

Abstract
Two striated muscle myosins, namely skeletal muscle myosin (SkM) and cardiac myosin (CM), may potentially contribute to physiologic mechanisms for regulation of thrombosis and hemostasis. Thrombin is generated from activation of prothrombin by the prothrombinase (IIase) complex comprising factor Xa, factor Va, and Ca++ ions located on surfaces where these factors are assembled. We discovered that SkM and CM, which are abundant motor proteins in skeletal and cardiac muscles, can provide a surface for thrombin generation by the prothrombinase complex without any apparent requirement for phosphatidylserine or lipids. These myosins can also provide a surface that supports the inactivation of factor Va by activated protein C/protein S, resulting in negative feedback downregulation of thrombin generation. While the physiologic significance of these reactions remains to be established for humans, substantive insights may be gleaned from murine studies. In mice, exogenously infused SkM and CM can promote hemostasis as they are capable of reducing tail cut bleeding. In a murine myocardial ischemia-reperfusion injury model, exogenously infused CM exacerbates myocardial infarction damage. Studies of human plasmas show that SkM antigen isoforms of different MWs circulate in human plasma, and they can be used to identify 3 plasma SkM phenotypes. A pilot clinical study showed that one SkM isoform pattern appeared to be linked to isolated pulmonary embolism. These discoveries enable multiple preclinical and clinical studies of SkM and CM which should provide novel mechanistic insights with potential translational relevance for the roles of CM and SkM in the pathobiology of hemostasis and thrombosis.
Keywords: myosin, blood coagulation, thrombin, factor X, prothrombinase, myocardial infarction
1. INTRODUCTION – DISCOVERY OF SKELETAL MUSCLE MYOSIN’S PROCOAGULANT ACTIVITY
Thrombosis represents a failure to regulate thrombin generation and contributes to morbidity and mortality in many patients, including those with acute myocardial infarction, ischemic stroke, venous thrombosis (VTE), cancer, and COVID-19. In spite of detailed knowledge about the coagulation pathways and recent additions of new arrows representing connections between plasma components and cellular factors, about contributions of blood cells to immunothrombosis, and about multiple platelet-dependent contributions to coagulation [1–11], new discoveries are needed to expand basic knowledge about mechanisms underlying blood clotting pathologies. Unprovoked VTE in younger patients can be driven by genetic variations that cause hypercoagulability, and we believe that significant discoveries may come from identifying rare genetic variants linked to unprovoked VTE. Therefore, when exome rare variant genotyping chips containing >300,000 SNPs first became available, we used this tool for a pilot study for discovery of VTE-linked genetic rare variants in the Scripps Venous Thrombosis registry (subjects < 55 years old). When rare variants in chr17p13.1 MYH genes (clustered skeletal muscle myosin (SkM) heavy chain genes [12]) were analyzed using collapsing methods, the results suggested their association with recurrent VTE (p = 2.70 ×10−16) [13]. This suggested association of recurrent VTE with rare variants in SkM genes led us to hypothesize that SkM is a potentially physiologic and/or pathologic procoagulant factor.
Because SkM was not known to affect any conventional coagulation pathway, plasma coagulation assays were initially used to probe for potential procoagulant effects of SkM. Those assays led to the discovery of SkM’s procoagulant activity which found to be based on its ability to provide a surface that binds factor (F) Xa and FVa to promote prothrombin activation [14]. Subsequently we found that SkM would also replace phospholipid surfaces and promote FVa inactivation by activated protein C (APC)/protein S [15]. Then, building on SkM’s procoagulant activity, we found that another striated muscle myosin, namely cardiac myosin (CM), was procoagulant similar to SkM by similar mechanisms and that CM could modify myocardial damage in murine myocardial ischemia reperfusion studies [16]. This review summarizes current knowledge regarding the procoagulant and anticoagulant properties of SkM and CM and also regarding some aspects of their mechanisms. This knowledge provides a basis for future research to enhance our understanding of how these abundant proteins might influence the regulation of thrombosis and hemostasis with potential new insights that might be translated to the clinic.
2. MYOSIN STRUCTURE AND NOMENCLATURE
Myosins are a large family of motor proteins sharing common features of ATP hydrolysis, actin binding, and potential for kinetic energy transduction. The conventional myosins consist of a dimer of heterotrimers, and they are very abundant and constitute primary contractile elements in the sarcomeres of skeletal muscle, cardiac muscle, and smooth muscle. SkM was originally isolated from muscle cells but is found throughout the body, even at low levels in plasma (4–25 nmol/L) [17, 18]. The “conventional” isoform of muscle myosin contains 6 polypeptides, specifically 2 heavy chains (HC) and 4 light chains [2 “essential” light chains (ELC) and 2 “regulatory” light chains (RLC)] (Figure 1A, 1B) [19–22]. Myosin can be split by proteases to yield light meromyosin (LMM) and heavy meromyosin (HMM), the latter of which can be further split into 2 subfragments designated S1 fragment and S2 fragment [23] (Figure 1A). Overall lengths of SkM and the S1 fragment are ~ 1,900 Å and ~ 190 Å, respectively. The S1 fragment contains two “head” regions, each of which has the N-terminus of the HC and the neck region. The HC N-terminal domain comprises subsections with well-defined functions for the myosin motor, including an HC “converter” domain, an upper 50K domain, and a lower 50 K domain. These domains provide, as labeled in the upper region of Figure 1B, a binding pocket for ATP and the actin binding site [19–22]. The polypeptides of three chains of SkM define the so-called “neck region”, as seen in the lower region of Figure 1B, where both the helix-rich ELC and RLC each wraps around an extended, bent helical segment of the HC. This neck region is ~ 70 Å long (lower region in Figure 1B) and, as described in detail below, provides at least part of myosin’s procoagulant surface [24].
Figure 1. Conventional Myosin Structure and Nomenclature.
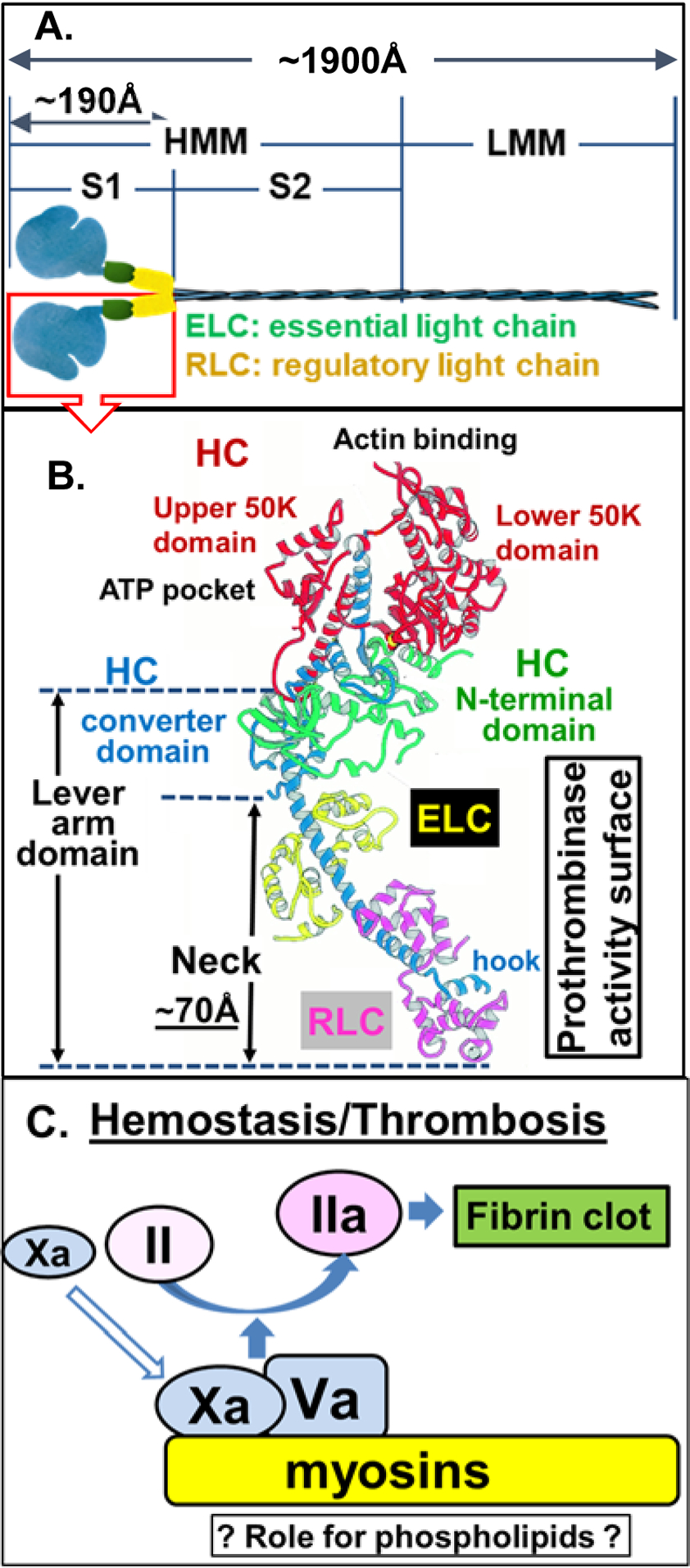
(A) Structure of conventional myosins which contain 6 polypeptides, 2 heavy chains (HC), and 4 light chains [2 ‘essential’ and 2 ‘regulatory’ light chains (ELC and RLC, respectively)]. Conventional myosin can be split into 1 light meromyosin (LMM) fragment and 1 heavy meromyosin (HMM) fragment; HMM can be further split into two subfragments, designated S1 and S2 subfragments [23]. (B) Structure of conventional myosin S1 domain containing a neck (N-terminal HC, ELC and RLC, the hypothesized procoagulant surface [24]), a converter domain (HC followed by neck), upper and lower 50 K domains that contain actin and ATP binding sites, in the N-terminal domain [19–22]. Structure shown in (B) is adopted with modifications from Rayment et al [21]. (C) Either SkM or CM (myosins) which is depicted on a yellow background can bind factors Xa and Va and thereby can enhance prothrombin activation on its surface. Kinetic studies indicate that the potency for SkM and CM preparations to enhance prothrombin activation is generally comparable to that of procoagulant phospholipid vesicles (Table 1). SkM preparations contain very low levels of phosphatidylserine [15], raising the queston of whether myosin-bound phospholipids may contribute to enhance SkM’s or CM’s procoagulant activity.
3. PROCOAGULANT ACTIVITIES OF SkM AND CM
3.1. SkM and procoagulant activity
Thrombogenicity of SkM was seen when fresh human blood was exposed ex vivo to myosin-coated surfaces or when exogenous SkM was added to human whole blood [14, 16, 25]. SkM dose-dependently increased tissue factor (TF)-induced thrombin generation (12.5–100 nM) both in platelet-rich and platelet-poor plasmas and in normal plasma, even in the presence of phospholipid vesicles [14, 16]. In purified prothrombinase (IIase) assays using only FXa and FVa, prothrombin and Ca++ ions, SkM (0.5 nM-2 μM) enhanced thrombin generation [14]. Direct binding of inactivated (i) FXai to immobilized SkM was observed using Bilayer interferometry [14]. The Kd for FXai binding to SkM was 46 nM without FVa, and FVa reduced Kd for FXai to ~ 1.0 nM. The overall concept that binding of FXa and FVa to these myosins enables prothrombin activation is depicted in Figure 1C. Kinetic studies that determined kcat values for prothrombin activation by myosin-bound FXa:FVa showed that the IIase enhancement potency of SkM was generally on the same order of magnitude as that for enhancement by phospholipid vesicles or platelets (Table 1) [14].
Table 1.
Comparison of kcat values for human prothrombinase activity
| Source | Prothrombinase complex surface | PL Conc. | FXa Conc. | FVa Conc. | |
|---|---|---|---|---|---|
| Moyer et al [52] | PC/PS (75/25) | 5.0 μM | 5.0 nM | 5.0 nM | 480 |
| Deguchi et al [14] | SkM | - | 0.2 nM | 5.0 nM | 490 |
| Zilberman-Rudenko et al [16] | CM | - | 0.2 nM | 5.0 nM | 371 |
| Deguchi et al [14] | PC/PS (80/20) | 20 μM | 0.2 nM | 5.0 nM | 396 |
| Krishnaswamy et al [51] | PC/PS (75/25) | 30 μM | 1 nM | 4.1 nM | 1,344 |
| Bunce et al [53] | Platelet (1×108/mL) | 0.1 nM | 10 nM | 560 |
kcat (Vmax/[FXa]) values for SkM, CM, phospholipids, and activated platelets are summarized. For comparisons in this Table, note that experimental conditions included modest variations, inter alia, in assay buffer contents (Tris vs. HEPES, 2 mM vs. 5 mM CaCl2 concentrations, etc.), different temperatures and different phospholipid (PC/PS) ratios. See references [14, 51–53] for details about differences in assay contents and conditions. This Table is modified from previous papers [14, 16]. Abbreviations: PL, phospholipid; FXa, Factor Xa; FVa, Factor Va; PC/PS, phosphatidylcholine/phosphatidylserine.
3.2. SkM’s “neck region” binds FXa to support IIase
Since trifluoperazine (TFP) inhibited SkM-supported IIase enhancement and TFP binds to the ELC in the neck region, we hypothesized that the ELC and possibly other polypeptides in the SkM neck region (Figures 1, 2) might bind FXa to promote IIase activity. Based on the premise that SkM amino acid sequences which bind FXa would be competitive inhibitors of SkM:FXa protein-protein interactions, we made and tested SkM-based synthetic peptides for inhibition of SkM’s enhancement of IIase activity. These efforts successfully identified some peptides that inhibited SkM-enhanced IIase activity (Figure 3A and 3B) and that bound FXa (Figure 3C), leading us to identify an hypothetical “IIase cofactor surface” (Figures 1B, 2B, 2C) [24] that includes ELC residues 129–138, RLC residues 133–162 and HC residues 816–835 in the neck region of SkM for IIase-enhancing activity. Further experiments using myosin-based peptides for RLC133–162, RLC130–145 and RLC152–164 helped to implicate RLC residues 152–164 as a key sequence within RLC133–162 for IIase-enhancing activity (unpublished data). Thus, we identified peptides with sequences of SkM’s HC, ELC and RLC in the neck region which inhibited SkM’s IIase activity, and these sequences are located, more or less, on one surface of myosin’s neck region [24]. It is interesting to keep in mind the overall dimensions for the IIase complex of FXa:FVa (Figure 2A) that is central for prothrombin activation and the dimensions of SkM’s neck region (Figure 2B). For phospholipid membrane-bound FXa, the distance from the membrane to the active site in the protease domain of FXa is approximately 70–75 Å (Figure 2A) [26, 27] which is close to the length of SkM neck region (~ 70Å) (Figure 2B, 2C) where it is very likely that FXa and FVa are interacting to promote prothrombin activation. The dimensions of FVa are greater than those of FXa, and the FXa:FVa complex overall dimensions exceed those of the neck region (Figure 2A, 2B). We have no clear picture for the orientation of either FXa or FVa bound to SkM’s neck region, but we speculate that the IIase complex, which is bound to the neck region, most likely extends up and/or down the HC structure (Figure 2B, 2C), implying that further studies of HC structures and peptides may give further mechanistic insights for SkM’s IIase activity.
Figure 2. Dimensions for space-filling structures of FXa:FVa complex and of SkM’s neck region.
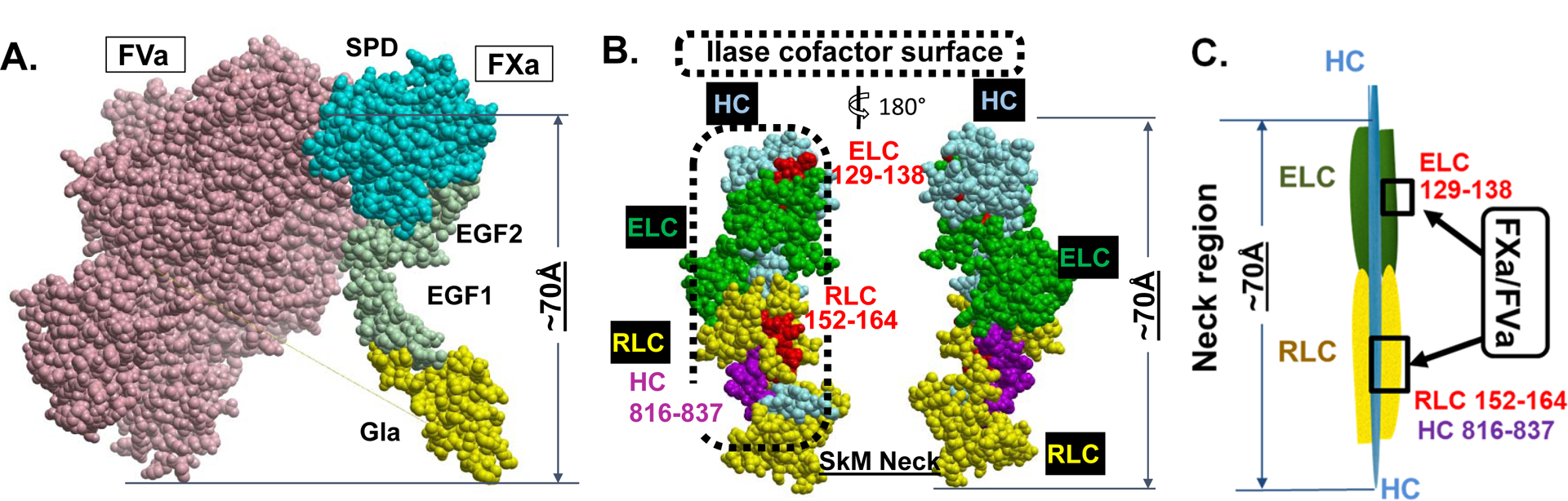
(A) 3-dimensional structure computer model for FXa/FVa complex (from Autin et al [27]). (B) Candidate SkM sequences in neck region that are hypothesized to form at least part of the IIase surface in space-filling format. Each structure is displayed as front and back views, related by a 180 degree rotation. (C) Simplified scheme and its length depicting the FXa/FVa interaction sites on the neck region of SkM.
Figure 3. Experimental data from studies indicating that SkM and CM proteins are essential for enhancing prothrombin activation.
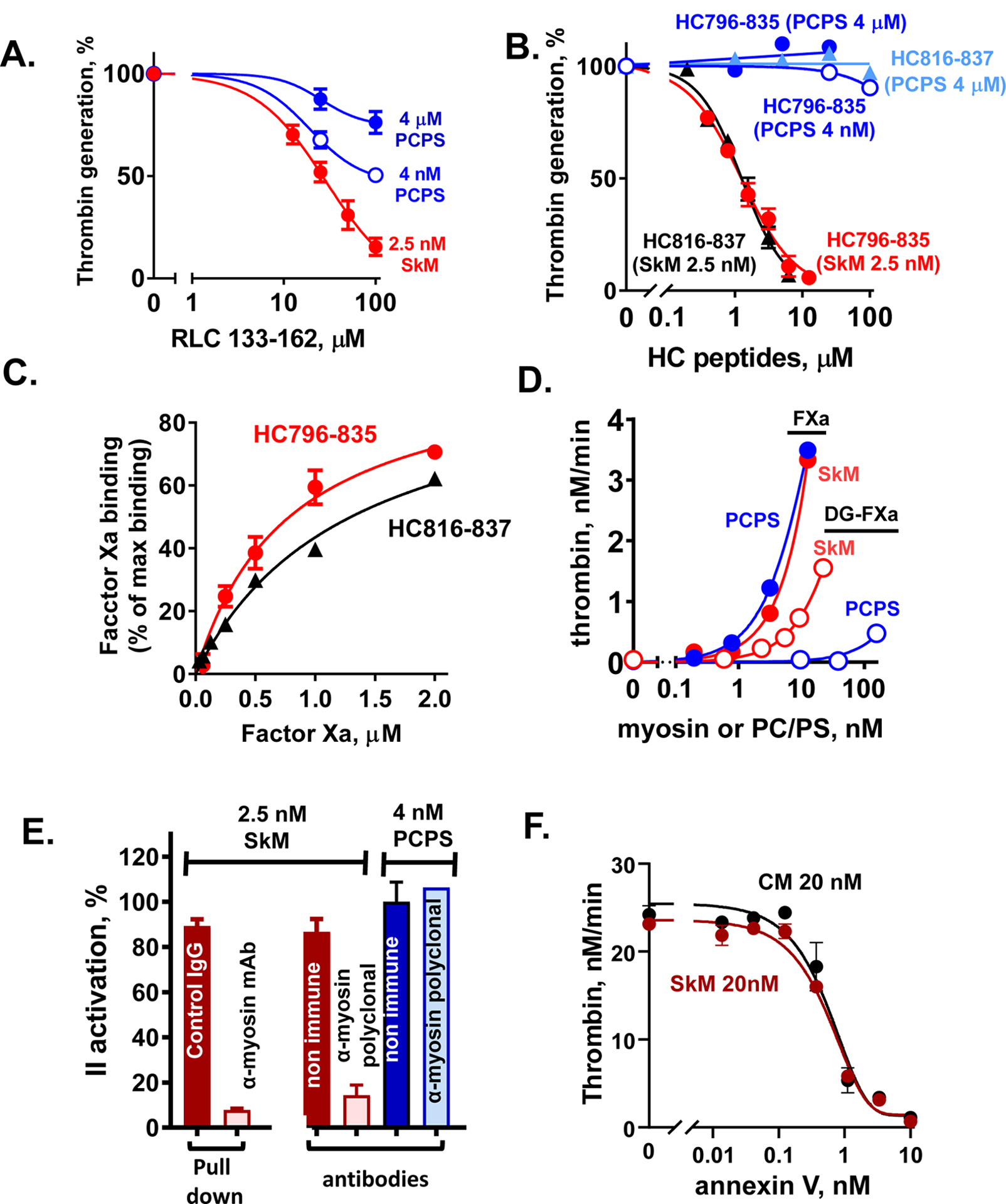
(A, B) Inhibition of myosin-enhanced or phospholipid vesicle-enhanced prothrombin activation by SkM-sequence based peptides (RLC133–162 (A) and HC796–835 and HC816–836 (B)) was studied in the presence of SkM (2.5 nM, final) or phospholipid vesicles (PC/PS, 80%/20% w/w) (4 μM or 4 nM, final) as described [24]. Thrombin generation was determined as described [24]. 100 % was the value thrombin generation for controls in the absence of added peptides. (C) The binding of FXa, as determined using FXa chromogenic activity assays, to immobilized HC peptides was tested as described [24]. Each value represents the mean [SD] of at least triplicate determinations. (D) The effect of varying concentrations of SkM or of PC/PS (80%/20%) vesicles on the initial rate of prothrombin activation by FXa or Gla domain-less FXa (DG-FXa) (0.2 nM, final) was determined as described [14]. (E) Anti-myosin antibodies were used to pull-down SkM from solution or to block myosin’s procoagulant activity, as described [14]. From left to right, 2 studies are depicted. First, immobilized mAb-MF20 removed > 90 % of SkM’s procoagulant activity from solution vs. control non-immune IgG. Second, polyclonal anti-myosin antibodies in solution blocked > 80% of SkM’s procoagulant activity vs. controls (two red bars), and there were no significant effects on the procoagulant activity of PC/PS vesicles by anti-myosin or non-immune IgG’s (two blue bars). (F) The effects of varying concentrations of annexin V on the initial rate of prothrombin activation by FXa/FVa in the presence of SkM (closed symbols) or CM (open symbols) (each at 20 nM final) were determined. Annexin V was incubated with FVa (2.4 nM, final) and FXa (0.2 nM, final) in Tris-buffered saline, 0.5% BSA plus 5 mM CaCl2 with SkM or CM, and thrombin generation was initiated by adding prothrombin (0.75 μM, final). After 5 min, quenching was made with EDTA (10 mM, final) and thrombin formation was quantified. For all studies described in this figure, as for our published studies [13–16, 24, 25], the SkM or CM preparation was dissolved in water and then immediately dialyzed into Tris buffer (pH 7.4) containing 0.6 M NaCl at 4°C. After dialysis, particles causing turbidity were removed by high speed centrifugation (21,130xg for 1 min) which removed visible aggregates.
3.3. CM and procoagulant activity
SkM and CM serve as motor proteins engaged in translating chemical signals into motion, and they are highly abundant in skeletal and cardiac muscles. The muscle myosin protein is a large complex structure, and SkM and CM are each a very similar dimer of heterotrimers. CM and SkM belong to the family of conventional striated muscle myosins, and their amino acid sequences are relatively highly conserved; e.g., generally CM has ~ 80% sequence identity to SkM [28, 29]. These similarities led us to evaluate the procoagulant properties of CM which are found to exhibit many procoagulant properties similar to those of SkM, as described above. Perfusion of fresh human blood over CM-coated surfaces caused extensive thrombus formation and fibrin deposition [16]. Addition of CM to blood enhanced fibrin formation [16, 25] and enhanced thrombin generation in whole blood, in platelet-rich and platelet-poor plasmas, and in mixtures of purified factors Xa and Va plus prothrombin [16]. The procoagulant potency of CM, based on kcat values for prothrombin activation by FXa:FVa, is generally similar to that of SkM [16] (Table 1). Moreover, direct binding studies with purified proteins showed that CM binds FXa, supporting the model that CM, like SkM, binds FXa:FVa to enhance prothrombin activation [16]. Quite remarkably, when CM was administered i.v. during murine ischemia reperfusion injury studies, CM exacerbated myocardial damage in terms of myocardial infarct volume and circulating troponin I levels, consistent with CM’s in vitro procoagulant activity [16] (see below for more discussion). This raises the speculation that various coronary pathologic processes that expose CM to blood may promote coagulation reactions which exacerbate cardiac muscle damage, suggesting that inhibitors which target CM’s procoagulant activity may reduce cardiac muscle damage under certain circumstances.
3.4. SkM’s IIase enhancement not due to contaminating phosphatidylserine vesicles
A recent study showed that annexin V, phospholipase A2, and lactadherin blocked SkM’s and CM’s enhancement of IIase [30]. Because these proteins are known to target phosphatidylserine (PS), the authors asserted, without any direct measurements of PS or other functional assays, that contaminating PS-vesicles cause and explain IIase enhancement by SkM and CM preparations, implying no role for the myosin protein. But annexin V and lactadherin are globular proteins that can bind not only PS but also many proteins (e.g., actin, elastin, integrins, amyloid β-peptide, protein kinase C, and multiple cell receptors) and other lipids [31–49]. Thus, there was reasonably an unfulfilled responsibility [30] for the authors to quantify PS and/or to show with reagents more specific for PS (e.g., anti-PS monoclonal antibody blockade) that contaminating PS was present in sufficient quantity to explain myosins’ enhancement of IIase.
Multiple observations, whose merits were not adequately recognized by Novakovic and Gilbert, indicate that the authors’ assertion for contaminating PS to be the causal explanation for myosin preparation’s IIase enhancement, to the exclusion of a central role for the myosin protein, was a gross oversimplification of the authors’ limited data [30]. First, the concentration of PS in the SkM preparation was determined [15]. The relatively small amount of PS present in a vial of SkM obtained by us or Gilbert from the manufacture (Cytoskeletal Inc), was approximately 0.90 μmol PS per 40. μmol SkM (Table 2)[15]. Clearly this amount of PS present in SkM preparations could not explain SkM’s procoagulant activity. For example, standardized purified prothrombinase reaction mixture assays show that 10 nM SkM enables formation of 3 nmol thrombin/min and 0.22 nM PS in 1.1 nM phosphatidylcholine (PC)/PS(20%) vesicles enabled formation of only 0.4 nmol thrombin/min (Figure 3D). Second, because the N-terminal gamma-carboxyglutamic acid (Gla) of FXa mediates its binding to PS-containing phospholipid vesicles, Gla-domainless (DG)-FXa was used to test for an essential role for PS in myosin’s enhancement of prothrombinase. DG-FXa has lost phospholipid vesicle enhancement of IIase (> 99% loss of activity) but DG-FXa retains ~ 30 % activity for SkM’s enhancement of prothrombin activation (Figure 3D), and DG-FXa still binds to SkM [14, 16]. Gilbert reported that DG-FXa had only 1 % of FXa activity [30] without any recognition or discussion of our published data. The assertion that contaminating PS-vesicles explain SkM’s enhancement of prothrombin activation is contradicted by the facts (Figure 3D and Table 2).
Table 2. Phosphatidylserine (PS) content of 40 μmol/L of skeletal muscle myosin (SkM).
SkM (from Cytoskeletal Inc.) was analyzed for PS content by liquid chromatography–mass spectrometry (AVANTI POLAR LIPIDS Inc., Alabaster AL). The SkM material that was submitted for analysis was reconstituted in water without further dialysis or centrifugation and a frozen sample was submitted. This same source for SkM and same handling procedures were used for recently published studies of SkM [30] (G. Gilbert, personal communication) implying that the recent report’s SkM had this same PS content as shown here.
| side chain s | ng/mL | PS, nM |
|---|---|---|
| 18:0 | 0.0 | 0.04 |
| 34:2 | 3.8 | 4.8 |
| 34:1 | 10.5 | 13.4 |
| 34:0 | 1.0 | 1.3 |
| 36:3 | 8.1 | 10.1 |
| 36:2 | 208.3 | 259.1 |
| 36:1 | 168.9 | 209.6 |
| 36:0 | 19.2 | 23.8 |
| 38:6 | 0.2 | 0.24 |
| 38:5 | 1.3 | 1.5 |
| 38:4 | 32.8 | 39.5 |
| 38:3 | 32.9 | 39.5 |
| 38:2 | 10.9 | 13.0 |
| 38:1 | 5.0 | 5.9 |
| 40:6 | 6.5 | 7.5 |
| 40:5 | 75.5 | 86.8 |
| 40:4 | 145.7 | 167.5 |
| 40:3 | 16.7 | 19.2 |
| total | 747.2 | 902.8 |
Data in peer-reviewed papers reporting biochemical studies using myosin-sequence based peptides [24] or the inhibitory effects of anti-myosin antibodies [14, 16] (and unpublished data) to show a central role for the myosin protein were given no experimental assessment although the testable hypothesis for the published peptide data was advanced that helical myosin peptides might inhibit SkM IIase activity by binding PS [30]. The SkM heavy chain peptides, HC796–835 and its truncated version HC816–837, inhibit SkM-enhanced IIase (Figure 3B) and directly bind FXa (Figure 3C). In contrast, these peptides do not inhibit PS-vesicle-enhanced IIase (Figure 3B). This implies that HC816–837 inhibits SkM-FXa protein-protein interactions, not phospholipid interactions with FXa or FVa or prothrombin. In key studies, SkM removal from solution using immobilized monoclonal anti-myosin antibody (MF20) reduced IIase enhancement activity >90% (Figure 3E). Other studies showed that anti-myosin polyclonal antibodies that do not block IIase activity of PC/PS vesicles do significantly inhibit SkM’s IIase enhancement by >80% (Figure 3E). Additionally, we found that an anti-PS monoclonal antibody blocked >90% PC/PS vesicle-supported IIase activity but did not significantly block SkM-supported IIase (manuscript submitted). Thus, many studies using SkM-based peptides and various anti-SkM antibodies imply that the myosin protein itself is the central component for SkM’s IIase enhancement, in strong contradiction of the assertion [30] that contaminating PS causes SkM’s IIase activity.
Nothing rules out the possibility that SkM or CM may involve myosin-bound procoagulant lipid(s) independent of PS (Figure 1C). One may speculate that myosin-bound lipid(s) themselves do not affect the coagulation system reactions while they may stabilize the myosin proteins. Lipids are present in some non-muscle myosin preparations and appear to stabilize myosin. If lipids bind to myosin in the neck region, which is the key region for the SkM’s IIase cofactor activity, the bound lipid might interfere with the interactions of myosins with clotting factors or might stabilize protein-protein interactions. We confirm that annexin V inhibits the prothrombinase enhancement by SkM and CM (Figure 3F), as reported [30]. Thus, driven by the effects of annexin V and lactadherin on SkM and CM, there are a number of open questions regarding their interactions and regarding interactions of SkM or CM with lipids that need to be addressed in the future.
The recent Novakovic and Gilbert paper asserted that tissue factor (TF) contamination likely explains SkM’s and CM’s prothrombotic activity when fresh flowing human blood is exposed to myosin-coated surfaces; however, that report contained no direct measurement of TF or any direct method (e.g., anti-TF antibodies) to block directly the putative contaminating TF [30]. The reported studies involved indirect assessment of TF activities using TF pathway inhibitor which were suggestive of TF. There may be trace amounts of TF in SkM (e.g., estimated to be ≤ 3.8 pmol TF/μmol, unpublished data). However, for SkM, we are certain that contaminating TF does not explain the prothrombotic activity of SkM when it is exposed to fresh flowing human blood because studies that were made in two different laboratories using anti-rabbit TF monoclonal antibody (RbTF7–3A5) [50] to inhibit thrombus formation showed this antibody did not substantially reduce SkM’s prothrombotic activity (unpublished data and personal communication, O.J.T. McCarty). Because we were aware of those data ruling out any major role for contaminating TF for SkM’s prothrombotic activity with flowing blood, we did not test CM for any influence of TF.
3.5. Coagulation and surfaces
Activated platelet surfaces, microparticles or phospholipid vesicles enhance IIase by promoting FXa:FVa complex assembly and by tethering prothrombin and its activation intermediates to reduce Km for prothrombin [51–54]. Other mechanisms may also enhance FXa’s IIase activity (e.g., allostery, conformation changes, etc.). Notably, studies of thrombin generation in vivo show that damaged endothelium, possibly more than platelet aggregates, is a major site for thrombin generation following vessel wall damage [3, 55]. Although phospholipids are clearly required for many types of IIase assembly, SkM could provide a coagulation reaction surface to assemble IIase components and enhance IIase with some reduction of Km [14]. In some cases where SkM is exposed to FXa, FVa, and prothrombin, IIase activity may be independent of phospholipids. Indeed, some snake venom IIase complexes need no phospholipids to be potent procoagulants [56]. Another challenge of the canonical IIase-phospholipid dogma was the finding that loss of membrane binding by the prothrombin substrate caused only a modest decrease in IIase efficacy [57]. Moreover, platelet IIase data fail to fit a simple IIase-phospholipid model, as platelet subpopulations differ in IIase activity for unexplained reasons [58–61]. Mechanistic studies show that phospholipid-enhanced clotting reactions involving substrates containing a Gla-domain (e.g., FX for the extrinsic Xase, FVIIa:tissue factor) do not require a pool of phospholipid-bound substrate [62]. Catalysis can seemingly be achieved when one substrate molecule at a time binds to binary enzyme:cofactor complexes [62]. Thus, we posit that the ternary complex of myosin:FXa:FVa can process one prothrombin substrate molecule at a time for successful thrombin generation.
3.6. Smooth muscle myosin and non-muscle myosin
Smooth muscle myosin and non-muscle myosin are distinct myosin molecular species that differ notably from SkM and CM. Non-muscle myosin is located in non-muscle cells, including platelets and plays a major role for regulation of cell morphology with influences on biochemical signaling [63]. Smooth muscle myosin and non-muscle myosin HC’s are generally similar to each other, e.g., on the order of 76%, and they use similar LC isomers. They have very rather limited identity with SkM, only ~ 40% in the HC. To date, there are no reported studies of the procoagulant activity of smooth muscle and non-muscle myosins, likely due to the unavailability of such intact myosins. It is interesting to speculate about their potential procoagulant activities based on sequences of neck region polypeptides for each type of myosin (Table 3). For CM which resembles SkM’s procoagulant activity, the sequence of the SkM peptide, HC 816–837, which directly binds FXa and which potently inhibits SkM’s IIase activity [24], is 64 % identical and very homologous. This contrasts sharply with the general lack of identity for smooth muscle and non-muscle myosins’ HC corresponding sequences (23 % and 18 %, respectively) compared to SkM (Table 3), suggesting smooth muscle myosin and non-muscle myosin do not have the same binding capability for FXa in the neck region and are unlikely to provide potent IIase activity. Nonetheless, studies of these myosins to affirm this speculation are needed.
Table 3.
Comparisons and identity for amino acid sequences of the neck region polypeptides for SkM, CM, smooth muscle myosin (SmM), and non-muscle myosin (NM) for SkM peptides that inhibit myosin procoagulant activity.

|
indicates identity for amino acid residues with SkM’s sequence. The SkM MYH2 peptide HC 816–837 potently inhibits SkM-enhancement of IIase and directly binds FXa. Abbreviations: MYH, myosin heavy chain; RLC, regulatory light chain; MYLPF, myosin phosphorylatable light chain; ELC, essential light chain; MYL, myosin light chain.
3.7. SkM and von Willebrand Factor
Flood et al reported that SkM binds von Willebrand factor but does not directly bind factor VIII [64]. Thus, by binding von Willebrand factor, SkM may help deliver FVIII to sites of injury where SkM is exposed on damaged tissue. The binding of von Willebrand factor to SkM could enhance blood clotting, especially the local generation of FXa that is required for SkM’s IIase activity. Moreover, von Willebrand factor that binds to activated platelets might potentially localize circulating SkM and its procoagulant activities on platelet surfaces; however, studies of these various mechanistic possibilities have not yet been reported.
4. SkM AND THE ACTIVATED PROTEIN C ANTICOAGULANT MECHANISM
SkM can serve as a surface for prothrombin activation by FXa and FVa in purified systems by binding FXa and FVa [14, 24]. Since activated protein C (APC) cleaves FVa on phospholipid surfaces, we assessed whether SkM could provide a surface to support FVa inactivation by APC and its cofactor protein S. In studies using purified proteins, SkM enhanced FVa inactivation by APC [15]. The potency of SkM for the enhancement of FVa inactivation was similar to that of an abundant amount of phospholipids (e.g., 25 μM phospholipids) [15]. Immunoblot studies of FVa cleavages revealed that SkM enhanced Arg506 cleavage in FVa by APC in the absence of protein S whereas, in the presence of protein S, SkM enhanced both Arg506 and Arg306 cleavages in FVa by APC [15]. In other studies of SkM’s potential effects on key coagulation reactions in purified systems, SkM did not support activation of protein C by thrombin ± thrombomodulin [15]. In summary, SkM can meaningfully help maintain the hemostatic balance because not only can it promote hemostasis which needs thrombin to prevent bleeding, but it also can promote negative feedback downregulation of thrombin generation to reduce risks for thrombosis by proteolytic inactivation of FVa (Figure 4). Further studies are needed to assess the physiologic relevance of these various counterbalancing influences of SkM on thrombin generation.
Figure 4.
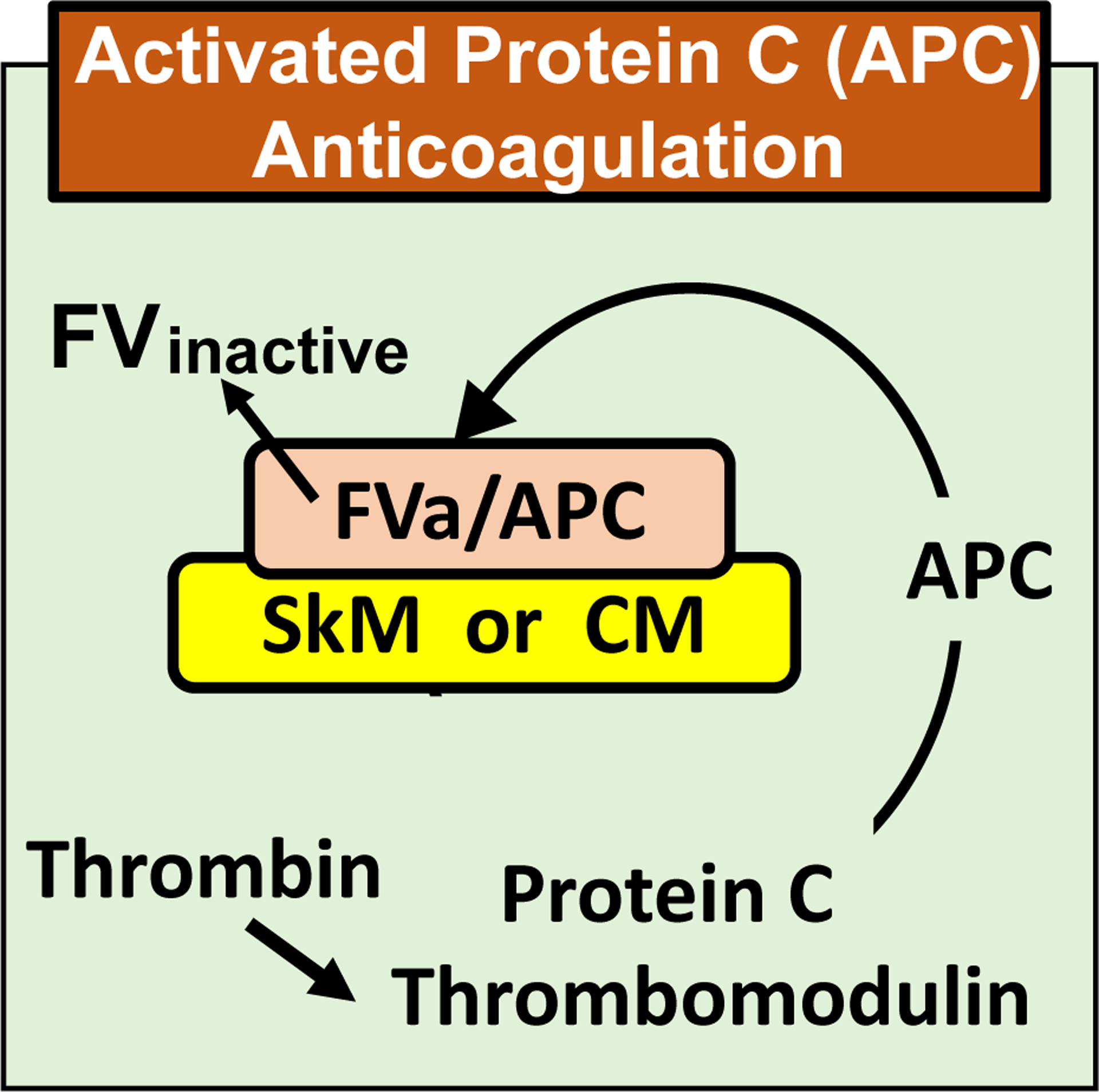
Scheme depicting the ability of myosin to bind FVa and enhance proteolytic inactivation of FVa by activated protein C (APC).
5. CARDIAC MYOSIN AND FIBRINOLYSIS
CM at high concentration was reported in one study to inhibit fibrinolysis by binding to fibrin and fibrinogen, and in another study CM, especially the tail region, were reported to promote fibrinolysis by serving as a cofactor for plasminogen activation by tissue plasminogen activator (tPA) [65, 66]. These two reports led us to characterize the effects of CM on tPA-induced clot lysis in extensive mechanistic studies [16]. Our studies on tPA-induced clot lysis showed that clotting factors which are needed for robust thrombin generation, i.e., prothrombin and factors X, V, IX, and VIII, are required for CM’s attenuation of tPA-induced clot lysis [16]. Notably, CM did not inhibit tPA-induced clot lysis in thrombin activatable fibrinolysis inhibitor (TAFI)-deficient plasma; however, reconstitution of TAFI-deficient with purified TAFI restored CM’s inhibition of clot lysis [16]. Addition of the carboxypeptidase inhibitor to the tPA-induced clot lysis assays blocked the effect of CM on clot lysis [16]. Thus, based on extensive data, the primary mechanism for CM’s inhibition of tPA-induced clot lysis, under the conditions studied, derives from the antifibrinolytic activity of TAFIa following robust thrombin generation which enables potent thrombin activation of TAFI.
6. CLINICAL ASSOCIATIONS OF SkM AND CM WITH PATHOBIOLOGY RELATED TO HEMOSTASIS AND THROMBOSIS
6.1. SkM isoforms in human plasma
SkM is present in plasma (4–25 nmol/L) [17, 18] and it is elevated in patients with muscle damage (e.g., rhabdomyolysis) [17, 18, 67, 68]. Plasma levels of SkM are frequently elevated in polymyositis and dermatomyositis patients [67], and polymyositis or dermatomyositis are associated with increased VTE risk [69]. However, since almost all previous studies concerning SkM focused primarily on muscle and not plasma SkM, there has not been clear information about what forms of SkM circulate in the blood. We detected three isoforms of SkM HC in plasma, namely, full length isoforms at 250–260 kDa, HMM at 160–170kDa, and subfragment S1 bands at 100 kDa. These findings of circulating full length SkM and other isoforms opened the door to explore the potential pathobiological contributions of SkM for VTE. We discovered three distinct phenotypes of SkM band molecular weight patterns in plasmas from different individuals, designated as follows: Type I for bands of full-length HC and of HMM plus small amounts of lesser bands including S1; Type II for bands primarily of full-length HC; and Type III for only S1 bands [70]. Although von Willebrand factor binds to SkM in purified reaction mixtures [64], it is not known whether it binds to any particular circulating SkM isoforms in human plasma.
6.2. SkM phenotypes in human plasma related to risk for pulmonary embolism
Pulmonary embolism (PE) is a major life-threatening form of VTE, and it is the third most common cause of cardiovascular death worldwide after stroke and heart attack [71, 72]. Identifying patients with high or low risk for PE occurrence is important in determining appropriate anticoagulant usage. Several clinical co-morbidities such as heart disease and cancer have been proposed as possible causes contributing to isolated PE. Our recent pilot study showed that 90% of Caucasian patients with isolated PE displayed the Type II phenotype [70], suggesting that the SkM plasma phenotype might become a useful biomarker for helping to identify risk for PE among VTE patients and eventually for possibly helping to determine appropriate VTE prophylaxis. At this time, replications with larger cohorts are required to validate the findings of this pilot study and testing different populations, e.g., African American, east Asian, etc., needs to be done.
6.3. SkM and thrombin generation in acute trauma induced coagulopathy plasmas
Acute trauma induced coagulopathy (TIC) is associated with derangements of the procoagulant and APC anticoagulant systems and of the fibrinolysis systems [73–80]. Multiple mechanism(s) for TIC have been suggested, but much remains to be clarified and translation of this knowledge to the clinic remains highly challenging. As a consequence of traumatic injury, exposure of blood to SkM, including SkM released into the circulation [18], could possibly augment the ability of blood to generate excessive thrombin or to manifest excess activities of the APC anticoagulant system [15], or to modify fibrinolysis by extensive activation of TAFI [16]. Our pilot study showed the different sensitivity of anti-myosin blocking antibodies neutralizing SkM’s procoagulant activity between trauma patient’s plasma and control plasma [14], suggesting that, myosin derived from trauma may contribute to plasma thrombin generation. However, there has never been any published study monitoring SkM plasma levels in relationship to TIC. Elucidation of new pathobiology insights into the association of TIC with circulating SkM is warranted, but admittedly this new concept that the procoagulant or anticoagulant activities of SkM may contribute to TIC pathophysiology is quite speculative.
7. IN VIVO ACTIVITIES OF SkM AND CM IN MURINE INJURY MODELS
7.1. Procoagulant activity of SkM and CM in murine tail bleeding model
The in vitro procoagulant activity of SkM and CM led us to seek in vivo proof of concept for SkM’s and CM’s procoagulant actions. Using a murine acquired hemophilia A model where mice received anti-FVIII Mab to increase bleeding following a tail cut injury, we showed that when SkM or CM is administered i.v. prior to tail cutting, both SkM and CM reduce bleeding (Figure 5A) [16]. Thus, SkM and CM in circulating blood can exert pro-hemostatic, i.e., procoagulant activity in vivo.
Figure 5. Myosin effects on in vivo hemostasis and thrombosis in murine injury models.
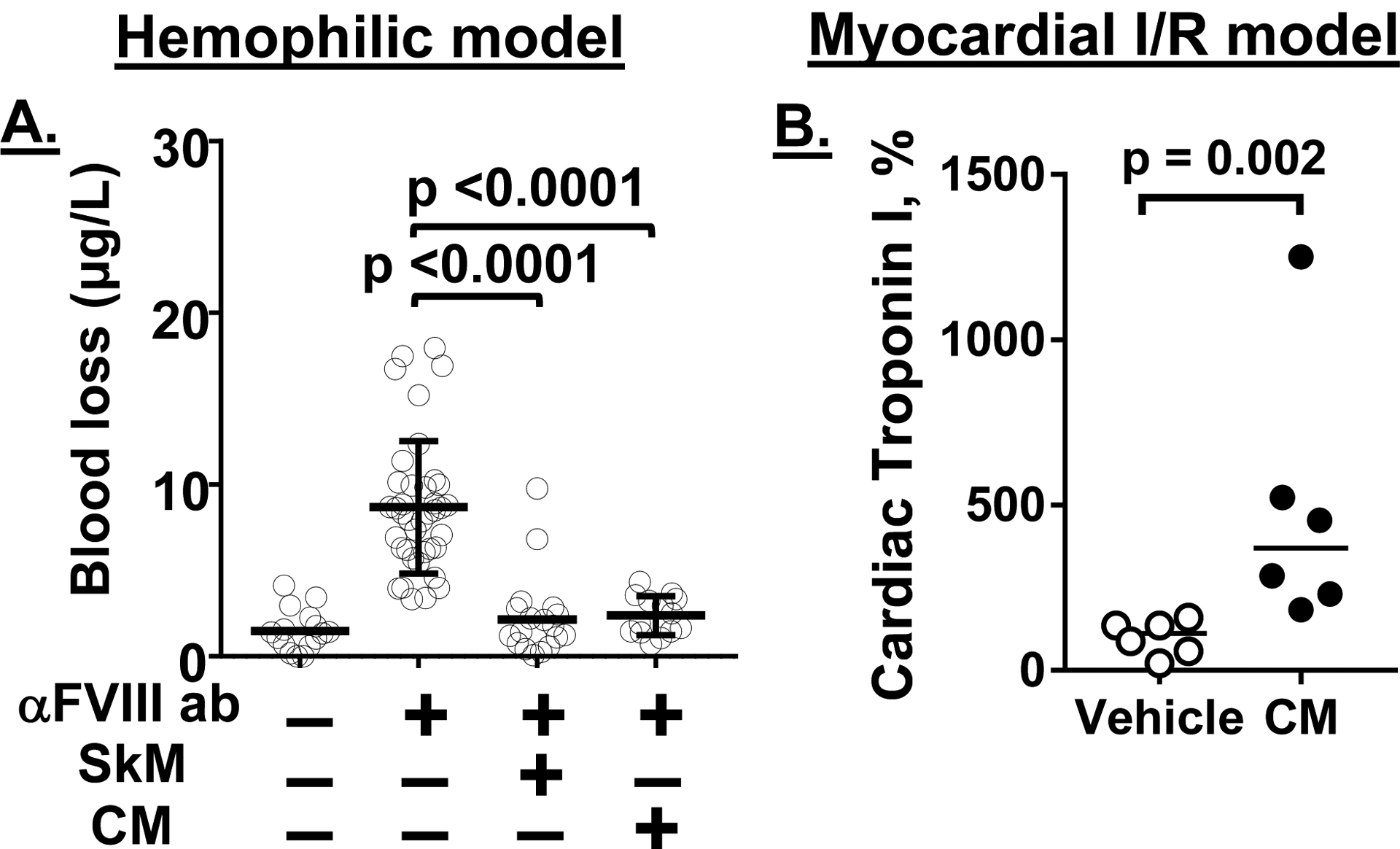
(A) Tail bleeding in acquired hemophilia A mice. wt-C57BL/6J mice were treated with anti-FVIII Mab to induce a bleeding tendency due to acquired hemophilia A. When SkM or CM (5.4 mg/kg) or vehicle was given i.v. 15 min before tail clipping and then tail bleeding after cutting was quantified, SkM and CM each very significantly reduced blood loss [13, 16]. (B) Murine myocardial ischemia reperfusion injury. When wt-C57BL/6J mice (n=6/group) subjected to myocardial ischemia reperfusion injury were given CM (5.4 mg/kg) or vehicle via intraarterial infusion at 15 min after initiation of reperfusion, serum cardiac troponin I levels measured at 3 hours after injury initiation were increased [16]. 100% was defined as the median of control vehicle group values. Bars show medians and p values were calculated by Mann-Whitney test.
For severe hemophilia A patients, there is significant inter-patient variation in bleeding phenotypes, and they can be sub-classified as having severe hemophilia A with greater or lesser bleeding clinical patterns; and currently, mechanisms for greater or lesser bleeding remain unclear [81–90]. The pro-hemostatic property of SkM in hemophilic mice leads to the speculation that differences in levels of plasma procoagulant SkM might influence hemostasis and bleeding in severe hemophilia A patients where procoagulant plasma myosin isoforms could reduce the risk of bleeding in severe hemophilia A patients. This speculation merits assessment in studies that evaluate the various SkM phenotypes among severe hemophilia A patients.
7.2. CM exacerbation of myocardial injury during murine myocardial ischemia reperfusion injury
Knowledge about any direct contributions of CM to coronary physiology in terms of the regulation of hemostasis, thrombosis or inflammation is seriously lacking. However, our first efforts for exploring potential roles for CM in coronary biology showed that CM can have a profound effect on murine myocardial ischemia reperfusion injury [16]. When infused early during the reperfusion phase of an ischemia reperfusion injury model, CM exacerbated myocardial injury as shown by CM-induced increased myocardial infarction volume and increased circulating troponin I levels (Figure 5B) [16]. Currently it is not yet clear what might be the contributions of endogenous CM to the myocardial ischemia reperfusion injury because informative studies of roles of endogenous CM have not yet been done. The deleterious effects of infused CM indicate it is very warranted to pursue studies of endogenous CM, its activities related to blood coagulation, and myocardial pathobiology in the future.
During myocardial infarction, CM, one of the major cardiomyocyte components, is released from infarcted tissue as are troponins and myosin binding protein C [91–95]. This implies that, following cardiac muscle tissue damage, the procoagulant activity of CM would be expressed when exposed to blood. Initially, this might beneficially promote thrombin’s pro-hemostatic activity to prevent or limit bleeding into the tissue and reduce damage. Subsequently, or alternatively, extended exposure of blood to CM might promote thrombin-driven inflammation and/or thrombosis [8, 11]. To rescue myocardium from ischemic obstruction and infarction, reperfusion is essential. However, reperfusion itself may cause injury, such as an additional myocardial infarction, namely, ischemia reperfusion injury which is accompanied with increased risk for morbidity and mortality [96, 97]. Ischemia and reperfusion cause damage not only in cardiomyocytes but also in the coronary circulation. Debris and release of soluble factors from the infarcted region are key factors causing ischemia reperfusion injury. Multiple reactions (e.g., microembolization, inflammation, platelet activation, etc.) can cause damage to the capillaries with microvascular obstruction and intramyocardial hemorrhage [96, 97]. During ischemia reperfusion injury, coagulation is activated with substantial thrombin generation [8, 98, 99]. Thrombin is central for the pathology of ischemia reperfusion injury, causing thrombus formation, inflammation and platelet activation [8, 11, 99]. It is possible that CM accelerates thrombin generation in the microvasculature, thereby contributing to accelerated development of ischemia reperfusion injury with microvascular thrombosis and inflammation.
The treatment of ischemia and reperfusion is a recognized target for improved therapies [96, 100]. Anti-ischemia/reperfusion injury drugs that target CM’s procoagulant actions could be a novel class of drugs that may improve microvascular perfusion and reduce vascular dysfunction. CM-targeted agents might reduce local thrombin generation and TAFI activation which could enhance fibrinolysis by inhibiting TAFI activation [16]. However, currently it is not yet clear what might be the contributions of endogenous CM to the myocardial ischemia reperfusion injury because no informative studies of roles of endogenous CM have appeared. Although it’s an attractive idea to develop and test CM-targeted anticoagulant compounds, the value for CM-targeted anticoagulants for reducing cardiac damage caused by myocardial infarction is only an interesting possibility whose merit will require much future research.
8. SUMMARY
Excessive or insufficient coagulation of blood that causes either thrombosis or bleeding, respectively, is central to morbidity and mortality in cardiovascular diseases and other pathologies. CM and SkM, which are abundant muscle motor proteins in the body, have the capacity to enhance thrombin generation as well as APC’s anticoagulant activity. In murine injury models, CM and SkM can exert pro-hemostatic activity and CM can affect myocardial infarction damage in myocardial ischemia reperfusion injury models. The potential contributions of SkM and CM to pathobiology in a variety of diseases where blood coagulation dysregulation is evident merits thoughtful future experimental investigations where the results may well have translational implications.
Essentials.
Striated muscle myosins can promote prothrombin activation by FXa or FVa inactivation by APC.
Cardiac myosin and skeletal muscle myosin are pro-hemostatic in murine tail cut bleeding models.
Infused cardiac myosin exacerbates myocardial injury caused by myocardial ischemia reperfusion.
Skeletal muscle myosin isoforms that circulate in human plasma can be grouped into 3 phenotypes.
ACKNOWLEDGMENTS
This work was supported, in part, by the National Institutes of Health grants HL133728 and HL142975 (J.H.G.).
Footnotes
CONFLICTS OF INTEREST
None of the authors has a conflict of interest to declare with regard to the content of this review.
References
- 1.Monroe DM, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol. 2006; 26: 41–8. 10.1161/01.Atv.0000193624.28251.83. [DOI] [PubMed] [Google Scholar]
- 2.Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008; 359: 938–49. 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 3.Flaumenhaft R. Thrombus formation reimagined. Blood. 2014; 124: 1697–8. 10.1182/blood-2014-06-579656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrnes JR, Wolberg AS. Red blood cells in thrombosis. Blood. 2017; 130: 1795–9. 10.1182/blood-2017-03-745349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weisel JW, Litvinov RI. Red blood cells: the forgotten player in hemostasis and thrombosis. J. Thromb. Haemost 2019; 17: 271–82. 10.1111/jth.14360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergmeier W, Antoniak S, Conway EM, Denis CV, George LA, Isermann B, Key NS, Krishnaswamy S, Lam WA, Lillicrap D, Liu J, Looney MR, Lopez JA, Maas C, Peyvandi F, Ruf W, Sood AK, Versteeg HH, Wolberg AS, Wong PC, Wood JP, Weiler H. Advances in Clinical and Basic Science of Coagulation: Illustrated abstracts of the 9th Chapel Hill Symposium on Hemostasis. Res Pract Thromb Haemost. 2018; 2: 407–28. 10.1002/rth2.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tillman BF, Gruber A, McCarty OJT, Gailani D. Plasma contact factors as therapeutic targets. Blood Rev. 2018; 32: 433–48. 10.1016/j.blre.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood. 2019; 133: 906–18. 10.1182/blood-2018-11-882993. [DOI] [PubMed] [Google Scholar]
- 9.Thalin C, Hisada Y, Lundstrom S, Mackman N, Wallen H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler Thromb Vasc Biol. 2019; 39: 1724–38. 10.1161/atvbaha.119.312463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker CJ, Smith SA, Morrissey JH. Polyphosphate in thrombosis, hemostasis, and inflammation. Res. Pract. Thromb. Haemost 2019; 3: 18–25. 10.1002/rth2.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petzold T, Massberg S. Thrombin: A Gas Pedal Driving Innate Immunity. Immunity. 2019; 50: 1024–6. 10.1016/j.immuni.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Yoon SJ, Seiler SH, Kucherlapati R, Leinwand L. Organization of the human skeletal myosin heavy chain gene cluster. Proc Natl Acad Sci U S A. 1992; 89: 12078–82. 10.1073/pnas.89.24.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deguchi H, Shukla M, Hayat M, Torkamani A, Elias DJ, Griffin JH. Novel exomic rare variants associated with venous thrombosis. Br J Haematol. 2020; 190: 783–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deguchi H, Sinha RK, Marchese P, Ruggeri ZM, Zilberman-Rudenko J, McCarty OJ, Cohen MJ, Griffin JH. Prothrombotic skeletal muscle myosin directly enhances prothrombin activation by binding factors Xa and Va. Blood. 2016; 128: 1870–8. 10.1182/blood-2016-03-707679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heeb MJ, Fernandez JA, Yamashita A, McDowell OR, Guo Z, Mosnier LO, Deguchi H, Griffin JH. Activated protein C anticoagulant activity is enhanced by skeletal muscle myosin. Haematologica. 2020. 105: e424–e427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zilberman-Rudenko J, Deguchi H, Shukla M, Oyama Y, Orje J, Guo Z, Wyseure T, Mosnier L, McCarty O, Ruggeri Z, Eckle T, Griffin J. Cardiac myosin promotes thrombin generation and coagulation in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2020; 40: 901–13. 10.1161/ATVBAHA.120.313990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerrero M, Guiu-Comadevall M, Cadefau JA, Parra J, Balius R, Estruch A, Rodas G, Bedini JL, Cusso R. Fast and slow myosins as markers of muscle injury. Br J Sports Med. 2008; 42: 581–4; discussion 4. 10.1136/bjsm.2007.037945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu T, Qian WJ, Gritsenko MA, Xiao W, Moldawer LL, Kaushal A, Monroe ME, Varnum SM, Moore RJ, Purvine SO, Maier RV, Davis RW, Tompkins RG, Camp DG 2nd, Smith RD. High dynamic range characterization of the trauma patient plasma proteome. Mol Cell Proteomics. 2006; 5: 1899–913. 10.1074/mcp.M600068-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sellers JR. Myosins: a diverse superfamily. Biochim Biophys Acta. 2000; 1496: 3–22. [DOI] [PubMed] [Google Scholar]
- 20.Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993; 261: 50–8. 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 21.Rayment I. The structural basis of the myosin ATPase activity. J Biol Chem. 1996; 271: 15850–3. 10.1074/jbc.271.27.15850. [DOI] [PubMed] [Google Scholar]
- 22.Brown JH, Kumar VS, O’Neall-Hennessey E, Reshetnikova L, Robinson H, Nguyen-McCarty M, Szent-Gyorgyi AG, Cohen C. Visualizing key hinges and a potential major source of compliance in the lever arm of myosin. Proc. Natl. Acad. Sci. U.S.A 2011; 108: 114–9. 10.1073/pnas.1016288107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Margossian SS, Lowey S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Meth Enzymol. 1982; 85 Pt B: 55–71. [DOI] [PubMed] [Google Scholar]
- 24.Deguchi H, Guo Z, Hayat M, Pflimlin E, Lear S, Shen W, Griffin JH. Molecular interaction site on procoagulant myosin for factor Xa-dependent prothrombin activation. J. Biol. Chem 2019; 294: 15176–81. 10.1074/jbc.AC119.010236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coleman JR, Moore EE, Zilberman-Rudenko J, Samuels JM, Cohen MJ, Silliman CC, Banerjee A, Sauaia A, Griffin JH, Deguchi H. Cardiac and Skeletal Muscle Myosin Exert Procoagulant Effects. Shock. 2019; 52: 554–5. 10.1097/shk.0000000000001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pomowski A, Ustok FI, Huntington JA. Homology model of human prothrombinase based on the crystal structure of Pseutarin C. Biol Chem. 2014; 395: 1233–41. 10.1515/hsz-2014-0165. [DOI] [PubMed] [Google Scholar]
- 27.Autin L, Steen M, Dahlback B, Villoutreix BO. Proposed structural models of the prothrombinase (FXa-FVa) complex. Proteins. 2006; 63: 440–50. 10.1002/prot.20848. [DOI] [PubMed] [Google Scholar]
- 28.Weiss A, Leinwand LA. The mammalian myosin heavy chain gene family. Annu Rev Cell Dev Biol. 1996; 12: 417–39. 10.1146/annurev.cellbio.12.1.417. [DOI] [PubMed] [Google Scholar]
- 29.Weiss A, Schiaffino S, Leinwand LA. Comparative sequence analysis of the complete human sarcomeric myosin heavy chain family: implications for functional diversity. J Mol Biol. 1999; 290: 61–75. 10.1006/jmbi.1999.2865. [DOI] [PubMed] [Google Scholar]
- 30.Novakovic V, Gilbert GE. Procoagulant Activities of Skeletal and Cardiac Muscle Myosin depend on contaminating phospholipid. Blood. 2020. 136: 2469–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfäffle M, Ruggiero F, Hofmann H, Fernández MP, Selmin O, Yamada Y, Garrone R, von der Mark K. Biosynthesis, secretion and extracellular localization of anchorin CII, a collagen-binding protein of the calpactin family. Embo J. 1988; 7: 2335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turnay J, Pfannmüller E, Lizarbe MA, Bertling WM, von der Mark K. Collagen binding activity of recombinant and N-terminally modified annexin V (anchorin CII). Journal of cellular biochemistry. 1995; 58: 208–20. 10.1002/jcb.240580210. [DOI] [PubMed] [Google Scholar]
- 33.Böhm BB, Wilbrink B, Kuettner KE, Mollenhauer J. Structural and functional comparison of anchorin CII (cartilage annexin V) and muscle annexin V. Arch Biochem Biophys. 1994; 314: 64–74. 10.1006/abbi.1994.1412. [DOI] [PubMed] [Google Scholar]
- 34.Tzima E, Trotter PJ, Orchard MA, Walker JH. Annexin V binds to the actin-based cytoskeleton at the plasma membrane of activated platelets. Exp Cell Res. 1999; 251: 185–93. 10.1006/excr.1999.4553. [DOI] [PubMed] [Google Scholar]
- 35.Tzima E, Trotter PJ, Orchard MA, Walker JH. Annexin V relocates to the platelet cytoskeleton upon activation and binds to a specific isoform of actin. Eur J Biochem. 2000; 267: 4720–30. 10.1046/j.1432-1327.2000.01525.x. [DOI] [PubMed] [Google Scholar]
- 36.Andersen MH, Berglund L, Rasmussen JT, Petersen TE. Bovine PAS-6/7 Binds αVβ5 Integrin and Anionic Phospholipids through Two Domains. Biochemistry. 1997; 36: 5441–6. [DOI] [PubMed] [Google Scholar]
- 37.Andersen MH, Graversen H, Fedosov SN, Petersen TE, Rasmussen JT. Functional Analyses of Two Cellular Binding Domains of Bovine Lactadherin. Biochemistry. 2000; 39: 6200–6. [DOI] [PubMed] [Google Scholar]
- 38.Larsson A, Peng S, Persson H, Rosenbloom J, Abrams WR, Wassberg E, Thelin S, Sletten K, Gerwins P, Westermark P. Lactadherin binds to elastin--a starting point for medin amyloid formation? Amyloid. 2006; 13: 78–85. 10.1080/13506120600722530. [DOI] [PubMed] [Google Scholar]
- 39.Silvestre JS, Thery C, Hamard G, Boddaert J, Aguilar B, Delcayre A, Houbron C, Tamarat R, Blanc-Brude O, Heeneman S, Clergue M, Duriez M, Merval R, Levy B, Tedgui A, Amigorena S, Mallat Z. Lactadherin promotes VEGF-dependent neovascularization. Nat Med. 2005; 11: 499–506. 10.1038/nm1233. [DOI] [PubMed] [Google Scholar]
- 40.Taylor MR, Couto JR, Scallan CD, Ceriani RL, Peterson JA. Lactadherin (formerly BA46), a membrane-associated glycoprotein expressed in human milk and breast carcinomas, promotes Arg-Gly-Asp (RGD)-dependent cell adhesion. DNA Cell Biol. 1997; 16: 861–9. 10.1089/dna.1997.16.861. [DOI] [PubMed] [Google Scholar]
- 41.Silva E, Frost D, Li L, Bovin N, Miller DJ. Lactadherin is a candidate oviduct Lewis X trisaccharide receptor on porcine spermatozoa. Andrology. 2017; 5: 589–97. 10.1111/andr.12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boddaert J, Kinugawa K, Lambert JC, Boukhtouche F, Zoll J, Merval R, Blanc-Brude O, Mann D, Berr C, Vilar J, Garabedian B, Journiac N, Charue D, Silvestre JS, Duyckaerts C, Amouyel P, Mariani J, Tedgui A, Mallat Z. Evidence of a role for lactadherin in Alzheimer’s disease. Am J Pathol. 2007; 170: 921–9. 10.2353/ajpath.2007.060664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ensslin MA, Shur BD. Identification of Mouse Sperm SED1, a Bimotif EGF Repeat and Discoidin-Domain Protein Involved in Sperm-Egg Binding. Cell. 2003; 114: 405–17. 10.1016/s0092-8674(03)00643-3. [DOI] [PubMed] [Google Scholar]
- 44.Andersen MH, Berglund L, Petersen TE, Rasmussen JT. Annexin-V binds to the intracellular part of the beta(5) integrin receptor subunit. Biochem Biophys Res Commun. 2002; 292: 550–7. 10.1006/bbrc.2002.6673. [DOI] [PubMed] [Google Scholar]
- 45.Ohsawa K, Imai Y, Ito D, Kohsaka S. Molecular cloning and characterization of annexin V-binding proteins with highly hydrophilic peptide structure. J Neurochem. 1996; 67: 89–97. 10.1046/j.1471-4159.1996.67010089.x. [DOI] [PubMed] [Google Scholar]
- 46.Bandorowicz-Pikula J, Buchet R, Pikula S. Annexins as nucleotide-binding proteins: facts and speculations. Bioessays. 2001; 23: 170–8. [DOI] [PubMed] [Google Scholar]
- 47.Kheifets V, Bright R, Inagaki K, Schechtman D, Mochly-Rosen D. Protein kinase C delta (deltaPKC)-annexin V interaction: a required step in deltaPKC translocation and function. J Biol Chem. 2006; 281: 23218–26. 10.1074/jbc.M602075200. [DOI] [PubMed] [Google Scholar]
- 48.Capila I, VanderNoot VA, Mealy TR, Seaton BA, Linhardt RJ. Interaction of heparin with annexin V. FEBS Lett. 1999; 446: 327–30. [DOI] [PubMed] [Google Scholar]
- 49.Doubell AF, Bester AJ, Thibault G. Annexins V and VI: major calcium-dependent atrial secretory granule-binding proteins. Hypertension. 1991; 18: 648–56. [DOI] [PubMed] [Google Scholar]
- 50.Speidel CM, Eisenberg PR, Ruf W, Edgington TS, Abendschein DR. Tissue factor mediates prolonged procoagulant activity on the luminal surface of balloon-injured aortas in rabbits. Circulation. 1995; 92: 3323–30. 10.1161/01.cir.92.11.3323. [DOI] [PubMed] [Google Scholar]
- 51.Krishnaswamy S, Church WR, Nesheim ME, Mann KG. Activation of human prothrombin by human prothrombinase. Influence of factor Va on the reaction mechanism. J Biol Chem. 1987; 262: 3291–9. [PubMed] [Google Scholar]
- 52.Moyer MP, Tracy RP, Tracy PB, van’t Veer C, Sparks CE, Mann KG. Plasma lipoproteins support prothrombinase and other procoagulant enzymatic complexes. Arterioscler Thromb Vasc Biol.. 1998; 18: 458–65. [DOI] [PubMed] [Google Scholar]
- 53.Bunce MW, Toso R, Camire RM. Zymogen-like factor Xa variants restore thrombin generation and effectively bypass the intrinsic pathway in vitro. Blood. 2011; 117: 290–8. 10.1182/blood-2010-08-300756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tracy PB, Eide LL, Mann KG. Human prothrombinase complex assembly and function on isolated peripheral blood cell populations. The Journal of biological chemistry. 1985; 260: 2119–24. [PubMed] [Google Scholar]
- 55.Ivanciu L, Krishnaswamy S, Camire RM. New insights into the spatiotemporal localization of prothrombinase in vivo. Blood. 2014; 124: 1705–14. 10.1182/blood-2014-03-565010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lechtenberg BC, Murray-Rust TA, Johnson DJ, Adams TE, Krishnaswamy S, Camire RM, Huntington JA. Crystal structure of the prothrombinase complex from the venom of Pseudonaja textilis. Blood. 2013; 122: 2777–83. 10.1182/blood-2013-06-511733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bradford HN, Orcutt SJ, Krishnaswamy S. Membrane binding by prothrombin mediates its constrained presentation to prothrombinase for cleavage. J Biol Chem. 2013; 288: 27789–800. 10.1074/jbc.M113.502005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haynes LM, Bouchard BA, Tracy PB, Mann KG. Prothrombin activation by platelet-associated prothrombinase proceeds through the prethrombin-2 pathway via a concerted mechanism. J Biol Chem. 2012; 287: 38647–55. 10.1074/jbc.M112.407791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bouchard BA, Williams JL, Meisler NT, Long MW, Tracy PB. Endocytosis of plasma-derived factor V by megakaryocytes occurs via a clathrin-dependent, specific membrane binding event. J Thromb Haemost. 2005; 3: 541–51. 10.1111/j.1538-7836.2005.01190.x. [DOI] [PubMed] [Google Scholar]
- 60.Tracy PB, Mann KG. Prothrombinase complex assembly on the platelet surface is mediated through the 74,000-dalton component of factor Va. Proc Natl Acad Sci USA. 1983; 80: 2380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wood JP, Silveira JR, Maille NM, Haynes LM, Tracy PB. Prothrombin activation on the activated platelet surface optimizes expression of procoagulant activity. Blood. 2011; 117: 1710–8. 10.1182/blood-2010-09-311035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shaw AW, Pureza VS, Sligar SG, Morrissey JH. The local phospholipid environment modulates the activation of blood clotting. J Biol Chem. 2007; 282: 6556–63. 10.1074/jbc.M607973200. [DOI] [PubMed] [Google Scholar]
- 63.Pecci A, Ma X, Savoia A, Adelstein RS. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene. 2018; 664: 152–67. 10.1016/j.gene.2018.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Flood VH, Slobodianuk TL, Keesler D, Lohmeier HK, Fahs S, Zhang L, Simpson P, Montgomery RR. von Willebrand factor binding to myosin assists in coagulation. Blood Adv. 2020; 4: 174–80. 10.1182/bloodadvances.2019000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kolev K, Tenekedjiev K, Ajtai K, Kovalszky I, Gombas J, Varadi B, Machovich R. Myosin: a noncovalent stabilizer of fibrin in the process of clot dissolution. Blood. 2003; 101: 4380–6. 10.1182/blood-2002-10-3227. [DOI] [PubMed] [Google Scholar]
- 66.Machovich R, Ajtai K, Kolev K, Owen WG. Myosin as cofactor and substrate in fibrinolysis. FEBS letters. 1997; 407: 93–6. [DOI] [PubMed] [Google Scholar]
- 67.Erlacher P, Lercher A, Falkensammer J, Nassonov EL, Samsonov MI, Shtutman VZ, Puschendorf B, Mair J. Cardiac troponin and beta-type myosin heavy chain concentrations in patients with polymyositis or dermatomyositis. Clin Chim Acta. 2001; 306: 27–33. [DOI] [PubMed] [Google Scholar]
- 68.Lofberg M, Tahtela R, Harkonen M, Somer H. Myosin heavy-chain fragments and cardiac troponins in the serum in rhabdomyolysis. Diagnostic specificity of new biochemical markers. Arch Neurol. 1995; 52: 1210–4. [DOI] [PubMed] [Google Scholar]
- 69.Chung WS, Lin CL, Sung FC, Lu CC, Kao CH. Increased risk of venous thromboembolism in patients with dermatomyositis/polymyositis: a nationwide cohort study. Thromb Res. 2014; 134: 622–6. 10.1016/j.thromres.2014.06.021. [DOI] [PubMed] [Google Scholar]
- 70.Deguchi TK, Deguchi H, Guo Z, Elias DJ, Griffin JH. Plasma skeletal muscle myosin phenotypes identified by immunoblotting are associated with pulmonary embolism occurrence in young adults. Thromb Res. 2020; 189: 88–92. 10.1016/j.thromres.2020.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Essien EO, Rali P, Mathai SC. Pulmonary Embolism. The Medical clinics of North America. 2019; 103: 549–64. 10.1016/j.mcna.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 72.Huisman MV, Barco S, Cannegieter SC, Le Gal G, Konstantinides SV, Reitsma PH, Rodger M, Vonk Noordegraaf A, Klok FA. Pulmonary embolism. Nat Rev Dis Primers 2018; 4: 18028 10.1038/nrdp.2018.28. [DOI] [PubMed] [Google Scholar]
- 73.Cohen MJ, Call M, Nelson M, Calfee CS, Esmon CT, Brohi K, Pittet JF. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann Surg. 2012; 255: 379–85. 10.1097/SLA.0b013e318235d9e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kutcher ME, Xu J, Vilardi RF, Ho C, Esmon CT, Cohen MJ. Extracellular histone release in response to traumatic injury: implications for a compensatory role of activated protein C. J Trauma Acute Care Surg. 2012; 73: 1389–94. 10.1097/TA.0b013e318270d595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cohen MJ, Kutcher M, Redick B, Nelson M, Call M, Knudson MM, Schreiber MA, Bulger EM, Muskat P, Alarcon LH, Myers JG, Rahbar MH, Brasel KJ, Phelan HA, del Junco DJ, Fox EE, Wade CE, Holcomb JB, Cotton BA, Matijevic N, Group PS. Clinical and mechanistic drivers of acute traumatic coagulopathy. J Trauma Acute Care Surg. 2013; 75: S40–7. 10.1097/TA.0b013e31828fa43d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Davenport RA, Brohi K. Cause of trauma-induced coagulopathy. Curr Opin Anaesthesiol. 2016; 29: 212–9. 10.1097/aco.0000000000000295. [DOI] [PubMed] [Google Scholar]
- 77.Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood. 2016; 128: 1043–9. 10.1182/blood-2016-01-636423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cohen MJ, Christie SA. Coagulopathy of Trauma. Crit Care Clin. 2017; 33: 101–18. 10.1016/j.ccc.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 79.Gando S, Otomo Y. Trauma-induced coagulopathy: The past, present, and future: A comment. J Thromb Hemost. 2019; 17: 1567–9. 10.1111/jth.14520. [DOI] [PubMed] [Google Scholar]
- 80.Cohen MJ. Translational approaches to coagulopathy after trauma: Towards targeted treatment. PLoS Med. 2017; 14: e1002359 10.1371/journal.pmed.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Konkle BA, Skinner M, Iorio A. Hemophilia trials in the twenty-first century: Defining patient important outcomes. Res Pract Thromb Haemost. 2019; 3: 184–92. 10.1002/rth2.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rendo P, Shafer F, Korth-Bradley JM, Sivamurthy K, Korin J. Factors that influence the bleeding phenotype in severe hemophilic patients. Blood Coagul Fibrinolysis. 2013; 24: 683–90. 10.1097/MBC.0b013e3283614210. [DOI] [PubMed] [Google Scholar]
- 83.Pavlova A, Oldenburg J. Defining severity of hemophilia: more than factor levels. Semin Thromb Hemost. 2013; 39: 702–10. 10.1055/s-0033-1354426. [DOI] [PubMed] [Google Scholar]
- 84.Ljung R. Aspects of prophylactic treatment of hemophilia. Thromb J. 2016; 14: 30 10.1186/s12959-016-0103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sorensen B, Auerswald G, Benson G, Elezovic I, Felder M, Lambert T, Morfini M, Remor E, Salaj P, Santagostino E, Salek SZ, Ljung R. Rationale for individualizing haemophilia care. Blood Coagul Fibrinolysis. 2015; 26: 849–57. 10.1097/mbc.0000000000000225. [DOI] [PubMed] [Google Scholar]
- 86.Nogami K, Shima M. Phenotypic heterogeneity of hemostasis in severe hemophilia. Semin Thromb Hemost. 2015; 41: 826–31. 10.1055/s-0034-1395349. [DOI] [PubMed] [Google Scholar]
- 87.Fischer K, Berntorp E. Targeting factor replacement therapy in severe hemophilia: which level is important? Semin Thromb Hemost. 2015; 41: 860–3. 10.1055/s-0035-1552562. [DOI] [PubMed] [Google Scholar]
- 88.Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014; 12: 1935–9. 10.1111/jth.12672. [DOI] [PubMed] [Google Scholar]
- 89.Carcao MD, van den Berg HM, Ljung R, Mancuso ME. Correlation between phenotype and genotype in a large unselected cohort of children with severe hemophilia A. Blood. 2013; 121: 3946–52, s1. 10.1182/blood-2012-11-469403. [DOI] [PubMed] [Google Scholar]
- 90.Franchini M, Montagnana M, Targher G, Veneri D, Zaffanello M, Salvagno GL, Manzato F, Lippi G. Interpatient phenotypic inconsistency in severe congenital hemophilia: a systematic review of the role of inherited thrombophilia. Semin Thromb Hemost. 2009; 35: 307–12. 10.1055/s-0029-1222609. [DOI] [PubMed] [Google Scholar]
- 91.Sonel A, Sasseen BM, Fineberg N, Bang N, Wilensky RL. Prospective study correlating fibrinopeptide A, troponin I, myoglobin, and myosin light chain levels with early and late ischemic events in consecutive patients presenting to the emergency department with chest pain. Circulation. 2000; 102: 1107–13. [DOI] [PubMed] [Google Scholar]
- 92.Lavin F, Kane M, Forde A, Gannon F, Daly K. Comparison of five cardiac markers in the detection of reperfusion after thrombolysis in acute myocardial infarction. Br Heart J. 1995; 73: 422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Isobe M, Nagai R, Yamaoki K, Nakaoka H, Takaku F, Yazaki Y. Quantification of myocardial infarct size after coronary reperfusion by serum cardiac myosin light chain II in conscious dogs. Circ Res. 1989; 65: 684–94. [DOI] [PubMed] [Google Scholar]
- 94.Trahern CA, Gere JB, Krauth GH, Bigham DA. Clinical assessment of serum myosin light chains in the diagnosis of acute myocardial infarction. Am J Cardiol. 1978; 41: 641–5. [DOI] [PubMed] [Google Scholar]
- 95.Jacquet S, Yin X, Sicard P, Clark J, Kanaganayagam GS, Mayr M, Marber MS. Identification of cardiac myosin-binding protein C as a candidate biomarker of myocardial infarction by proteomics analysis. Mol Cell Proteomics. 2009; 8: 2687–99. 10.1074/mcp.M900176-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ibanez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015; 65: 1454–71. 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 97.Hausenloy DJ, Chilian W, Crea F, Davidson SM, Ferdinandy P, Garcia-Dorado D, van Royen N, Schulz R, Heusch G. The coronary circulation in acute myocardial ischaemia/reperfusion injury: a target for cardioprotection. Cardiovasc Res. 2019; 115: 1143–55. 10.1093/cvr/cvy286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ten Cate H, Hackeng TM, Garcia de Frutos P. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thromb Haemost 2017; 117: 1265–71. 10.1160/th17-02-0079. [DOI] [PubMed] [Google Scholar]
- 99.Raivio P, Lassila R, Petaja J. Thrombin in myocardial ischemia-reperfusion during cardiac surgery. Ann Thorac Surg. 2009; 88: 318–25. 10.1016/j.athoracsur.2008.12.097. [DOI] [PubMed] [Google Scholar]
- 100.Olie RH, van der Meijden PEJ, Ten Cate H. The coagulation system in atherothrombosis: Implications for new therapeutic strategies. Res Pract Thromb Haemost. 2018; 2: 188–98. 10.1002/rth2.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]