

Initially, the Sigma-1 receptor (S1R) was incorrectly categorized as one of the opioid receptors. But it has since become clear that S1R is a non-opioid non-phencyclidine receptor with molecular chaperone activity. S1R is a 223 amino-acid long protein that shares no clear homology with any other known mammalian proteins, its closest homolog being the fungal ERG2 sterol isomerase. Even its pharmacological counterpart, the Sigma-2 receptor that was only recently cloned, is genetically distinct. S1R is more likely considered as an atypical ligand-modulated chaperone protein. S1R is widely expressed in many organs including brain where its function has mainly been explored. In the brain, S1R is found in neurons as well in astrocytes, microglia or oligodendrocytes. While S1R regulates important glial functions like inflammatory response or supply of neurotrophic factors, this perspective article mostly focuses on the various roles of S1R in neurons. At cellular level, S1R resides mainly as a transmembrane protein in the endoplasmic reticulum (ER) and more particularly in the vicinity of mitochondria. At this subdomain, called mitochondrial associated ER-membranes (MAM), S1R regulates calcium exchange between the two organelles through inositol triphosphate receptor type 3 (IP3R type 3). S1R prevents proteosomal degradation of IP3R but also activates its opening for calcium efflux by dissociating IP3R from the scaffolding protein ankyrin G (Su et al., 2016). As a consequence, increased calcium influx in mitochondria boosts production of the nicotinamide adenine dinucleotide cofactor, stimulates the respiratory complex 1 activity and hence increases ATP biosynthesis (Figure 1). MAM are particularly enriched in cholesterol and supply mitochondria with this precursor for steroidogenesis. S1R can promote cholesterol transfer by interaction with steroidogenic acute regulatory protein and voltage dependant anion channel, both facilitating escort of ER-derived cholesterol across the mitochondrial outer membrane (Su et al., 2016) (Figure 1). Moreover, in normal condition, cognate co-chaperone GRP78/Bip is associated to S1R and the ER stress sensor, inositol requiring enzyme 1 (IRE1), keeping them inactive. But when facing stress condition, GRP78/Bip dissociates from S1R and IRE1. Then S1R can stabilize the proper folding of IRE1 and promotes long lasting dimerization and trans-autophosphorylation of IRE1 (Su et al., 2016). This leads to activation of its ribonuclease function and the subsequent expression of the spliced/active transcription factor X-box binding protein 1, which regulates nuclear production of antioxidant proteins and chaperone proteins. On the other hand, over-expression of S1R or binding of S1R by exogenous or putative endogenous agonists can promote its interaction with ion channels, receptors and kinases, and then finely tunes neuronal excitability and plasticity (Su et al., 2016). Among endogenous agonists are neuroactive steroids such as sulfate esters of pregnenolone or dehydroepiandrosterone, the hallucinogenic N,N-dimethyltryptamine and choline. Nevertheless, it is unclear whether endogenous agonists have a true physiological role through S1R in vivo. Finally, upon stimulation by cocaine, S1R protein can translocate to the nuclear envelope, where it binds Emerin to regulate gene transcription by recruiting chromatin-remodelling factors (Su et al., 2016).
Figure 1.
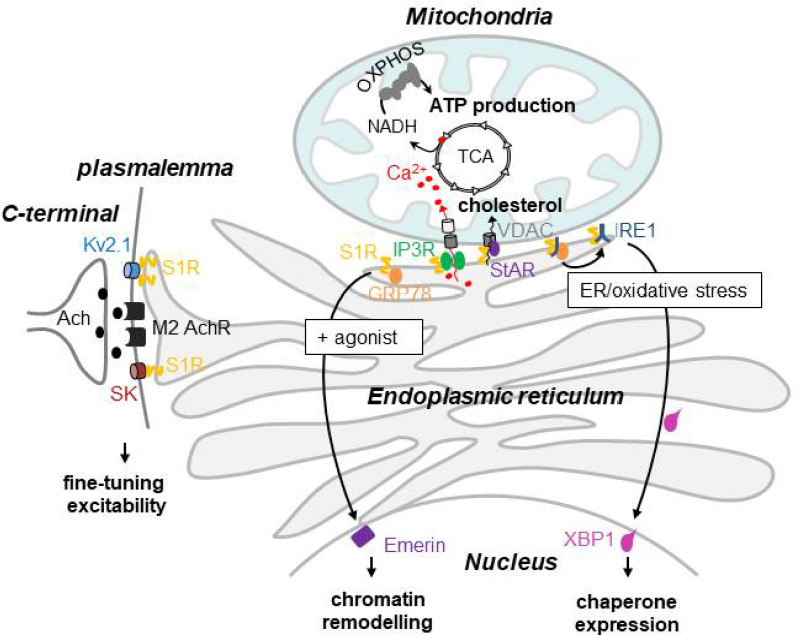
Putative neuroprotective functions of Sigma-1 receptor in motor neurons.
In motor neurons Sigma-1 receptor (S1R) is enriched at the vicinity of presynaptic cholinergic C-terminals and interacts with potassium channels, Kv2.1 and SK, to fine-tune neuronal excitability. S1R is also a main resident of mitochondrial associated endoplasmic reticulum (ER) membranes where it modulates calcium transfer from ER to mitochondria through inositol triphosphate receptor (IP3R). As a consequence, this boosts nicotinamide adenine dinucleotide cofactor (NADH) levels through the tricarboxylic acid cycle (TCA), which is indispensable for ATP production by oxidative phosphorylation (OXPHOS). S1R also facilitates cholesterol escort from ER to mitochondria by interaction with steroidogenic acute regulatory protein (StAR) and voltage dependent anion channel (VDAC). During a cellular stress or when S1R is activated, S1R dissociates from 78 kDa glucose-regulated protein (GRP78) and stabilizes inositol requiring enzyme 1 (IRE1) dimers. Dimerization of IRE1 leads to the expression of active spliced X-box binding protein 1 (XBP1), a transcription factor that induces ER stress response. When activated, S1R can also translocate to the nucleus on Emerin and regulates transcriptional activity.
S1R dysregulation is involved in many diseases such as drug addiction, schizophrenia, depression and neurodegenerative diseases and ligands activating S1R are anti-amnesic, anti-depressant and neuroprotective (Su et al., 2016). In the last decade, a direct link has emerged between S1R and motor neuron diseases. Recessive mutations in S1R are a cause of distal hereditary motor neuropathy (dHMN), a disorder characterized by distal amyotrophy and weakness of lower limbs. To date, three truncating mutations (p.Gly31_Ala50del; p.Gln80*; p.Asp188Profs*69) and three point mutations (p.Glu138Gln; p.Glu150Lys; p. Leu65Gln) were identified in several families with dHMN. Juvenile cases of amyotrophic lateral sclerosis (ALS) were also linked to a missense mutation (p.Glu102Gln) and a frameshift mutation (p.Leu95Profs*29) in S1R. ALS patients show muscle weakness, progressive paralysis and spasticity due to motor neuron degeneration. The majority of ALS patients are sporadic while only 10% are due to ALS gene mutation. So far, more than 20 genes have been identified as the cause of ALS pathology.
An important question that has been raised is why S1R dysfunction is particularly detrimental to motoneurons. Despite being expressed throughout the brain, it is worth mentioning that the highest levels of S1R are found in spinal and brainstem motor neurons (Mavlyutov et al., 2015). In these neuronal cells, S1R presents a particular distribution mainly in the ER at the postsynaptic subsurface cisternae of cholinergic C-terminals (Mavlyutov et al., 2015). Presynaptic C-terminal boutons originate from spinal cholinergic interneurons that increase motoneuron activity for organized pattern of motor behavior like locomotion. The C-terminal postsynaptic membrane is particularly enriched in M2 muscarinic acetylcholine and voltage-gated potassium Kv2.1 channel and calcium-activated potassium SK channel. Activation of Kv2.1 and SK channels hyperpolarize motor neuron and thereby modulate their excitability. Thus, it has been proposed that S1R might protect motoneurons by activating those channels and thereby reducing excitability. Consistent to this, excitability of motor neuron is higher in mice knocked out for S1R (Mavlyutov et al., 2015). While molecular mechanisms by which S1R activates Kv2.1 and/or SK channels are still unknown, physical proximity of postsynaptic plasmalemma and ER cisternae at C-terminals makes possible direct interaction between S1R and its targets. S1R might also indirectly modulate activities of Kv2.1 and SK channels through calcium-dependent pathways. An intriguing observation is that indole(ethyl)amine N-methyltransferase, the enzyme that converts tryptophan into the S1R agonist N,N-dimethyltryptamine, co-localizes with S1R at C-terminals. Further studies are needed to understand whether this co-localization is of importance. Prause et al. (2013) studied the distribution of S1R in alpha-motor neurons of some sporadic and familial ALS patients. In the ALS context, S1R is found abnormally accumulated in enlarged C-terminals and ER structures. Aberrant accumulation was also observed in the spinal cord of ALS transgenic mice expressing mutant superoxide dismutase 1 (SOD1G93A) or fibroblasts from familial ALS patient with mutation in vesicle-associated membrane protein-associated protein B. Altogether these observations lead to propose that mislocalization of S1R from C-terminal postsynaptic membrane may contribute to motor neuron dysfunction. Unfortunately, to our knowledge the impact of S1R mutations on its subcellular localisation at C-terminals has not yet been investigated. Changes in excitability may not be the only pathomechanism. In cell cultures, S1R that carries dHMN or ALS mutations is prone to mislocalize out of MAM and to form cytoplasmic clusters (Tagashira et al., 2014; Gregianin et al., 2016; Watanabe et al., 2016). S1R function at MAM may thus be strongly altered, deregulating mitochondrial calcium and energy metabolism. Accordingly, while cells transfected with wild-type S1R show enhanced mitochondrial calcium mobilization and ATP production, they are found decreased when S1R carries ALS or dHMN mutations (Tagashira et al., 2014; Gregianin et al., 2016; Watanabe et al., 2016). Only recently, a study from our group demonstrated in vivo, using Drosophila genetics, that the expression of human ALS S1R (e.g. S1RGlu102Gln) leads to locomotor decline associated to ATP depletion and mitochondrial fragmentation, two hallmarks of mitochondrial dysfunction (Couly et al., 2020). Motor neurons may be particularly vulnerable to mitochondrial ATP depletion as they present long axons and require high energy levels for action potential and synaptic transmission. In the future, it would be of interest to address the impact in vivo of the other S1R mutations on mitochondrial functioning. Another consequence of S1R dysfunction is the deregulation of ER stress response to unfolded protein accumulation. Both dHMN and ALS mutations enhance ER stress markers such as GRP78/Bip (Li et al., 2015; Dreser et al., 2016) but also exacerbate vulnerability of cells to ER stressors like tunicamycin or thapsigagin (Tagashira et al., 2014; Gregianin et al., 2016). Eventually, prolonged ER stress may result in a vicious cycle with accumulation of RNA-binding proteins like Tar-DNA binding protein (TDP43) or Fused in sarcoma, both of them also directly involved in ALS pathogenesis (Tagashira et al., 2014; Dreser et al., 2016). Finally, a possible alteration at the nuclear levels remains to be explored since a study reported an accumulation of Emerin with mutant clusters of ALS mutant S1RGlu102Gln (Dreser et al., 2016).
Considering the importance of S1R in motor neuron functioning and integrity, it is tempting to propose S1R as a therapeutic target. Especially, as a large number of exogenous S1R agonists or modulators are available. To date, studies on ALS were focused on murine models expressing mutant SOD1. Administration of the S1R agonist PRE-084 ameliorated motor neuron survival and motor performance, and extended survival of SOD1G93A mice by more than 15% (Mancuso et al., 2012). Another agonist, SA4503, extended the lifespan but failed to prevent locomotor decline onset of diseased SOD1G93A mice (Ono et al., 2014). The selective S1R agonist Pridopidine reduced mutant SOD1 aggregation and preserved motor neuron integrity (Ionescu et al., 2019). In all these studies, beneficial effects of S1R agonists were associated to activation of signalling pathways including protein kinase C, AKT or ERK (Mancuso et al., 2012; Ono et al., 2014; Ionescu et al., 2019). Unfortunately, the impact on MAM and mitochondria function was not investigated, despite the importance of S1R in ER-mitochondria crosstalk. Another concern is that mutations in SOD1 are linked to only 15–20% of familial cases (< 2% of all ALS cases). Observations made on models expressing mutant SOD1 do not allow extrapolating to the rest of ALS conditions. Moreover, although S1R agonists developed to date in clinical trials are rather devoid of side effects, the drug activity at other receptors and pathways must be addressed. It is thus necessary to provide a more direct evidence of the therapeutic value of S1R. In our recent study, we provided the proof of concept that S1R may serve as a therapeutic target in ALS (Couly et al., 2020). Drosophila models expressing TDP43 were chosen since TDP43 deregulation and mislocalization are involved in familial cases but also in almost all sporadic patients. Overexpressing human S1R ameliorates locomotion of flies expressing TDP43, at least in part, by increasing mitochondrial ATP production. Moreover, S1R overexpressing flies exhibit enhanced resistance to oxidative stress. At present, further analysis should be pursued to fully dissect the neuroprotective properties of S1R in vivo and especially in vertebrate models.
In conclusion, progress has been made in the last decade to understand the role of S1R in motor diseases. However, most of the studies aiming at understanding S1R functions were rarely performed on motor neurons. More attention must now be put on MAM, mitochondrial metabolism and ER stress but also on S1R activity in the nucleus. More than ever, the therapeutic value of S1R that can be targeted using genetic and pharmacological approaches must be generalized on other models of ALS and dHMN.
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Couly S, Khalil B, Viguier V, Roussel J, Maurice T, Liévens JC. Sigma-1 receptor is a key genetic modulator in amyotrophic lateral sclerosis. Hum Mol Genet. 2020;13:529–540. doi: 10.1093/hmg/ddz267. [DOI] [PubMed] [Google Scholar]
- 2.Dreser A, Vollrath JT, Sechi A, Johann S, Roos A, Yamoah A, Katona I, Bohlega S, Wiemuth D, Tian Y, Schmidt A, Vervoorts J, Dohmen M, Beyer C, Anink J, Aronica E, Troost D, Weis J, Goswami A. The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 2017;24:1655–1671. doi: 10.1038/cdd.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gregianin E, Pallafacchina G, Zanin S, Crippa V, Rusmini P, Poletti A, Fang M, Li Z, Diano L, Petrucci A, Lispi L, Cavallaro T, Fabrizi GM, Muglia M, Boaretto F, Vettori A, Rizzuto R, Mostacciuolo ML, Vazza G. Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum Mol Genet. 2016;25:3741–3753. doi: 10.1093/hmg/ddw220. [DOI] [PubMed] [Google Scholar]
- 4.Ionescu A, Gradus T, Altman T, Maimon R, Saraf Avraham N, Geva M, Hayden M, Perlson E. Targeting the sigma-1 receptor via pridopidine ameliorates central features of ALS pathology in a SOD1(G93A) model. Cell Death Dis. 2019;10:210. doi: 10.1038/s41419-019-1451-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X, Hu Z, Liu L, Xie Y, Zhan Y, Zi X, Wang J, Wu L, Xia K, Tang B, Zhang R. A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology. 2015;84:2430–2437. doi: 10.1212/WNL.0000000000001680. [DOI] [PubMed] [Google Scholar]
- 6.Mancuso R, Oliván S, Rando A, Casas C, Osta R, Navarro X. Sigma-1R agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics. 2012;9:814–826. doi: 10.1007/s13311-012-0140-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mavlyutov TA, Guo LW, Epstein ML, Ruoho AE. Role of the sigma-1 receptor in amyotrophic lateral sclerosis (ALS) J Pharmacol Sci. 2015;127:10–16. doi: 10.1016/j.jphs.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ono Y, Tanaka H, Takata M, Nagahara Y, Noda Y, Tsuruma K, Shimazawa M, Hozumi I, Hara H. SA4503, a sigma-1 receptor agonist, suppresses motor neuron damage in in vitro and in vivo amyotrophic lateral sclerosis models. Neurosci Lett. 2014;559:174–178. doi: 10.1016/j.neulet.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Prause J, Goswami A, Katona I, Roos A, Schnizler M, Bushuven E, Dreier A, Buchkremer S, Johann S, Beyer C, Deschauer M, Troost D, Weis J. Altered localization, abnormal modification and loss of function of Sigma receptor-1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:1581–1600. doi: 10.1093/hmg/ddt008. [DOI] [PubMed] [Google Scholar]
- 10.Su TP, Su TC, Nakamura Y, Tsai SY. The sigma-1 receptor as a pluripotent modulator in living systems. Trends Pharmacol Sci. 2016;37:262–278. doi: 10.1016/j.tips.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tagashira H, Bhuiyan MS, Shioda N, Fukunaga K. Fluvoxamine rescues mitochondrial Ca2+ transport and ATP production through sigma(1)-receptor in hypertrophic cardiomyocytes. Life Sci. 2014;95:89–100. doi: 10.1016/j.lfs.2013.12.019. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe S, Ilieva H, Tamada H, Nomura H, Komine O, Endo F, Jin S, Mancias P, Kiyama H, Yamanaka K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol Med. 2016;8:1421–1437. doi: 10.15252/emmm.201606403. [DOI] [PMC free article] [PubMed] [Google Scholar]