

Abstract
The rhesus macaque (Macaca mulatta) is the most widely studied nonhuman primate (NHP) in biomedical research. We present an updated reference genome assembly (Mmul_10, contig N50 = 46 Mbp), increasing the sequence contiguity 120-fold and annotate it using 6.5 million full-length transcripts, thus improving our understanding of gene content, isoform diversity, and repeat organization. With the improved assembly of segmental duplications, we discover novel lineage-specific genes and expand gene families that are potentially informative in studies of evolution and disease susceptibility. Whole-genome sequence data from 853 captive rhesus macaques identifies polymorphism in 85.7 million single-nucleotide and 10.5 million indel variants, including potentially damaging variants in genes associated with human autism and developmental delay, providing a framework for developing non-invasive NHP models of human disease.
Summary:
A compendium of rhesus macaque genome variation
A detailed understanding of nonhuman primate (NHP) genome evolution is key to recognizing the origins of human traits and identifying putative disease genes. Evolutionary analyses of a diverse range of NHP genomes, spanning the breadth of primate phylogeny from great apes to prosimians, have begun to uncover the genetic basis of this phenotypic and biochemical diversity. Comparisons among species reveal lineage-specific changes in retroelements, the death and birth of duplicated genes, including the segmental duplications (SDs) underlying them, and functionally significant, sometimes deleterious, mutations in genes associated with human disease (1) (2) (3) (4) (5). Collectively, these studies are beginning to illuminate the history and novel mechanisms of molecular and phenotypic adaptation.
Rhesus macaques play a unique and critical role in both evolutionary comparisons and biomedical research. While the great apes (chimpanzees, bonobos, gorillas and orangutans) are phylogenetically closer to humans, the rhesus macaque is an essential model for a wide variety of studies related to infectious disease, neurobiology, developmental psychology, and other elements of primate (including human) biological function (6). This significance is highlighted by the role that rhesus macaque models have played in our understanding of AIDS pathogenesis and prevention strategies such as pre-exposure prophylaxis regimes (7), the development of highly effective Ebola vaccines (8), and the striking results obtained by editing genes related to risk for autism (9).
In 2007, the first whole-genome analysis of the rhesus macaque genome revealed both fundamental genetic similarities as well as interesting differences from the human genome (2). By combining our new rhesus reference assembly with the high-quality genomes now available for the great apes (5), more extensive reconstruction of human genome evolution is possible, such as gene structural changes that are unique to humans and apes. Early analyses suggested that macaques showed reduced SD content and complexity when compared to human although a final determination required a higher quality genome assembly (2). Similarly, initial analyses reported an expanded and more complex major histocompatibility complex (MHC) loci in macaques but the organization of such loci has been difficult to resolve (10). Finally, macaques provide valuable models for diseases or processes that would not be adequately modeled in rodents(11–13). Naturally occurring variation, which is higher among macaques (3) (4), has been leveraged to develop improved genetic models of Mendelian disorders (14) (15), complex disease (16) (17), and a hereditary form of cancer(11). To improve our understanding of rhesus macaque genetic diversity and its future translational implications, we annotate this new macaque reference by extensively characterizing genomic variation among 853 Indian- and Chinese-origin rhesus macaques from US research colonies. Consequently, this work provides a roadmap for naturally occurring mutations and disease models.
Results
Sequencing and assembly.
We generated long-read sequence data (~66-fold single-molecule, real-time [SMRT] sequence coverage) and assembled the genome of a female Indian-origin rhesus macaque (2.9 Gbp) using a series of genomic methods followed by extensive manual curation (18). This reference (Mmul_10, GenBank accession GCA_003339765.3) consists of 20 autosomes and the X chromosome; for completeness, we added a previous bacterial artificial chromosome (BAC)-based representation of the Y chromosome (19). Only 3.3% (97 Mbp) of the assembled macaque genome remains unassigned to scaffolds and is highly repetitive (>85% repeat-masked bases >10 kbp in length). In total, the new macaque reference is represented by 2,979 scaffolds with contig and scaffold N50 lengths of 46 and 82 Mbp, respectively (Table 1). This resulted in a highly contiguous and accurate assembly (Fig. 1) owing to extensive manual curation, gap filling, and unique sequence directionality assessments (18). Overall contiguity increased by 120- and 5.6-fold when compared to the previous Indian (Mmul_8.0.1) and Chinese rhesus macaque (rheMacS) (20) assemblies, respectively (Table 1). The underlying raw sequence data and other data are available at NCBI (Table S1).
Table 1.
Macaque genome assembly comparisons.
| Genus species | Assembly version | N50 contig (Mbp) | Total size (Mbp) | Total contigs | Chromosome gaps1 | Unplaced bases (Mbp) | Protein-coding genes | % Missing genes3 |
|---|---|---|---|---|---|---|---|---|
| Macaca mulatta | Mmul_8.0.1 | 0.107 | 2,835 | 348,579 | 62,231 | 82 | 21,574 | 3.8 |
| Macaca mulatta | rheMacS_1.02 | 8 | 3,031 | 4,713 | 2,862 | 82 | 20,389 | 1.8 |
| Macaca mulatta | Mmul_10 | 46 | 2,936 | 3,182 | 203 | 97 | 21,120 | 0.4 |
Figure 1. Rhesus macaque genome assembly quality and contiguity.
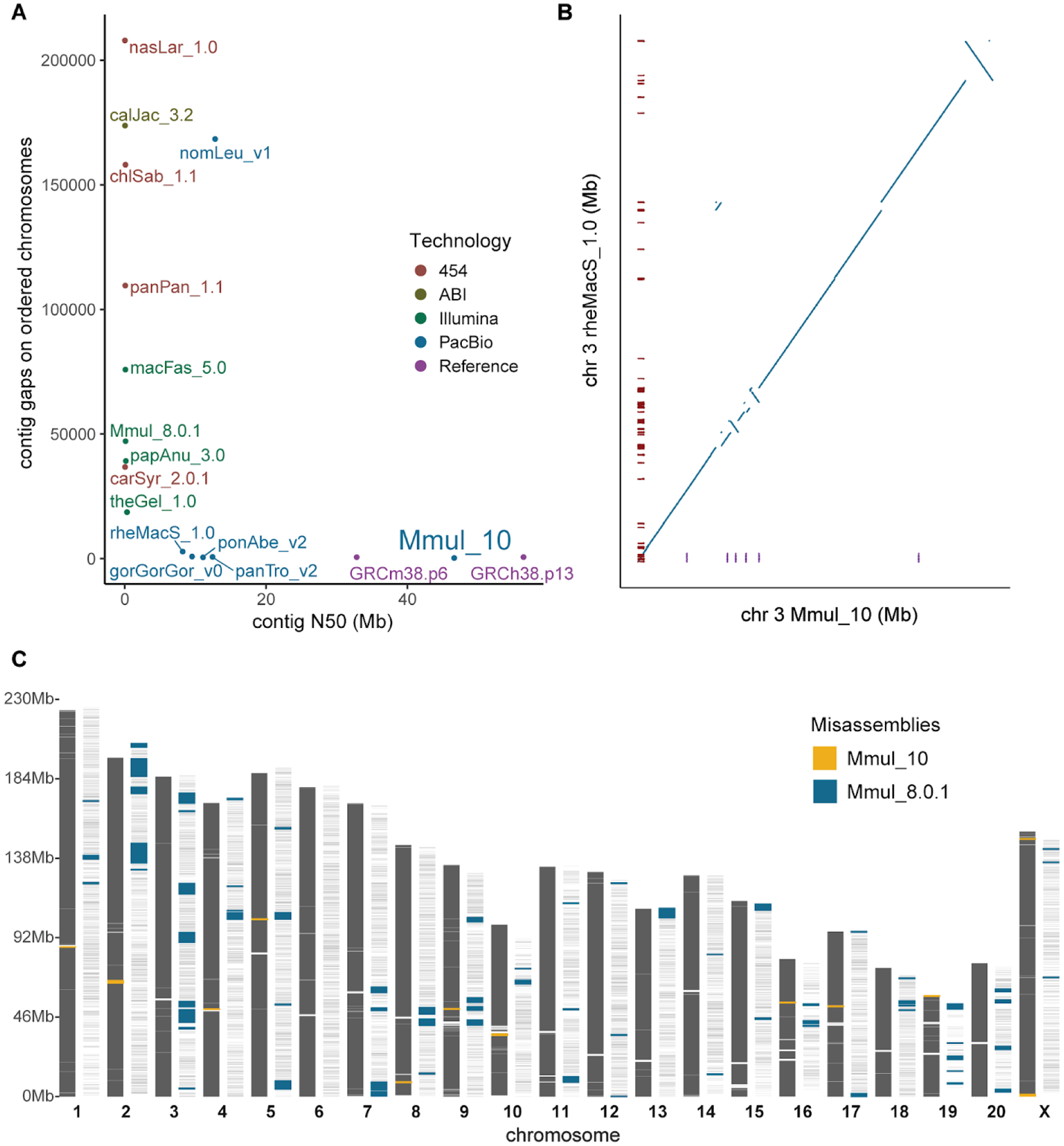
(A) The number of gaps and contig N50 lengths are compared among mammalian genomes (color-coded based on sequencing technology). The contiguity of macaque (Mmul_10) is comparable to human (GRCh38) and mouse (GRCm38.p6) reference genomes. (B) The number of gaps (red ticks) are compared against a synteny plot of Chinese (rheMacS) and Indian (Mmul_10) macaque chromosome 3 assemblies. (C) Comparison of potential orientation misassemblies based on Strand-seq analysis (18). Mmul_10 shows far fewer (yellow; n = 13) inversions when compared to an earlier macaque assembly, Mmul_8.0.1 (blue; n = 82), predicting 34 times less misoriented bases; 99.7% of gaps (white rectangles) in the earlier assembly are now closed.
Quality assessment.
We performed a series of analyses to evaluate the quality of the Mmul_10 reference assembly. For example, we assessed accuracy and contiguity using macaque BAC-end sequence (BES) data estimating values of 99.7% and 92.5%, respectively, consistent with allelic variation among individual macaques. Detailed analyses of even complex regions such as the MHC genes (10) and the killer immunoglobulin-like receptor (KIR) gene families (21) show that the majority of these loci were assembled accurately (Tables S2 and S3; (18)). Using orthogonal sequencing datasets from the same sample, we estimate an overall assembly sequence accuracy of one error every 20,000 base pairs.
Over 99.7% of the gaps present in the previous Indian-origin rhesus macaque genome assembly are now closed. Eleven of the 20 macaque autosomes are now represented as two scaffolds separated only by an unassembled centromere. While there is high overall synteny with Mmul_8.0.1, rheMacS, and macFas_5.0 (chromosome 3 [Figs. 1B; S1]), there are 39 large orientation errors identified in the original Mmul_8.0.1 assembly that our assembly is able to correct (22) (chromosomes 13 [Fig. S2] and 2 [Fig. S3]). In order to develop a more accurate assembly for this species, we investigated other potential orientation issues by generating Strand-seq data from an unrelated macaque (18), compared it to the two Indian macaque assemblies, and identified “homozygous inversions” as potential errors in orientation. In total, we detected 82 (130,115,998 bp) potentially misoriented regions in Mmul_8.0.1 in contrast to 13 (3,800,615 bp) in the new reference assembly (Fig. 1C; Tables S4 and S5). Importantly, these data indicate that the number of misoriented genes has been reduced from 4.83% to only 0.13% in Mmul_10. Many of these unresolved regions, not surprisingly, map to structurally diverse and complex immune gene families, such as the MHC and KIR regions, where in the latter a short homozygous inversion is predicted (Fig. S4). The Strand-seq analysis also detected one chimeric scaffold where a terminal part of chromosomal scaffold CM014356.1 belongs to the beginning of chromosomal scaffold CM014355.1 (Fig. S5), which has now been corrected.
Gene annotation.
We assessed the completeness of gene annotation by applying benchmarking universal single-copy ortholog (BUSCO) scores, which measure the representation of highly conserved mammalian genes (23). We find that 99.6% of BUSCO genes are annotated with only 0.1% missing—an improvement over the Chinese macaque assembly (rheMacS) where 1.8% are missing (20) (Table S6). Analyzing a curated set of 6,422 Macaca mulatta RefSeq transcripts (NCBI) shows an average coverage of 99.8% confirming the high degree of completeness. Predicted protein-coding gene comparisons within each annotation pipeline, NCBI and Ensembl, for rhesus macaque and human show consistency (Table S7). In rhesus macaque, NCBI and Ensembl produced similar outcomes: 21,121 and 21,748 genes, respectively (Table S7). We note a substantial improvement in ncRNA identification in Mmul_10, with 8,720 new ncRNAs (Table S7), especially lncRNA (Tables S8 and S9; (18)). Many of the missing genes map to more complex regions of the genome or duplicated genes that have expanded or contracted differentially when compared to human (Tables S10 and S11).
In an effort to further define macaque-specific transcript and protein isoform diversity, we generated and sequenced 6.5 million full-length cDNAs from macaque brain, induced pluripotent stem cell (iPSC) lines, and testes (Table S12). We applied the Comparative Annotation Toolkit (CAT) (24) to annotate 17,838 protein-coding and 31,873 noncoding macaque genes. The set includes 83,692 protein-coding isoforms, of which 5,353 were identified from Iso-Seq data as potentially novel isoforms (Fig. S6). A total of 980 genes have frameshifting indels disrupting all isoforms; RNA-seq-based cleanup reduced the proportion of isoforms with frameshifting indels from 16.5% to 4.9%. The final CAT gene set includes 80,248 protein-coding transcripts aligned in a 1–1 fashion, with the remaining 3,444 paralogous isoforms being resolved with alignment metrics. We note that 550 gene structures are split over multiple contigs (Table S13) and 827 protein-coding genes show evidence of being part of gene families exhibit reduced copy number in this assembly relative to human, with 473 of those showing a 2 to 1 relationship and 295 being 3 to 1 in human when compared to macaque (Table S10). In contrast, 967 protein-coding genes show evidence of gene family expansion in macaque, with 711 copied once and 116 copied twice in rhesus when compared to humans (Table S11).
Novel exon adaptation in macaque.
CAT predicted 2,880 novel transcripts that did not arise from any previously annotated transcript in the input human annotation. Of these, 2,812 maintain open reading frames. From this set, we manually curate 84 novel exons with Iso-Seq support (Fig. 2; Table S14). We searched the translated protein sequence in the Pfam 32.0 database (25) using this set and found significant homology to notable protein families. For example, three transcripts (Rhesus_T0212625, Rhesus_T0212626, and Rhesus_T0212627) correspond to a novel gene model (Rhesus_G0055137 on chromosome 9:95,447,150–95,611,700) that shares homology with human CYP2C18 protein and has abundant Iso-Seq transcript support across multiple macaque tissues (Fig. 2A). The predicted mRNA sequence for tropoelastin (ELN) has two identifiable exons near the C-terminus of the protein with Iso-Seq support from six tissues for three alternatively spliced isoforms of ELN. These exons are shared to the base of the mammalian tree, but not found in humans, great or lesser apes, suggesting a unique loss of these exons in the hominoid lineage (Fig. 2B).
Figure 2. Novel genes and gene models.
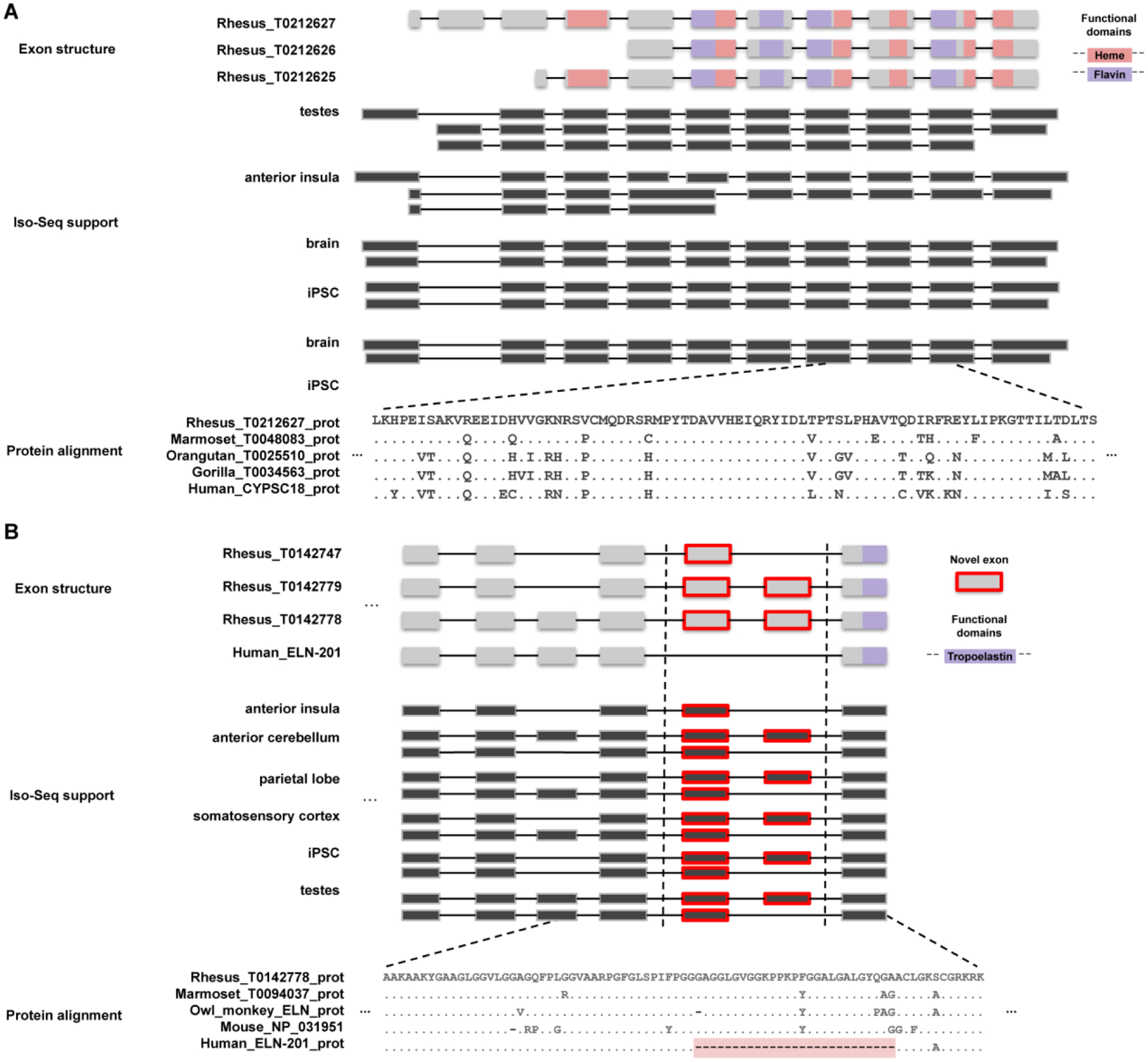
(A) A novel gene model with homology to the cytochrome p450 protein family is predicted by the AugustusPB mode of the CAT. The gene structure and protein domain architecture of three isoforms are shown (top). The predictions arose from supporting Iso-Seq reads from five tissues (middle). Orthologous novel genes are also predicted in marmoset, orangutan, and gorilla assemblies; a protein alignment (bottom) of those genes along with a human CYP2C18 protein is shown. (B) Two macaque isoforms in ELN (tropoelastin) are predicted by the AugustusPB mode of CAT and are supported by macaque Iso-Seq data but differ significantly from human by two exons. The gene structure and functional domains for the last seven exons of this gene are shown (top), along with a comparison to a human transcript model. These two protein-encoding exons are also observed in marmoset, owl monkey, and mouse, but not in apes, as a result of an ape-specific deletion (bottom) that changed the gene structure of tropoelastin.
We explored additional exon adaptations that were potentially unique to rhesus macaque. The first is a deletion of 64 bases in the macaque genome affecting MYO3A (Fig. S7A). This deletion leads to a new isoform of MYO3A with altered exon structure and is supported by Iso-Seq data from multiple tissues. Second, is an isoform of GAS8 where a novel exon leads to a frameshift in downstream exons creating a premature stop codon that is alternatively spliced as confirmed by Iso-Seq (Fig. S7B). The third example is a rhesus-specific 6,250 base-pair insertion in DCHS2 that introduces a novel exon to one of the DCHS2 isoforms (Fig. S7C). Once again, this novel DCHS2 exon is supported by Iso-Seq reads from testes tissue where it is alternatively spliced. However, the Iso-Seq transcripts do not support the original CAT gene annotation predictions as a whole. Rather than a predicted exon skipping event, the novel exon appears to correspond to an alternate first exon of the gene. Additional experimental work will be required to determine the functional impact of these genic differences.
Segmental duplication analyses.
We analyzed Mmul_10 for recent duplications (18) and identified 111.5 Mbp of assembled SDs (≥1 kbp and ≥90% sequence identity). In principle this represents a >3-fold improvement compared to the analysis of the first macaque assembly (2) where only 32 Mbp were characterized as SDs. Despite this improvement, 54% of the duplicated base pairs remain unlocalized (Figs. S8 and S9). Nevertheless, the vast majority of those assigned to a chromosome are clustered or distributed interchromosomally among pericentromeric and subtelomeric regions (Fig. S10). Only 8% (755/9,475) of the pairwise alignments are intrachromosomal and separated by at least 1 Mbp (Fig. S10), for example, the collapsed SD of NXF2 on the X chromosome (Fig. S11).
We identified 276 regions of collapsed duplications (26) corresponding to 9.1 Mbp of the genome (Table S15) and estimate that these correspond to 41.8 Mbp of SDs not yet properly integrated into the macaque assembly. FISH analyses classified the majority of these (74%) as pericentromeric based on signals mapping to either side of the centromere. In order to resolve the sequence of these collapses, we applied a graph-based approach (26) (27) to resolve SDs based on clustering and assembling reads using diagnostic paralogous sequence variants. This method resolved 168 of the 276 collapses into 531 distinct contigs representing 19.8 Mbp of SD sequence (contig N50 = 37.4 kbp). Among these are highly accurate sequence contigs corresponding to recently expanded rhesus macaque gene families, including MHC, olfactory receptor, and zinc finger genes. We have deposited these contigs into GenBank under BioProject PRJNA662298 as a resource where they may be used to improve gene annotation. For example, we identified nine assembly collapses (20–92 kbp) corresponding to ZNF669 genes in the Mmul_10 assembly (Table S15). Segmental Duplication Assembler (SDA) assembles 53 contigs from these collapses generating an additional 1.9 Mbp of assembled sequence (N50 = 36.5 kbp) and identified three contigs where ZNF669 Iso-Seq mapped with higher identity than the original Mmul_10 assembly (Fig. 3A–B). Translation of the full-length cDNA confirmed open reading frames and duplicated gene models that had been missed by our initial annotation of the genome (Fig. 3C). FISH analysis of a large-insert BAC corresponding to one of the loci confirmed a cluster of ZNF669 genes mapping to chromosome 6 as well as several additional duplicated loci of this gene family distributed throughout the macaque genome (Fig. 3D).
Figure 3. Macaque ZNF669 gene family expansion.
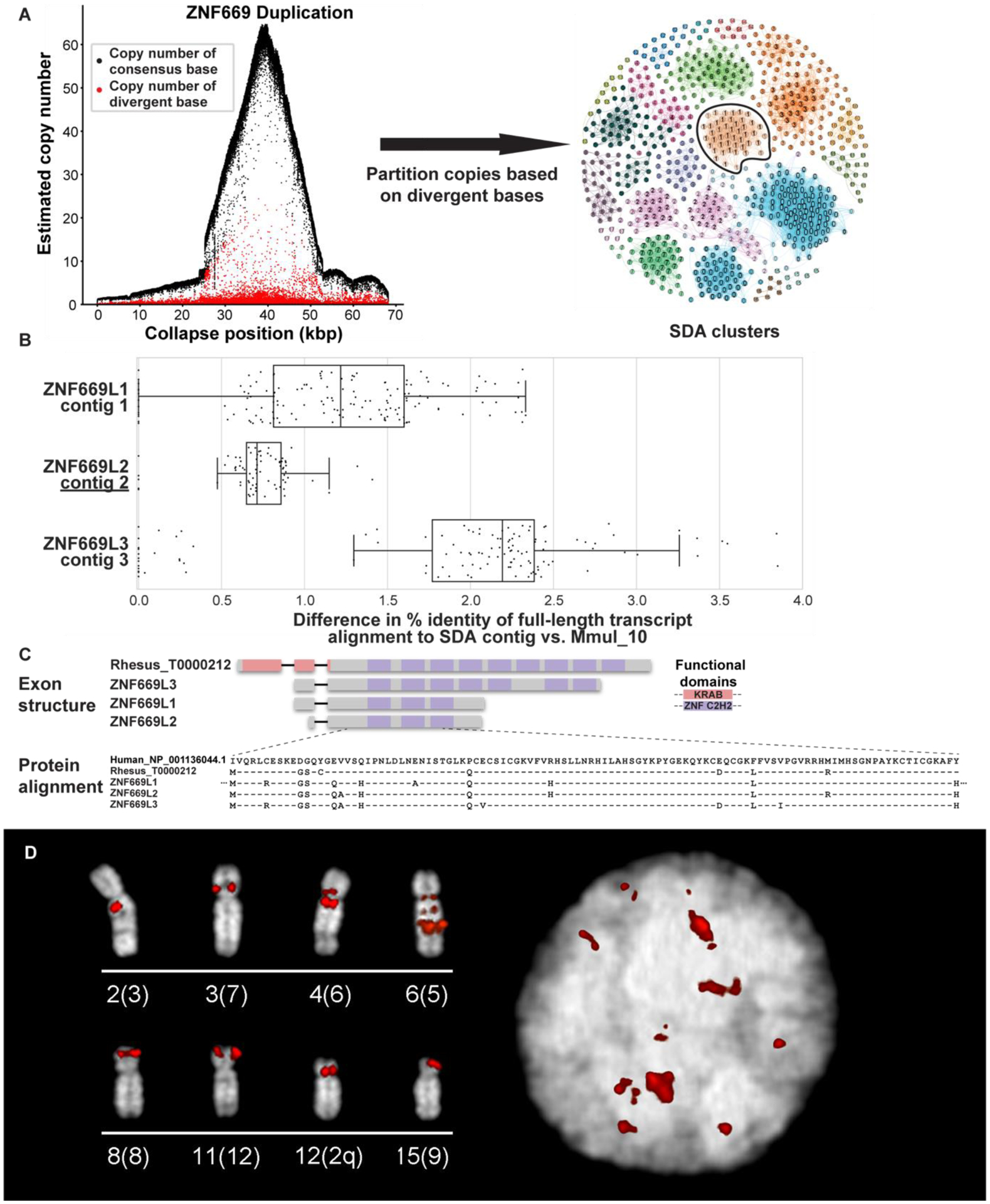
(A) A 68 kbp region of collapsed assembly corresponding to the ZNF669 gene family as indicated by the excess read depth and increased number of paralogous sequence variants (PSVs, red dots) that are diverged when compared to the consensus sequence (black dots). The highly identical copies were, thus, unresolved in Mmul_10 and predicted to be present in about 50 copies in macaque (left). Segmental Duplication Assembler (SDA) partitions the long reads into 19 distinct paralog clusters (colored and numbered) based on shared PSVs and assembled these clusters into 18 contigs. Vertices reflect individual PSVs and edges represent long-read sequences that contain both of the connected PSVs (right). SDA partitioned and assembled the remaining ZNF669 collapses into 35 additional contigs. The outlined PSV cluster corresponds to contig 2 in panel B. (B) Mapping of FLNC transcripts shows they align better to SDA-resolved contigs than the original assembly. (C) Annotation of these genes shows that these three contigs encode a highly expanded ZNF669 gene family where there is FLNC data supporting complete open reading frames that differ by only a few amino acids. (D) FISH with BAC CH250–540H16 as a probe corresponding to a ZNF669 locus demonstrate interchromosomal duplications (red) on interphase nucleus (right) and metaphase chromosomes (left), labeled by chromosome (human syntenic chromosome in parentheses).
Repetitive sequence analyses.
Overall, fewer mobile element insertions (MEIs) were identified in Mmul_10 when compared to Mmul_8.0.1((18); Table S16), including lineage-specific elements across various retrotransposon classes (Table S16). Despite these reductions, subfamily network analyses for both Alu (Fig. S12) and LINE 1 elements show increases in the number and connectivity of younger subfamilies, particularly full-length L1 (Fig. 4A–B). Notably, there is an increase in full-length potentially active L1 elements (n = 6,892 vs. n = 4,380) in Mmul_10 compared to Mmul_8.0.1 (Table S16). Full-length L1 elements are less fragmented and more likely to be assigned to chromosomes as opposed to being mapped to unlocalized contigs (Fig. 4C). Similarly, the new assembly moves 8,291 unlocalized Alu elements to specific chromosomal assignments (Fig. 4D). Interestingly, 33% of potentially full-length endogenous repeat elements (LTR > 7 kbp in length) now map to different chromosomal locations in the newer assembly (Fig. S13) consistent with improvements in the sequence resolution and integration of longer and potentially active repeat elements.
Figure 4. Full-length LINE1 analyses.
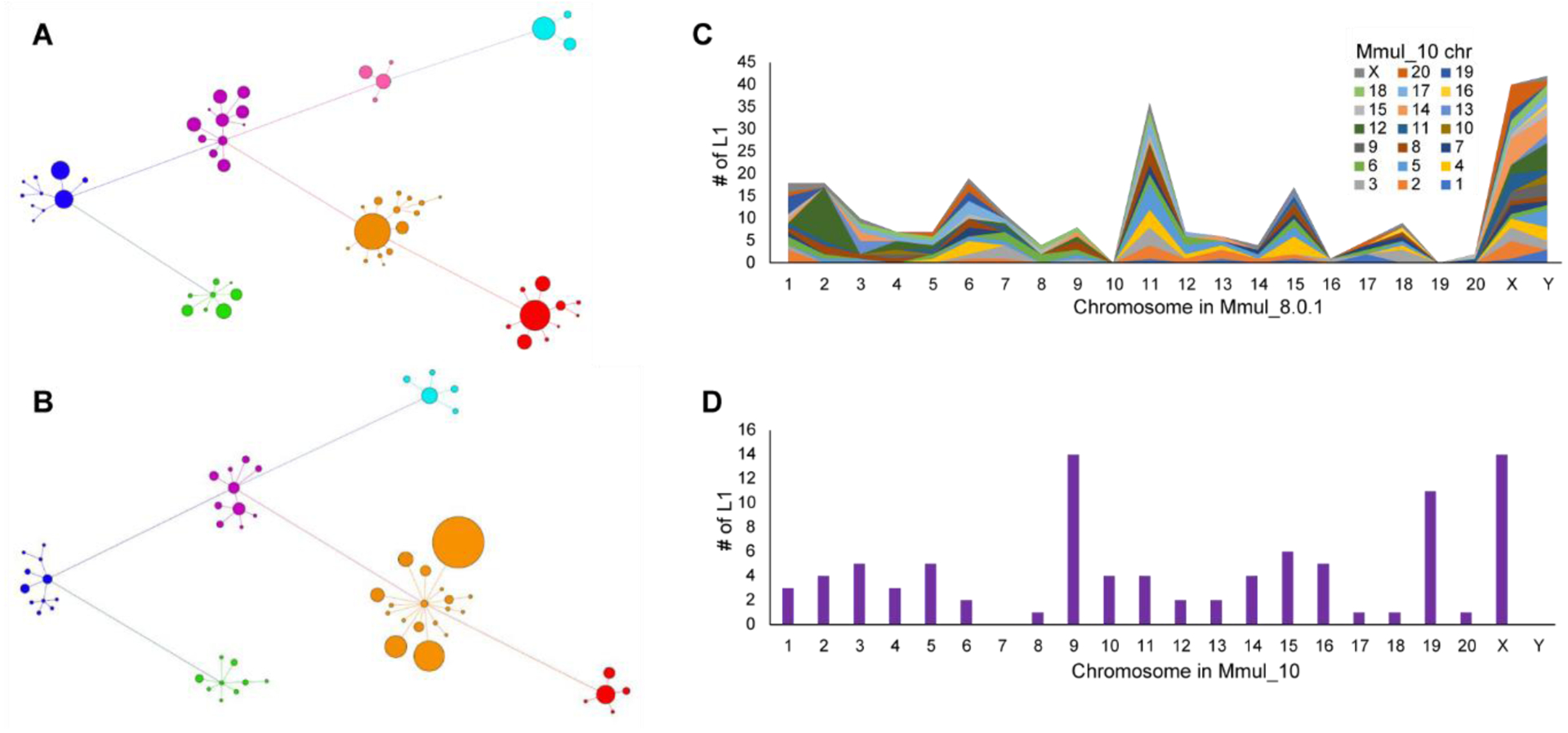
A LINE1 subfamily network analysis comparing (A) an earlier macaque assembly, Mmul_8.0.1 (61 subfamilies), to (B) the new assembly, Mmul_10 (58 subfamilies) (18). Related subfamilies are connected by lines and clustered by color: L1RS37 (purple), L1PA7/8 (blue), L1PA6 (green), L1RS36 (pink), L1RS2 (red), L1RS10/16/21 (orange), and L1RS25 (teal). The size of each node corresponds to the relative number of LINE1 elements. There is an increase in annotated younger elements (orange) although the number of subfamilies has decreased the L1RS36 cluster as a result of reassignment based on a higher-quality assembly. (C) The plot depicts the number of full-length L1 elements (y-axis) that have been assigned to a new chromosome in Mmul_10 (key) when compared to Mmul_8.0.1 (x-axis). (D) A similar analysis depicting the number of full-length LINE1 elements previously unplaced (n = 92) but now assigned to a chromosomal location in Mmul_10.
Recurrent deletions in full-length L1RS elements.
Given the better representation of full-length repeats, we searched for systematic changes in the 5’ UTR (untranslated region) structure of the primate-specific L1PA subfamily from which L1 rhesus-specific (L1RS) retroelements originate (28). The 5’ UTR of these selfish elements are often targeted by host factors, e.g., KRAB zinc finger (KZNF) proteins, which repress their transcription and, as a result, are differentially expanded across primate lineages (29). We mapped 20,541 full-length L1RS elements to the human L1PA5 consensus sequence (UCSC Repeat Browser) and identified specific coverage drops that accrue and persist in subsets of L1RS elements (28). Using these deletion patterns and subfamily designations, we propose an order for the evolution of these 5’ UTR deletion events (Fig. 5A) and suggest a reclassification of L1RS nomenclature. Although older families are typically assigned a higher number, the appearance of coverage drops establishes that the L1RS16 family predates the L1RS21 family (Fig. 5B). Furthermore, the results indicate that after the human–rhesus divergence, at least three different regions of the L1RS 5’ UTR were altered. A comparative analysis of these L1RS elements in the genomes of other Old World monkeys (OWMs; rhesus, crab-eating macaque, baboon, and golden snub-nosed monkey with human as an outgroup) supports this adaptive model (Fig. 5B), as all OWM species display changes at these positions, although the actual sequence changes and size of the region varies, suggesting that these events recur or have been refined at different points along the OWM phylogenetic tree (Fig. S14). Two of these three sites (Site 1 and Site 3) also overlap changes observed in active human-specific L1 elements supporting the hypothesis that the deletions result from independent recurrent parallel evolution in the primates. The remaining site (Site 2) is restricted to OWM as no coverage drop is observed at this site in young full-length L1 human elements. The existence of a deletion at this site in all OWMs suggests that a repressive factor was present in the OWM common ancestor and highlights the uniqueness of L1RS2 transcript diversity (Fig. 5C).
Figure 5. Evolution of L1RS elements.
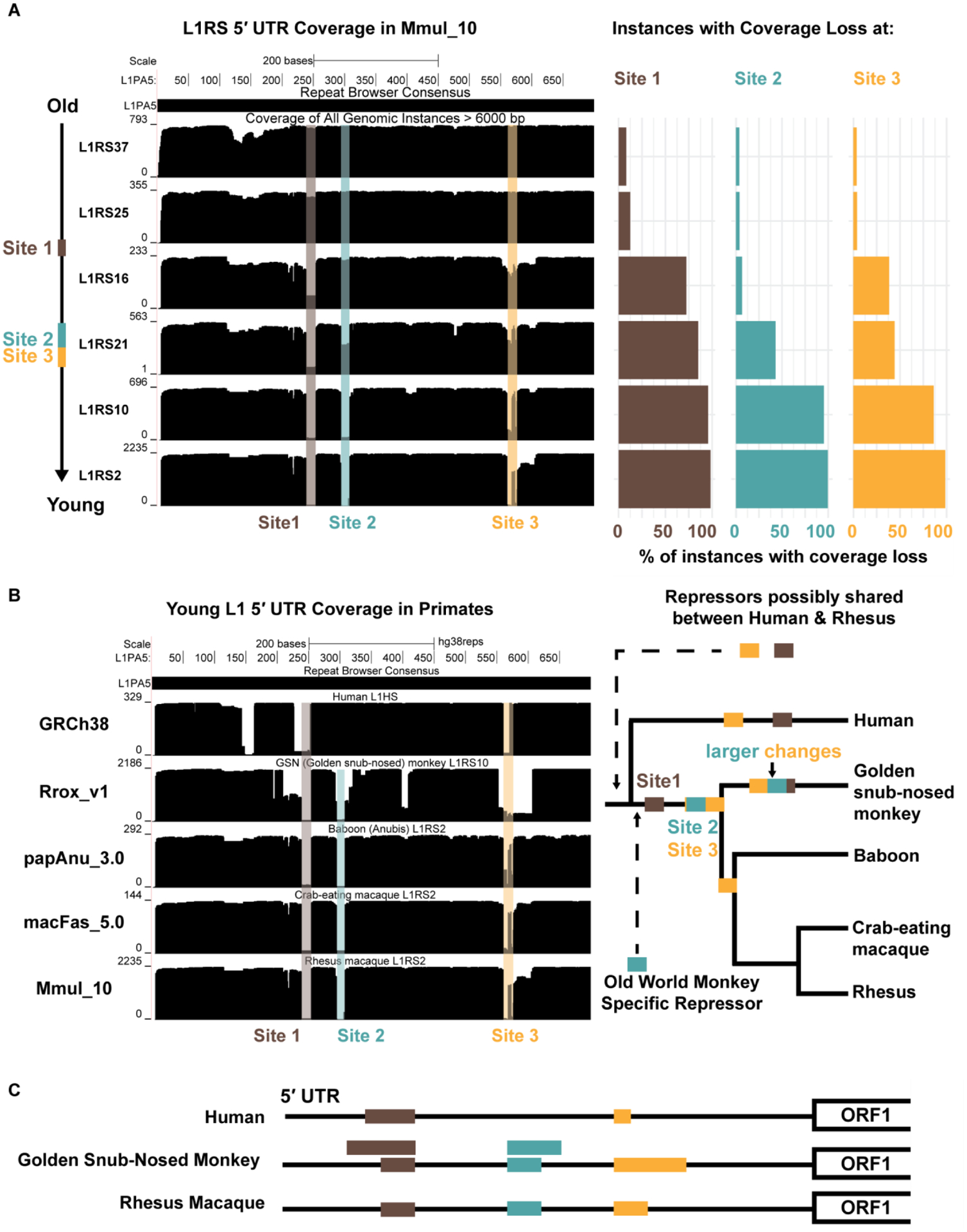
(A) (Left) All full-length L1RS elements (>6000 nt, top schematic) were grouped by families and mapped to a consensus version of L1PA5 (the ancestral LINE-1 element from which they derive) with the first 700 nt (red) of the 5’ UTR analyzed further. Site 1 (brown) experiences a coverage drop that is found in the majority of L1RS16 and younger families. Coverage drops at Site 2 (blue) and Site 3 (yellow) occur in the L1RS21 family at nearly the same time. (Right) Percentage of individual instances that do not map to the L1PA5 consensuses for each L1RS family. Coverage drops are not found in old L1RS elements but found in nearly all young elements, suggesting a fitness advantage for the changes at each site. (B) (Left) All full-length elements (>6000 nt) of the youngest L1RS families in four OWM genomes (L1RS10 in Rrox_v1/rhiRox1 [golden snub-nosed monkey] and L1RS2 in Panu_3.0/papAnu4 [baboon], Macaca_fascicularis_5.0/macFas5 [crab-eating macaque], and Mmul_10/rheMac10) were aligned to the L1PA5 consensus to generate coverage plots. The youngest human L1 (L1HS) was also aligned to L1PA5 as an outgroup. Drops in coverage (Site 1, Site 2 and Site 3) were seen in OWM, although golden snub-nosed monkeys (Rrox_v1/rhiRox1) display distinct patterns from other OWM suggesting convergent but distinct changes in the 5’ UTR, possibly to escape repressive elements. (Right) An evolutionary model for shared and convergent changes in L1RS elements. Site 1 changes are shared amongst all OWM while Site 2 and 3 changes experience similar but not exact changes in Rrox_v1/rhiRox1 compared to other OWM. Coverage drops at Sites 1 and 3 are also observed in human while Site 2 changes are OWM specific. (C) Schematic of Site 1, 2, and 3 (brown, blue, yellow) changes on the L1 5’ UTR in representative lineages: human, golden snub-nosed monkey, and rhesus. Rhesus macaque and golden snub-nosed monkey have identical coverage drops at Sites 1 and 2 that arose in the OWM common ancestor; golden snub-nosed monkeys also experience larger changes (larger bars) spanning these sites that most likely occurred after the Colobinae divergence as they are not observed in rhesus. Humans experience a unique coverage drop at Site 1 larger than rhesus but smaller than the large golden snub-nosed monkey-specific changes. All three species experience unique changes resulting in differing length elements at Site 3.
Macaque genetic diversity among research populations.
The US research colonies of rhesus macaques were founded primarily with animals imported from India decades ago, although a much smaller number of Chinese-origin rhesus macaque have been added to some colonies over time. It is not possible in all cases to trace the exact geographic origins of the US research population, but sufficient information is available to identify many animals as derived from either Indian- or Chinese-origin founders. We generated whole-genome sequence (WGS) data for 850 rhesus macaques from captive US research colonies and three wild-caught Chinese samples, including 133 previously published samples (3). The majority of the samples (n = 810) were designated as Indian origin whereas the remaining individuals were of Chinese or suspected admixed origin. Most samples were sequenced to at least 20-fold coverage (n = 764; average 33.69-fold) with the remaining (n = 89) sequenced to an average of 8.58-fold for the detection of single-nucleotide variants (SNVs) and indels (Figs. S15 and S16). SNVs and indels were identified based on mapping reads to Mmul_10 (18) with an overall 2.12 Ts/Tv ratio consistent with prior studies (3). We identified 85.7 million SNVs, including 21.3 million singletons in addition to 10.5 million indels (Table 2), creating the most extensive collection of segregating genetic variants for any NHP species (Table S17). By comparison, a recent study of 929 human genomes from 54 diverse global populations, sequenced to average 35-fold coverage, identified just 67.3 million SNVs (30). Thus, the research rhesus macaques were more than twice as diverse per individual as humans with the average macaque carrying 9.7 million SNVs.
Table 2.
Summary of macaque genetic variation.
| Type | Total variants | Singletons | Multiallelic variants |
|---|---|---|---|
| All SNVs | 85,721,160 | 21,270,272 | 3,160,393 |
| All indels | 10,501,197 | 2,956,760 | 1,875,826 |
| Protein-coding SNVs | 790,377 | 222,766 | 23,335 |
| Protein-coding indels | 33,823 | 13,227 | 3,663 |
| LGD SNVs2 | 20,400 | 6,570 | 1,026 |
| LGD indels3 | 26,774 | 10,330 | 3,051 |
SNV and indel protein-coding classifications based on Ensembl Variant Effect Predictor (VEP)
Likely gene disruptive (LGD) SNVs
LGD indels defined as VEP consequences splice_acceptor_variant, splice_donor_variant, stop_gained, stop_lost, start_lost
A principal component analysis (PCA) of the SNV genotypes readily discriminated between the Chinese and Indian rhesus macaques (Fig. 6A). Thirty-one individuals that were initially identified as Indian origin show some degree of Chinese rhesus admixture, although the extent of admixture varies considerably (Fig. 6A). The free-ranging Cayo Santiago rhesus macaque population (Caribbean Primate Research Center) shows a gradient of variation with respect to other Indian rhesus macaques (PC2)—likely a consequence of a genetic bottleneck since its initial founding in 1938 on the Puerto Rican Island of Cayo Santiago (31). Consistent with this observation, the Cayo macaque population shows lower heterozygosity and larger runs of homozygosity when compared to other National Primate Research Center (NPRC) populations (Figs. S17–19). Interestingly, a preliminary analysis shows that linkage disequilibrium (LD) decays more rapidly among unrelated Indian rhesus macaques than in a subset of the human African population, but at greater physical distance (>50 kbp) the macaques retain higher linkage disequilibrium (Fig. S20).
Figure 6. Rhesus macaque population structure and developing macaque models of disease.
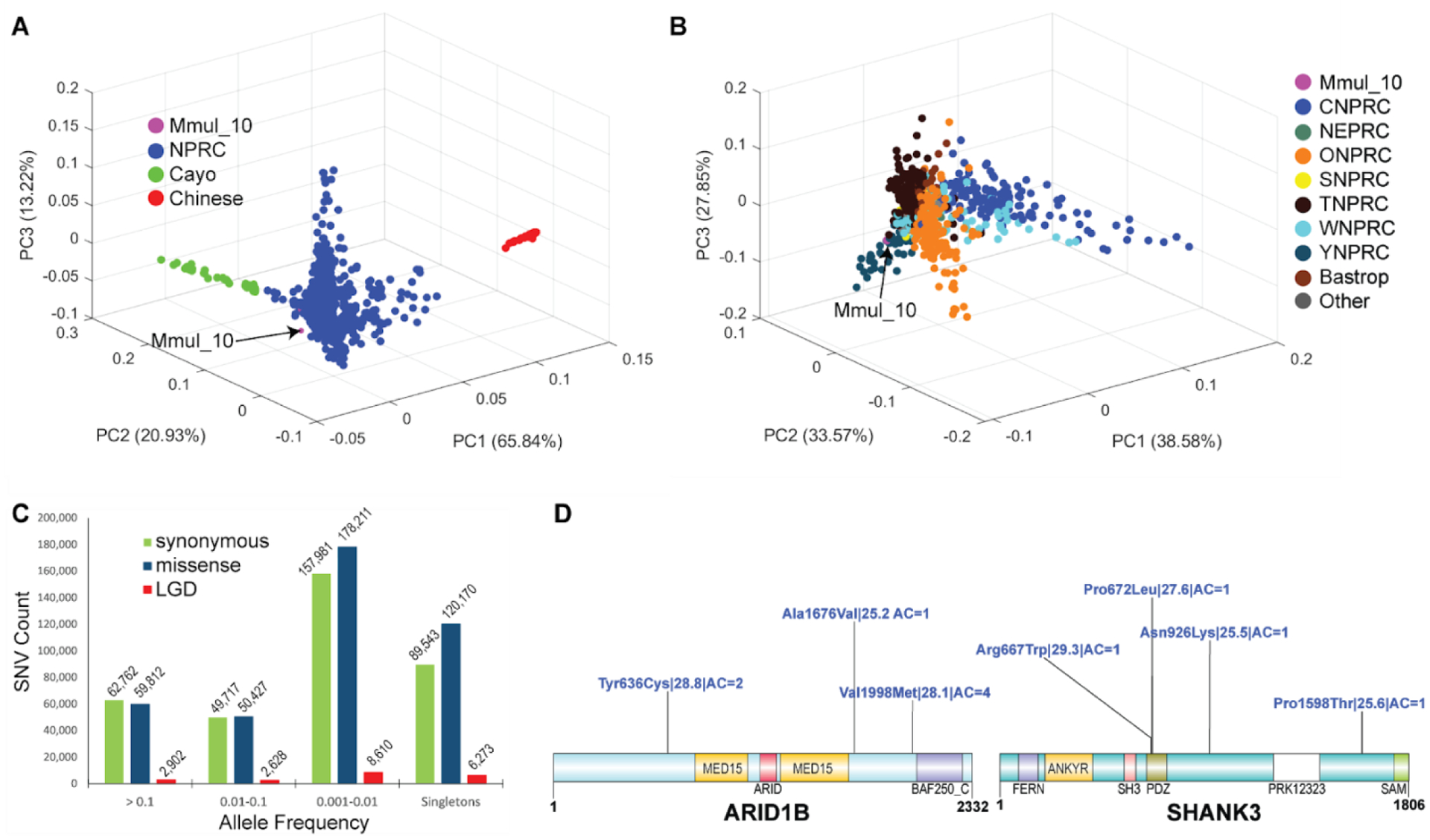
(A) A 3D principal component analysis (PCA) based of SNVs filtered for missing call rates > 0.05 or major allele frequency (MAF) < 0.1 from sequencing 853 macaque genomes shows clear separation of Chinese (PC1) genomes (red) and a gradient for Cayo macaques (green) with respect to other Indian macaques (PC2). (B) A PCA excluding Chinese and Cayo populations comparing 771 macaques from different NPRCs. The Cattell–Nelson–Gorsuch (CNG) screen test retained the top three principal components in both PCAs and the percent variance explained calculations are based on those three components. (C) Allele frequency distribution of likely gene-disruptive (LGD) including splice acceptor, splice donor, stop gained, stop loss and start loss variants (red) and missense (blue) variants compared to synonymous changes (green). (D) Genes implicated in human neurodevelopmental disorders (NDDs) showing naturally occurring putatively damaging variants in macaque orthologs. A schematic of damaging missense (blue) variants (CADD >= 25) for NDD genes: MBD5, ARID1B, and SHANK3. For each variant, we indicate the amino acid change| CADD score| allele count. All potentially deleterious mutations are low frequency.
We repeated the PCA excluding both the Cayo and Chinese macaque populations (Fig. 6B). In this analysis macaque genetic variation from most primate research centers was indistinguishable with the exception of Oregon National Primate Center (ONPRC), Yerkes Primate Research Center (YPRC), and California National Primate Research Centers (CNPRC) for which subsets of individuals appear genetically distinct (Fig. 6B). A population structure analysis shows that the CNPRC macaques have a somewhat greater admixture with Chinese macaques than other populations on the basis of the genomes analyzed here (Figs. S21 and S22). In addition, within some of the research populations (e.g., YNPRC, Cayo, etc.), genetically distinct subgroups of animals are identified with some consistent substructure.
Analysis of variants for functional changes.
Macaques are important models of human genetic disease (3) (4) so we investigated both common and rare variants from our samples that affect the sequences of protein-coding genes. In total, we identified 85.7M (million) SNVs of which 3.2M were multiallelic and 21.3M were singletons, as well as 10.5 indels (Table 2). For protein-coding sequences we find 790,377 SNVs and 33,823 indels (Table 2). Based on Variant Effect Predictor annotations, we identify 408,496 missense and 20,400 likely gene-disruptive (LGD) mutations (Table S18, Table 2). Of all variant classes, the LGD mutations occur at the lowest frequency (0.001—0.01) consistent with a more deleterious effect on phenotype (Fig. 6C). An assessment of homozygous-damaging mutations shows considerable overlap among the NPRC colonies for more common variants while a smaller subset is unique to each (Fig. S23). We generated a summary distribution of all missense variant counts per gene and normalized by the gene length as defined by the number of protein-coding bases in the gene, excluding introns and UTRs (Fig. S24).
To illustrate the biological potential of the macaque genetic diversity, we identified naturally occurring macaque mutations in orthologs of human genes implicated in autism and developmental delay (32) (33). In humans, de novo deleterious mutations in these genes are thought to be dominant and have a large effect, but mouse models often do not recapitulate the complexity of neurobehavioral features of human disease (32). We considered all missense and LGD mutations such as frameshift, stop, and splice site mutations that would disrupt protein-coding sequence. We further classified the genes on the basis of their intolerance to gene-disruptive mutation (pLI or the probability of being loss-of-function intolerant) (Figs. 6D, S25 and S26). The pLI score has been widely adopted and is based on the number of observed versus expected protein-truncating variants in a population. The closer the pLI score is to 1, the more intolerant to variation the gene is predicted to be. Revisiting gene annotation (based on the Iso-Seq resource) shows that while some of these correspond to changes in gene annotation between species, other changes represent viable candidates to establish new models of neurodevelopmental disease (e.g., severe missense mutations in ARID1B). Neurodevelopmental delay genes are significantly depleted for missense variants when compared to all genes (p = 8.883e-41; Fig. S25A). Remarkably, we identified nine genes with candidate deleterious mutations in macaque that are intolerant to mutation in humans and where de novo mutations in human are associated with neurodevelopmental disorders (Table S19). Homozygous LGD variants segregating in rhesus macaque research centers offer even more opportunities for exploring their biological relevance among NHPs (Table S20).
Finally, structural variation differences also present an opportunity to develop new rhesus macaque disease models as well as enhance our understanding of fixed and polymorphic changes between species. For example, we identified ~301,000 insertions and 241,000 deletions shared between the Indian- and Chinese-origin macaques (Fig. S27; Tables S21 and S22). Of these 542,000 insertion or deletion events, only a small fraction (1.68%) are predicted to affect genes (Table S22). Among the 87,227 structural variants unique to either subspecies, we predict that 1,614 may affect genes although validation in addition to genotyping and genome assembly of more individuals (especially wild caught) will be required to establish fixed differences between the Chinese and Indian macaque genomes.
Discussion
The rhesus macaque is arguably the most important NHP for biomedical research and is a key species in the study of primate evolution. The resources we developed and present here will significantly advance both areas by providing new biological insights and an improved framework for future gene-based disease research. In this new assembly we have corrected indel errors and misassemblies, properly represented inverted sequences, and flagged several remaining orientation issues. The various orthogonal technologies employed in the data production process, coupled with detailed manual curation, led to dramatically improved gene annotation, eliminated 99.7% of gaps and reduced misorientations when compared to the previous Indian-origin macaque assembly (2).
This new reference genome identifies previously unknown genes and genes whose intron/exon structure differs in comparison to humans, such as the 3.1 kbp Alu-mediated deletion that removed two protein-encoding exons from tropoelastin (ELN) relative to the human and ape orthologs (Fig. 2). Similar to finishing efforts for the mouse and human assemblies (34) (35), most of the novel macaque and OWM genes occur among duplicated gene families (Fig. 3). Indeed, our estimate of recent SDs has nearly doubled for this assembly (130–140 Mbp) and is beginning to approximate that of human (5–6%) (36) (37). Unlike human, however, where a large fraction of the SDs are interspersed along chromosomal arms, most macaque duplications appear either clustered or pericentromeric, bracketing the centromeres of macaque chromosomes. Similar to SDs, repeat content, especially for full-length elements, has facilitated comparative analyses identifying recurrent deletion events in the 5’ UTR of L1 elements in human and multiple OWM lineages as a potential adaptive response helping to evade KRAB-ZNF repression. It is tempting to speculate that expansion of OWM (including rhesus macaque) ZNF genes such as ZNF669 are part of an ongoing arms race to suppress new rhesus L1 subfamilies (38). While the assembly we generated is superior by most metrics, we recognize that centromeres, acrocentric regions, and some of the largest SDs still remain unresolved or unassigned.
Using this new macaque reference, we establish an extensive SNV resource that will facilitate future genetic analyses of biomedical research colonies. Our catalog of thousands of naturally occurring common missense variants and identification of other rare macaque mutations may help the discovery of new models of disease, such as those implicated in autism and human neurodevelopmental disorders (32) (33). These naturally occurring mutations provide an opportunity to develop noninvasive models of human disease without the expense of CRISPR-engineering of embryos (9). These models may be particularly powerful in relation to phenotypes that are not readily reproduced in non-primate knockout models (12) and for evaluating the effect of genetic variation on the efficacy of treatments prior to human trials.
Methods summary
A single female rhesus macaque of Indian origin (AG07107) was sequenced, assembled, and manually curated with a physical map, proximity ligation, and Strand-seq sequence data. Full-length cDNA was prepared from various tissue sources and used to annotate the genome assembly with three independent gene annotation pipelines: NCBI, Ensembl, and CAT. A whole-genome analysis comparison coupled with read-depth assessment was used to estimate the proportion of SDs. We used interphase and metaphase FISH on a female rhesus macaque lymphoblast cell line to validate SD order and orientations. All repeat elements were quantified using RepeatMasker for comparisons among rhesus macaque assemblies. To estimate rhesus macaque genetic diversity, we sequenced 853 animals across nine US rhesus macaque research colonies using standard Illumina sequencing instruments. We used a compendium of best practices to call sequence variants for SNVs, insertions, deletions, and structural variants. To classify the potential impact of SNVs, we aligned all to the human genome and focused our analysis on those that cause loss of function by stop-gain, start-lost, splice-donor or -acceptor base changes and missense variants that alter amino acid coding of the protein. A subset of these SNVs were compared to human genes known to harbor rare de novo deleterious variants that have been implicated in human neurodevelopmental disorders.
Supplementary Material
Acknowledgements:
We thank staff at the NPRCs involved in the preparation of biomaterials used for WGS; Sam Peterson (OHSU) for preparation of mRNA used in the generation of Iso-Seq datasets; and specifically, the UC Davis Primate Research Center for the rhesus macaque fetal brain material used for Iso-Seq data production. We would like to thank the Baylor College of Medicine Human Genome Sequencing Center production teams, especially Donna Muzny and Harshavardhan Doddapaneni, for their work on macaque diversity sequencing, and the Baylor Human Genome Sequencing Center, the Texas Advanced Computing Center, and Rice University for allowing us to use their computational resources for mapping and variant calling. The cell line MMU1 used for Strand-seq and FISH analyses was obtained from the Department of Comparative Genetics and Refinement, Biomedical Primate Research Centre (BPRC), Netherlands (courtesy of Ronald E. Bontrop). Alex Pollen (UCSF) and Sam Peterson (OHSU) for preparation of mRNA used in the generation of Iso-Seq datasets; and specifically, the UC Davis Primate Research Center for the rhesus macaque fetal brain material and specific tissues from developmentally staged samples used for Iso-Seq data production.
Funding: This work was supported, in part, by the National Institutes of Health (NIH) grants: HG002385 and 1U24HG009081 to E.E.E.; U01HG010961, U41HG010972, R01HG010485, 2U41HG007234, U01HL137183, 5U54HG007990 and 5T32HG008345-04 to B.P.; R01MH081884, R01MH046729 and P50MH100031 to N.H.K.; R01GM59290 to M.A.B.; by a subagreement from European Molecular Biology Laboratory with funds provided by agreement number 2U41HG007234-05 from National Institute of Health, NHGRI. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH, NHGRI, or European Molecular Biology Laboratory. Rhesus WGS from primate research centers was supported by NIH grant 5R24OD011173 to J.R.; R24OD021324 grant that supported sequencing of the ONPRC cohort to B.F.; P51-OD011106 grant that supported sequencing of the WNPRC cohort as well as support for D.O., J.R., N.K., D.A., and J.K.; CNPRC base grant (P51OD011107) and BBA grant OD010962 to J.P.C.; YNPRC Pilot Research Project Program to Z.A.K-B. and its base grant P51OD011132; TNPRC grants P51OD011104, U42OD024282 and U42OD010568; 1R01HG010329 to S.R.S.; R01HG002939 to M.A.B. provided support for repeat analyses. E.E.E. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: The authors declare no competing financial interests.
Data availability: All data is available in the main text or the supplementary materials. The Mmul_10 genome assembly is available in the NCBI assembly archive under accession number GCF_003339765.1.
References and notes
- 1.Bailey JA, Eichler EE, Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet 7, 552–564 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Rhesus Macaque Genome S et al. , Evolutionary and biomedical insights from the rhesus macaque genome. Science 316, 222–234 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Xue C et al. , The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences. Genome Res 26, 1651–1662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bimber BN et al. , Whole genome sequencing predicts novel human disease models in rhesus macaques. Genomics 109, 214–220 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kronenberg ZN et al. , High-resolution comparative analysis of great ape genomes. Science 360, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogers J, Gibbs RA, Comparative primate genomics: emerging patterns of genome content and dynamics. Nat Rev Genet 15, 347–359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Rompay KKA, Tackling HIV and AIDS: contributions by non-human primate models. Lab Anim (NY) 46, 259–270 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Feldmann H, Feldmann F, Marzi A, Ebola: Lessons on Vaccine Development. Annu Rev Microbiol 72, 423–446 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y et al. , Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature 570, 326–331 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Daza-Vamenta R, Glusman G, Rowen L, Guthrie B, Geraghty DE, Genetic divergence of the rhesus macaque major histocompatibility complex. Genome Res 14, 1501–1515 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dray BK et al. , Mismatch repair gene mutations lead to lynch syndrome colorectal cancer in rhesus macaques. Genes Cancer 9, 142–152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao BY, Zhang J, Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc Natl Acad Sci U S A 105, 6987–6992 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seok J et al. , Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 110, 3507–3512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson SM et al. , Bardet-Biedl Syndrome in rhesus macaques: A nonhuman primate model of retinitis pigmentosa. Exp Eye Res 189, 107825 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moshiri A et al. , A nonhuman primate model of inherited retinal disease. J Clin Invest 129, 863–874 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers J et al. , CRHR1 genotypes, neural circuits and the diathesis for anxiety and depression. Mol Psychiatry 18, 700–707 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abbott DH, Rogers J, Dumesic DA, Levine JE, Naturally Occurring and Experimentally Induced Rhesus Macaque Models for Polycystic Ovary Syndrome: Translational Gateways to Clinical Application. Med Sci (Basel) 7, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warren WC, Sequence diversity analyses of an improved rhesus macaque genome enhances its biomedical utility. Supplemental Materials, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes JF et al. , Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature 483, 82–86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He Y et al. , Long-read assembly of the Chinese rhesus macaque genome and identification of ape-specific structural variants. Nat Commun 10, 4233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook JG et al. , Single haplotype analysis demonstrates rapid evolution of the killer immunoglobulin-like receptor (KIR) loci in primates. Genome Res 15, 25–35 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Catacchio CR et al. , Inversion variants in human and primate genomes. Genome Res 28, 910–920 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seppey M, Manni M, Zdobnov EM, BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol Biol 1962, 227–245 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Fiddes IT et al. , Comparative Annotation Toolkit (CAT)-simultaneous clade and personal genome annotation. Genome Res 28, 1029–1038 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finn RD et al. , The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44, D279–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vollger MR et al. , Long-read sequence and assembly of segmental duplications. Nat Methods 16, 88–94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaisson MJ, Mukherjee S, Kannan S, Eichler EE, Resolving multicopy duplications de novo using polyploid phasing. Res Comput Mol Biol 10229, 117–133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandes JD et al. , The UCSC repeat browser allows discovery and visualization of evolutionary conflict across repeat families. Mob DNA 11, 13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imbeault M, Helleboid PY, Trono D, KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Bergstrom A et al. , Insights into human genetic variation and population history from 929 diverse genomes. Science 367, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Widdig A et al. , Low incidence of inbreeding in a long-lived primate population isolated for 75 years. Behav Ecol Sociobiol 71, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coe BP et al. , Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet 51, 106–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satterstrom FK et al. , Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584 e523 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Church DM et al. , Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol 7, e1000112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.C. International Human Genome Sequencing, Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE, Segmental duplications: organization and impact within the current human genome project assembly. Genome Res 11, 1005–1017 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailey JA et al. , Recent segmental duplications in the human genome. Science 297, 1003–1007 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Fernandes JD, Haeussler Maximilian, Armstrong Joel, Tigyi Kristof, Gu Joshua, Filippi Natalie, Pierce Jessica, Thisner Tiffany, Angulo Paola, Katzman Sol, Paten Benedict, Haussler David, Salama Sofie R, KRAB Zinc Finger Proteins coordinate across evolutionary time scales to battle retroelements. bioRxiv, (2018). [Google Scholar]
- 39.Browning BL, Browning SR, A Unified Approach to Genotype Imputation and Haplotype-Phase Inference for Large Data Sets of Trios and Unrelated Individuals. The American Journal of Human Genetics 84, 210–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chin J, FALCON: experimental PacBio diploid assembler. https://github.com/PacificBiosciences/falcon/tree/v0.1.3 (2014).
- 41.Lam ET et al. , Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotechnol 30, 771–776 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deschamps S et al. , A chromosome-scale assembly of the sorghum genome using nanopore sequencing and optical mapping. Nat Commun 9, 4844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Putnam NH et al. , Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res 26, 342–350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lazar NH et al. , Epigenetic maintenance of topological domains in the highly rearranged gibbon genome. Genome Res 28, 983–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaharia M, Bolosky WJ, Curtis K, Fox A, Patterson D, Shenker S, Stoica I, Karp RM, Sittler T, Faster and More Accurate Sequence Alignment with SNAP. arXiv 5572v1, (2011). [Google Scholar]
- 46.Marcais G et al. , MUMmer4: A fast and versatile genome alignment system. PLoS Comput Biol 14, e1005944 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Falconer E et al. , DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat Methods 9, 1107–1112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marchetto MCN et al. , Differential L1 regulation in pluripotent stem cells of humans and apes. Nature 503, 525–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dougherty ML et al. , Transcriptional fates of human-specific segmental duplications in brain. Genome Res 28, 1566–1576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Armstrong J, Fiddes IT, Diekhans M, Paten B, Whole-Genome Alignment and Comparative Annotation. Annu Rev Anim Biosci 7, 41–64 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stanke M, Diekhans M, Baertsch R, Haussler D, Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Stanke M, Steinkamp R, Waack S, Morgenstern B, AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res 32, W309–312 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Konig S, Romoth LW, Gerischer L, Stanke M, Simultaneous gene finding in multiple genomes. Bioinformatics 32, 3388–3395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pruitt KD, Tatusova T, Brown GR, Maglott DR, NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 40, D130–135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zerbino DR et al. , Ensembl 2018. Nucleic Acids Res 46, D754–D761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koren S et al. , Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pertea M et al. , CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol 19, 208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wheeler TJ et al. , Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res 41, D70–82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kent WJ, BLAT--the BLAST-like alignment tool. Genome Res 12, 656–664 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Batzer MA et al. , Standardized nomenclature for Alu repeats. J Mol Evol 42, 3–6 (1996). [DOI] [PubMed] [Google Scholar]
- 62.H. S Bastian M, Jacomy M, paper presented at the AAAI Conference on Weblogs and Social Media, 2009. [Google Scholar]
- 63.Jurka J et al. , Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110, 462–467 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Han K et al. , Mobile DNA in Old World monkeys: a glimpse through the rhesus macaque genome. Science 316, 238–240 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Fernandes JD, Armando Zamudio-Hurtado W Kent James, Haussler David, Salama Sofie R., Haeussler Maximilian, The UCSC Repeat Browser allows discovery and visualization of evolutionary conflict across repeat families. bioRxiv November 27, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Durbin R, Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McLaren W et al. , The Ensembl Variant Effect Predictor. Genome Biol 17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang C, Dong SS, Xu JY, He WM, Yang TL, PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Lek M et al. , Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.P. Biosciences (https://github.com/PacificBiosciences/pbsv, 2018).
- 71.Sedlazeck FJ et al. , Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chapman JA et al. , Meraculous: de novo genome assembly with short paired-end reads. PLoS One 6, e23501 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edgar RC, MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ventura M et al. , Evolutionary formation of new centromeres in macaque. Science 316, 243–246 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Sanders AD, Falconer E, Hills M, Spierings DCJ, Lansdorp PM, Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat Protoc 12, 1151–1176 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Karl JA et al. , Major histocompatibility complex class I haplotype diversity in Chinese rhesus macaques. G3 (Bethesda) 3, 1195–1201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shortreed CG et al. , Characterization of 100 extended major histocompatibility complex haplotypes in Indonesian cynomolgus macaques. Immunogenetics, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caskey JR et al. , MHC genotyping from rhesus macaque exome sequences. Immunogenetics 71, 531–544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bruijnesteijn J et al. , Nomenclature report for killer-cell immunoglobulin-like receptors (KIR) in macaque species: new genes/alleles, renaming recombinant entities and IPD-NHKIR updates. Immunogenetics 72, 37–47 (2020). [DOI] [PubMed] [Google Scholar]
- 80.Iskow RC et al. , Regulatory element copy number differences shape primate expression profiles. Proc Natl Acad Sci U S A 109, 12656–12661 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.