- de Vries S. J.; van Dijk A. D. J.; Krzeminski M.; van Dijk M.; Thureau A.; Hsu V.; Wassenaar T.; Bonvin A. M. J. J. HADDOCK versus HADDOCK: New Features and Performance of HADDOCK2.0 on the CAPRI Targets. Proteins: Struct., Funct., Bioinf. 2007, 69, 726–733. 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- Eichenberger A. P.; Allison J. R.; Dolenc J.; Geerke D. P.; Horta B. A. C.; Meier K.; Oostenbrink C.; Schmid N.; Steiner D.; Wang D.; van Gunsteren W. F. GROMOS++ Software for the Analysis of Biomolecular Simulation Trajectories. J. Chem. Theory Comput. 2011, 7, 3379–3390. 10.1021/ct2003622. [DOI] [PubMed] [Google Scholar]
- de Ruiter A.; Oostenbrink C. Protein–Ligand Binding from Distancefield Distances and Hamiltonian Replica Exchange Simulations. J. Chem. Theory Comput. 2013, 9, 883–892. 10.1021/ct300967a. [DOI] [PubMed] [Google Scholar]
- Xue L. C.; Rodrigues J. P.; Kastritis P. L.; Bonvin A. M.; Vangone A. PRODIGY: A Web Server for Predicting the Binding Affinity of Protein-Protein Complexes. Bioinformatics 2016, 32, 3676–3678. 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- Kastritis P. L.; Rodrigues J. P. G. L. M.; Folkers G. E.; Boelens R.; Bonvin A. M. J. J. Proteins Feel More Than They See: Fine-Tuning of Binding Affinity by Properties of the Non-Interacting Surface. J. Mol. Biol. 2014, 426, 2632–2652. 10.1016/j.jmb.2014.04.017. [DOI] [PubMed] [Google Scholar]
- Vangone A.; Bonvin A. M. Contacts-Based Prediction of Binding Affinity in Protein-Protein Complexes. eLife 2015, 4, e07454 10.7554/elife.07454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin J.; Bahadur R. P.; Chakrabarti P. Protein–Protein Interaction and Quaternary Structure. Q. Rev. Biophys. 2008, 41, 133–180. 10.1017/s0033583508004708. [DOI] [PubMed] [Google Scholar]
- Li X.; Beeson W. T.; Phillips C. M.; Marletta M. A.; Cate J. H. D. Structural Basis for Substrate Targeting and Catalysis by Fungal Polysaccharide Monooxygenases. Structure 2012, 20, 1051–1061. 10.1016/j.str.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittl R.; Kracher D.; Burgstaller D.; Haltrich D.; Ludwig R. Production of Four Neurospora crassa Lytic Polysaccharide Monooxygenases in Pichia Pastoris Monitored by a Fluorimetric Assay. Biotechnol. Biofuels 2012, 5, 79. 10.1186/1754-6834-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadek A.; Kavan D.; Felice A. K. G.; Ludwig R.; Halada P.; Man P. Structural Insight into the Calcium Ion Modulated Interdomain Electron Transfer in Cellobiose Dehydrogenase. FEBS Lett. 2015, 589, 1194–1199. 10.1016/j.febslet.2015.03.029. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Vahidi S.; Sowole M. A. Mass Spectrometry Methods for Studying Structure and Dynamics of Biological Macromolecules. Anal. Chem. 2014, 86, 213–232. 10.1021/ac4039306. [DOI] [PubMed] [Google Scholar]
- Sowole M. A.; Konermann L. Effects of Protein–Ligand Interactions on Hydrogen/Deuterium Exchange Kinetics: Canonical and Noncanonical Scenarios. Anal. Chem. 2014, 86, 6715–6722. 10.1021/ac501849n. [DOI] [PubMed] [Google Scholar]
- Lindgren A.; Larsson T.; Ruzgas T.; Gorton L. Direct Electron Transfer between the Heme of Cellobiose Dehydrogenase and Thiol Modified Gold Electrodes. J. Electroanal. Chem. 2000, 494, 105–113. 10.1016/s0022-0728(00)00326-0. [DOI] [Google Scholar]
- Nicholson R. S. Theory and Application of Cyclic Voltammetry for Measurement of Electrode Reaction Kinetics. Anal. Chem. 1965, 37, 1351–1355. 10.1021/ac60230a016. [DOI] [Google Scholar]
- Bowden E. F.; Hawkridge F. M.; Blount H. N. Interfacial Electrochemistry of Cytochrome c at Tin Oxide, Indium Oxide, Gold, and Platinum Electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1984, 161, 355–376. 10.1016/s0022-0728(84)80193-x. [DOI] [Google Scholar]
- Raices M.; Paifer E.; Cremata J.; Montesino R.; Ståhlberg J.; Divne C.; Szabó I. J.; Henriksson G.; Johansson G.; Pettersson G. Cloning and Characterization of a CDNA Encoding a Cellobiose Dehydrogenase from the White Rot Fungus Phanerochaete chrysosporium. FEBS Lett. 1995, 369, 233–238. 10.1016/0014-5793(95)00758-2. [DOI] [PubMed] [Google Scholar]
- Hallberg B. M.; Bergfors T.; Bäckbro K.; Pettersson G.; Henriksson G.; Divne C. A New Scaffold for Binding Haem in the Cytochrome Domain of the Extracellular Flavocytochrome Cellobiose Dehydrogenase. Structure 2000, 8, 79–88. 10.1016/s0969-2126(00)00082-4. [DOI] [PubMed] [Google Scholar]
- Diêp Lê K. H.; Lederer F.; Golinelli-Pimpaneau B. Structural Evidence for the Functional Importance of the Heme Domain Mobility in Flavocytochrome b2. J. Mol. Biol. 2010, 400, 518–530. 10.1016/j.jmb.2010.05.035. [DOI] [PubMed] [Google Scholar]
- Vaaje-Kolstad G.; Westereng B.; Horn S. J.; Liu Z.; Zhai H.; Sørlie M.; Eijsink V. G. H. An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. 10.1126/science.1192231. [DOI] [PubMed] [Google Scholar]
- Kadek A.; Kavan D.; Marcoux J.; Stojko J.; Felice A. K. G.; Cianférani S.; Ludwig R.; Halada P.; Man P. Interdomain Electron Transfer in Cellobiose Dehydrogenase Is Governed by Surface Electrostatics. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 157–167. 10.1016/j.bbagen.2016.11.016. [DOI] [PubMed] [Google Scholar]
- Harreither W.; Sygmund C.; Augustin M.; Narciso M.; Rabinovich M. L.; Gorton L.; Haltrich D.; Ludwig R. Catalytic Properties and Classification of Cellobiose Dehydrogenases from Ascomycetes. Appl. Environ. Microbiol. 2011, 77, 1804–1815. 10.1128/aem.02052-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. M.; Transue W. J.; Meier K. K.; Kelemen B.; Solomon E. I. Kinetic Analysis of Amino Acid Radicals Formed in H2O2-Driven CuI LPMO Reoxidation Implicates Dominant Homolytic Reactivity. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 11916–11922. 10.1073/pnas.1922499117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradisi A.; Johnston E. M.; Tovborg M.; Nicoll C. R.; Ciano L.; Dowle A.; McMaster J.; Hancock Y.; Davies G. J.; Walton P. H. Formation of a Copper(II)–Tyrosyl Complex at the Active Site of Lytic Polysaccharide Monooxygenases Following Oxidation by H2O2. J. Am. Chem. Soc. 2019, 141, 18585–18599. 10.1021/jacs.9b09833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chylenski P.; Bissaro B.; Sørlie M.; Røhr Å. K.; Várnai A.; Horn S. J.; Eijsink V. G. H. Lytic Polysaccharide Monooxygenases in Enzymatic Processing of Lignocellulosic Biomass. ACS Catal. 2019, 9, 4970–4991. 10.1021/acscatal.9b00246. [DOI] [Google Scholar]
- Petrović D. M.; Várnai A.; Dimarogona M.; Mathiesen G.; Sandgren M.; Westereng B.; Eijsink V. G. H. Comparison of Three Seemingly Similar Lytic Polysaccharide Monooxygenases from Neurospora crassa Suggests Different Roles in Plant Biomass Degradation. J. Biol. Chem. 2019, 294, 15068–15081. 10.1074/jbc.RA119.008196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loose J. S. M.; Forsberg Z.; Kracher D.; Scheiblbrandner S.; Ludwig R.; Eijsink V. G. H.; Vaaje-Kolstad G. Activation of Bacterial Lytic Polysaccharide Monooxygenases with Cellobiose Dehydrogenase. Protein Sci. 2016, 25, 2175–2186. 10.1002/pro.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K.; Stecher G.; Peterson D.; Filipski A.; Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harreither W.; Felice A. K. G.; Paukner R.; Gorton L.; Ludwig R.; Sygmund C. Recombinantly Produced Cellobiose Dehydrogenase from Corynascus thermophilus for Glucose Biosensors and Biofuel Cells. Biotechnol. J. 2012, 7, 1359–1366. 10.1002/biot.201200049. [DOI] [PubMed] [Google Scholar]
- Weis R.; Luiten R.; Skranc W.; Schwab H.; Wubbolts M.; Glieder A. Reliable High-Throughput Screening with Pichia Pastoris by Limiting Yeast Cell Death Phenomena. FEMS Yeast Res. 2004, 5, 179–189. 10.1016/j.femsyr.2004.06.016. [DOI] [PubMed] [Google Scholar]
- McIlvaine T. C. A Buffer Solution for Colorimetric Comparison. J. Biol. Chem. 1921, 49, 183–186. [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud S.; Wilkins M. R.; Appel R. D.; Bairoch A.. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker J. M., Ed.; Humana Press: Totowa, NJ, 2005; pp 571–607. [Google Scholar]
- Nicholson R. S.; Shain I. Theory of Stationary Electrode Polarography. Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems. Anal. Chem. 1964, 36, 706–723. 10.1021/ac60210a007. [DOI] [Google Scholar]
- Matsuda H. Zur Theorie der Wechselspannungs-Polarographie. Z. für Elektrochem. Berichte Bunsenges. für Phys. Chem. 1958, 62, 977–989. 10.1002/bbpc.19580620909. [DOI] [Google Scholar]
- Ševčík A. Oscillographic Polarography with Periodical Triangular Voltage. Collect. Czech. Chem. Commun. 1948, 13, 349–377. 10.1135/cccc19480349. [DOI] [Google Scholar]
- Randles J. E. B. A Cathode Ray Polarograph Part II.—The Current-Voltage Curves. Trans. Faraday Soc. 1948, 44, 327–338. 10.1039/tf9484400327. [DOI] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. The SWISS-MODEL Workspace: A Web-Based Environment for Protein Structure Homology Modelling. Bioinformatics 2006, 22, 195–201. 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Benkert P.; Biasini M.; Schwede T. Toward the Estimation of the Absolute Quality of Individual Protein Structure Models. Bioinformatics 2011, 27, 343–350. 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M.; Bienert S.; Waterhouse A.; Arnold K.; Studer G.; Schmidt T.; Kiefer F.; Cassarino T. G.; Bertoni M.; Bordoli L.; Schwede T. SWISS-MODEL: Modelling Protein Tertiary and Quaternary Structure Using Evolutionary Information. Nucleic Acids Res. 2014, 42, W252–W258. 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid N.; Christ C. D.; Christen M.; Eichenberger A. P.; van Gunsteren W. F. Architecture, Implementation and Parallelisation of the GROMOS Software for Biomolecular Simulation. Comput. Phys. Commun. 2012, 183, 890–903. 10.1016/j.cpc.2011.12.014. [DOI] [Google Scholar]
- Schmid N.; Eichenberger A. P.; Choutko A.; Riniker S.; Winger M.; Mark A. E.; van Gunsteren W. F. Definition and Testing of the GROMOS Force-Field Versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Poger D.; Van Gunsteren W. F.; Mark A. E. A New Force Field for Simulating Phosphatidylcholine Bilayers. J. Comput. Chem. 2010, 31, 1117–1125. 10.1002/jcc.21396. [DOI] [PubMed] [Google Scholar]
- Olsson M. H. M.; Søndergaard C. R.; Rostkowski M.; Jensen J. H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Søndergaard C. R.; Olsson M. H. M.; Rostkowski M.; Jensen J. H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- Dolinsky T. J.; Nielsen J. E.; McCammon J. A.; Baker N. A. PDB2PQR: An Automated Pipeline for the Setup of Poisson–Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 10037–10041. 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank R. E.; Holst M. A New Paradigm for Parallel Adaptive Meshing Algorithms. SIAM Rev. 2003, 45, 291–323. 10.1137/s003614450342061. [DOI] [Google Scholar]
- Holst M. Adaptive Numerical Treatment of Elliptic Systems on Manifolds. Adv. Comput. Math. 2001, 15, 139–191. 10.1023/a:1014246117321. [DOI] [Google Scholar]
- Holst M.; Saied F. Multigrid Solution of the Poisson—Boltzmann Equation. J. Comput. Chem. 1993, 14, 105–113. 10.1002/jcc.540140114. [DOI] [Google Scholar]
- Holst M. J.; Saied F. Numerical Solution of the Nonlinear Poisson–Boltzmann Equation: Developing More Robust and Efficient Methods. J. Comput. Chem. 1995, 16, 337–364. 10.1002/jcc.540160308. [DOI] [Google Scholar]
- Kastritis P. L.; Rodrigues J. P. G. L. M.; Folkers G. E.; Boelens R.; Bonvin A. M. J. J. Proteins Feel More Than They See: Fine-Tuning of Binding Affinity by Properties of the Non-Interacting Surface. J. Mol. Biol. 2014, 426, 2632–2652. 10.1016/j.jmb.2014.04.017. [DOI] [PubMed] [Google Scholar]
- Vangone A.; Bonvin A. M. Contacts-Based Prediction of Binding Affinity in Protein–Protein Complexes. eLife 2015, 4, e07454 10.7554/elife.07454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L. C.; Rodrigues J. P.; Kastritis P. L.; Bonvin A. M.; Vangone A. PRODIGY: A Web Server for Predicting the Binding Affinity of Protein–Protein Complexes. Bioinformatics 2016, 32, 3676–3678. 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- Lê S.; Josse J.; Husson F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Software 2008, 25, 1. 10.18637/jss.v025.i01. [DOI] [Google Scholar]
- Revelle W. R.Psych: Procedures for Personality and Psychological Research, 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.