

Abstract
Bcl-xL is a pro-survival protein of the Bcl2 family found in the mitochondrial membrane. Bcl-xL supports growth, development, and maturation of neurons, and it also prevents neuronal death during neurotoxic stimulation. This article reviews the mechanisms and upstream signaling that regulate the activity and abundance of Bcl-xL. Our team and others have reported that oxidative stress is a key regulator of intracellular Bcl-xL balance in neurons. Oxidative stress regulates synthesis, degradation, and activity of Bcl-xL and therefore neuronal function. During apoptosis, pro-apoptotic Bcl2 proteins such as Bax and Bak translocate to and oligomerize in the mitochondrial membrane. Formation of oligomers causes release of cytochrome c and activation of caspases that lead to neuronal death. Bcl-xL binds directly to pro-apoptotic Bcl2 proteins to block apoptotic signaling. Although anti-apoptotic roles of Bcl-xL have been well documented, an increasing number of studies in recent decades show that protein binding partners of Bcl-xL are not limited to Bcl2 proteins. Bcl-xL forms a complex with F1Fo ATP synthase, DJ-1, DRP1, IP3R, and the ryanodine receptor. These proteins support physiological processes in neurons such as growth and development and prevent neuronal damage by regulating mitochondrial ATP production, synapse formation, synaptic vesicle recycling, neurotransmission, and calcium signaling. However, under conditions of oxidative stress, Bcl-xL undergoes proteolytic cleavage thus lowering the abundance of functional Bcl-xL in neurons. Additionally, oxidative stress alters formation of Bcl-xL-mediated multiprotein complexes by regulating post-translational phosphorylation. Finally, oxidative stress regulates transcription factors that target the Bcl-x gene and alter accessibility of microRNA to mRNA influencing mRNA levels of Bcl-xL. In this review, we discussed how Bcl-xL supports the normal physiology of neurons, and how oxidative stress contributes to pathology by manipulating the dynamics of Bcl-xL production, degradation, and activity.
Key Words: ATP, Bcl-xL, F1Fo ATP synthase, mitochondria, neuroprotection, oxidative stress
Bcl-xL and Neuroprotection
Bcl-xL is an anti-apoptotic protein found on the mitochondrial membrane. Bcl-xL prevents mitochondrially-dependent death processes by blocking oligomerization of pro-death proteins including Bax and Bak. Although Bcl-xL is present in a broad range of cell types, the role of Bcl-xL in brain cells has been an emphasis of research in recent decades. Bcl-xL is necessary for embryonic development of the brain (Chen et al., 2011; Fogarty et al., 2019). Depletion of Bcl-xL leads to apoptosis of postmitotic neurons and impairs developmental neurogenesis (Fogarty et al., 2019). Conditional knock out of Bcl-xL causes loss of the upper layer of cortical neurons and the CA1–CA3 regions of hippocampal neurons, which eventually leads to neurobehavioral abnormalities (Nakamura et al., 2016). Application of Bcl-xL shRNA in vitro impairs the extension and arborization of primary hippocampal neurites (Park et al., 2015) which may hinder formation of neuronal interconnections. We have shown that impaired neurite outgrowth in neurons lacking Bcl-xL was reversed by depletion of death receptor 6 (DR6) (Park et al., 2015). DR6 is a tumor necrosis factor receptor widely expressed in developing neurons. Binding between DR6 and its ligand, the N-terminus of amyloid precursor protein, activates caspases causing axonal degeneration (Nikolaev et al., 2009). In addition to Bcl-xL, other Bcl2 proteins such as Puma and Bax are required during axon degeneration (Cusack et al., 2013; Geden and Deshmukh, 2016; Simon et al., 2016). Bcl-xL is reported to enhance function of the F1Fo ATP synthase and improve neuronal ATP production (Alavian et al., 2011; Chen et al., 2011), thus Bcl-xL-mediated energy metabolism may favor metabolically demanding processes during neuronal development and growth. Overexpression of Bcl-xL increases recruitment of mitochondria in presynaptic neurons, induces the formation of synapses, and enhances the frequency and amplitude of synaptic currents to promote synaptic function (Li et al., 2008).
In addition to its role in the development and growth of neurons, Bcl-xL also exhibits protective properties against brain-associated diseases, particularly cerebral ischemia; its expression is often found to be increased during stroke and cardiac arrest (Wu et al., 2003). Levels of protection vary by experimental design. However, strategies to retain Bcl-xL have shown a range of 60–90% neuroprotection in both in vitro and in vivo models. Bcl-xL attenuates ischemia-mediated brain injury (Wu et al., 2003). Inhibiting the proteolytic cleavage of Bcl-xL prevents ischemia-induced neuronal loss in CA1 hippocampal neurons in rodents (Miyawaki et al., 2008; Ofengeim et al., 2012). In vivo delivery of a Bcl-xL fusion protein protects the brain in mice against focal ischemic injury (Cao et al., 2002). Consistent results showing Bcl-xL protection were also found for in vitro models of excitotoxicity (Park et al., 2017) and oxidative stress (Park et al., 2019); these models are known to mimic the effects of cerebral ischemia. In this review, PubMed was searched to retrieve the articles published between 2000–2020.
Bcl-xL functions via formation of a multiprotein complex with its binding partners (Figure 1). Besides the well-known pro-apoptotic Bcl2 family proteins, Bcl-xL binds to various proteins which regulate neuronal function. Lys 87 in the BH3 domain of Bcl-xL interacts with ryanodine receptor 3, a calcium channel found in the brain, and interaction between Bcl-xL and ryanodine receptor 3 inhibits calcium release in primary hippocampal neurons (Vervliet et al., 2015). Bcl-xL shows high affinity to the C-terminal site of inositol 1, 4, 5-triphosphate receptor (IP3R) in the endoplasmic reticulum near the calcium channel pore (Monaco et al., 2012). Since calcium is an important second messenger that triggers both neurotransmission and neurotoxicity, Bcl-xL may act as a switch to control neuronal calcium signaling. Bcl-xL binds directly to the β-subunit of F1Fo ATP synthase and enhances efficiency of ATP synthesis (Alavian et al., 2011) and prevents opening of the mitochondrial permeability transition pore in primary neurons (Alavian et al., 2014). Bcl-xL also interacts with DJ-1, a protein encoded by the PARK7 gene (Chen et al., 2019). Our recent study showed that DJ-1 undergoes protein-protein interaction with F1Fo ATP synthase to support neurite outgrowth of dopaminergic neurons (Chen et al., 2019). Bcl-xL may interact with various protein binding partners of F1Fo ATP synthaze to govern neuronal energy metabolism. The BH2 domain of Bcl-xL is required to bind Drp1, and a Bcl-xL-Drp1 complex is found bound to clathrin-coated pits supporting the maintenance of endocytic vesicles responsible for re-uptake of neurotransmitters in neurons (Li et al., 2013).
Figure 1.
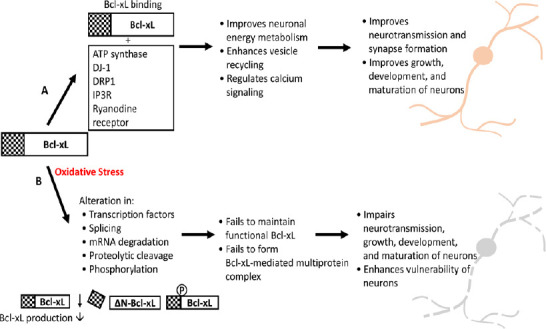
Regulation of Bcl-xL in neurons during normal versus oxidative stress.
(A) Bcl-xL binds to non-Bcl2 family proteins such as F1Fo ATP synthase, DJ-1, DRP1, IP3R, and the ryanodine receptor. Formation of a multiprotein complex improves neurotransmission, synapse formation, and normal neuronal growth and development via enhancing mitochondrial energy metabolism, synaptic vesicle recycling, and intracellular cell signaling. (B) During oxidative stress, levels of Bcl-xL mRNA change via regulation of transcription factors, splicing, and microRNA-mediated mRNA degradation. Oxidative stress further controls the abundance of Bcl-xL protein levels by influencing post-translational modification like phosphorylation and proteolytic cleavage. Together, oxidative stress alters maintenance of functional Bcl-xL in neurons impairing formation of Bcl-xL-mediated protein complexes. This may interfere with normal neuronal activity or enhance vulnerability of neurons challenged by oxidative stress.
Oxidative Stress and Cleavage of Bcl-xL
Given Bcl-xL’s significant role in protecting the brain during physiological and pathological processes, it is important to understand the mechanisms of upstream signaling that regulate activity and abundance of Bcl-xL. Recently, we reported that oxidative stress is a key regulator of intracellular Bcl-xL balance in neurons. Primary hippocampal neurons challenged with hydrogen peroxide have significantly increased post-translational cleavage of Bcl-xL (Park et al., 2019). Hydrogen peroxide increases both the activity and abundance of caspase 3, the key protease which cleaves full length Bcl-xL to its N-terminal truncated form ΔN-Bcl-xL (Park et al., 2019). Accumulation of ΔN-Bcl-xL is known to cause mitochondrial dysfunction including abnormally increased mitochondrial ion channel activity and conductance, mitochondrial membrane potential loss, and impaired ATP production, all of which contribute to neuronal death (Jonas et al., 2004; Ofengeim et al., 2012; Park et al., 2017; Park and Jonas, 2017).
Since mitochondria consume oxygen for oxidative phosphorylation, mitochondria play a major role in generating reactive oxygen species (ROS) (Murphy, 2009). Studies show that ROS cause an increase in the protein level and activity of caspase 3 (Park et al., 2019). In addition, caspase 3 is reported to translocate to mitochondria, where Bcl-xL is localized (Chandra and Tang, 2003). Therefore, ROS are an important upstream control of Bcl-xL and ΔN-Bcl-xL balance due to their regulation of caspase 3, and application of antioxidants may be effective in manipulating Bcl-xL and ΔN-Bcl-xL profiles in neurons (Park et al., 2019). Although caspase 3 has been frequently studied for its role to cleave Bcl-xL during brain disease, other proteases such as calpains may also contribute to Bcl-xL cleavage during events that raise intracellular calcium levels (Gil-Parrado et al., 2002; Liu et al., 2009). Application of calpain inhibitors rescues neurons against oxidative stress-mediated death (See and Loeffler, 2001; D’Orsi et al., 2012). Additionally, Dho et al. (2013) reported that asparagine residues in Bcl-xL undergo deamination, and inhibition of deamination at Asn 52 and 66 prevents calpain-dependent Bcl-xL degradation. Since Asp 61 and 76 are a target of caspase 3 (Clem et al., 1998; Basanez et al., 2001; Ofengeim et al., 2012), the calpain-mediated cleavage product of Bcl-xL may exhibit ΔN-Bcl-xL-like neurotoxic effects. It would be of interest to investigate how other Bcl-xL cleavage products influence neuronal survival under ROS challenge in the future.
Oxidative Stress and Phosphorylation of Bcl-xL
The activity of Bcl-xL is influenced by phosphorylation and dephosphorylation. Amino acid residues of Bcl-xL including Ser 49, Ser 62, Thr 69, and Ser 73 undergo phosphorylation (Upreti et al., 2008; Arena et al., 2013; Baruah et al., 2016; Megyesi et al., 2016). In particular, Ser 62 is subject to various kinases and has been proposed as an important site for regulation of neuronal function. PTEN-induced kinase 1 (PINK1), a mitochondrial serine/threonine-protein kinase, directly binds and phosphorylates Bcl-xL (Arena et al., 2013). PINK1-dependent phosphorylation of Ser 62 causes Bcl-xL to be resistant to cleavage at Asp 61 and thus inhibits formation of ΔN-Bcl-xL (Arena et al., 2013). Studies have shown that ROS are regulators of PINK1. CCCP-induced oxidative stress causes PINK1 dependent mitophagy (Xiao et al., 2017). Mutations in PINK1 increase the accumulation of ROS, alter mitochondrial morphology, and trigger mitochondrially-dependent apoptosis in dopaminergic SH-SY5Y cells in vitro (Yuan et al., 2010). In vivo models using PINK1 knock out mice show increased lipid peroxidation and depletion of antioxidant pools and associated mitochondrial dysfunction (Billia et al., 2011). The Ser 62 residue on Bcl-xL also has a role in cyclin-dependent kinase-1 (Cdk1)-mediated phosphorylation during oxidative stress (Veas-Perez de Tudela et al., 2015). Phosphorylation of Bcl-xL at Ser 62 dissociates Bcl-xL from the Bcl-xL-F1Fo ATP synthase complex, decreasing activity of the F1Fo ATP synthase (Veas-Perez de Tudela et al., 2015). Assembly of the F1Fo ATP synthase influences mitochondrial ATP production (Alavian et al., 2011), thus cyclin-dependent kinase-1-mediated phosphorylation of Bcl-xL may contribute to the efficiency of the electron transport chain controlling mitochondrial ROS homeostasis. In contrast, depletion of cJun N-terminal kinase shows no effect on Bcl-xL phosphorylation in cerebellar granule neurons (Xu et al., 2011). Thus, identification of kinases responsible for phosphorylation at specific sites of Bcl-xL and investigating their impact on intracellular redox is an important area for future study.
Oxidative Stress and Transcription of Bcl-xL
In addition to oxidative stress regulating post-translational modification of Bcl-xL, treatment with H2O2 has been shown to decrease Bcl-xL mRNA expression in vitro, leading to a decline in Bcl-xL protein level (Wang et al., 2015; Wu et al., 2016). Although there are limited studies explaining the underlying mechanisms, oxidative stress may also govern post-transcriptional regulation of Bcl-xL. MicroRNAs (miRNA) are short non-coding RNAs that bind to the target mRNA inhibiting its translation. Wang et al. (2015) found that ROS causes oxidative modification of miR-184, and oxidized miR-184 interacts with Bcl-xL mRNA, suppressing Bcl-xL protein levels. Due to miR-184’s high level in the brain (Danis et al., 2016; Mendes-Silva et al., 2019), preventing miR-184 oxidation may be an effective therapeutic strategy to protect the brain against ROS-associated pathology.
Oxidative stress may also contribute to splicing of Bcl-xL pre-mRNA. The bcl-x gene is spliced into one of two isoforms: anti-apoptotic Bcl-xL, or pro-apoptotic Bcl-xS (Stevens and Oltean, 2019). Bcl-xS is structurally similar to Bcl-xL but contains only a BH3, BH4, and transmembrane domain. Bcl-xS undergoes dimerization in mitochondria (Lindenboim et al., 2001) causing intrinsic apoptosis. Due to its unique chemical structure containing BH4, but lacking BH1 and BH2 domains, Bcl-xS-mediated apoptosis is independent from mechanisms induced by other pro-apoptotic Bcl2 proteins (Lindenboim et al., 2000). However, both Bcl-xS and ΔN-Bcl-xL production occur during apoptosis in a caspase dependent manner (Willimott et al., 2011), and Bcl-xS binds directly to Bcl-xL (Lindenboim et al., 2001). Therefore, conditions that enhance Bcl-xS contribute to limiting the pro-survival function of Bcl-xL.
CUG-binding protein 1 (CUGBP1) is a key splicing factor responsible for regulating Bcl-xL/Bcl-xS ratio: overexpression of CUGBP1 increases the proportion of Bcl-xL whereas depletion of CUGBP1 decreases Bcl-xL (Xiao et al., 2012). Xiao et al. (2012) showed that rat oligodendrocyte progenitor cells treated with C2-ceramide decreased CUGBP1 in the nuclear fraction, and hippocampal and cortical tissues collected from neonatal rat challenged with ischemia decreased the Bcl-xL/Bcl-xS ratio. Although this study did not emphasize a direct role of ROS in Bcl-x gene splicing, both the C2-ceramide model in vitro and the ischemia model in vivo are known to enhance ROS-associated neurotoxicity (France-Lanord et al., 1997; Ten and Starkov, 2012).
Conversely, another study showed that ROS may lead to increases in Bcl-xL mRNA. C57BL/6 mice that were treated with bilateral common carotid artery occlusion had increased Bcl-xL mRNA levels 24 hours after ischemia, and this led to increased Bcl-xL protein levels 48 hours after ischemia (Wu et al., 2003). Since depletion of Bcl-xL is well documented to increase the vulnerability of neurons, these results suggest that there is a protective molecular response against ischemia by modifying the transcription of Bcl-xL. One mechanism may be through regulation of transcription factors. Nuclear factor kappa B (NF-κB) is well-established as a transcription factor that targets the bcl-x gene (Chen et al., 2000). It is known that over-accumulation of mitochondrial ROS can stimulate NF-κB to target genes that promote survival, including bcl-x (Chen et al., 2000; Morgan and Liu, 2011). Lanzillotta et al. (2013) research group demonstrated this connection in a rodent model of ischemia. Mutation of the NF-κB binding site on Bcl-xL’s promoter region led to downregulation of the bcl-x gene, indicating that NF-κB is indeed an important regulator of transcription following ischemia. Similarly, nuclear factor erythroid 2-related factor 2 (Nrf2) may be involved in a protective response to ROS insult. Nrf2 is a transcription factor that is known for its role in antioxidant systems against ROS (Ryoo and Kwak, 2018). Nrf2 remains inactive and bound to Keap1; under conditions of oxidative stress or by phosphorylation, Nrf2 dissociates from Keap1 and translocates to the nucleus, where it then binds to antioxidant response elements of target genes (Wu et al., 2013; Deshmukh et al., 2017; Wen et al., 2018). Bcl-x contains an antioxidant response element, resulting in direct binding of Nrf2. The reverse of transcriptional enhancement of Bcl-xL was demonstrated by an siRNA-mediated decrease in Nrf2, leading to subsequent reduction in Bcl-xL mRNA (Niture and Jaiswal, 2013). Through activation of transcription factors such as NF-κB and Nrf2, ROS can influence the transcription of Bcl-xL. However, it remains unclear what additional factors may lead to Bcl-xL mRNA upregulation versus downregulation.
Conclusion
Bcl-xL is a key protein that enhances neuronal function by regulating energy metabolism, neurotransmission, and the survival or death of neuroprogenitors and post-mitotic neurons. The activity and abundance of Bcl-xL is controlled by oxidative stress: ROS control the post-translational modification of Bcl-xL including its proteolytic cleavage and residue phosphorylation (Figure 1). These modifications alter the availability of functional Bcl-xL and its activity. It has been shown that ROS also play a part in the transcriptional regulation of Bcl-xL (Figure 1). Therefore, understanding ROS-mediated Bcl-xL alterations are important to further elucidate mechanisms of ROS-associated brain disease, and approaches that manipulate ROS may be an effective way to manipulate Bcl-xL function and abundance in the brain.
Additional file: Open peer review reports 1 (84.4KB, pdf) and 2 (86.1KB, pdf) .
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Elizabeth D. Kirby, Ohio State University, USA; Hospital Universitario Central de Asturias, Spain.
P-Reviewers: Kirby ED, Fernandez-Vega I; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, Rahner C, McNay E, Shore GC, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arena G, Gelmetti V, Torosantucci L, Vignone D, Lamorte G, De Rosa P, Cilia E, Jonas EA, Valente EM. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013;20:920–930. doi: 10.1038/cdd.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baruah PS, Beauchemin M, Hebert J, Bertrand R. Dynamic Bcl-xL (S49) and (S62) Phosphorylation/dephosphorylation during mitosis prevents chromosome instability and aneuploidy in normal human diploid fibroblasts. PLoS One. 2016;11:e0159091. doi: 10.1371/journal.pone.0159091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basanez G, Zhang J, Chau BN, Maksaev GI, Frolov VA, Brandt TA, Burch J, Hardwick JM, Zimmerberg J. Pro-apoptotic cleavage products of Bcl-xL form cytochrome c-conducting pores in pure lipid membranes. J Biol Chem. 2001;276:31083–31091. doi: 10.1074/jbc.M103879200. [DOI] [PubMed] [Google Scholar]
- 6.Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. 2011;108:9572–9577. doi: 10.1073/pnas.1106291108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao G, Pei W, Ge H, Liang Q, Luo Y, Sharp FR, Lu A, Ran R, Graham SH, Chen J. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J Neurosci. 2002;22:5423–5431. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandra D, Tang DG. Mitochondrially localized active caspase-9 and caspase-3 result mostly from translocation from the cytosol and partly from caspase-mediated activation in the organelle. Lack of evidence for Apaf-1-mediated procaspase-9 activation in the mitochondria. J Biol Chem. 2003;278:17408–17420. doi: 10.1074/jbc.M300750200. [DOI] [PubMed] [Google Scholar]
- 9.Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol Cell Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen R, Park HA, Mnatsakanyan N, Niu Y, Licznerski P, Wu J, Miranda P, Graham M, Tang J, Boon AJW, Cossu G, Mandemakers W, Bonifati V, Smith PJS, Alavian KN, Jonas EA. Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis. 2019;10:469. doi: 10.1038/s41419-019-1679-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, O’Rourke B, Ko YH, Pedersen PL, Kaczmarek LK, Jonas EA, Hardwick JM. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–276. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clem RJ, Cheng EH, Karp CL, Kirsch DG, Ueno K, Takahashi A, Kastan MB, Griffin DE, Earnshaw WC, Veliuona MA, Hardwick JM. Modulation of cell death by Bcl-XL through caspase interaction. Proc Natl Acad Sci U S A. 1998;95:554–559. doi: 10.1073/pnas.95.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cusack CL, Swahari V, Hampton Henley W, Michael Ramsey J, Deshmukh M. Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat Commun. 2013;4:1876. doi: 10.1038/ncomms2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Orsi B, Bonner H, Tuffy LP, Dussmann H, Woods I, Courtney MJ, Ward MW, Prehn JH. Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J Neurosci. 2012;32:1847–1858. doi: 10.1523/JNEUROSCI.2345-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danis B, van Rikxoort M, Kretschmann A, Zhang J, Godard P, Andonovic L, Siegel F, Niehusmann P, Hanon E, Delev D, von Lehe M, Kaminski RM, Pfeifer A, Foerch P. Differential expression of miR-184 in temporal lobe epilepsy patients with and without hippocampal sclerosis - Influence on microglial function. Sci Rep. 2016;6:33943. doi: 10.1038/srep33943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deshmukh P, Unni S, Krishnappa G, Padmanabhan B. The Keap1-Nrf2 pathway: promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys Rev. 2017;9:41–56. doi: 10.1007/s12551-016-0244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dho SH, Deverman BE, Lapid C, Manson SR, Gan L, Riehm JJ, Aurora R, Kwon KS, Weintraub SJ. Control of cellular Bcl-xL levels by deamidation-regulated degradation. PLoS Biol. 2013;11:e1001588. doi: 10.1371/journal.pbio.1001588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fogarty LC, Flemmer RT, Geizer BA, Licursi M, Karunanithy A, Opferman JT, Hirasawa K, Vanderluit JL. Mcl-1 and Bcl-xL are essential for survival of the developing nervous system. Cell Death Differ. 2019;26:1501–1515. doi: 10.1038/s41418-018-0225-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.France-Lanord V, Brugg B, Michel PP, Agid Y, Ruberg M. Mitochondrial free radical signal in ceramide-dependent apoptosis: a putative mechanism for neuronal death in Parkinson’s disease. J Neurochem. 1997;69:1612–1621. doi: 10.1046/j.1471-4159.1997.69041612.x. [DOI] [PubMed] [Google Scholar]
- 20.Geden MJ, Deshmukh M. Axon degeneration: context defines distinct pathways. Curr Opin Neurobiol. 2016;39:108–115. doi: 10.1016/j.conb.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gil-Parrado S, Fernandez-Montalvan A, Assfalg-Machleidt I, Popp O, Bestvater F, Holloschi A, Knoch TA, Auerswald EA, Welsh K, Reed JC, Fritz H, Fuentes-Prior P, Spiess E, Salvesen GS, Machleidt W. Ionomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. A probable role for Bcl-2 family members. J Biol Chem. 2002;277:27217–27226. doi: 10.1074/jbc.M202945200. [DOI] [PubMed] [Google Scholar]
- 22.Jonas EA, Hickman JA, Chachar M, Polster BM, Brandt TA, Fannjiang Y, Ivanovska I, Basanez G, Kinnally KW, Zimmerberg J, Hardwick JM, Kaczmarek LK. Proapoptotic N-truncated BCL-xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proc Natl Acad Sci U S A. 2004;101:13590–13595. doi: 10.1073/pnas.0401372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lanzillotta A, Pignataro G, Branca C, Cuomo O, Sarnico I, Benarese M, Annunziato L, Spano P, Pizzi M. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol Dis. 2013;49:177–189. doi: 10.1016/j.nbd.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Alavian KN, Lazrove E, Mehta N, Jones A, Zhang P, Licznerski P, Graham M, Uo T, Guo J, Rahner C, Duman RS, Morrison RS, Jonas EA. A Bcl-xL-Drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat Cell Biol. 2013;15:773–785. doi: 10.1038/ncb2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, Bossy-Wetzel E, Bennett MV, Pypaert M, Hickman JA, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:2169–2174. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindenboim L, Borner C, Stein R. Bcl-x(S) can form homodimers and heterodimers and its apoptotic activity requires localization of Bcl-x(S) to the mitochondria and its BH3 and loop domains. Cell Death Differ. 2001;8:933–942. doi: 10.1038/sj.cdd.4400888. [DOI] [PubMed] [Google Scholar]
- 27.Lindenboim L, Yuan J, Stein R. Bcl-xS and Bax induce different apoptotic pathways in PC12 cells. Oncogene. 2000;19:1783–1793. doi: 10.1038/sj.onc.1203495. [DOI] [PubMed] [Google Scholar]
- 28.Liu Z, Wang S, Zhou H, Yang Y, Zhang M. Na+/H+ exchanger mediates TNF-alpha-induced hepatocyte apoptosis via the calpain-dependent degradation of Bcl-xL. J Gastroenterol Hepatol. 2009;24:879–885. doi: 10.1111/j.1440-1746.2008.05715.x. [DOI] [PubMed] [Google Scholar]
- 29.Megyesi J, Tarcsafalvi A, Seng N, Hodeify R, Price PM. Cdk2 phosphorylation of Bcl-xL after stress converts it to a pro-apoptotic protein mimicking Bax/Bak. Cell Death Discov. 2016 doi: 10.1038/cddiscovery.2015.66. doi:101038/cddiscovery201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mendes-Silva AP, Fujimura PT, Silva J, Teixeira AL, Vieira EM, Guedes PHG, Barroso LSS, Nicolau MS, Ferreira JDR, Bertola L, Nicolau ES, Tolentino-Araujo GT, Berlezzi C, Rodrigues TS, Borges LDF, Gomes MS, Amaral LRD, Bonetti AM, Ueira-Vieira C, Diniz BS. Brain-enriched MicroRNA-184 is downregulated in older adults with major depressive disorder: A translational study. J Psychiatr Res. 2019;111:110–120. doi: 10.1016/j.jpsychires.2019.01.019. [DOI] [PubMed] [Google Scholar]
- 31.Miyawaki T, Mashiko T, Ofengeim D, Flannery RJ, Noh KM, Fujisawa S, Bonanni L, Bennett MV, Zukin RS, Jonas EA. Ischemic preconditioning blocks BAD translocation, BclxL cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:4892–4897. doi: 10.1073/pnas.0800628105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monaco G, Beckers M, Ivanova H, Missiaen L, Parys JB, De Smedt H, Bultynck G. Profiling of the Bcl-2/Bcl-X(L)-binding sites on type 1 IP(3) receptor. Biochem Biophys Res Commun. 2012;428:31–35. doi: 10.1016/j.bbrc.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 33.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakamura A, Swahari V, Plestant C, Smith I, McCoy E, Smith S, Moy SS, Anton ES, Deshmukh M. Bcl-xL is essential for the survival and function of differentiated neurons in the cortex that control complex behaviors. J Neurosci. 2016;36:5448–5461. doi: 10.1523/JNEUROSCI.4247-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Niture SK, Jaiswal AK. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic Biol Med. 2013;57:119–131. doi: 10.1016/j.freeradbiomed.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ofengeim D, Chen YB, Miyawaki T, Li H, Sacchetti S, Flannery RJ, Alavian KN, Pontarelli F, Roelofs BA, Hickman JA, Hardwick JM, Zukin RS, Jonas EA. N-terminally cleaved Bcl-xL mediates ischemia-induced neuronal death. Nat Neurosci. 2012;15:574–580. doi: 10.1038/nn.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park HA, Jonas EA. DeltaN-Bcl-xL, a therapeutic target for neuroprotection. Neural Regen Res. 2017;12:1791–1794. doi: 10.4103/1673-5374.219033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park HA, Licznerski P, Alavian KN, Shanabrough M, Jonas EA. Bcl-xL Is Necessary for Neurite Outgrowth in Hippocampal Neurons. Antioxid Redox Signal. 2015;22:93–108. doi: 10.1089/ars.2013.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park HA, Licznerski P, Mnatsakanyan N, Niu Y, Sacchetti S, Wu J, Polster BM, Alavian KN, Jonas EA. Inhibition of Bcl-xL prevents pro-death actions of DeltaN-Bcl-xL at the mitochondrial inner membrane during glutamate excitotoxicity. Cell Death Differ. 2017;24:1963–1974. doi: 10.1038/cdd.2017.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park HA, Mnatsakanyan N, Broman K, Davis AU, May J, Licznerski P, Crowe-White KM, Lackey KH, Jonas EA. Alpha-tocotrienol prevents oxidative stress-mediated post-translational cleavage of Bcl-xL in primary hippocampal neurons. Int J Mol Sci. 2019 doi: 10.3390/ijms21010220. doi: 103390/ijms21010220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryoo IG, Kwak MK. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol Appl Pharmacol. 2018;359:24–33. doi: 10.1016/j.taap.2018.09.014. [DOI] [PubMed] [Google Scholar]
- 44.See V, Loeffler JP. Oxidative stress induces neuronal death by recruiting a protease and phosphatase-gated mechanism. J Biol Chem. 2001;276:35049–35059. doi: 10.1074/jbc.M104988200. [DOI] [PubMed] [Google Scholar]
- 45.Simon DJ, Pitts J, Hertz NT, Yang J, Yamagishi Y, Olsen O, Tesic Mark M, Molina H, Tessier-Lavigne M. Axon degeneration gated by retrograde activation of somatic pro-apoptotic signaling. Cell. 2016;164:1031–1045. doi: 10.1016/j.cell.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stevens M, Oltean S. Modulation of the apoptosis gene Bcl-x function through alternative splicing. Front Genet. 2019;10:804. doi: 10.3389/fgene.2019.00804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ten VS, Starkov A. Hypoxic-ischemic injury in the developing brain: the role of reactive oxygen species originating in mitochondria. Neurol Res Int. 2012;2012:542976. doi: 10.1155/2012/542976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Upreti M, Galitovskaya EN, Chu R, Tackett AJ, Terrano DT, Granell S, Chambers TC. Identification of the major phosphorylation site in Bcl-xL induced by microtubule inhibitors and analysis of its functional significance. J Biol Chem. 2008;283:35517–35525. doi: 10.1074/jbc.M805019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veas-Perez de Tudela M, Delgado-Esteban M, Maestre C, Bobo-Jimenez V, Jimenez-Blasco D, Vecino R, Bolanos JP, Almeida A. Regulation of Bcl-xL-ATP synthase interaction by mitochondrial cyclin B1-cyclin-dependent kinase-1 determines neuronal survival. J Neurosci. 2015;35:9287–9301. doi: 10.1523/JNEUROSCI.4712-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vervliet T, Lemmens I, Vandermarliere E, Decrock E, Ivanova H, Monaco G, Sorrentino V, Nadif Kasri N, Missiaen L, Martens L, De Smedt H, Leybaert L, Parys JB, Tavernier J, Bultynck G. Ryanodine receptors are targeted by anti-apoptotic Bcl-XL involving its BH4 domain and Lys87 from its BH3 domain. Sci Rep. 2015;5:9641. doi: 10.1038/srep09641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang JX, Gao J, Ding SL, Wang K, Jiao JQ, Wang Y, Sun T, Zhou LY, Long B, Zhang XJ, Li Q, Liu JP, Feng C, Liu J, Gong Y, Zhou Z, Li PF. Oxidative modification of miR-184 enables it to target Bcl-xL and Bcl-w. Mol Cell. 2015;59:50–61. doi: 10.1016/j.molcel.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Wen Z, Hou W, Wu W, Zhao Y, Dong X, Bai X, Peng L, Song L. 6’-O-Galloylpaeoniflorin attenuates cerebral ischemia reperfusion-induced neuroinflammation and oxidative stress via PI3K/Akt/Nrf2 activation. Oxid Med Cell Longev. 2018;2018:8678267. doi: 10.1155/2018/8678267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Willimott S, Merriam T, Wagner SD. Apoptosis induces Bcl-XS and cleaved Bcl-XL in chronic lymphocytic leukaemia. Biochem Biophys Res Commun. 2011;405:480–485. doi: 10.1016/j.bbrc.2011.01.057. [DOI] [PubMed] [Google Scholar]
- 54.Wu C, Fujihara H, Yao J, Qi S, Li H, Shimoji K, Baba H. Different expression patterns of Bcl-2, Bcl-xl, and Bax proteins after sublethal forebrain ischemia in C57Black/Crj6 mouse striatum. Stroke. 2003;34:1803–1808. doi: 10.1161/01.STR.0000077255.15597.69. [DOI] [PubMed] [Google Scholar]
- 55.Wu GJ, Wang W, Lin YL, Liu SH, Chen RM. Oxidative stress-induced apoptotic insults to rat osteoblasts are attenuated by nitric oxide pretreatment via GATA-5-involved regulation of Bcl-X L gene expression and protein translocation. Arch Toxicol. 2016;90:905–916. doi: 10.1007/s00204-015-1491-z. [DOI] [PubMed] [Google Scholar]
- 56.Wu J, Li Q, Wang X, Yu S, Li L, Wu X, Chen Y, Zhao J, Zhao Y. Neuroprotection by curcumin in ischemic brain injury involves the Akt/Nrf2 pathway. PLoS One. 2013;8:e59843. doi: 10.1371/journal.pone.0059843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiao B, Goh JY, Xiao L, Xian H, Lim KL, Liou YC. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J Biol Chem. 2017;292:16697–16708. doi: 10.1074/jbc.M117.787739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiao Q, Ford AL, Xu J, Yan P, Lee KY, Gonzales E, West T, Holtzman DM, Lee JM. Bcl-x pre-mRNA splicing regulates brain injury after neonatal hypoxia-ischemia. J Neurosci. 2012;32:13587–13596. doi: 10.1523/JNEUROSCI.2617-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011;25:310–322. doi: 10.1101/gad.1984311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan XL, Guo JF, Shi ZH, Xiao ZQ, Yan XX, Zhao BL, Tang BS. R492X mutation in PTEN-induced putative kinase 1 induced cellular mitochondrial dysfunction and oxidative stress. Brain Res. 2010;1351:229–237. doi: 10.1016/j.brainres.2010.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.