

Abstract
Background
Previous studies have revealed that mutations of Spalt Like Transcription Factor 1 (SALL1) are responsible for Townes-Brocks syndrome (TBS), a rare genetic disorder that is characterized by an imperforate anus, dysplastic ears, thumb malformations and other abnormalities, such as hearing loss, foot malformations, renal impairment with or without renal malformations, genitourinary malformations, and congenital heart disease. In addition, the protein tyrosine phosphatase receptor type Q (PTPRQ) gene has been identified in nonsyndromic hearing loss patients with autosomal recessive or autosomal dominant inherited patterns.
Methods
A Chinese family with TBS and hearing loss was enrolled in this study. The proband was a two-month-old girl who suffered from congenital anal atresia with rectal perineal fistula, ventricular septal defect, patent ductus arteriosus, pulmonary hypertension (PH), and finger deformities. The proband’s father also had external ear deformity with deafness, toe deformities and PH, although his anus was normal. Further investigation found that the proband’s mother presented nonsyndromic hearing loss, and the proband’s mother’s parents were consanguine married. Whole-exome sequencing and Sanger sequencing were applied to detect the genetic lesions of TBS and nonsyndromic hearing loss.
Results
Via whole-exome sequencing and Sanger sequencing of the proband and her mother, we identified a novel heterozygous mutation (ENST00000251020: c.1428_1429insT, p. K478QfsX38) of SALL1 in the proband and her father who presented TBS phenotypes, and we also detected a new homozygous mutation [ENST00000266688: c.1057_1057delC, p. L353SfsX8)] of PTPRQ in the proband’s mother and uncle, who suffered from nonsyndromic hearing loss. Both mutations were located in the conserved sites of the respective protein and were predicted to be deleterious by informatics analysis.
Conclusions
This study confirmed the diagnosis of TBS at the molecular level and expanded the spectrum of SALL1 mutations and PTPRQ mutations. Our study may contribute to the clinical management and genetic counselling of TBS and hearing loss.
Keywords: Townes-brocks syndrome, Hearing loss, SALL1 mutation, PTPRQ mutation
Background
Townes-Brocks syndrome (TBS, OMIM: #107480) is a congenital genetic disorder characterized by the triad of atresia of the anus, dysplasia of the external ears and thumb deformity [1, 2]. Secondary features of the syndrome include hearing loss, foot deformity, renal insufficiency with or without renal malformations, urogenital malformations, and congenital heart disease (CHD) [3, 4]. TBS was first reported in 1972 by Townes and Brocks, who described a family with congenital anorectal malformations (ARMs), polydactyly of the thumb, and other skeletal abnormalities (metatarsal fusion, missing thumb bones, and multifinger deformity) [5]. At present, patients who are diagnosed with TBS should have at least two of the following three major features: an imperforate anus (84% of the cases), abnormally shaped ears (87% of the cases) and thumb malformations (89% of the cases) [6]
In 1998, Kohlhase et al. first identified two different heterozygous mutations of Spalt Like Transcription Factor 1 (SALL1) in a family with three cases of TBS in two generations and in an unrelated family with a sporadic case of TBS [7]. Since then, more than ninety mutations of SALL1 have been detected in TBS patients, and most were nonsense mutations and frameshift mutations. SALL1, a strong transcriptional repressor, is regulated by the sonic hedgehog pathway, which plays a crucial role in the development of multiple organs [8, 9]. In addition, some authors proposed the involvement of SALL1 in the regulation of higher-order chromatin structures and hypothesized that the protein may be a component of a distinct heterochromatin-dependent silencing process [10, 11]. Recently, studies have found that SALL1 can interact with ciliogenesis suppressors CCP110 and CEP97 and mutations in SALL1 may disrupt the formation and function of cilia [1, 12].
Hearing loss or deafness (prevalence rate of 0.1%) is the most common sensory disorder and affects millions worldwide [13]. In terms of physical function and social movement, the quality of life of hearing loss patients is poor. Fifty percent of these cases are estimated to be caused by genetic factors. Previous studies have shown that mutations in protein tyrosine phosphatase receptor type Q (PTPRQ) may lead to autosomal recessive nonsyndromic hearing loss [14]. More than thirty mutations of PTPRQ have been identified in patients with hearing loss. Meanwhile, a recent study also found that the heterozygous c.6881G > A transition (NM_001145026.1) in the last coding exon of PTPRQ may lead to autosomal dominant nonsyndromic hearing loss [15, 16].
In this study, we identified a novel heterozygous mutation (ENST00000251020: c.1428_1429insT, p. K478QfsX38) of SALL1 and a new homozygous mutation (ENST00000266688: c.1057_1057delC, p. L353SfsX8) of PTPRQ in a Chinese family that included two TBS patients and another two individuals with hearing loss by whole-exome sequencing.
Methods
Subjects
The family, including ten persons, was investigated in this study (Fig. 1a). Peripheral blood samples of all these family members were collected and applied to isolate genomic DNA by a Universal Genomic DNA Extraction Kit (Solarbio, D2100) as we previously described [17]. In addition, 200 unrelated local healthy people were also enrolled to serve as normal controls. This study was approved by the Ethics Committee of Hunan Children’s Hospital, Changsha, China, and performed in accordance with the principles enshrined in the Declaration of Helsinki. The patients/participants provided their written informed consent to participate in this study.
Fig. 1.

Clinical data for the family. a Pedigree of this family. Square indicates male, circle indicates female, and arrow indicates the proband. b Proband’s X-ray with anorectal deformity and fistula. c X-rays of the proband’s hands with a multifinger phenotype. d External ear deformity of the proband’s father. e Toe deformities of the proband’s father
Whole-exome sequencing
We selected the proband (III-3) and her mother (II-3) to perform whole-exome sequencing. Exome capture and next-generation sequencing were conducted by the Novogene Bioinformatics Institute (Beijing, China). One microgram of qualified genomic DNA from each person was captured by the Agilent SureSelect Human All Exon kit V5 (Agilent Technologies, Inc., Santa Clara, USA) and sequenced using an Illumina HiSeq 4000 (Illumina Inc., San Diego, USA) [17]. Briefly, genomic DNA was randomly carved by a Covaris S220 sonicator (Covaris, Inc., Woburn, USA). Then, the fragmented DNAs underwent three enzymatic steps: end repair, A-tailing and adapter ligation. The adapter-ligated DNA fragments were amplified with Herculase II Fusion DNA Polymerase (Agilent). Finally, the exomes in the precapture libraries were captured by a SureSelect capture library kit (Agilent) [17]. After DNA quality assessment, the captured DNA library was used for next-generation sequencing on the Illumina HiSeq 4000 platform. After demultiplexing, we used Burrows Wheeler alignment to map the paired-end sequences to the human genome (NCBI Build 37/hg19). Downstream processing was carried out using the Genome Analysis Toolkit (GATK) as well as Varscan2 and Picard, and variant calls were performed with the GATK Haplotype Caller. Variant annotation referred to Ensemble release 82, and filtering was conducted by ANNOVAR Documentation [18].
Nonsynonymous SNPs or frameshift-causing INDELs with an alternative allele frequency > 0.005 in the NHLBI Exome Sequencing Project Exome Variant Server (ESP6500), dbSNP147 (http://www.ncbi.nlm.nih.gov/projects/SNP/index.html), the 1000 Genomes project (http://www.1000genomes.org/), and the ExAC database or in-house exome databases of Novogene (2500 exomes) were kicked before further analysis. Then, the SNVs and INDELs predicted by SIFT (http://sift.jcvi.org/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) and MutationTaster (http://www.mutationtaster.org/) to be damaging remained were filtered. Finally, mutations were found in two affected members (I-2 and III-4) but were absent in the healthy individual (II-2) [18].
Mutation validation and cosegregation analysis
After the filtering process, all mutations found for this family were validated by Sanger sequencing. The primer sequences were as follows: SALL1: forward, 5′-GGTACACATGGGCACTCACA-3′; reverse, 5′-GCCACCATAGGTCGCATTCT-3′, and PTPRQ: forward, 5′-TGTTGTCTTTGGCTCTGTACTT-3′; reverse, 5′-GTTCTTACCATCTGGTGGAGTG-3′. The sequences of the polymerase chain reaction (PCR) products were determined using the ABI 3100 Genetic Analyser (ABI, Foster City, CA, USA) [19].
Results
Clinical data
The proband (III-3), a two-month-old girl, was from the mountainous area of southern China and suffered from congenital anal atresia with rectal perineal fistula (Fig. 1b), ventricular septal defect, patent ductus arteriosus, pulmonary hypertension (PS), and finger deformities (Fig. 1c). A family history survey found that the proband’s father (II-4) also suffered from external ear deformity with deafness (Fig. 1d), toe deformities (Fig. 1e) and PS, although his anus was normal. Both the proband (III-3) and her father (II-4) were suspected of having TBS.
During the family history investigation, we found another interesting truth: both the proband’s mother (II-3) and uncle (II-2) were isolated deafness patients and their parents (I-1 and I-2) were consanguine married. No other deafness patients were detected. The proband (III-3) underwent surgical treatment of CHD as well as anoplasty surgery for ARM after three months.
Genetic analysis
Different genetic lesions may underlie a family with hearing loss and congenital malformations. The proband (III-3) (Additional file 1) and her mother (II-3) (Additional file 2) to perform whole-exome sequencing analysis. After data screening, we detected a novel heterozygous mutation (ENST00000251020: c.1428_1429insT, p. K478QfsX38) of SALL1 in the proband (III-3) (Fig. 2a) and a new homozygous mutation (ENST00000266688: c.1057_1057delC, p. L353SfsX8) of PTPRQ in the proband’s mother (II-3) (Fig. 2b). No other meaningful mutations were identified. Further cosegregation analysis found that the proband’s father (II-4) carried the heterozygous mutation of SALL1 (Fig. 2a) and both the proband (III-3) and her father (II-4) were confirmed to have TBS. Sanger sequencing further found that the proband’s uncle (II-2) carried a homozygous mutation in PTPRQ while the proband’s grandfather (I-1), grandmother (I-2) and proband (III-3) carried a heterozygous mutation (ENST00000266688: c.1057_1057delC, p. L353SfsX8) of PTPRQ (Fig. 2b). Both novel mutations (SALL1: c.1428_1429insT and PTPRQ: c.1057_1057delC) were located in the conserved sites of the respective protein and predicted to be deleterious by informatics analysis. Both mutations were also absent in our 200 local control cohorts as well as other public databases, such as ExAC (http://exac.broadinstitute.org) and genomAD (https://gnomad.broadinstitute.org). Moreover, ACMG guidelines [20] indicate that both mutations are pathogenic.
Fig. 2.
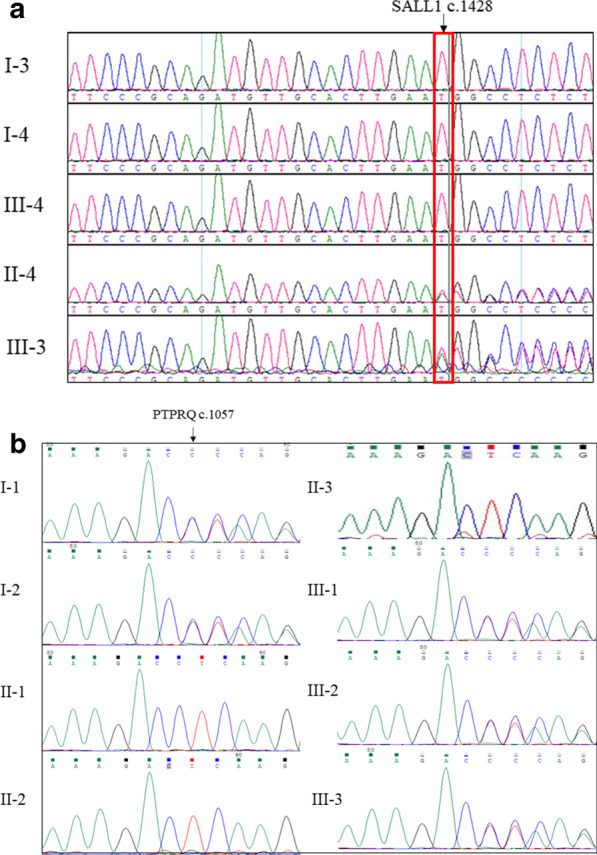
Genetic analysis of the family. a Sanger DNA sequencing chromatogram demonstrates the heterozygosity for a SALL1 mutation (c.1428_1429insT, p. K478QfsX38) in the proband and her father. b Sanger DNA sequencing chromatogram demonstrates the homozygosity for a PTPRQ mutation (c.1057_1057delC, p. L353SfsX8) in the proband’ mother and uncle
Discussion
SALL1
The current research confirms that TBS syndrome is an autosomal dominant genetic disease. The heterozygous mutation of the zinc finger protein transcription regulation gene SALL1 at 16q12.1 is the molecular basis of the disease. In 2005, Botzenhart et al. reported that most of the SALL1 gene mutations occurred in or within the 5-prime encoding the first double zinc finger region among the 35 identified genetic mutations [21]. The SALL1 gene contains three determined exons, with 11 WT1-binding sites and 1 SIX1-binding site. The 5 prime number flanking regions do not contain TATA or CAAT boxes, although they are rich in GC and contain more GC boxes [22]. In addition to the SALL1 gene, recent studies found that mutations in the Dishevelled Binding Antagonist Of Beta Catenin 1 (DACT1) gene located on chromosome 14q23 may also lead to TBS, which is called Townes-Brocks syndrome-2 (TBS2; 617466) [23].
Some families with Townes-Brocks syndrome have shown increased clinical severity over generations, thus presenting genetic anticipation. Sudo et al. reported an antedating Japanese family in which a four-year-old female proband was heterozygous for a frameshift mutation in the SALL1 gene, the first generation of affected patients (the proband’s grandfather and great aunt) showed polydactyly and deafness, the second generation (the proband’s mother and uncle) had kidney and anal abnormalities, and the third generation (the proband) showed the most serious symptoms of anteriorly placed and stenosed anus and ventricular septal defects (VSDs) [2]. Researchers noted that the severity of the phenotype with each succeeding generation was an anticipation-like increase in this family and showed that similar increases in clinical severity had been observed in other TBS families, frequently in patients who inherited the disease from their mothers.
In our study, we identified a novel heterozygous mutation (ENST00000251020: c.1428_1429insT, p. K478QfsX38) of SALL1 in a two-generation family with TBS. The first generation (II-4) only showed toe deformities, deafness, external ear deformities and PS, although more serious complications, including multiple CHDs and ARMs, were present in the second generation (III-3). At present, this proband (III-3) has no sensorineural deafness and only shows mild conductive deafness, which is related to otitis media that was present during the examination. Moreover, the child may not show sensorineural deafness; thus, follow-up review is still needed in the future.
PTPRQ
PTPRQ plays an important role in inner ear development and hair cell differentiation and maturation [24, 25] and is necessary for the normal maturation of shaft connectors and developing hair bundles in the mammalian cochlea. There are four PTPRQ isotypes, and each isotype is composed of a transmembrane domain, fibronectin type 3 domain and phosphatase domain [26]. Previous studies suggested that PTPRQ and myosin VI can form a complex, which may dynamically maintain the organization coated on the surface of the stereocilia base cells and may help maintain the overall structure of the stereocilia bundles [27]. PTPRQ knockout mice have significantly longer hair strands and suffer from the loss and fusion of hair cell static cilia. Researchers have also observed a simultaneous loss of function of the vestibular system in these mice [24]. Vestibular evoked potentials do not occur in most PTPRQ gene knockout mice, and subtle but obvious defects of evoked potentials are detected in mutant mice [24]. Goodyear et al. pointed out that the lack of PTPRQ in mice causes the apical membrane of hair cells to separate from the potential actin cytoskeleton and leads to the fusion of stereocilia [28]. PTPRQ mutations may alter the morphology and stereocilia and further lead to the gradual loss of cochlear hair cells and subsequent deafness [28].
Previous studies have proven that homozygous mutation of PTPRQ may lead to nonsyndromic hearing loss. In 2010, Arg457Gly (currently Arg239Gly) and Tyr497X (currently Tyr279X) were first identified by Schraders et al. in deaf families from Morocco and the Netherlands. Hashem et al. found that the cause of DFNB84 in Palestinian deaf families was Gln429X in PTPRQ [26]. In 2015, Qing Sang et al. identified c.1617insT (p. L8fsX18) and c.2714delA (p. E909fsX922) as new compound heterozygous mutations in the PTPRQ gene in a Kazakh family from Xinjiang, which was the first report of a PTPRQ gene mutation in China [29]. In the last coding exon (exon 45) of the PTPRQ gene, Eisenberger et al. identified a heterozygous c.6881G>A transition, which resulted in a p. W2294X substitution in affected members in four generations of German families in which the age at onset of hearing loss ranged from early childhood to the third decade, thus confirming that the PTPRQ gene was also a new autosomal dominant nonsyndromic hearing loss gene [16].
In this study, we identified a new homozygous mutation (ENST00000266688: c.1057_1057delC, p. L353SfsX8) of PTPRQ in a consanguineous family with nonsyndromic hearing loss. This family came from the remote Wuling Mountains in southern China, which is a gathering place for ethnic minorities and economically suppressed, where intimate marriages often occur. Close marriages greatly increase the probability that future generations will suffer from deafness. Alleles from a common ancestor reached a homozygous state in the mother (II-3) and uncle (II-2) of the proband (III-3).
Conclusions
In summary, we identified a novel heterozygous mutation of SALL1 (ENST00000251020: c.1428_1429insT, p. K478QfsX38) and a new homozygous mutation of PTPRQ (ENST00000266688: c.1057_1057delC, p. L353SfsX8) in a Chinese family with TBS and nonsyndromic hearing loss. Our study not only confirmed the diagnosis of TBS at the molecular level but also expanded the spectrum of SALL1 mutations and PTPRQ mutations. These findings may contribute to the clinical management and genetic counselling of TBS and hearing loss.
Supplementary information
Additional file 1. The sequencing data of III-3.
Additional file 2. The sequencing data of II-3.
Acknowledgements
We thank all subjects for participating in this study. We thank Dr. Shuai Guo from the University of Texas MD Anderson Cancer Center, USA, for editing the language.
Abbreviations
- SALL1
Spalt-like transcription factor 1
- TBS
Townes-Brocks syndrome
- CHD
Congenital heart disease
- PTPRQ
Protein tyrosine phosphatase receptor type Q
- PH
Pulmonary hypertension
- ARM
Congenital anorectal malformations
- VSD
Ventricular septal defect
Authors’ contributions
GY drafted the manuscript; ZT conceived and designed the experiments; NY and YY revised the manuscript; and XD and JL managed the patients. All authors have read and approved the manuscript.
Funding
None.
Availability of data and materials
The data used to support the findings of this study are included within the article.
Ethics approval and consent to participate
Written informed consent was obtained from all of the adult participants and legal guardians of minor participants. The study was approved by the Ethics Committee of Hunan Children's Hospital (HCHLL-2018-43).
Consent for publication
Written consent was obtained from all the participants or their guardians for the publication of this study.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
2/3/2021
Additional file 2 has been correctly linked.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12920-021-00871-9.
References
- 1.Bozal-Basterra L, Martin-Ruiz I, Pirone L, Liang Y, Sigurethsson JO, Gonzalez-Santamarta M, Giordano I, Gabicagogeascoa E, de Luca A, Rodriguez JA, et al. Truncated SALL1 impedes primary cilia function in Townes-Brocks syndrome. Am J Hum Genet. 2018;102(2):249–265. doi: 10.1016/j.ajhg.2017.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sudo Y, Numakura C, Abe A, Aiba S, Matsunaga A, Hayasaka K. Phenotypic variability in a family with Townes-Brocks syndrome. J Hum Genet. 2010;55(8):550–551. doi: 10.1038/jhg.2010.64. [DOI] [PubMed] [Google Scholar]
- 3.Eker HK, Balasar O. Variable expressivity of renal involvement in a further family with Townes-Brocks syndrome. Clin Dysmorphol. 2015;24(1):24–25. doi: 10.1097/MCD.0000000000000054. [DOI] [PubMed] [Google Scholar]
- 4.Elkaiali L, Ratliff K, Oueis H. Dental treatment considerations for children with complex medical histories: a case of Townes-Brock Syndrome. J Mich Dent Assoc. 2016;98(1):32–34. [PubMed] [Google Scholar]
- 5.Townes PL, Brocks ER. Hereditary syndrome of imperforate anus with hand, foot, and ear anomalies. J Pediatr. 1972;81(2):321–326. doi: 10.1016/S0022-3476(72)80302-0. [DOI] [PubMed] [Google Scholar]
- 6.Kohlhase J. Townes-Brocks syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews((R)). Seattle (WA); 1993. [PubMed]
- 7.Kohlhase J, Wischermann A, Reichenbach H, Froster U, Engel W. Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat Genet. 1998;18(1):81–83. doi: 10.1038/ng0198-81. [DOI] [PubMed] [Google Scholar]
- 8.Kiefer SM, McDill BW, Yang J, Rauchman M. Murine Sall1 represses transcription by recruiting a histone deacetylase complex. J Biol Chem. 2002;277(17):14869–14876. doi: 10.1074/jbc.M200052200. [DOI] [PubMed] [Google Scholar]
- 9.Liberalesso PBN, Cordeiro ML, Karuta SCV, Koladicz KRJ, Nitsche A, Zeigelboim BS, Raskin S, Rauchman M. Phenotypic and genotypic aspects of Townes-Brock syndrome: case report of patient in southern Brazil with a new SALL1 hotspot region nonsense mutation. BMC Med Genet. 2017;18(1):125. doi: 10.1186/s12881-017-0483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Netzer C, Rieger L, Brero A, Zhang CD, Hinzke M, Kohlhase J, Bohlander SK. SALL1, the gene mutated in Townes-Brocks syndrome, encodes a transcriptional repressor which interacts with TRF1/PIN2 and localizes to pericentromeric heterochromatin. Hum Mol Genet. 2001;10(26):3017–3024. doi: 10.1093/hmg/10.26.3017. [DOI] [PubMed] [Google Scholar]
- 11.Lopes Novo C, Rugg-Gunn P. Crosstalk between pluripotency factors and higher-order chromatin organization. Nucleus. 2016;7(5):447–452. doi: 10.1080/19491034.2016.1248013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bozal-Basterra L, Gonzalez-Santamarta M, Muratore V, Bermejo-Arteagabeitia A, Da Fonseca C, Barroso-Gomila O, Azkargorta M, Iloro I, Pampliega O, Andrade R, et al. LUZP1, a novel regulator of primary cilia and the actin cytoskeleton, is a contributing factor in Townes-Brocks Syndrome. Elife. 2020;9:e55957. doi: 10.7554/eLife.55957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korver AM, Smith RJ, Van Camp G, Schleiss MR, Bitner-Glindzicz MA, Lustig LR, Usami SI, Boudewyns AN. Congenital hearing loss. Nat Rev Dis Primers. 2017;3:16094. doi: 10.1038/nrdp.2016.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shahin H, Rahil M, Abu Rayan A, Avraham KB, King MC, Kanaan M, Walsh T. Nonsense mutation of the stereociliar membrane protein gene PTPRQ in human hearing loss DFNB84. J Med Genet. 2010;47(9):643–645. doi: 10.1136/jmg.2009.075697. [DOI] [PubMed] [Google Scholar]
- 15.Ozieblo D, Sarosiak A, Leja ML, Budde BS, Tacikowska G, Di Donato N, Bolz HJ, Nurnberg P, Skarzynski H, Oldak M. First confirmatory study on PTPRQ as an autosomal dominant non-syndromic hearing loss gene. J Transl Med. 2019;17(1):351. doi: 10.1186/s12967-019-2099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenberger T, Di Donato N, Decker C, Delle Vedove A, Neuhaus C, Nurnberg G, Toliat M, Nurnberg P, Murbe D, Bolz HJ. A C-terminal nonsense mutation links PTPRQ with autosomal-dominant hearing loss, DFNA73. Genet Med. 2018;20(6):614–621. doi: 10.1038/gim.2017.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan Z, Zeng H, Xu Z, Tian Q, Gao X, Zhou C, Zheng Y, Wang J, Ling G, Wang B, et al. Identification of ANKDD1B variants in an ankylosing spondylitis pedigree and a sporadic patient. BMC Med Genet. 2018;19(1):111. doi: 10.1186/s12881-018-0622-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang W, Li D, Wei S, Guo T, Wang J, Luo H, Yang Y, Tan Z. Whole-exome sequencing identifies a novel CCDC151 mutation, c.325G>T (p.E109X), in a patient with primary ciliary dyskinesia and situs inversus. J Hum Genet. 2019;64(3):249–252. doi: 10.1038/s10038-018-0540-x. [DOI] [PubMed] [Google Scholar]
- 19.Fan LL, Chen YQ, Huang H, Jin JY, Li JJ, Tan ZP. Whole-exome sequencing reveals doubly novel heterozygous Myosin Binding Protein C and Titin mutations in a Chinese patient with severe dilated cardiomyopathy. Cardiol Young. 2018;28(12):1410–1414. doi: 10.1017/S1047951118001403. [DOI] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Botzenhart EM, Green A, Ilyina H, Konig R, Lowry RB, Lo IF, Shohat M, Burke L, McGaughran J, Chafai R, et al. SALL1 mutation analysis in Townes-Brocks syndrome: twelve novel mutations and expansion of the phenotype. Hum Mutat. 2005;26(3):282. doi: 10.1002/humu.9362. [DOI] [PubMed] [Google Scholar]
- 22.Chai L, Yang J, Di C, Cui W, Kawakami K, Lai R, Ma Y. Transcriptional activation of the SALL1 by the human SIX1 homeodomain during kidney development. J Biol Chem. 2006;281(28):18918–18926. doi: 10.1074/jbc.M600180200. [DOI] [PubMed] [Google Scholar]
- 23.Webb BD, Metikala S, Wheeler PG, Sherpa MD, Houten SM, Horb ME, Schadt EE. Heterozygous pathogenic variant in DACT1 causes an autosomal-dominant syndrome with features overlapping Townes-Brocks syndrome. Hum Mutat. 2017;38(4):373–377. doi: 10.1002/humu.23171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodyear RJ, Jones SM, Sharifi L, Forge A, Richardson GP. Hair bundle defects and loss of function in the vestibular end organs of mice lacking the receptor-like inositol lipid phosphatase PTPRQ. J Neurosci. 2012;32(8):2762–2772. doi: 10.1523/JNEUROSCI.3635-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nayak G, Goodyear RJ, Legan PK, Noda M, Richardson GP. Evidence for multiple, developmentally regulated isoforms of Ptprq on hair cells of the inner ear. Dev Neurobiol. 2011;71(2):129–141. doi: 10.1002/dneu.20831. [DOI] [PubMed] [Google Scholar]
- 26.Schraders M, Oostrik J, Huygen PL, Strom TM, van Wijk E, Kunst HP, Hoefsloot LH, Cremers CW, Admiraal RJ, Kremer H. Mutations in PTPRQ are a cause of autosomal-recessive nonsyndromic hearing impairment DFNB84 and associated with vestibular dysfunction. Am J Hum Genet. 2010;86(4):604–610. doi: 10.1016/j.ajhg.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakaguchi H, Tokita J, Naoz M, Bowen-Pope D, Gov NS, Kachar B. Dynamic compartmentalization of protein tyrosine phosphatase receptor Q at the proximal end of stereocilia: implication of myosin VI-based transport. Cell Motil Cytoskeleton. 2008;65(7):528–538. doi: 10.1002/cm.20275. [DOI] [PubMed] [Google Scholar]
- 28.Goodyear RJ, Legan PK, Wright MB, Marcotti W, Oganesian A, Coats SA, Booth CJ, Kros CJ, Seifert RA, Bowen-Pope DF, et al. A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J Neurosci. 2003;23(27):9208–9219. doi: 10.1523/JNEUROSCI.23-27-09208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sang Q, Mei H, Kuermanhan A, Feng R, Guo L, Qu R, Xu Y, Li H, Jin L, He L, et al. Identification of a novel compound heterozygous mutation in PTPRQ in a DFNB84 family with prelingual sensorineural hearing impairment. Mol Genet Genomics. 2015;290(3):1135–1139. doi: 10.1007/s00438-014-0979-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. The sequencing data of III-3.
Additional file 2. The sequencing data of II-3.
Data Availability Statement
The data used to support the findings of this study are included within the article.